
Hydrogen Bonding: Between Strengthening the Crystal Packing and Improving Solubility of Three Haloperidol Derivatives

Abstract
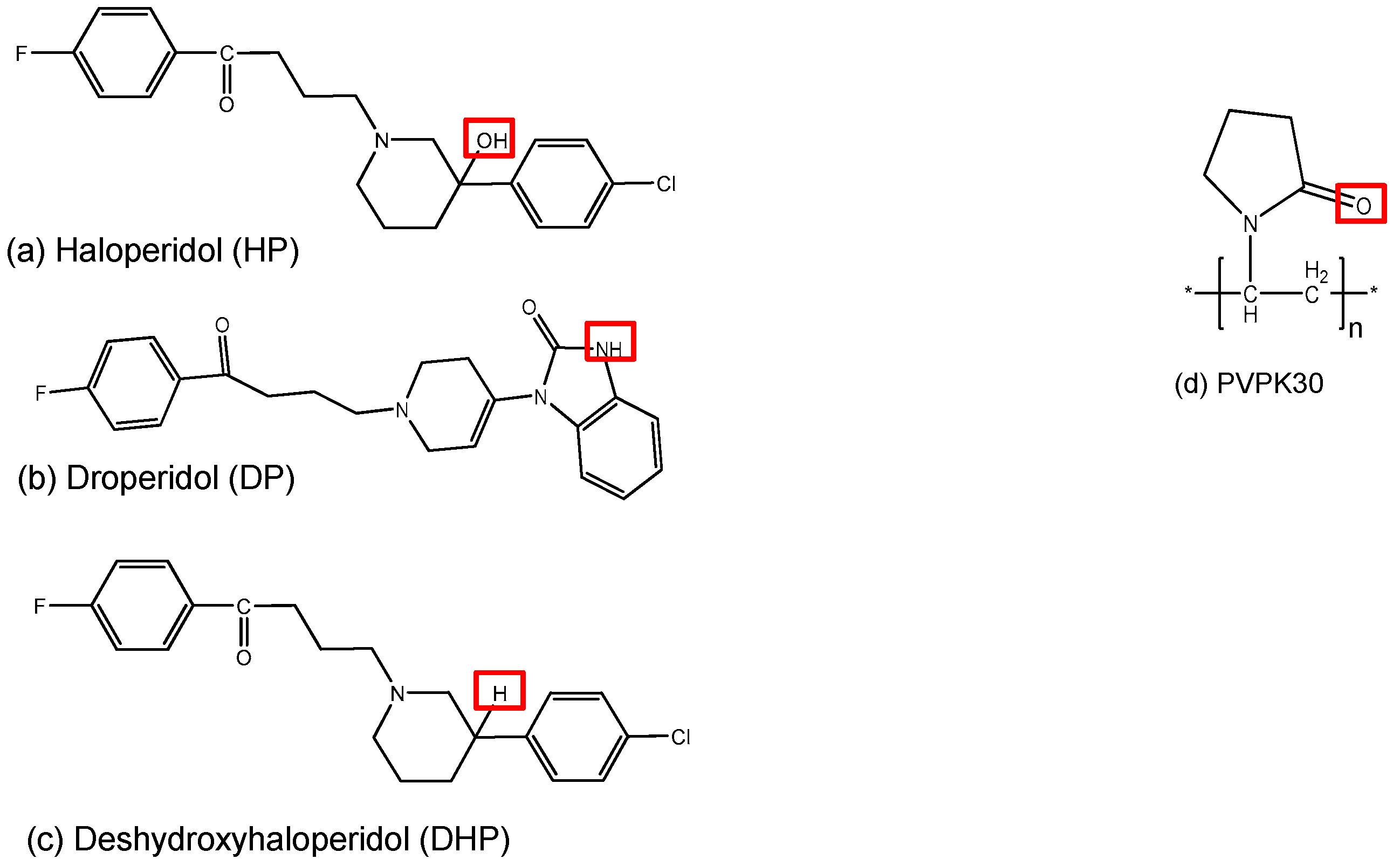
:1. Introduction
2. Results
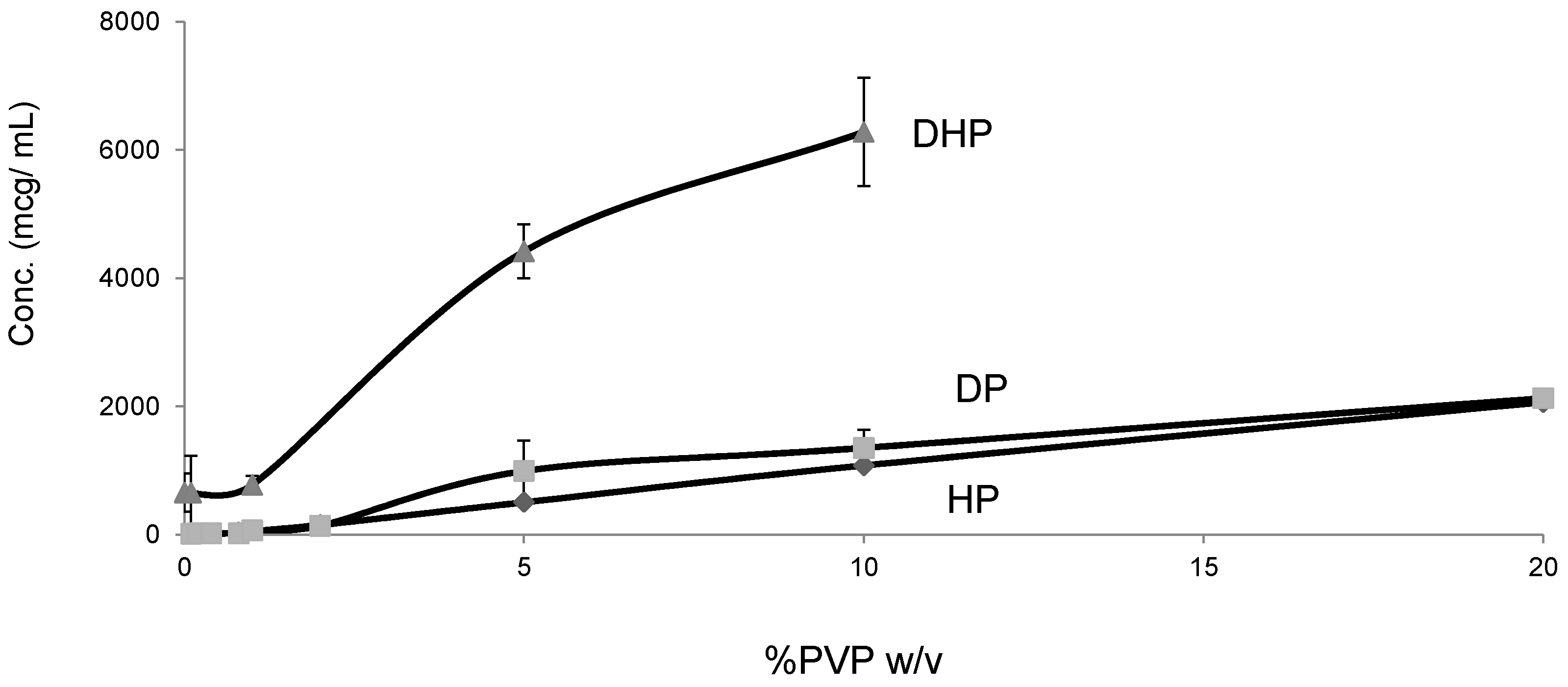
2.1. Determination of Solubility Parameters
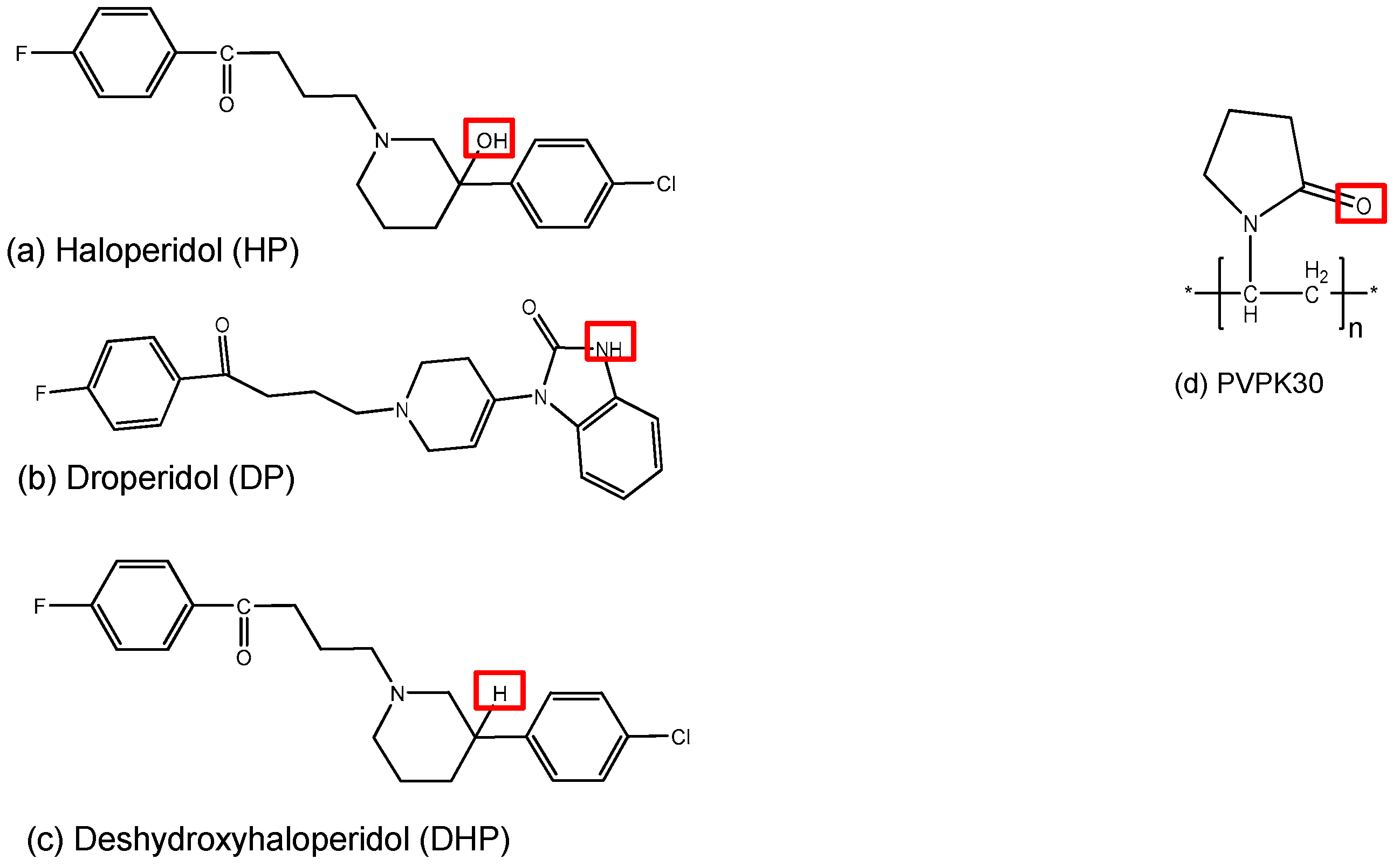
2.2. DSC Characterization of the Drug Molecules
2.3. Solid Dispersions Preparation
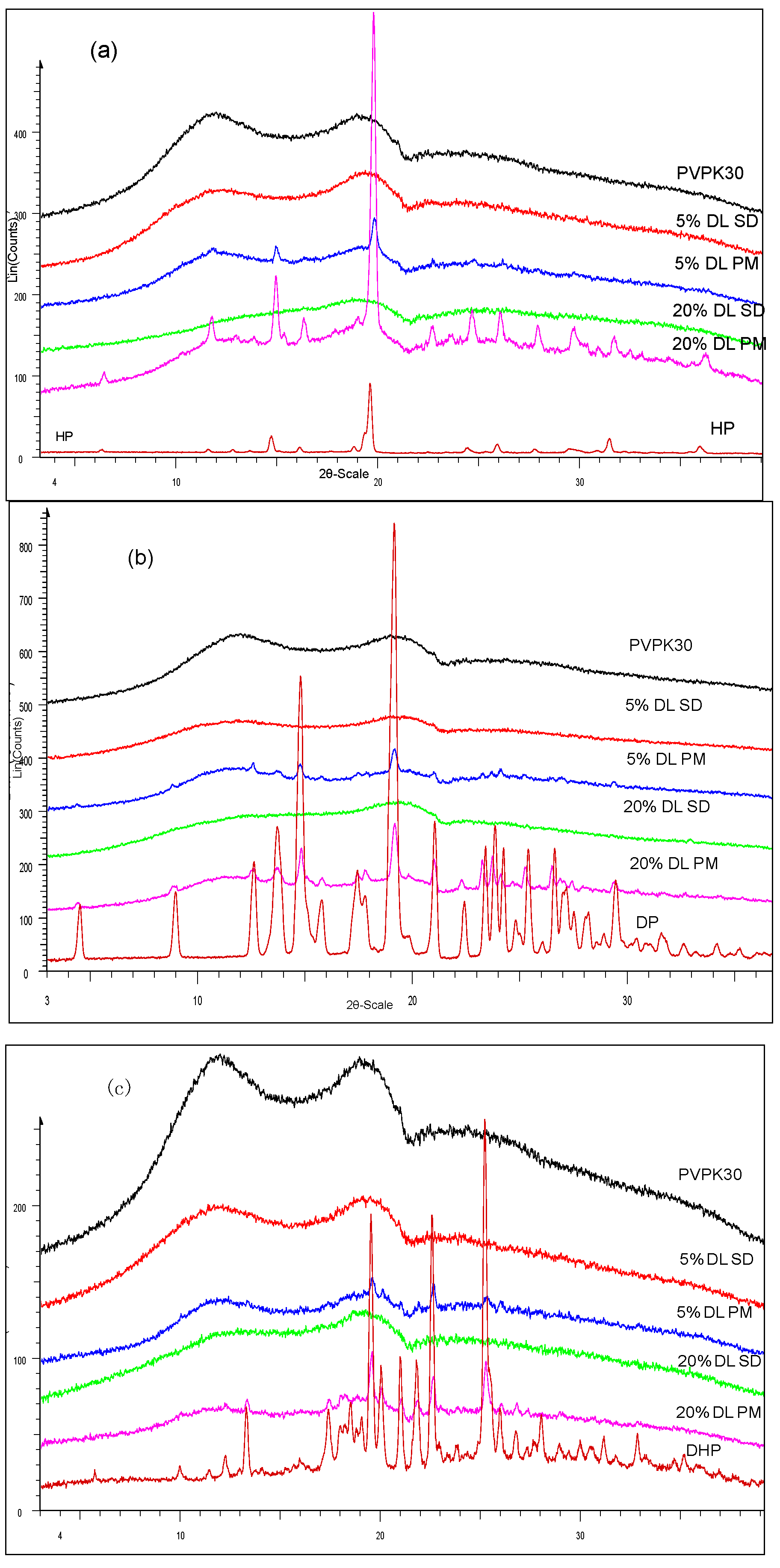
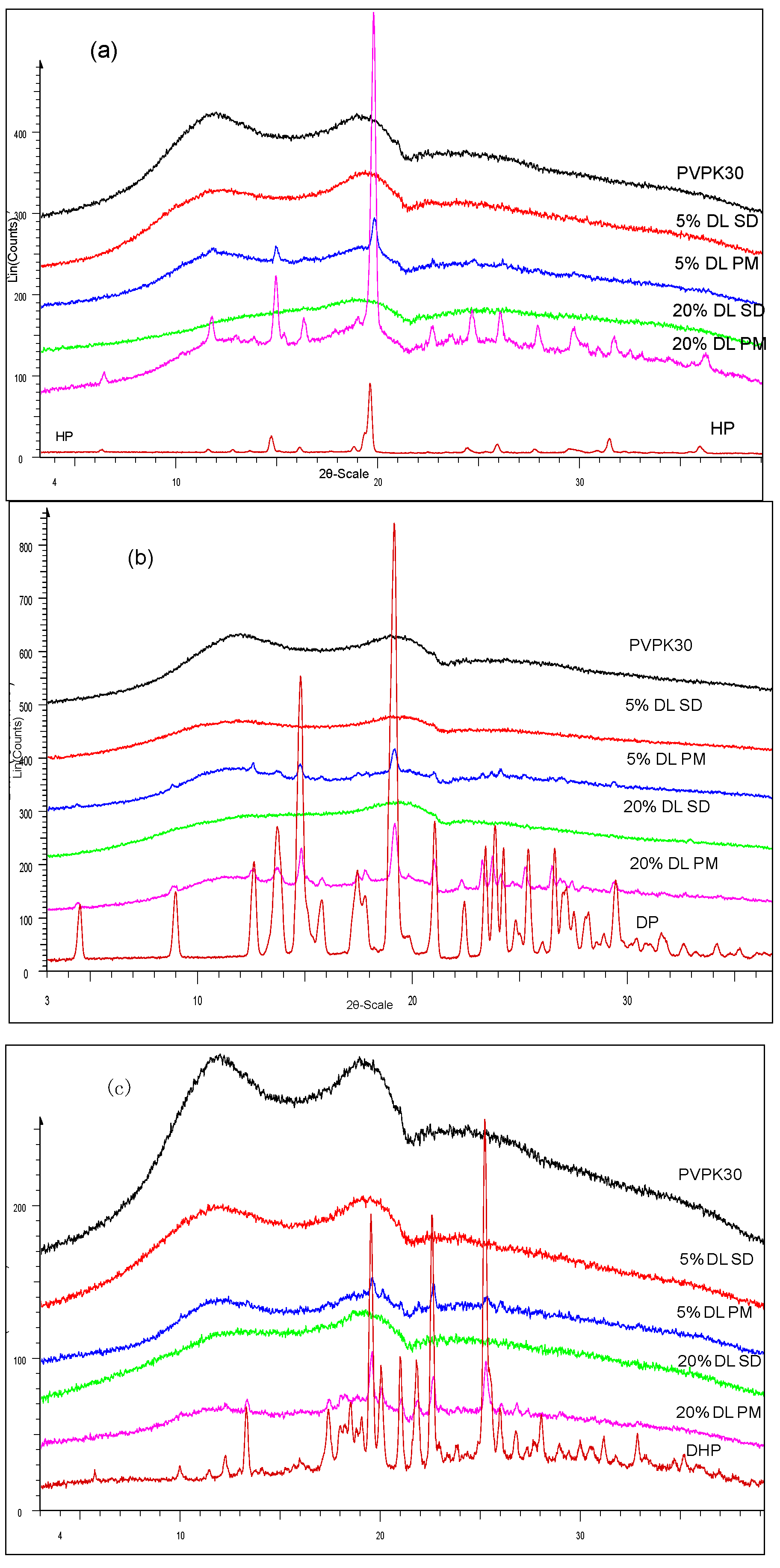
2.3.1. XRPD Characterization of the Solid Dispersions
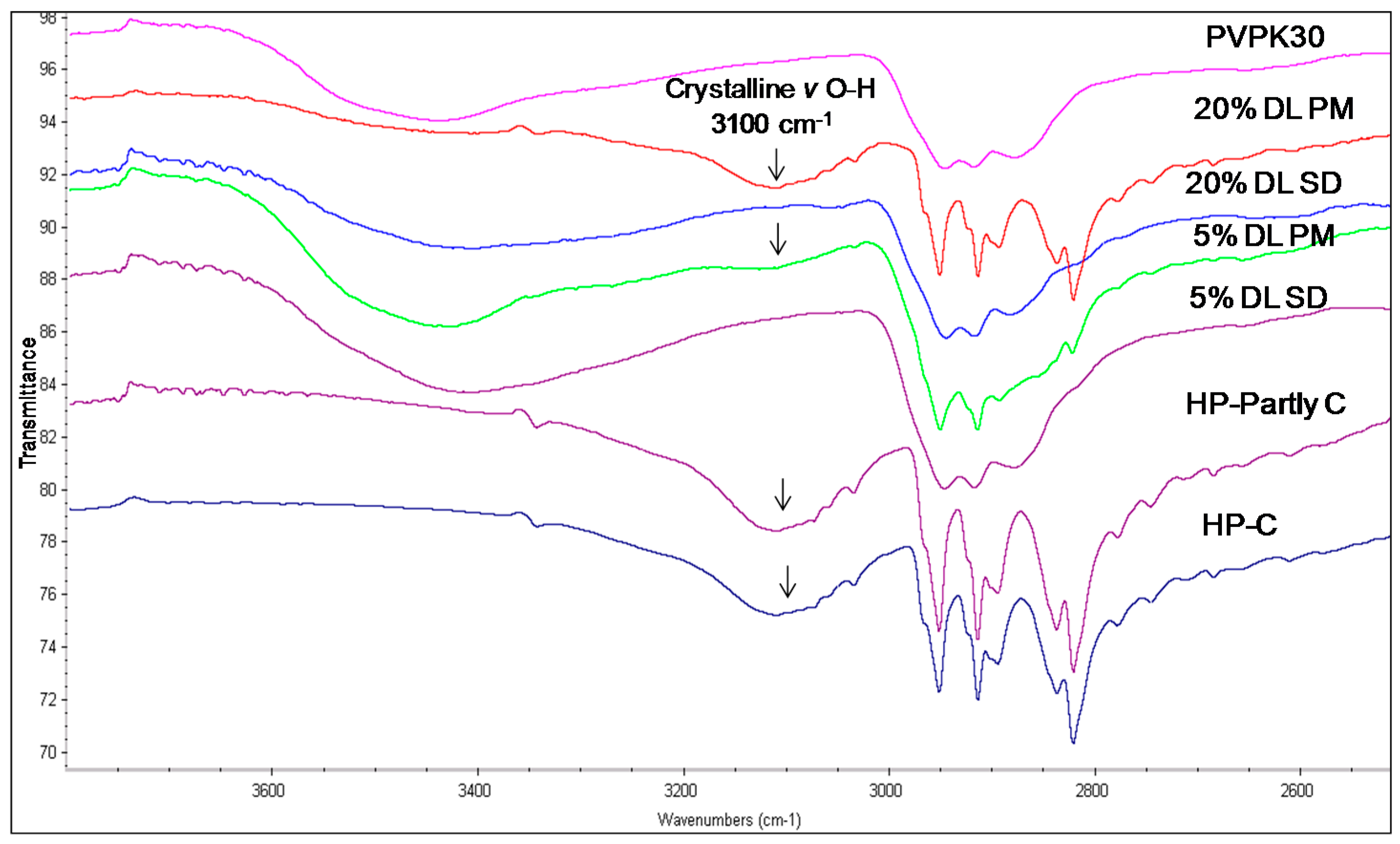
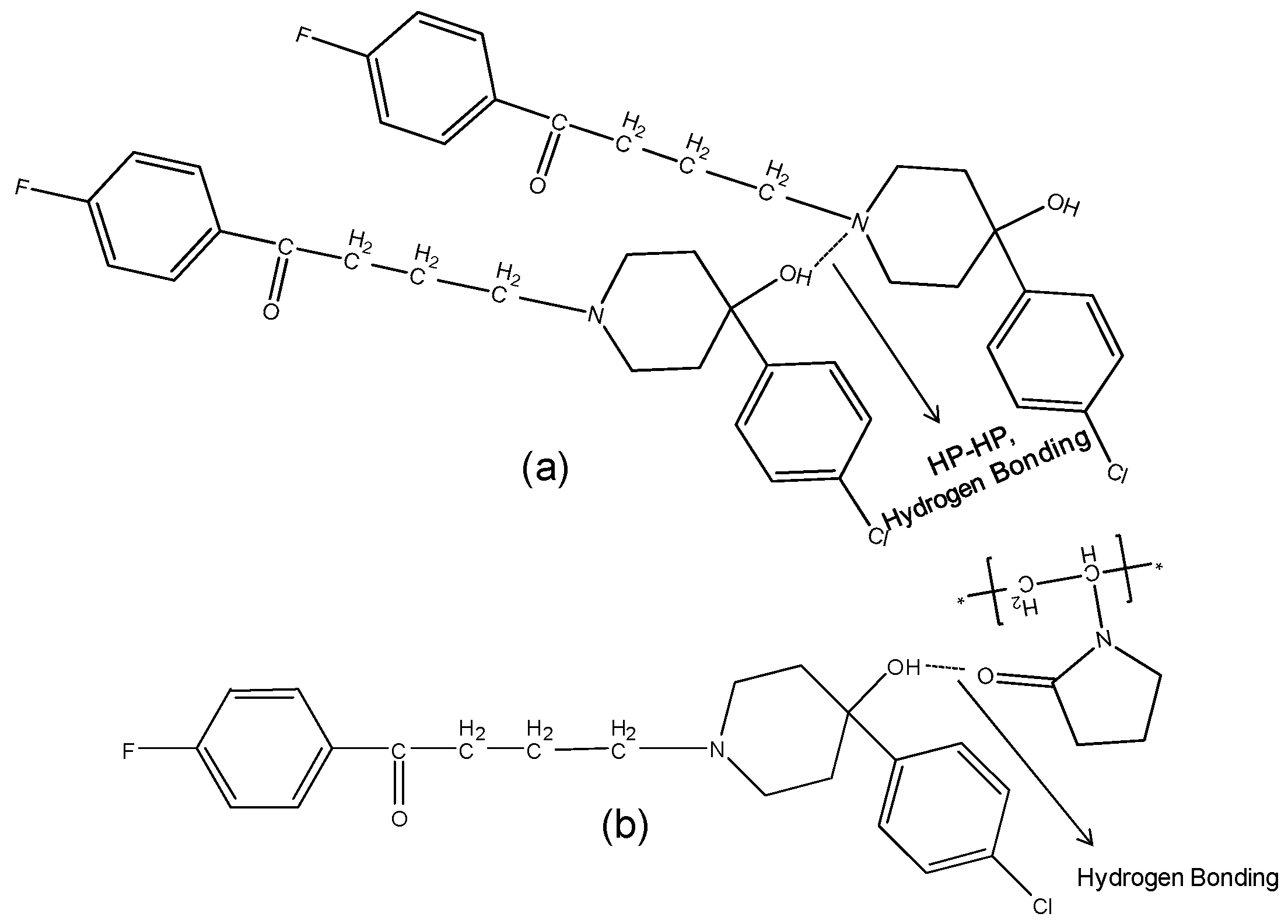
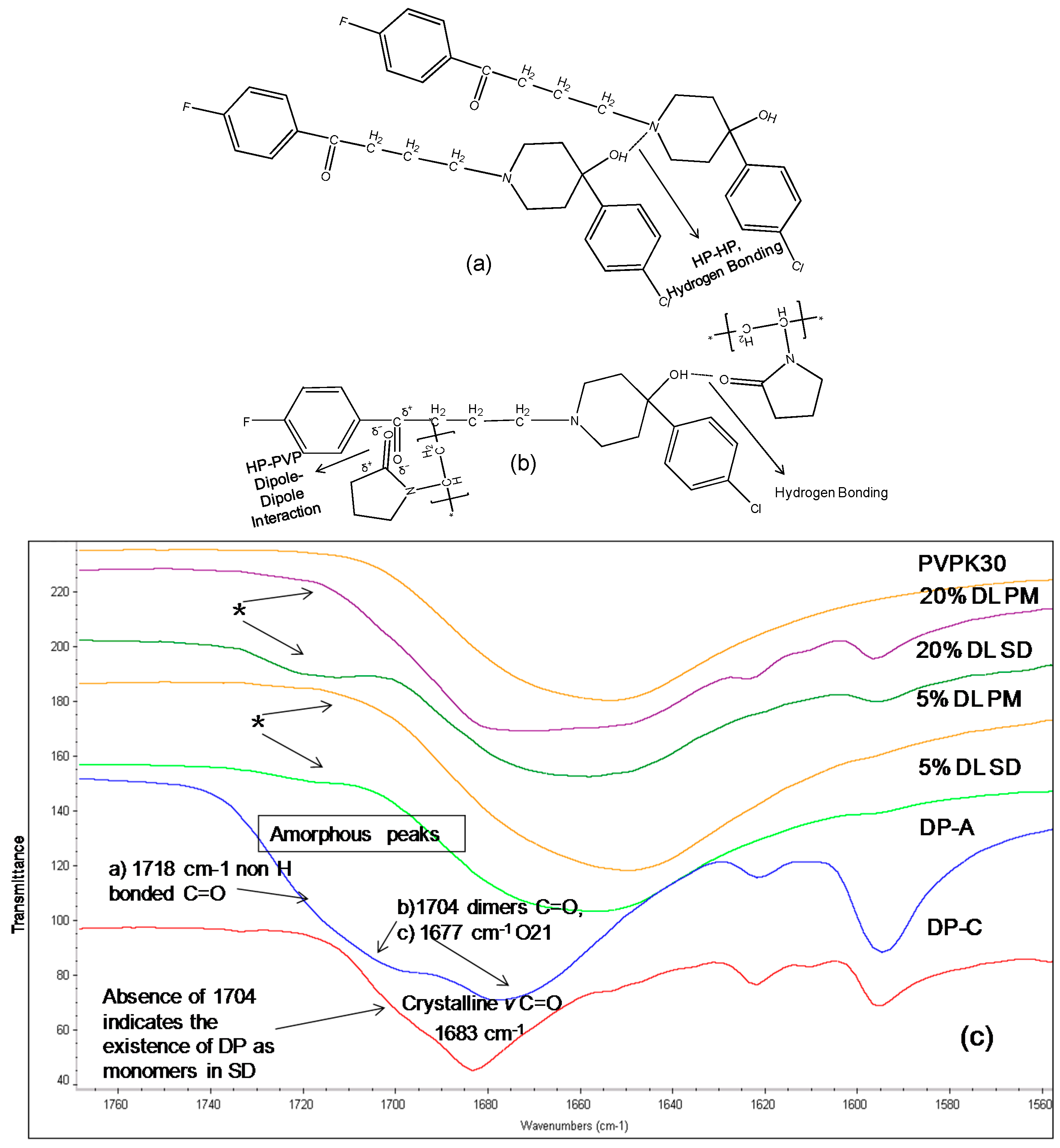
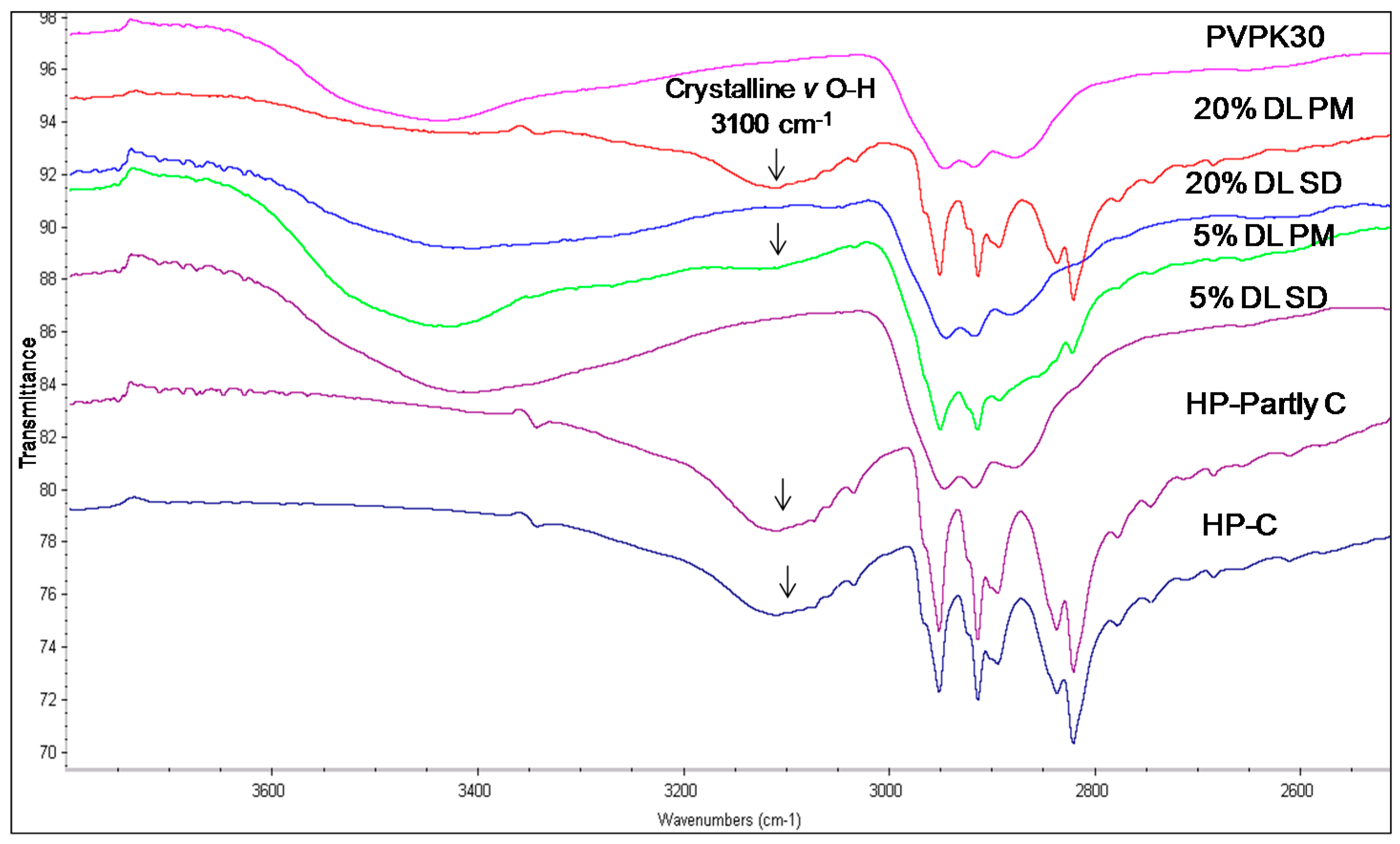
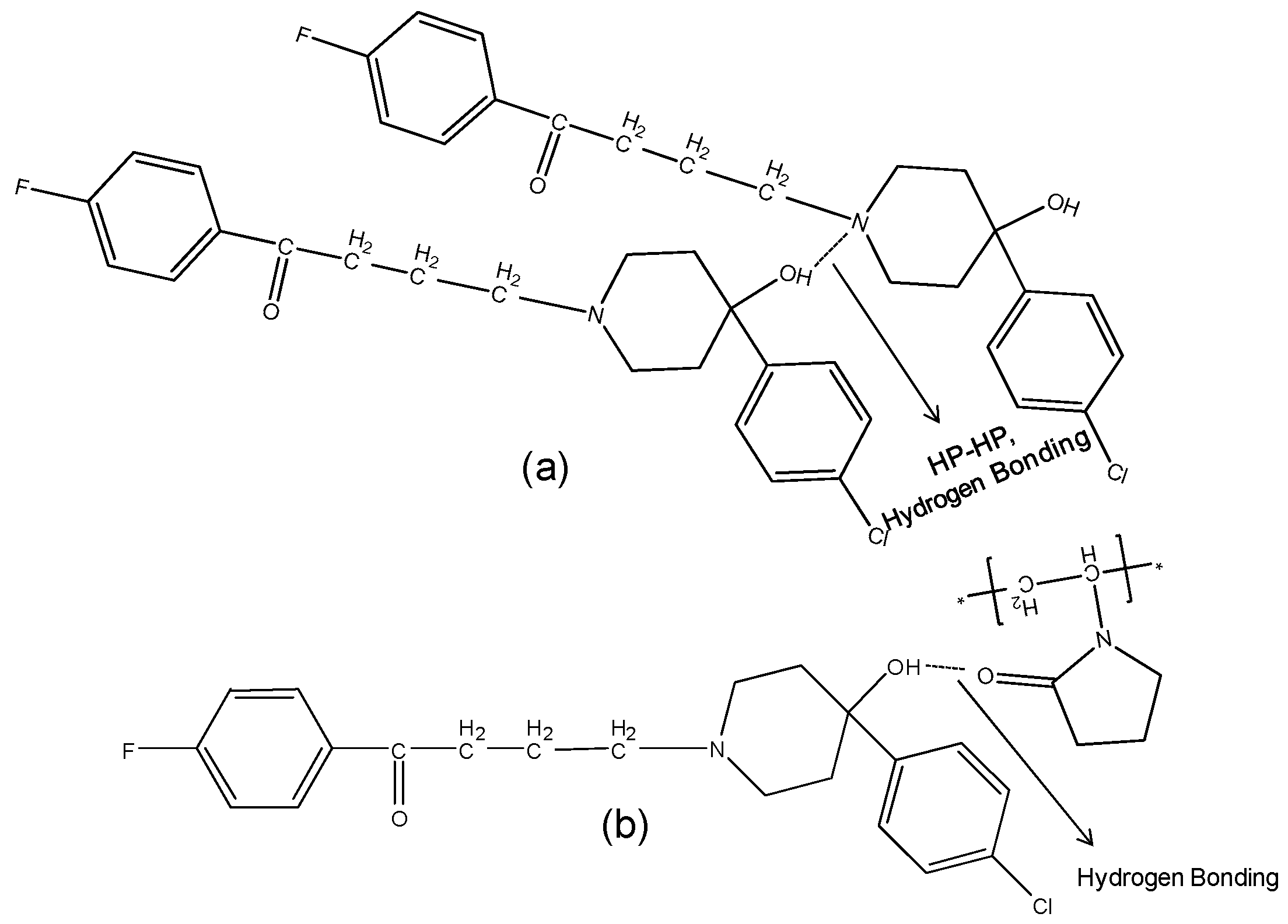
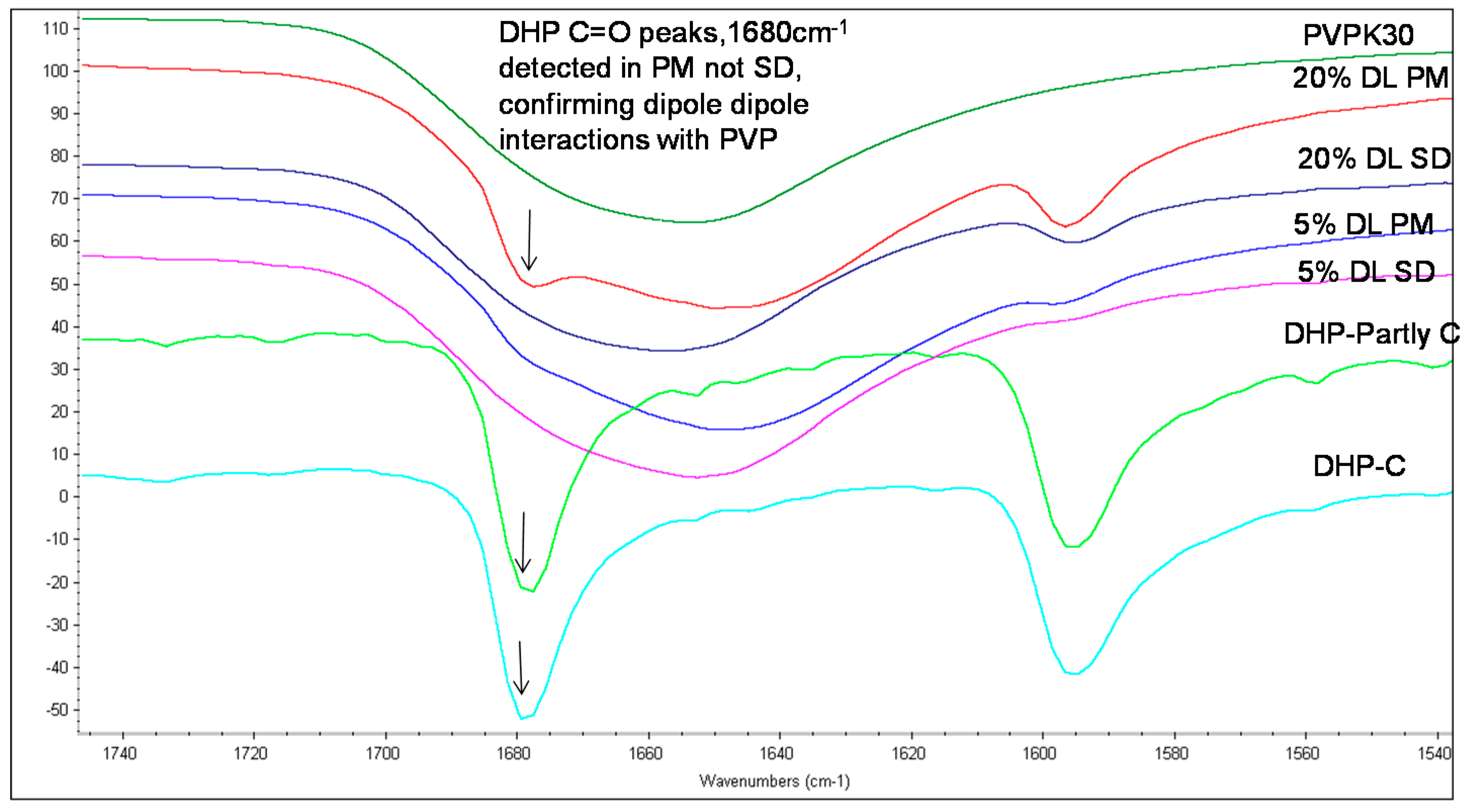
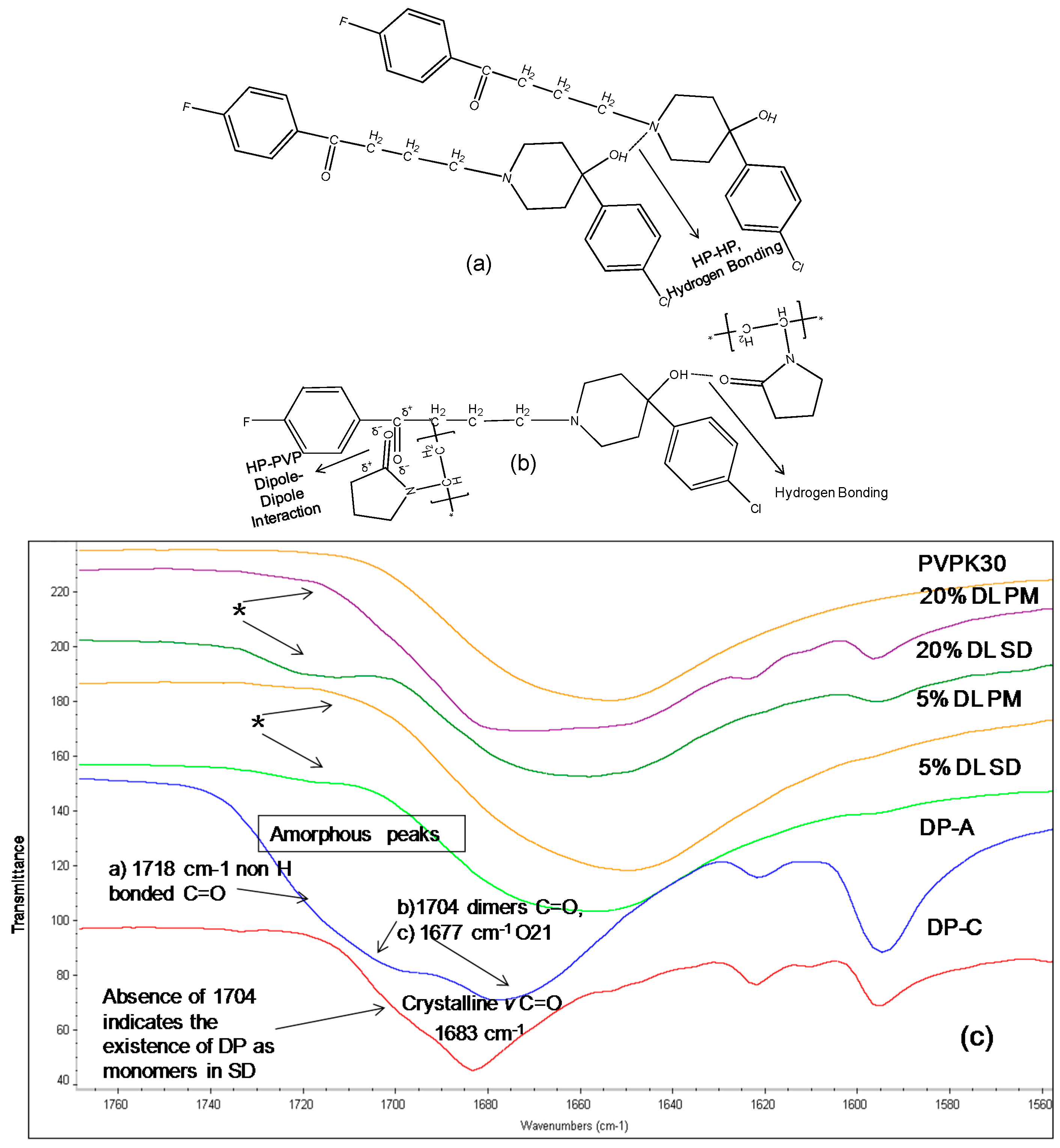
2.3.2. IR Characterization of the Solid Dispersions
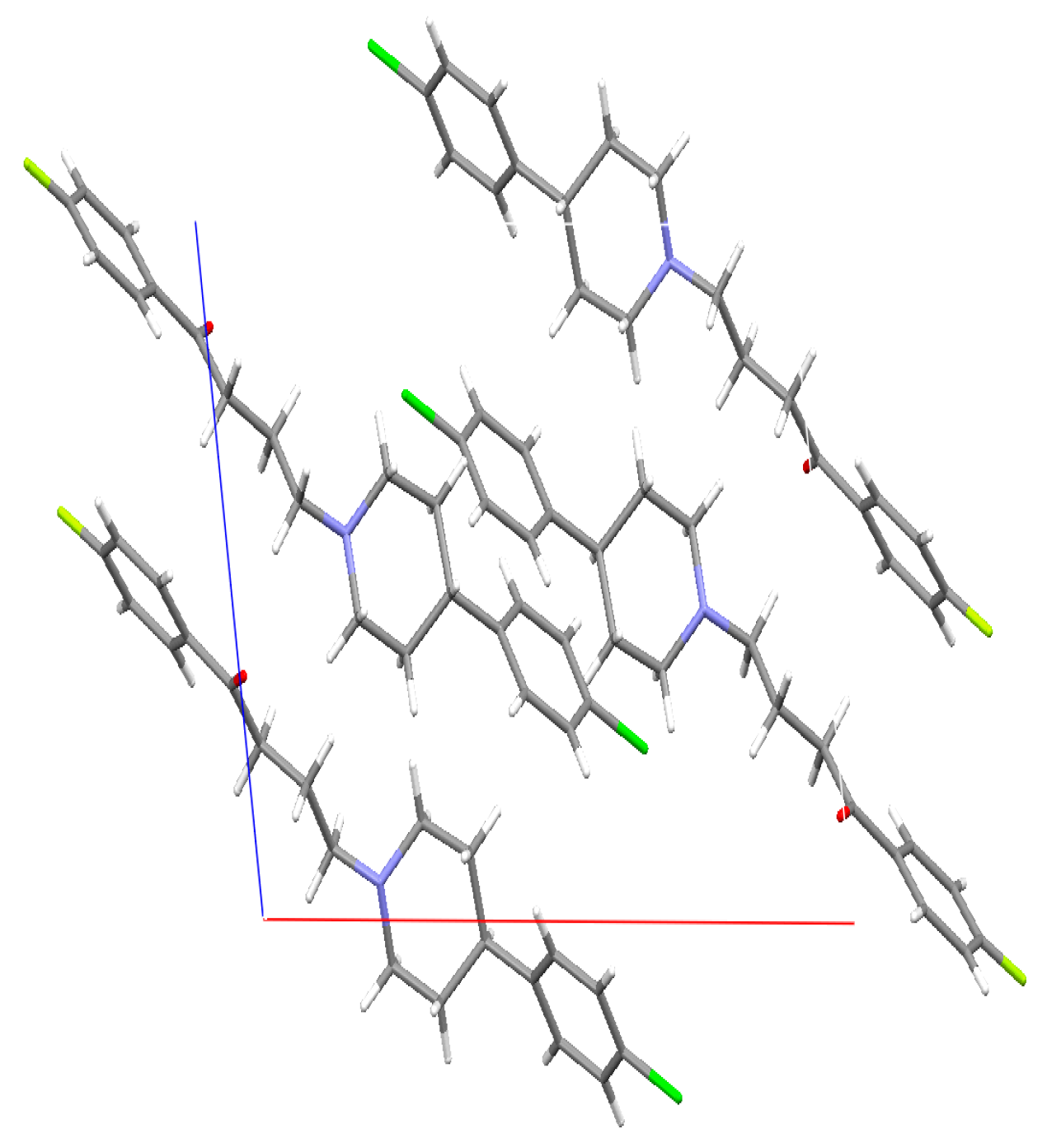
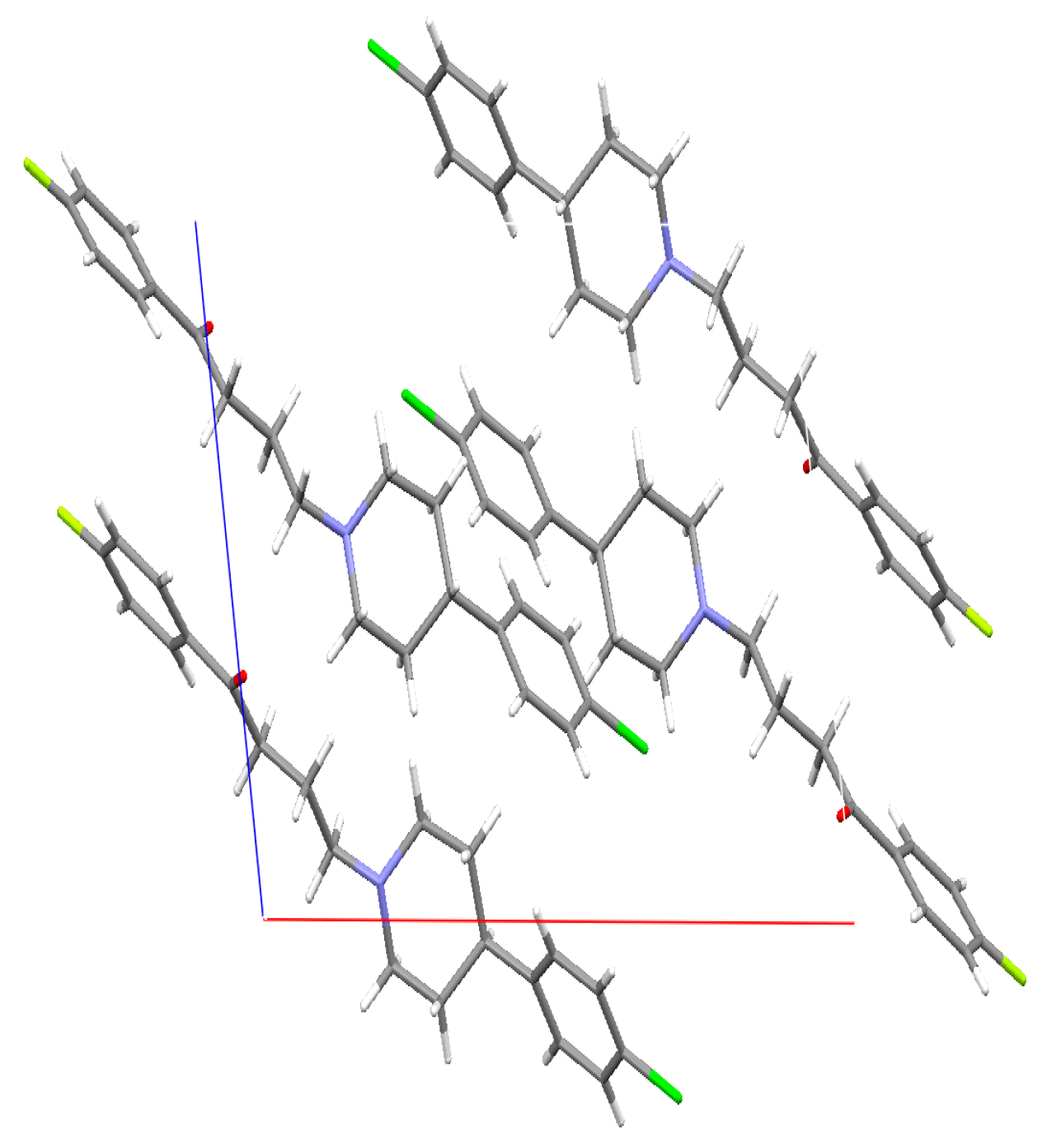
2.4. Single Crystal X-ray
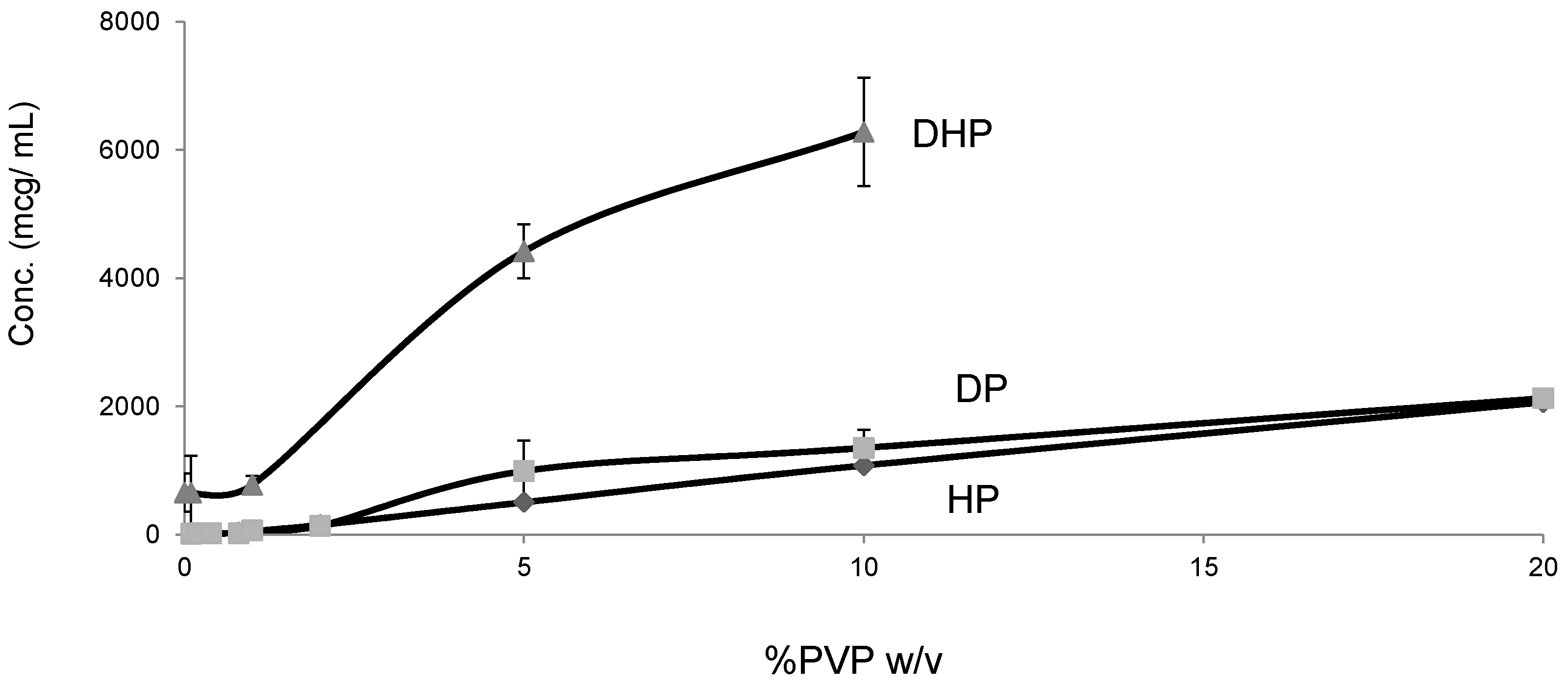
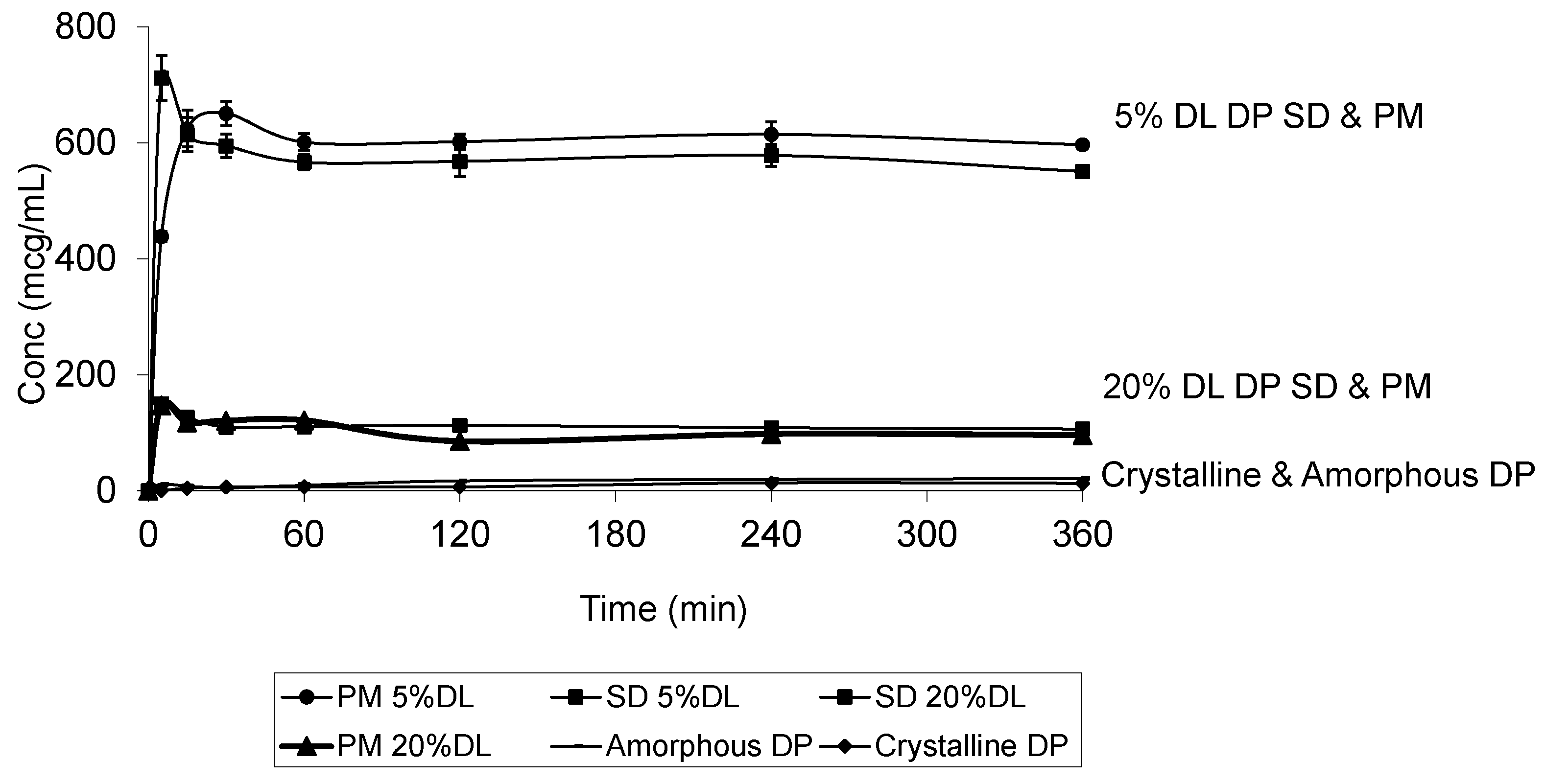
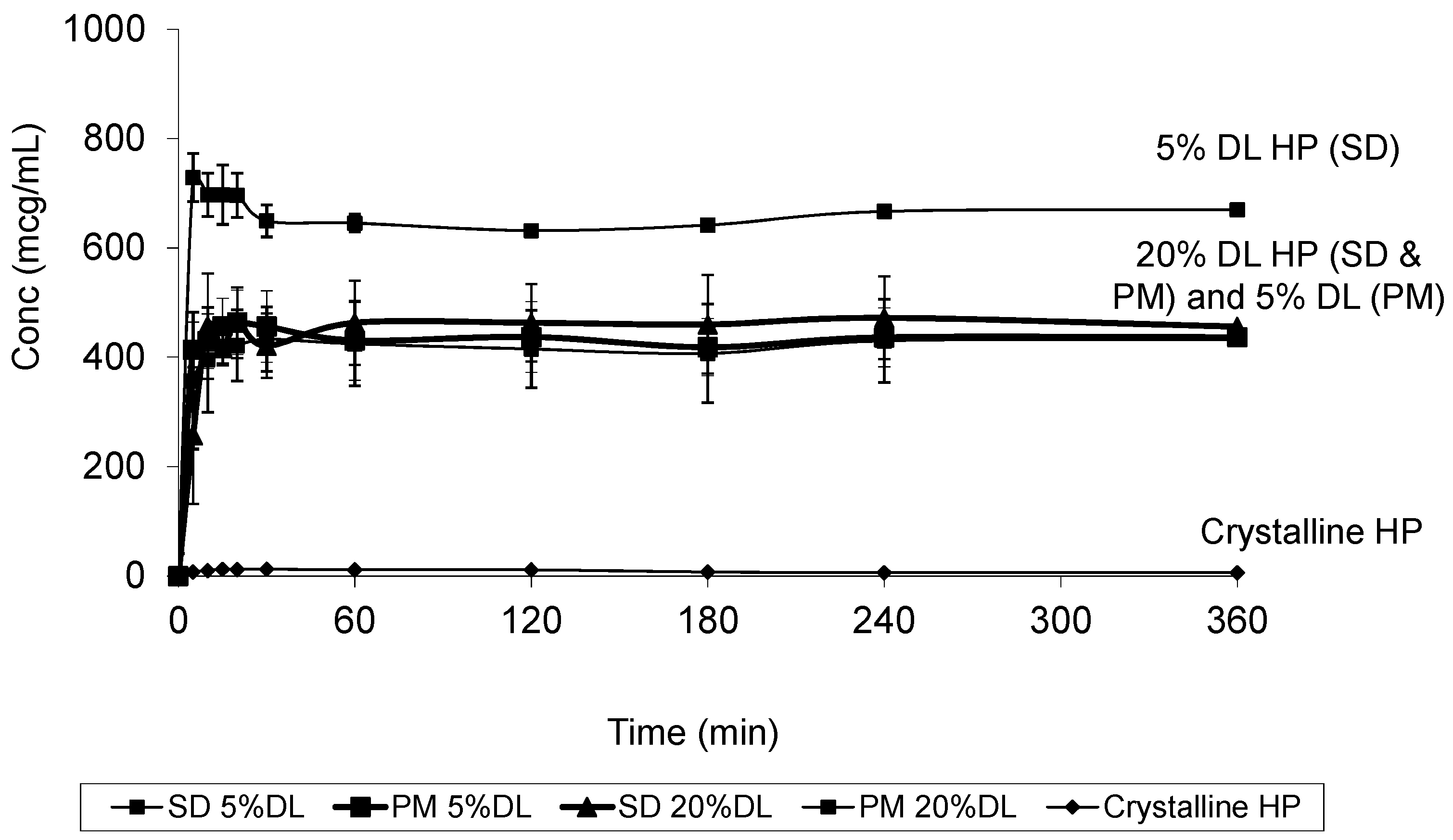
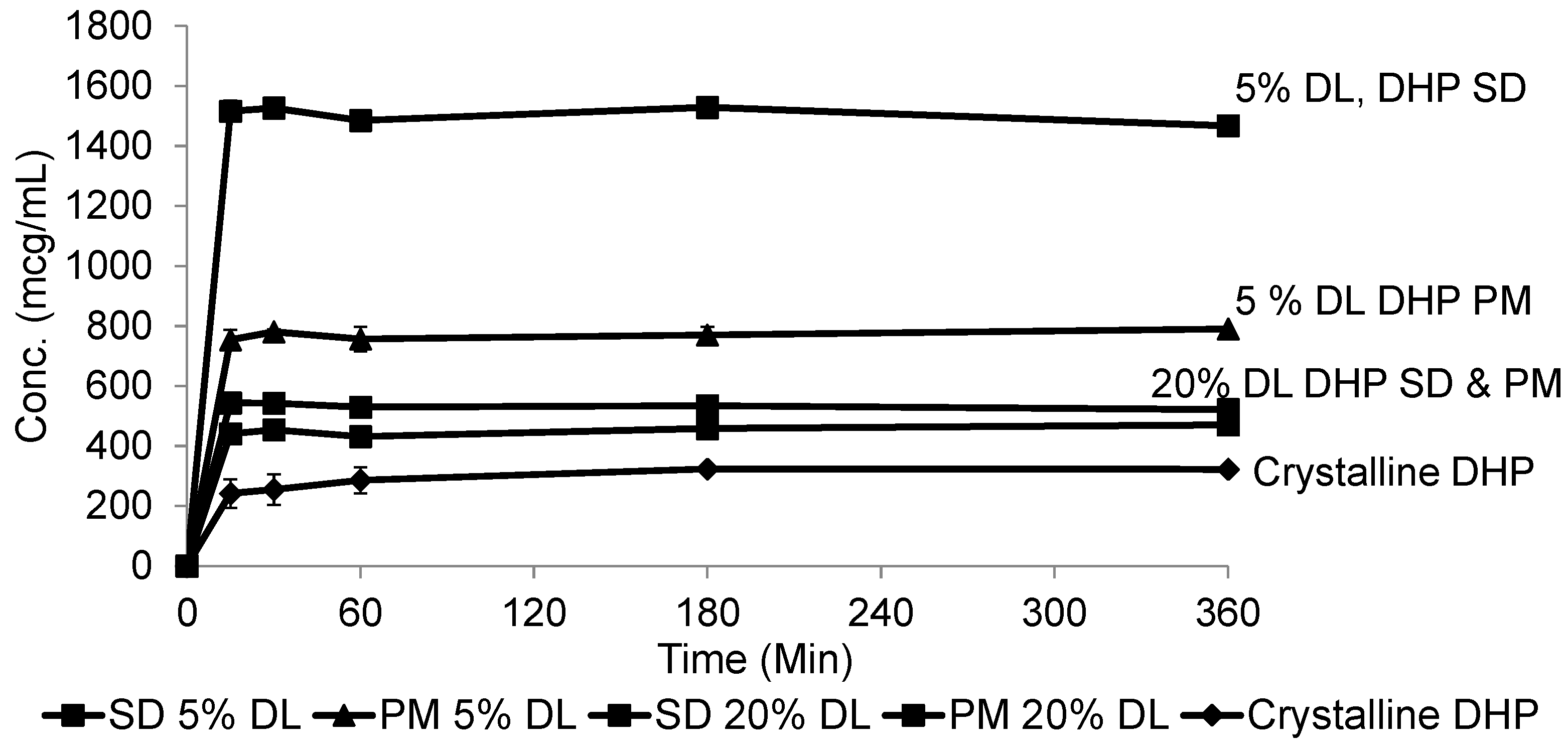
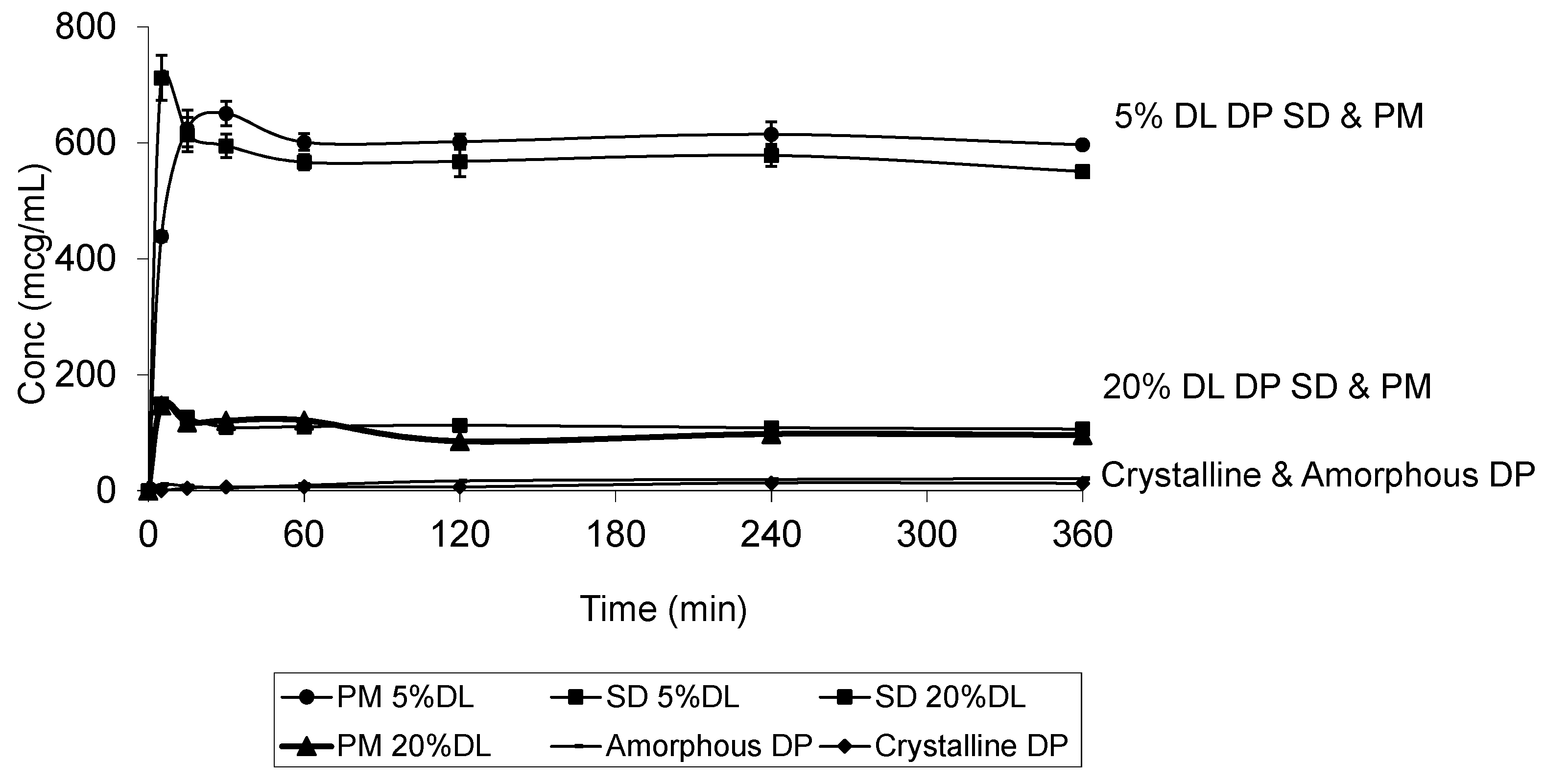
2.5. Solubility Studies
3. Materials and Methods
3.1. General Information
3.2. Preparation of Solid Dispersions and Physical Mixtures
3.3. Solubility Studies
3.4. Single Crystal Structure Determination
3.5. X-ray Power Diffraction
3.6. Dissolution Test
3.7. Thermal Analysis
3.8. Solubility Parameter Calculations
3.9. Fourier Transform Infrared Spectroscopy (FTIR)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ∆Ev | Energy of vaporization |
| Vm | Molar Volume |
| δ | Difference |
| °C | Degree Celsius |
| % | Percent |
| HP | Haloperidol |
| DP | Droperidol |
| DHP | Deshydroxy-haloperidol |
| DL | Drug Loading |
| XRPD | X ray Powder Diffraction |
| DSC | Differential Scanning Calorimetry |
| FTIR | Fourier Transform Infrared Spectroscopy |
References
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Phatak, S.S.; Stephan, C.C.; Cavasotto, C.N. High-throughput and in silico screenings in drug discovery. Expert Opin. Drug Discov. 2009, 4, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Park, J. Solubility enhancers for oral drug delivery: Can chemical structure manipulation be avoided? Am. J. Drug Deliver. 2004, 2, 113–130. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Kumar Kakumanu, V.; Bansal, A.K. Enthalpy relaxation studies of celecoxib amorphous mixtures. Pharm. Res. 2002, 19, 1873–1878. [Google Scholar] [CrossRef]
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Serajuddln, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]
- Qian, F.; Huang, J.; Hussain, M.A. Drug-polymer solubility and miscibility: Stability consideration and practical challenges in amorphous solid dispersion development. J. Pharm. Sci. 2010, 99, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- Sethia, S.; Squillante, E. Solid dispersions: Revival with greater possibilities and applications in oral drug delivery. Crit. Rev. Ther. Drug Carr. Syst. 2003, 20, 215–247. [Google Scholar] [CrossRef]
- Ford, J.L.; Rubinstein, M.H. Ageing of indomethacin-polyethylene glycol 6000 solid dispersion. Pharm. Acta Helv. 1979, 54, 353–358. [Google Scholar] [PubMed]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Bloch, D.W.; Elegakey, M.A.; Speiser, P.P. Solid dispersion of chlorthalidone in urea phase diagram and dissolution characteristics. Pharm. Acta Helv. 1982, 57, 231–235. [Google Scholar] [PubMed]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Acrins, A.; Arajis, R.; Belakovs, S.; Orola, L.; Veidis, M. The Crystal and Molecular Structure of a Polymorph and a Pseudo-polymorph of Droperidol. J. Chem. Crystallogr. 2008, 38, 169–174. [Google Scholar] [CrossRef]
- Ksendzova, G.A.; Polozov, G.I.; Skornyakov, I.V.; Sorokin, V.L.; Tolstorozhev, G.B.; Shadyro, O.I.; Yakunin, A.A. Manifestation of intramolecular hydrogen bonds in the IR spectra of bioactive aminophenols. Opt. Spec. 2007, 102, 551–556. [Google Scholar] [CrossRef]
- Reed, L.L.; Schaefer, J.P. The Crystal and Molecular Structure of Haloperidol, a Potent Psychotropic Drug. Acta Crystallogr. 1973, 29, 1886–1890. [Google Scholar] [CrossRef]
- Rumondor, A.C.F.; Marsac, P.J.; Stanford, L.A.; Taylor, L.S. Phase behavior of poly(vinylpyrrolidone) containing amorphous solid dispersions in the presence of moisture. Mol. Pharm. 2009, 6, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Destri, G.L.; Marrazzo, A.; Rescifina, A.; Punzo, F. How molecular interactions affect crystal morphology: The case of haloperidol. J. Pharm. Sci. 2011, 100, 4896–4906. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.A.; Van Eerdenbrugh, B.; Taylor, L.S. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef] [PubMed]
- Prasanna, M.D.; Row, T.N.G. Weak interactions involving organic fluorine: analysis of structural motifs in Flunazirine and Haloperidol. J. Mol. Struct. 2001, 562, 55–61. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Parthasarathy, R. The nature of halogen halogen interactions: Are short halogen contacts due to specific attractive forces or due to close packing of nonspherical atoms? J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Berzinš, A.; Skarbulis, E.; Rekis, T.; Actinš, A. On the formation of droperidol solvates: Characterization of structure and properties. Cryst. Growth Des. 2014, 14, 2654–2664. [Google Scholar] [CrossRef]
- Wegiel, L.A.; Zhao, Y.; Mauer, L.J.; Edgar, K.J.; Taylor, L.S. Curcumin amorphous solid dispersions: the influence of intra and intermolecular bonding on physical stability. Pharm. Dev. Technol. 2014, 19, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; York, P.; Rowe, R.C. The use of solubility parameters in pharmaceutical dosage form design. Int. J. Pharm. 1997, 148, 1–21. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds HP, DP and DHP are not availble from the authors.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solubility Parameter Comparison | ||||||
|---|---|---|---|---|---|---|
| Polymer/Drugs | Hansen’s 3D Method | Van Krevelen Method | Hoy’s Method | Average | Difference (δ) | Classification |
| PVPK30 | 21.57 | 22.18 | 22.95 | 22.23 | ||
| Haloperidol | 23.09 | 25.15 | 22.47 | 23.57 | 1.34 | Miscible |
| Droperidol | 23.63 | 25.09 | 23.39 | 24.04 | 1.80 | Miscible |
| Deshydroxyhaloperidol | 20.90 | 22.68 | 21.69 | 21.76 | 0.48 | Miscible |
| Drug | M.p., Tm °C (Cycle 1) | Enthalpy Fusion ∆H (J/g) (Cycle 1) | Tg (°C) (Cycle 1) | Recrys. Temperature Tc (°C) (Cycle 2) | Enthalpy Recrys ∆H (J/g) (Cycle 2) | Recrys Temperature Tc (°C)/Melting Point Tm (°C) (Cycle 3) | Enthalpy Recrys ∆H (J/g)/Enthalpy Fusion ∆H (J/g) (Cycle 3) | TGA (°C) | * Calculated Tg (°C) |
|---|---|---|---|---|---|---|---|---|---|
| HP | 151.2 ± 1.7 | 148 ± 1.2 | - | 91.5 ± 1.7 | 100.3 ± 3.3 | 150.8 ± 0.9 | 136.4 ± 1.2 | >240 | 23.8 |
| DP | 145.2 ± 1.1/ 152.1 ± 0.2 | 109 ± 6.8 | 34.5 ± 0.9 (Tg) 103.6 ± 4.9 (Tc) 149.1 ± 1.4 (Tm) | 60.8 ± 1.7 (∆H, recrys) 64.8 ± 15.6 (∆H, melting) | >270 | - | |||
| DHP | 90.4 ± 2.8 | 77 ± 1.7 | - | 21.2 ± 5 | 51.1 ± 3.3 | 78.9 ± 1.2/ 90.3 ± 0.6 | 11 ± 7.6/ 15.1 ± 7.4 | >200 | *-18.55 |
| Drug/Polymer | Functional Group | Crystalline (cm−1) | Amorphous | Solid Dispersion | Physical Mixture (cm−1) |
|---|---|---|---|---|---|
| Haloperidol | H-bonded hydroxyl (OH) | 3100 | - | Disappears | 3100 |
| Carbonyl (C=O) | 1680 | - | Disappears | 1680 | |
| Deshydroxyhaloperidol | Carbonyl (C=O) | 1680 | - | Disappears | 1680 |
| Droperidol | Carbonyl (C=O) | 1683 | 1677 (O21) | Disappears | 1683 |
| 1704 (O1 in dimer) | * Disappears | - | |||
| 1718 (O1 non H-bonded) | 1720 | - |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saluja, H.; Mehanna, A.; Panicucci, R.; Atef, E. Hydrogen Bonding: Between Strengthening the Crystal Packing and Improving Solubility of Three Haloperidol Derivatives. Molecules 2016, 21, 719. https://doi.org/10.3390/molecules21060719
Saluja H, Mehanna A, Panicucci R, Atef E. Hydrogen Bonding: Between Strengthening the Crystal Packing and Improving Solubility of Three Haloperidol Derivatives. Molecules. 2016; 21(6):719. https://doi.org/10.3390/molecules21060719
Chicago/Turabian StyleSaluja, Hardeep, Ahmed Mehanna, Riccardo Panicucci, and Eman Atef. 2016. "Hydrogen Bonding: Between Strengthening the Crystal Packing and Improving Solubility of Three Haloperidol Derivatives" Molecules 21, no. 6: 719. https://doi.org/10.3390/molecules21060719
APA StyleSaluja, H., Mehanna, A., Panicucci, R., & Atef, E. (2016). Hydrogen Bonding: Between Strengthening the Crystal Packing and Improving Solubility of Three Haloperidol Derivatives. Molecules, 21(6), 719. https://doi.org/10.3390/molecules21060719