
Antiproliferative Fate of the Tetraploid Formed after Mitotic Slippage and Its Promotion; A Novel Target for Cancer Therapy Based on Microtubule Poisons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
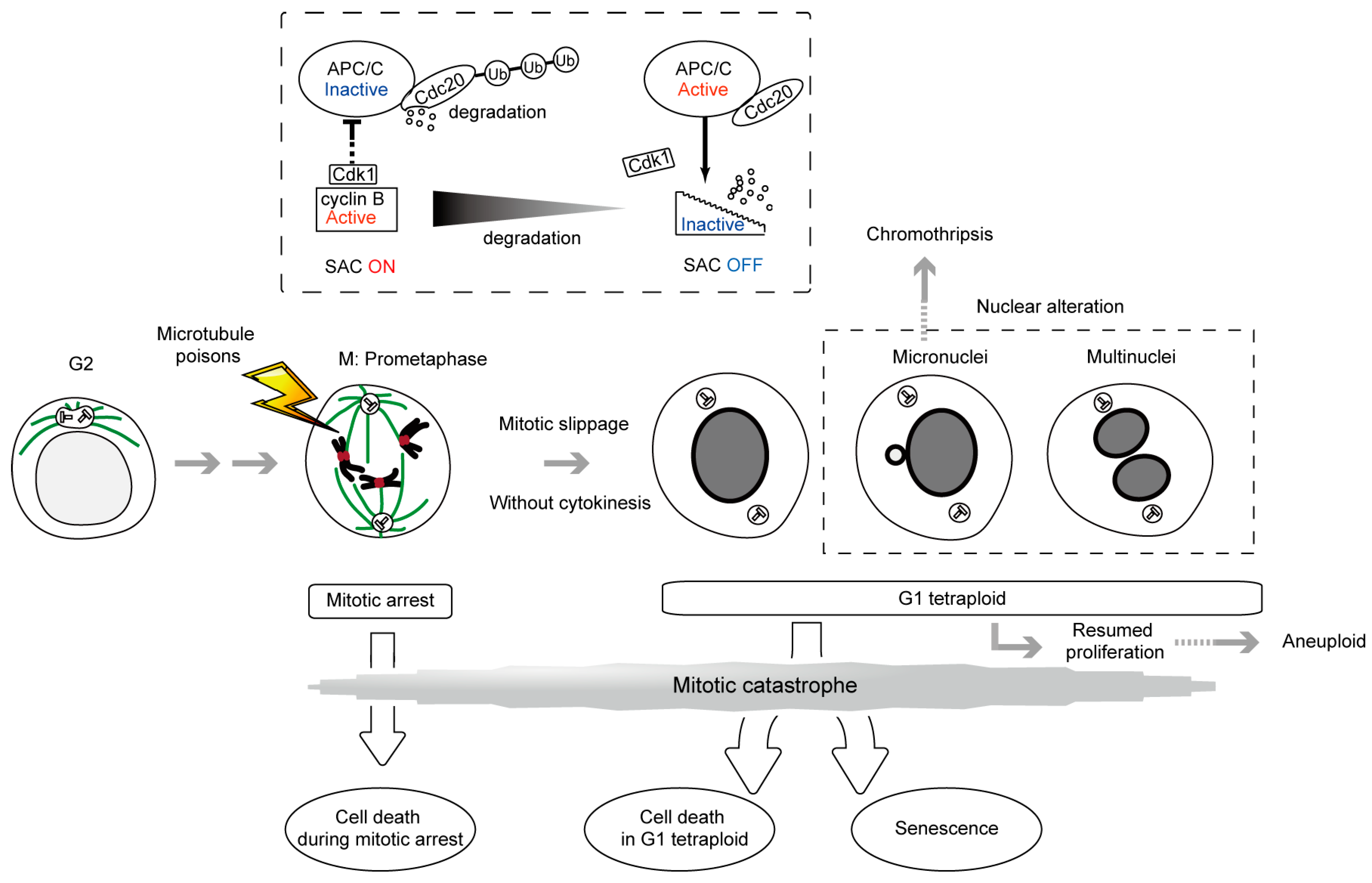
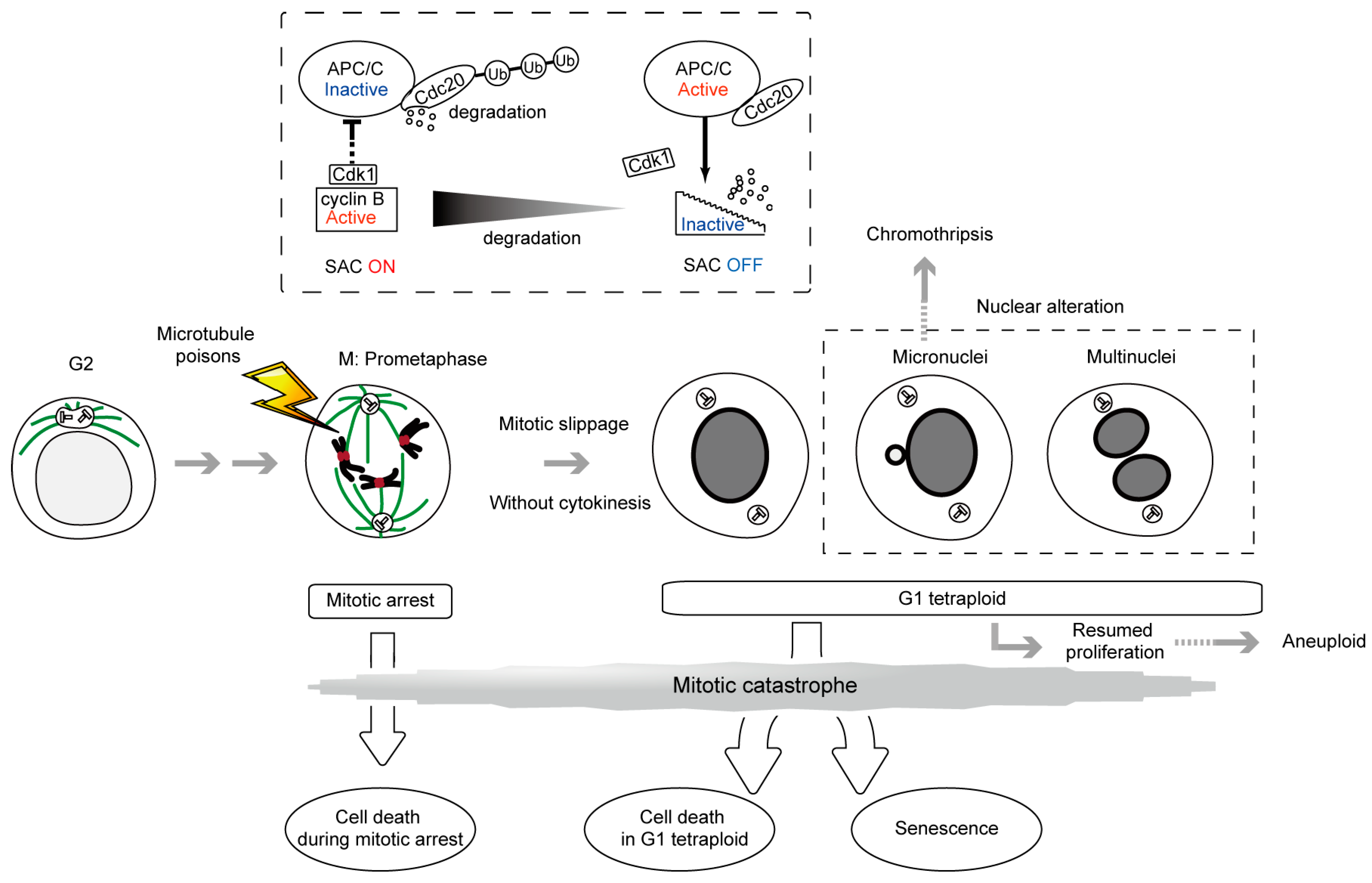
2. SAC, a ‘Wait Anaphase Signal’ in the Metaphase-Anaphase Transition and Cell Death during Mitotic Arrest
3. Antiproliferative Fate after Mitotic Slippage as another Target of Cancer Therapy by Microtubule Poisons
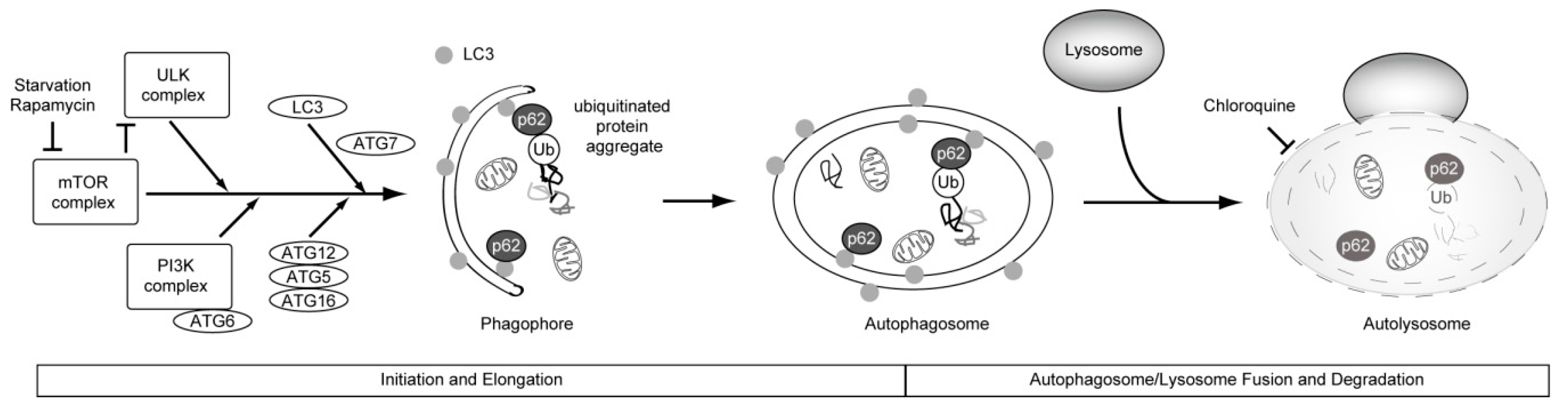
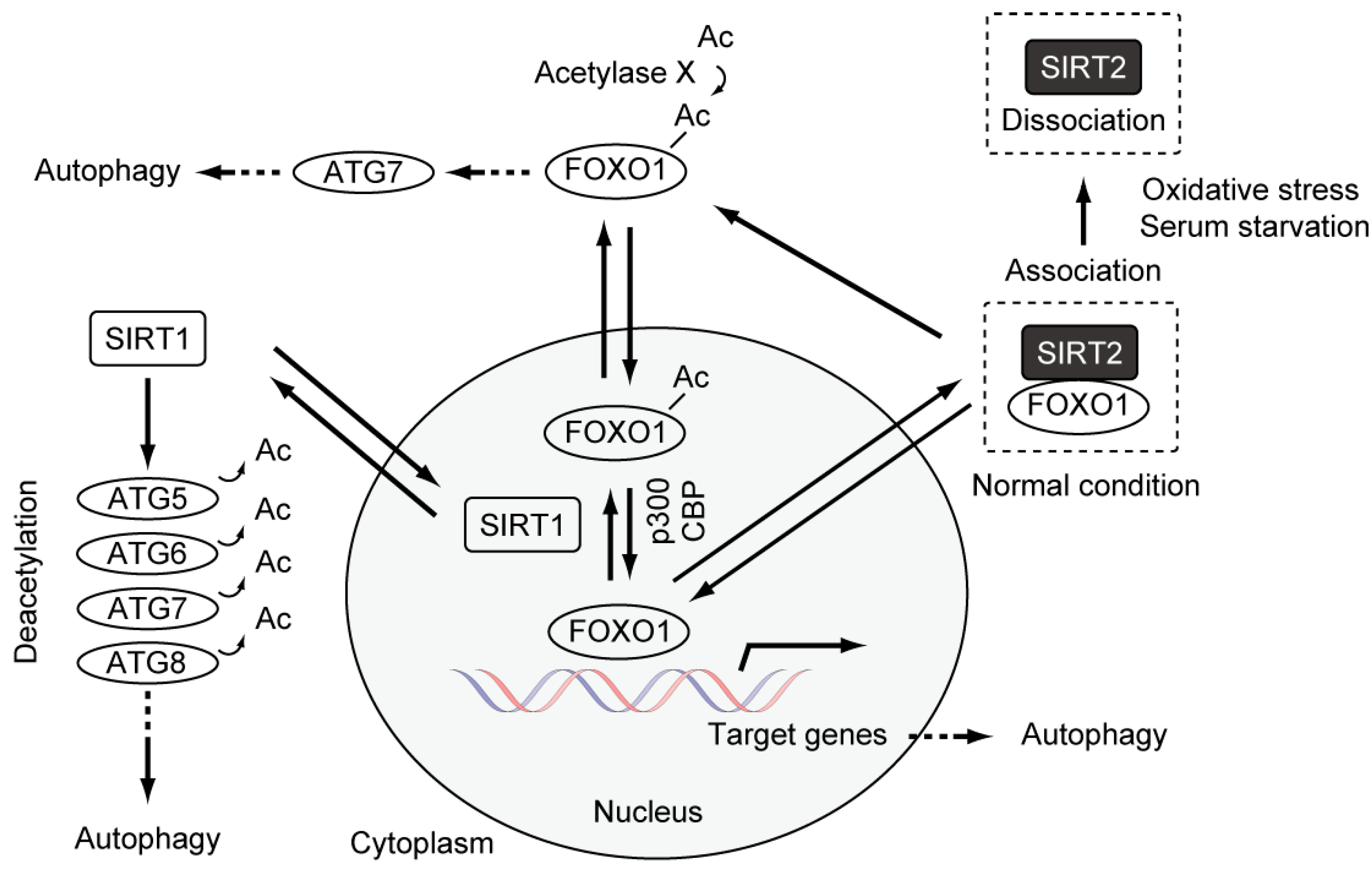
4. Causal Association between Basal Autophagy and Antiproliferative Fate after Mitotic Slippage
5. Autophagy and Resistance to Anti-Cancer Drugs
6. Potential Candidate Strategies to Improve the Efficacy of Microtubule Poisons in Cancer Therapy, Focusing on Antiproliferative Fate after Mitotic Slippage
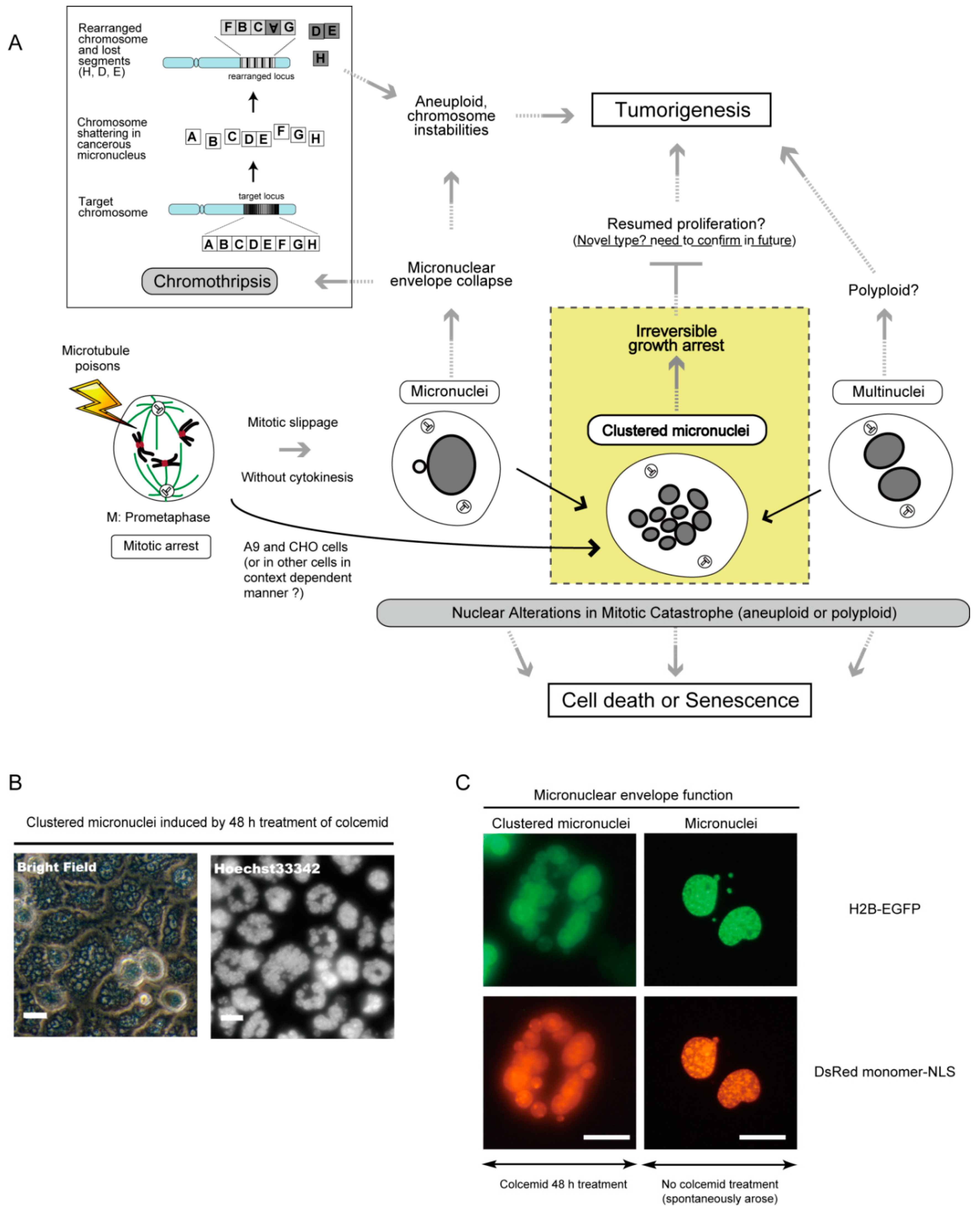
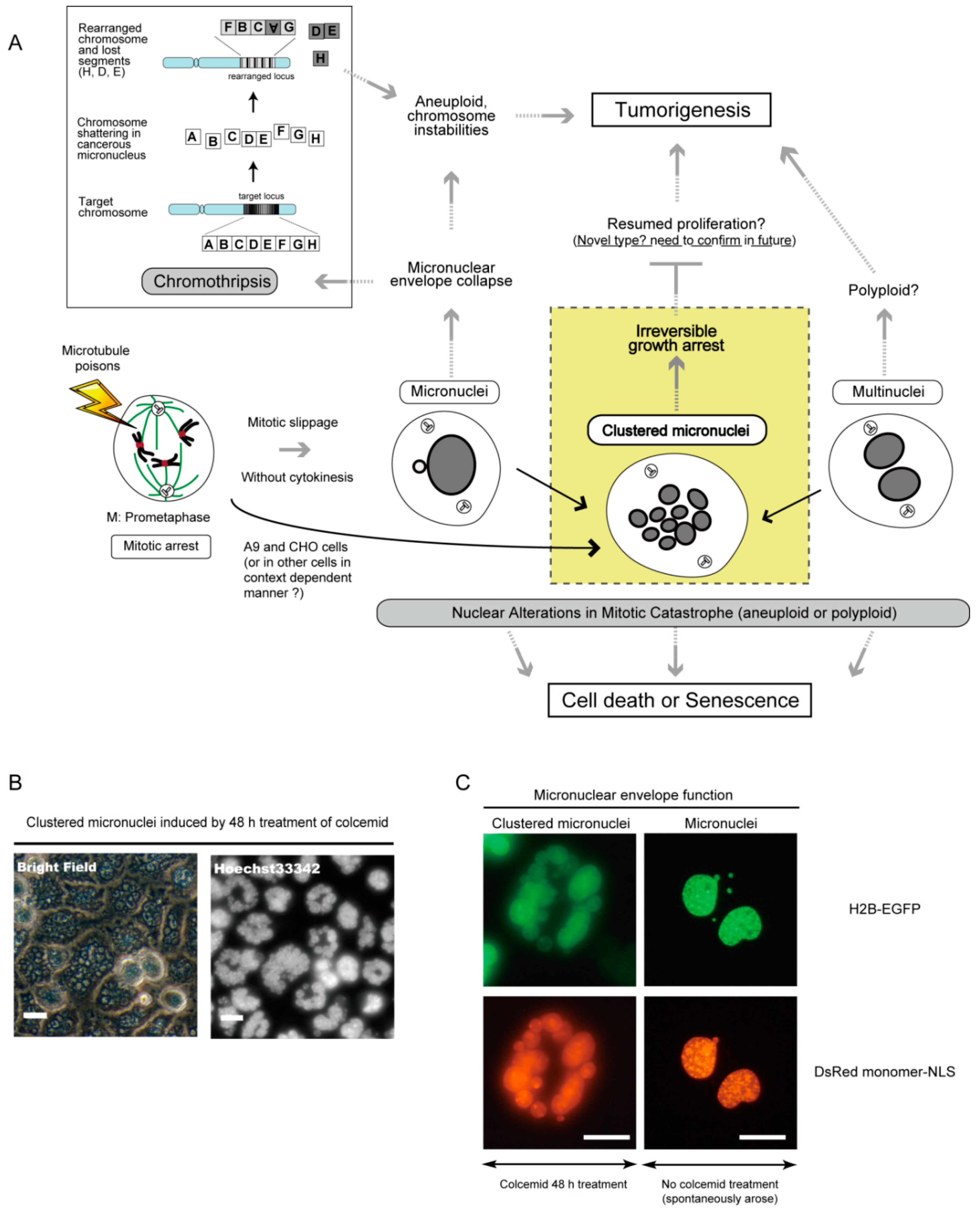
7. Micronucleus Formation as a Morphological Feature of Mitotic Catastrophe and the Link to Genomic Instability
8. Clustered Micronuclei Formation as a Possible New Type of Nuclear Alteration in Mitotic Catastrophe
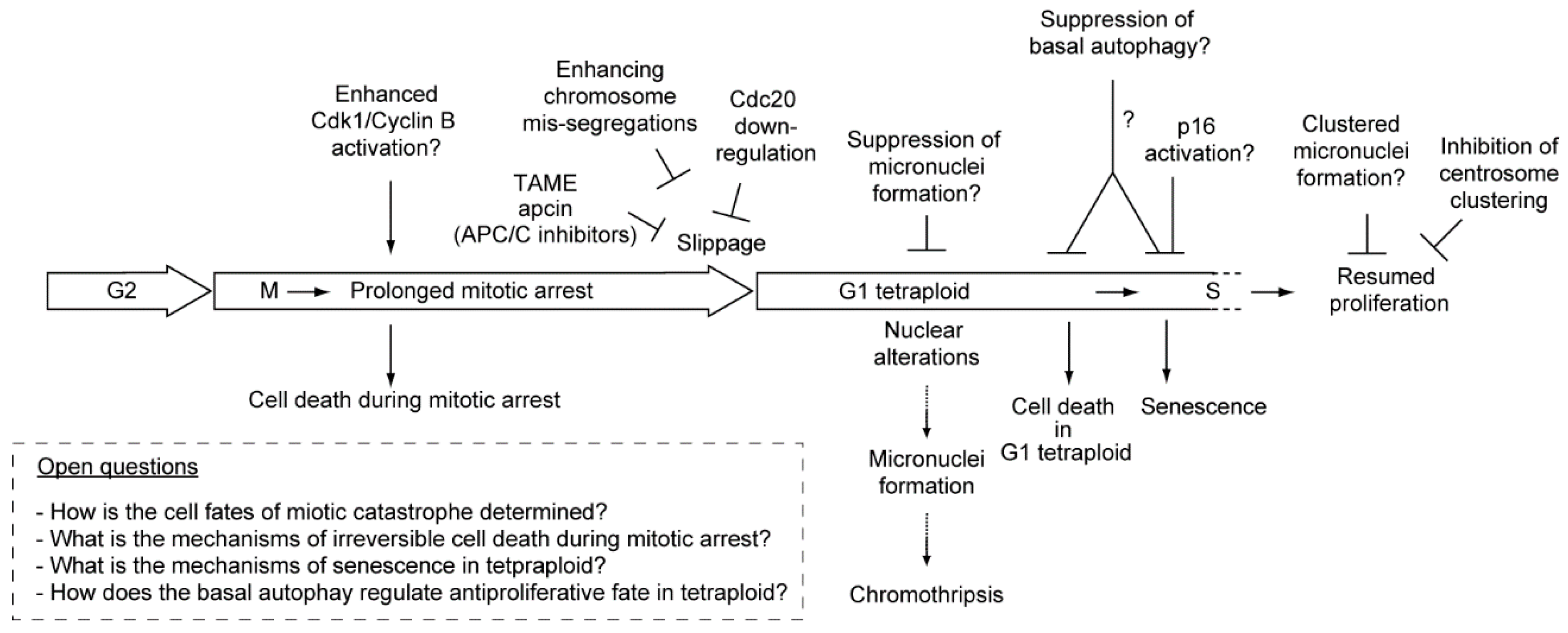
9. Conclusions and Future Prospects
Acknowledgments
Conflicts of Interest
Abbreviations
| APC/C | Anaphase promoting complex/cyclosome |
| SAC | Spindle assembly checkpoint |
| TAME | Tosyl- l-arginine methyl ester |
References
- Matson, D.R.; Stukenberg, P.T. Spindle poisons and cell fate: A tale of two pathways. Mol. Interv. 2011, 11, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Manchado, E.; Guillamot, M.; Malumbres, M. Killing cells by targeting mitosis. Cell Death Differ. 2012, 19, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Mc Gee, M.M. Targeting the Mitotic Catastrophe Signaling Pathway in Cancer. Mediat. Inflamm. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhou, Y.; Shi, J. Post-slippage multinucleation renders cytotoxic variation in anti-mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle 2014, 13, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- King, R.W. When 2 + 2 = 5: The origins and fates of aneuploid and tetraploid cells. Biochim. Biophys. Acta 2008, 1786, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Jallepalli, P.V. Chromothripsis: Chromosomes in crisis. Dev. Cell 2012, 23, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Fournier, R.E. A general high-efficiency procedure for production of microcell hybrids. Proc. Natl. Acad. Sci. USA 1981, 78, 6349–6353. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Uno, N.; Uno, K.; Mizoguchi, Y.; Komoto, S.; Kazuki, Y.; Nanba, E.; Inoue, T.; Oshimura, M. Recurrent micronucleation through cell cycle progression in the presence of microtubule inhibitors. Cell Struct. Funct. 2015, 40, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Lara-Gonzalez, P.; Westhorpe, F.G.; Taylor, S.S. The Spindle Assembly Checkpoint. Curr. Biol. 2012, 22, R966–R980. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.Y.; Kim, S.; Yu, H.T. Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem. Sci. 2013, 38, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, S.; Gorbsky, G.J. Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 2015, 16, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.B.; Pearson, C.G.; Yen, T.J.; Howell, B.J.; Salmon, E.D. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol. Biol. Cell 2001, 12, 1995–2009. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Overlack, K.; Primorac, I.; Vleugel, M.; Krenn, V.; Maffini, S.; Hoffmann, I.; Kops, G.J.; Musacchio, A. A molecular basis for the differential roles of Bub1 and BubR1 in the spindle assembly checkpoint. Elife 2015, 4, e05269. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumie, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Zhivotovsky, B. The unpredictable caspase-2: What can it do? Trends Cell Biol. 2010, 20, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Sakurikar, N.; Eichhorn, J.M.; Chambers, T.C. Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. J. Biol. Chem. 2012, 287, 39193–39204. [Google Scholar] [CrossRef] [PubMed]
- Harley, M.E.; Allan, L.A.; Sanderson, H.S.; Clarke, P.R. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010, 29, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Clarke, P.R. Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol. Cell 2007, 26, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, J.M.; Kothari, A.; Chambers, T.C. Cyclin B1 overexpression induces cell death independent of mitotic arrest. PLoS ONE 2014, 9, e113283. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. Cancer cells display intra- and interline variation profound following prolonged exposure to antimitotic drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Sudo, T.; Nitta, M.; Saya, H.; Ueno, N.T. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 2004, 64, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Terrano, D.T.; Upreti, M.; Chambers, T.C. Cyclin-dependent kinase 1-mediated Bcl-xL/Bcl-2 phosphorylation acts as a functional link coupling mitotic arrest and apoptosis. Mol. Cell Biol. 2010, 30, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Shi, J.; Orth, J.D.; Mitchison, T.J. Evidence that Mitotic Exit Is a Better Cancer Therapeutic Target Than Spindle Assembly. Cancer Cell 2009, 16, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Wolthuis, R.; Clay-Farrace, L.; van Zon, W.; Yekezare, M.; Koop, L.; Ogink, J.; Medema, R.; Pines, J. Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol. Cell 2008, 30, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Sigoillot, F.; Gaur, S.; Choi, S.; Pfaff, K.L.; Oh, D.C.; Hathaway, N.; Dimova, N.; Cuny, G.D.; King, R.W. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 2010, 18, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. [Google Scholar] [CrossRef] [PubMed]
- Isham, C.R.; Bossou, A.R.; Negron, V.; Fisher, K.E.; Kumar, R.; Marlow, L.; Lingle, W.L.; Smallridge, R.C.; Sherman, E.J.; Suman, V.J.; et al. Pazopanib Enhances Paclitaxel-Induced Mitotic Catastrophe in Anaplastic Thyroid Cancer. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Huck, J.J.; Zhang, M.; Mettetal, J.; Chakravarty, A.; Venkatakrishnan, K.; Zhou, X.; Kleinfield, R.; Hyer, M.L.; Kannan, K.; Shinde, V.; et al. Translational exposure-efficacy modeling to optimize the dose and schedule of taxanes combined with the investigational Aurora A kinase inhibitor MLN8237 (alisertib). Mol. Cancer Ther. 2014, 13, 2170–2183. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Cleveland, D.W. Beyond genetics: Surprising determinants of cell fate in antitumor drugs. Cancer Cell 2008, 14, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Hiratsuka, M.; Osaki, M.; Yamada, H.; Kishimoto, I.; Yamaguchi, S.; Nakano, S.; Katoh, M.; Ito, H.; Oshimura, M. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 2007, 26, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Rouble, A.N.; Storey, K.B. Characterization of the SIRT family of NAW-dependent protein deacetylases in the context of a mammalian model of hibernation, the thirteen-lined ground squirrel. Cryobiology 2015, 71, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Tang, B.L. Sirtuins’ modulation of autophagy. J. Cell. Physiol. 2013, 228, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Chen, W.Y. Sorting out functions of sirtuins in cancer. Oncogene 2014, 33, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Narayan, N.; Lee, I.H.; Borenstein, R.; Sun, J.; Wong, R.; Tong, G.; Fergusson, M.M.; Liu, J.; Rovira, I.I.; Cheng, H.L.; et al. The NAD-dependent deacetylase SIRT2 is required for programmed necrosis. Nature 2012, 492, 199–204. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, R.M.; Sarkander, J.; Kazantsev, A.G.; Outeiro, T.F. SIRT2 as a Therapeutic Target for Age-Related Disorders. Front. Pharmacol. 2012, 3, 82. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Tahimic, C.G.; Kurimasa, A.; et al. Proteomics-based identification of differentially expressed genes in human gliomas: Down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Tighe, A.; Johnson, V.L.; Albertella, M.; Taylor, S.S. Aneuploid colon cancer cells have a robust spindle checkpoint. EMBO Rep. 2001, 2, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Nitta, M.; Kobayashi, O.; Honda, S.; Hirota, T.; Kuninaka, S.; Marumoto, T.; Ushio, Y.; Saya, H. Spindle checkpoint function is required for mitotic catastrophe induced by DNA-damaging agents. Oncogene 2004, 23, 6548–6558. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Svenning, S.; Johansen, T. Selective autophagy. Essays Biochem. 2013, 55, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Lippai, M.; Low, P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed. Res. Int. 2014, 2014, 832704. [Google Scholar] [CrossRef] [PubMed]
- Papackova, Z.; Cahova, M. Important role of autophagy in regulation of metabolic processes in health, disease and aging. Physiol. Res. 2014, 63, 409–420. [Google Scholar] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Li, X.; Zhang, P.; Chen, W.D.; Zhang, H.L.; Li, D.D.; Deng, R.; Qian, X.J.; Jiao, L.; Ji, J.; et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat. Commun. 2015, 6, 7215. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Molkentin, J.D.; Yutzey, K.E. FoxO Transcription Factors Promote Autophagy in Cardiomyocytes. J. Biol. Chem. 2009, 284, 28319–28331. [Google Scholar] [CrossRef] [PubMed]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Invest. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011, 25, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.X.; Chiu, J.J.; Liu, G.S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Nakayama, Y.; Li, Y.; Matsumori, H.; Takahashi, H.; Kojima, H.; Wanibuchi, H.; Katoh, M.; Oshimura, M. SIRT2 knockdown increases basal autophagy and prevents postslippage death by abnormally prolonging the mitotic arrest that is induced by microtubule inhibitors. FEBS J. 2014, 281, 2623–2637. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Weidberg, H.; Gonen, C.; Wilder, S.; Elazar, Z.; Oren, M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 2010, 107, 18511–18516. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Xu, W.; Liu, D.; Shen, S.; Lu, Y.; Zhang, L.; Wang, H. Autophagy promotes BCG-induced maturation of human dendritic cells. Acta Biochim. Biophys. Sin. 2010, 42, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, N.; Faried, A.; Tsutsumi, S.; Kuwano, H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Eur. J. Cancer 2010, 46, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Gong, F.; Chen, Y.; Jiang, Y.; Liu, J.; Yu, M.; Zhang, S.; Wang, M.; Xiao, G.; Liao, H. Autophagy promotes paclitaxel resistance of cervical cancer cells: Involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis. 2014, 5, e1367. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhou, L.; Chen, Z.; Nice, E.C.; Huang, C. Stress Management by Autophagy: Implications for Chemoresistance. Int. J. Cancer 2016, 139, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Chen, N.; Zhao, F.; Wang, X.J.; Kong, B.H.; Zheng, W.X.; Zhang, D.D. High Levels of Nrf2 Determine Chemoresistance in Type II Endometrial Cancer. Cancer Res. 2010, 70, 5486–5496. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, N.F.; Tian, W.; Wu, T.D.; Sun, Z.; Lau, A.; Chapman, E.; Fang, D.Y.; Zhang, D.D. USP15 Negatively Regulates Nrf2 through Deubiquitination of Keap1. Mol. Cell 2013, 51, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Harder, B.G.; Wong, P.K.; Lang, J.E.; Zhang, D.D. Oxidative stress, mammospheres and Nrf2-new implication for breast cancer therapy? Mol. Carcinog. 2015, 54, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.F.; Wang, X.Y.; Fu, Z.Q.; Peng, Q.H.; Zhang, J.Y.; Ye, F.; Fu, Y.F.; Zhou, C.Y.; Lu, W.G.; Cheng, X.D.; et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy 2015, 11, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Shi, W. Autophagy regulates resistance of non-small cell lung cancer cells to paclitaxel. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Lissa, D.; Senovilla, L.; Rello-Varona, S.; Vitale, I.; Michaud, M.; Pietrocola, F.; Boileve, A.; Obrist, F.; Bordenave, C.; Garcia, P.; et al. Resveratrol and aspirin eliminate tetraploid cells for anticancer chemoprevention. Proc. Natl. Acad. Sci. USA 2014, 111, 3020–3025. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. p53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9819–9824. [Google Scholar] [CrossRef] [PubMed]
- Margolis, R.L.; Lohez, O.D.; Andreassen, P.R. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J. Cell. Biochem. 2003, 88, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, J.E.; Bregerie, O.; Robert, A.; Debey, P.; Brechot, C.; Desdouets, C. Liver cell polyploidization: A pivotal role for binuclear hepatocytes. J. Biol. Chem. 2003, 278, 19095–19101. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Johmura, Y.; Shimada, M.; Misaki, T.; Naiki-Ito, A.; Miyoshi, H.; Motoyama, N.; Ohtani, N.; Hara, E.; Nakamura, M.; Morita, A.; et al. Necessary and Sufficient Role for a Mitosis Skip in Senescence Induction. Mol. Cell 2014, 55, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Hoffelder, D.R.; Luo, L.; Burke, N.A.; Watkins, S.C.; Gollin, S.M.; Saunders, W.S. Resolution of anaphase bridges in cancer cells. Chromosoma 2004, 112, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D. Classification of chromosome segregation errors in cancer. Chromosoma 2008, 117, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Terradas, M.; Martin, M.; Tusell, L.; Genesca, A. Genetic activities in micronuclei: Is the DNA entrapped in micronuclei lost for the cell? Mutat. Res. 2010, 705, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Kirsch-Volders, M.; Natarajan, A.T.; Surralles, J.; Crott, J.W.; Parry, J.; Norppa, H.; Eastmond, D.A.; Tucker, J.D.; Thomas, P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 2011, 26, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Utani, K.; Kohno, Y.; Okamoto, A.; Shimizu, N. Emergence of Micronuclei and Their Effects on the Fate of Cells under Replication Stress. PLoS ONE 2010, 5, e10089. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; Kumar, Y. Cancer cell micronucleus: an update on clinical and diagnostic applications. APMIS 2013, 121, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Storchova, Z.; Kloosterman, W.P. The genomic characteristics and cellular origin of chromothripsis. Curr. Opin. Cell Biol. 2016, 40, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Jones, D.T.; Zapatka, M.; Stutz, A.M.; Zichner, T.; Weischenfeldt, J.; Jager, N.; Remke, M.; Shih, D.; Northcott, P.A.; et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012, 148, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Bargiela-Iparraguirre, J.; Prado-Marchal, L.; Pajuelo-Lozano, N.; Jimenez, B.; Perona, R.; Sanchez-Perez, I. Mad2 and BubR1 modulates tumourigenesis and paclitaxel response in MKN45 gastric cancer cells. Cell Cycle 2014, 13, 3590–3601. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.A.; Monroe, M.M.; Alves, M.V.O.; Patel, A.A.; Katsonis, P.; Fitzgerald, A.L.; Neskey, D.M.; Frederick, M.J.; HyeokWoo, S.; Caulin, C.; et al. Wee-1 Kinase Inhibition Overcomes Cisplatin Resistance Associated with High-Risk TP53 Mutations in Head and Neck Cancer through Mitotic Arrest Followed by Senescence. Mol. Cancer Ther. 2015, 14, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Sadaie, M.; Dillon, C.; Narita, M.; Young, A.R.; Cairney, C.J.; Godwin, L.S.; Torrance, C.J.; Bennett, D.C.; Keith, W.N.; Narita, M. Cell-based screen for altered nuclear phenotypes reveals senescence progression in polyploid cells after Aurora kinase B inhibition. Mol. Biol. Cell 2015, 26, 2971–2985. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu, H.; Olsson, M.; Tamm, C.; Heidari, N.; Orrenius, S.; Zhivotovsky, B. DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ. 2008, 15, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, T.V.; Sorokina, I.V.; Gogvadze, V.; Zhivotovsky, B. Mitotic catastrophe and cancer drug resistance: A link that must to be broken. Drug Resist. Updates 2016, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Breitkreutz, I.; Anderhub, S.; Ronnest, M.H.; Leber, B.; Larsen, T.O.; Weiz, L.; Konotop, G.; Hayden, P.J.; Podar, K.; et al. GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo. Cancer Res; 2012, 72, 5374–5385. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, E.; Fielding, A.B.; Kannan, N.; Balgi, A.; Eaves, C.J.; Roberge, M.; Dedhar, S. Identification of novel small molecule inhibitors of centrosome clustering in cancer cells. Oncotarget 2013, 4, 1763–1776. [Google Scholar] [CrossRef] [PubMed]
- Chavali, P.L.; Chandrasekaran, G.; Barr, A.R.; Tatrai, P.; Taylor, C.; Papachristou, E.K.; Woods, C.G.; Chavali, S.; Gergely, F. A CEP215-HSET complex links centrosomes with spindle poles and drives centrosome clustering in cancer. Nat. Commun. 2016, 7, 11005. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.; Kops, G.J.P.L.; Medema, R.H. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19108–19113. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakayama, Y.; Inoue, T. Antiproliferative Fate of the Tetraploid Formed after Mitotic Slippage and Its Promotion; A Novel Target for Cancer Therapy Based on Microtubule Poisons. Molecules 2016, 21, 663. https://doi.org/10.3390/molecules21050663
Nakayama Y, Inoue T. Antiproliferative Fate of the Tetraploid Formed after Mitotic Slippage and Its Promotion; A Novel Target for Cancer Therapy Based on Microtubule Poisons. Molecules. 2016; 21(5):663. https://doi.org/10.3390/molecules21050663
Chicago/Turabian StyleNakayama, Yuji, and Toshiaki Inoue. 2016. "Antiproliferative Fate of the Tetraploid Formed after Mitotic Slippage and Its Promotion; A Novel Target for Cancer Therapy Based on Microtubule Poisons" Molecules 21, no. 5: 663. https://doi.org/10.3390/molecules21050663
APA StyleNakayama, Y., & Inoue, T. (2016). Antiproliferative Fate of the Tetraploid Formed after Mitotic Slippage and Its Promotion; A Novel Target for Cancer Therapy Based on Microtubule Poisons. Molecules, 21(5), 663. https://doi.org/10.3390/molecules21050663