
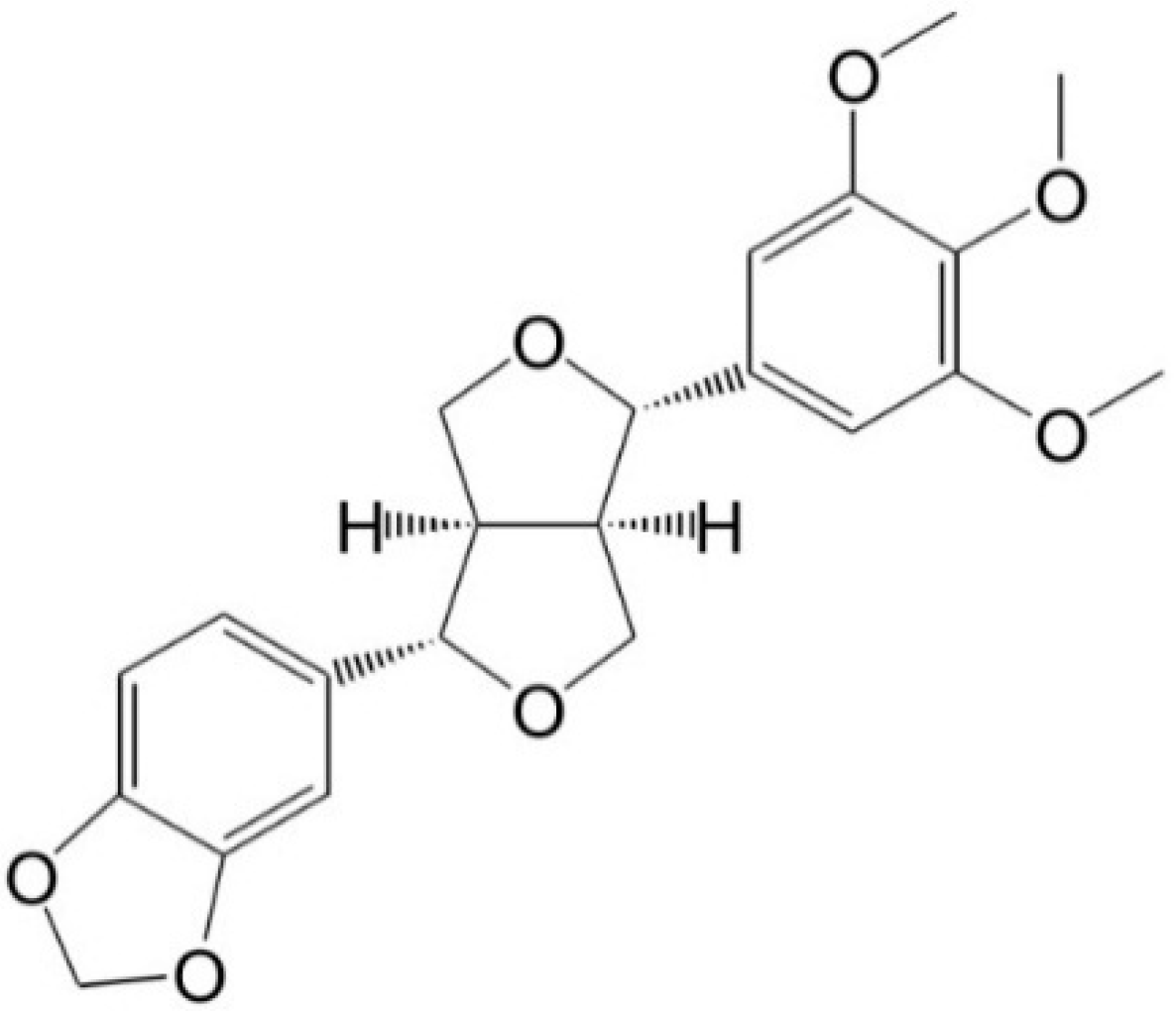
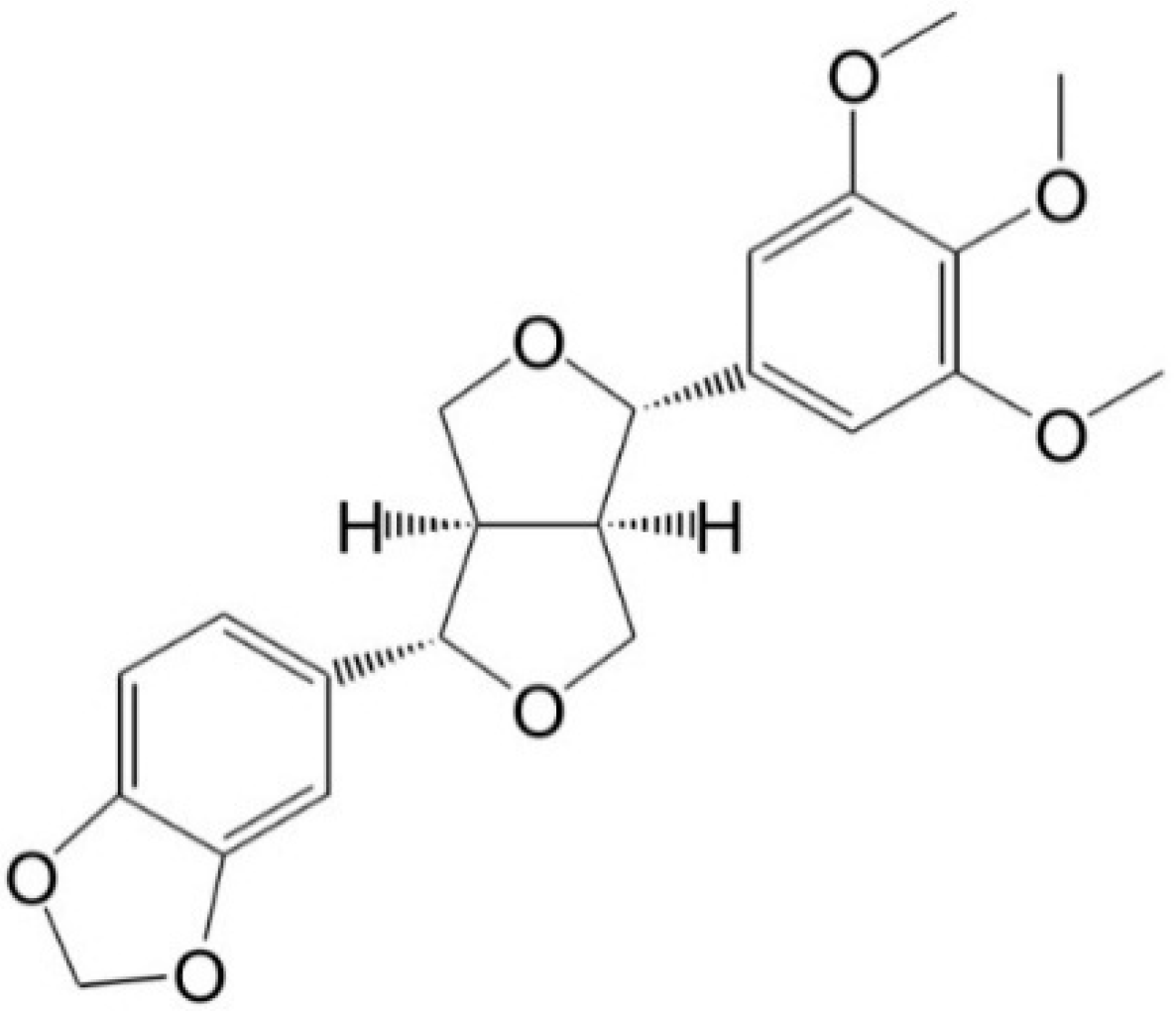
Inhibitory Effects of Aschantin on Cytochrome P450 and Uridine 5′-diphospho-glucuronosyltransferase Enzyme Activities in Human Liver Microsomes




Abstract
:1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. Materials and Reagents
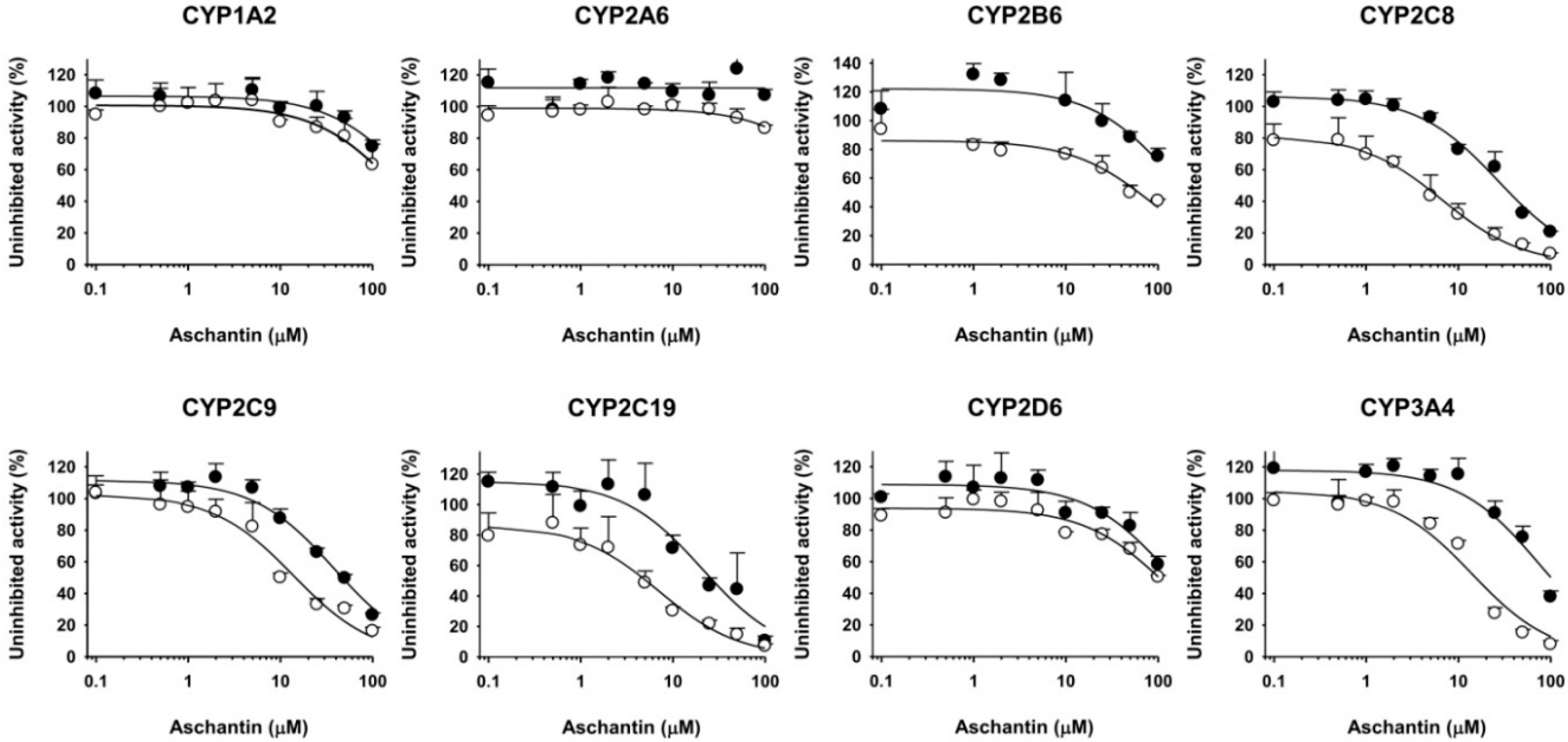
3.2. Inhibitory Effects of Aschantin on the Activities of Seven Major CYPs in Human Liver Microsomes
3.3. Inhibitory Effects of Aschantin on CYP2B6 in Human Liver Microsomes
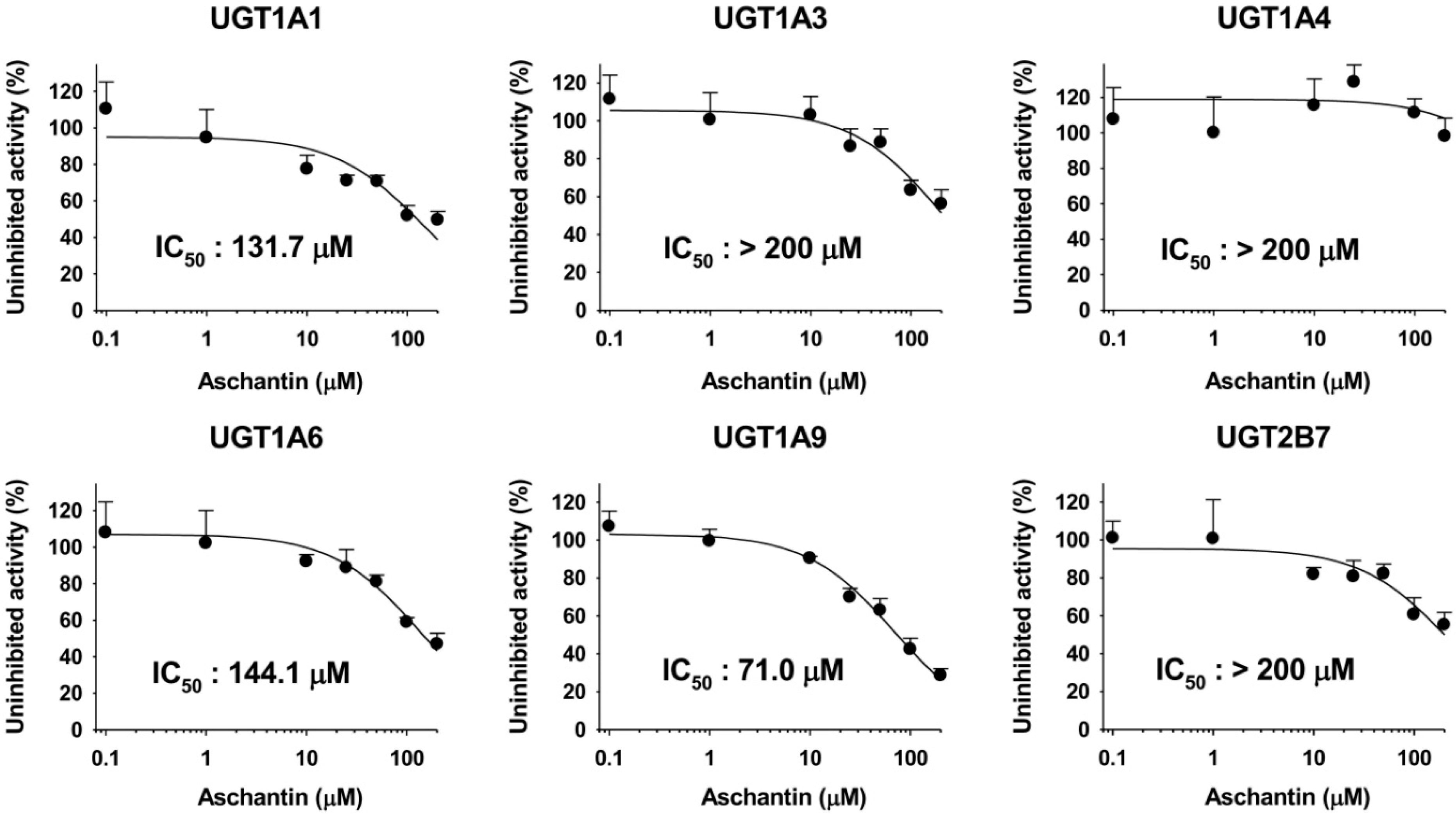
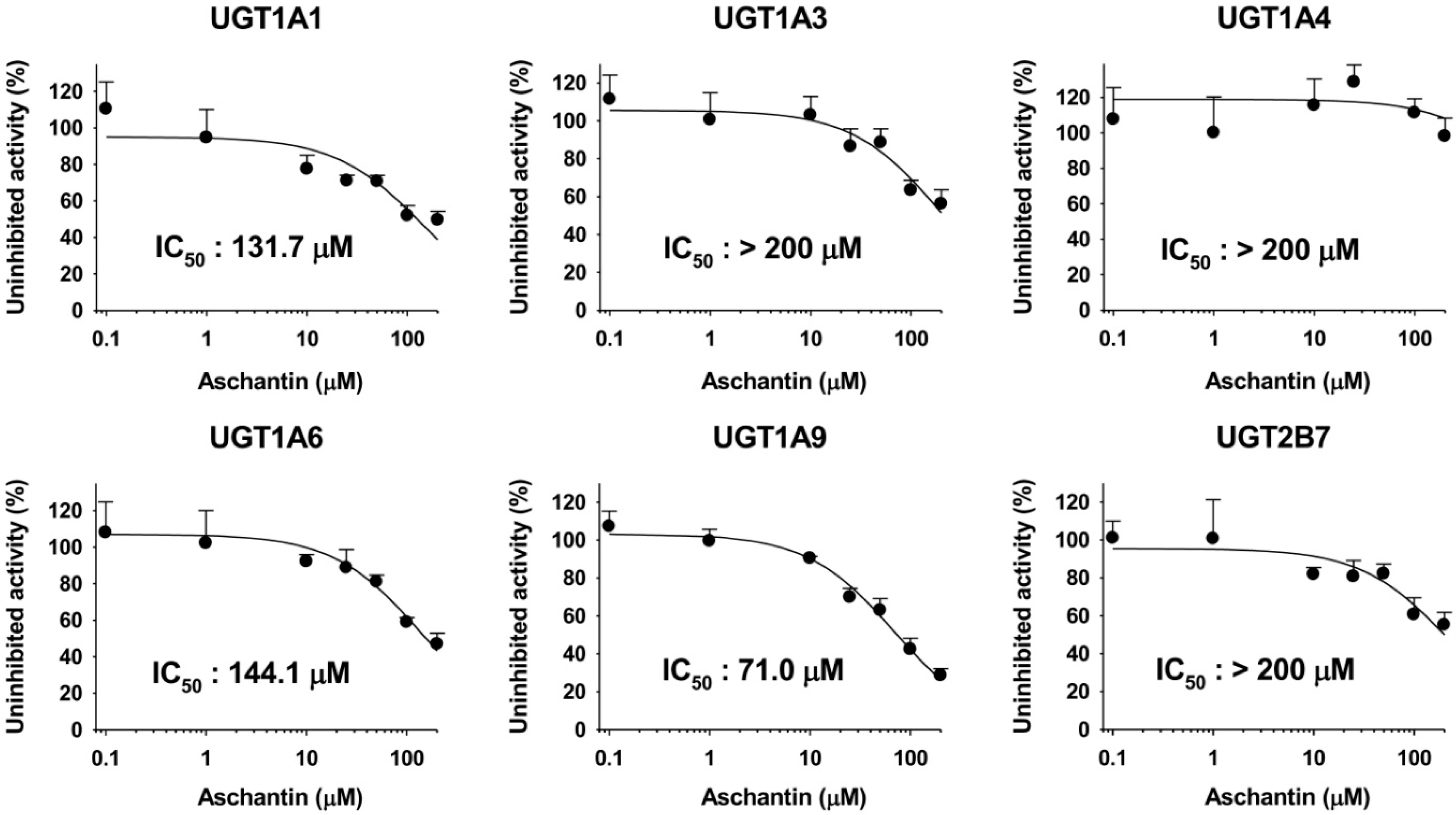
3.4. Inhibitory Effects of Aschantin on the Activities of Six Major UGTs in Human Liver Microsomes
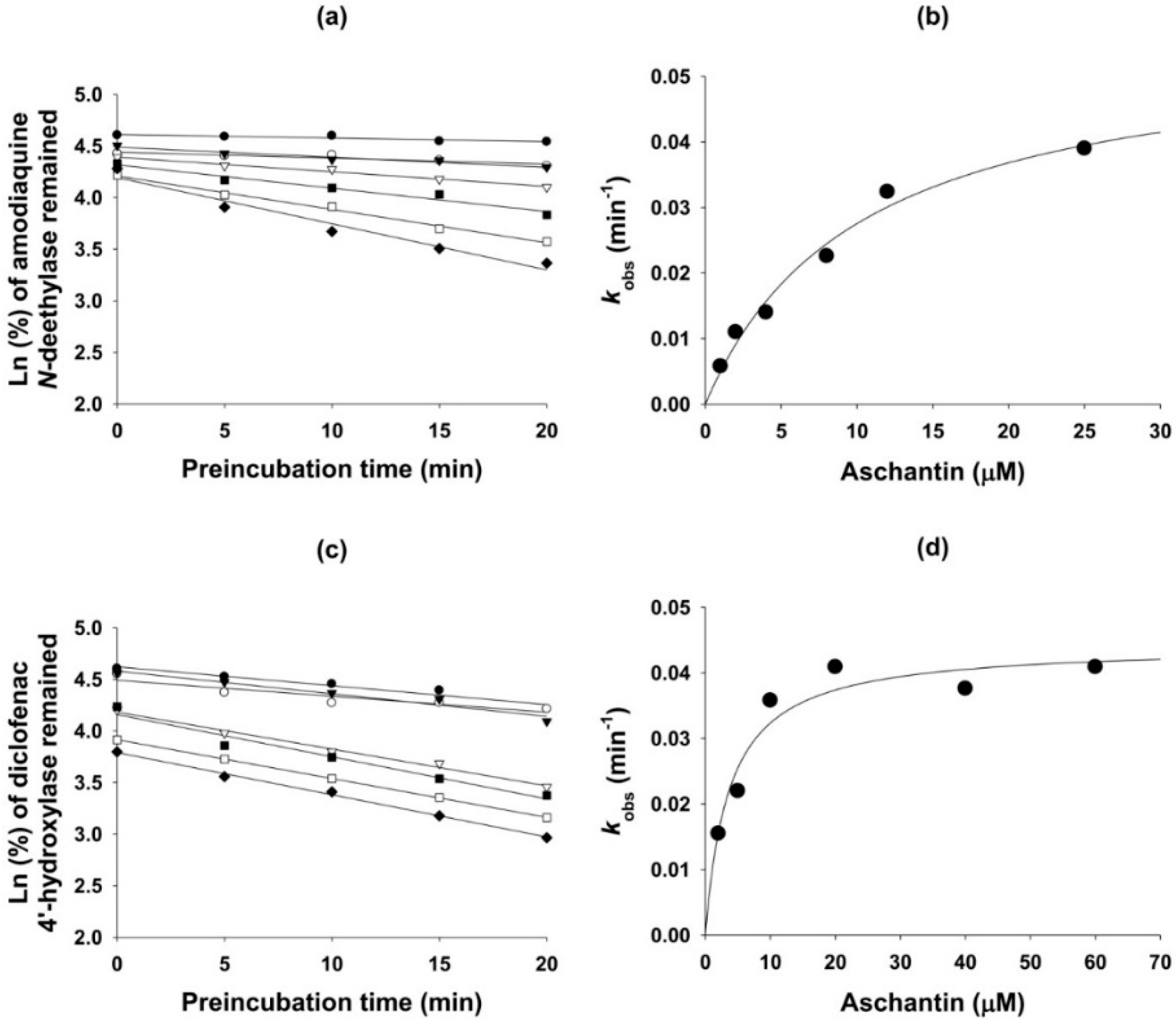
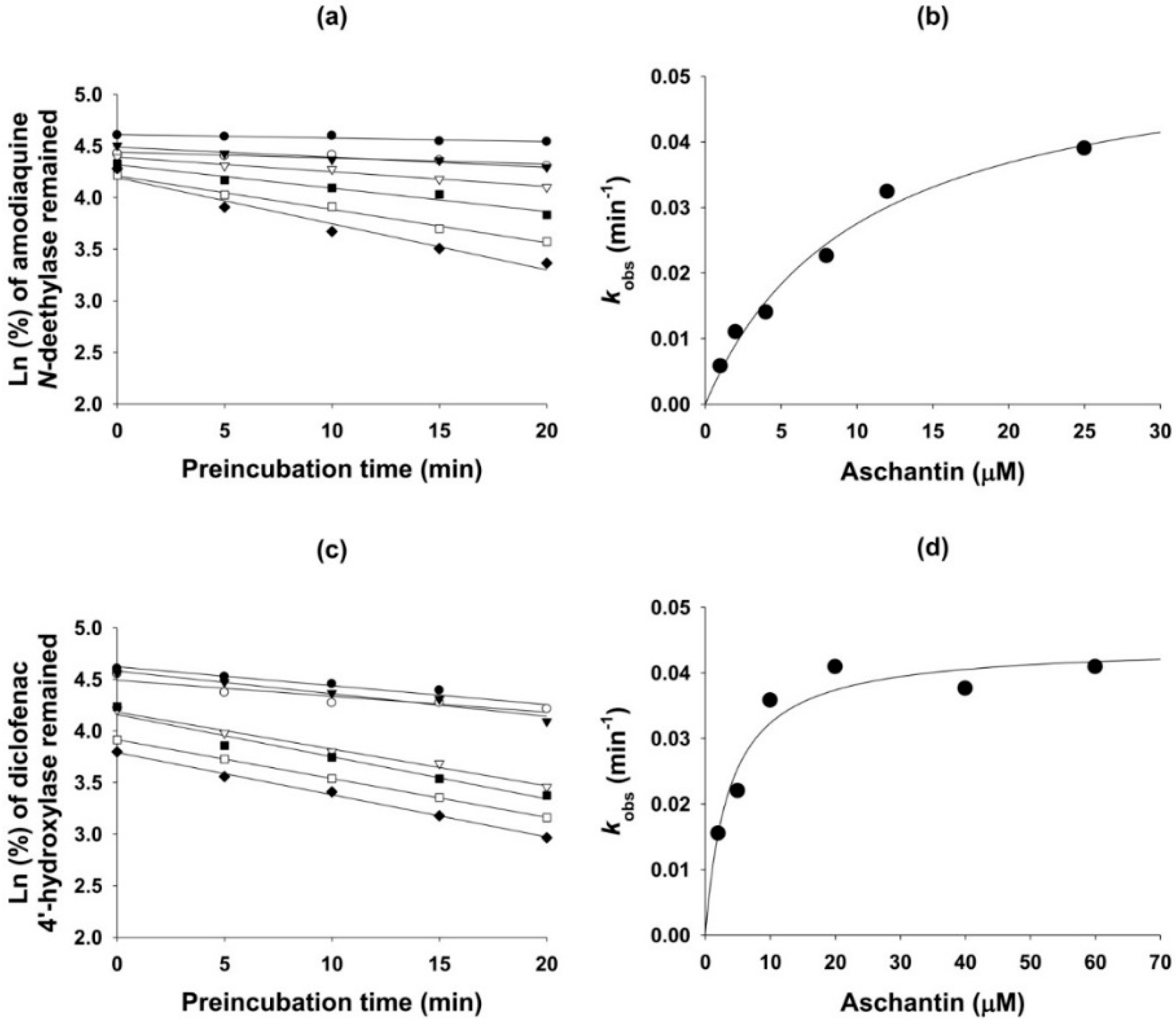
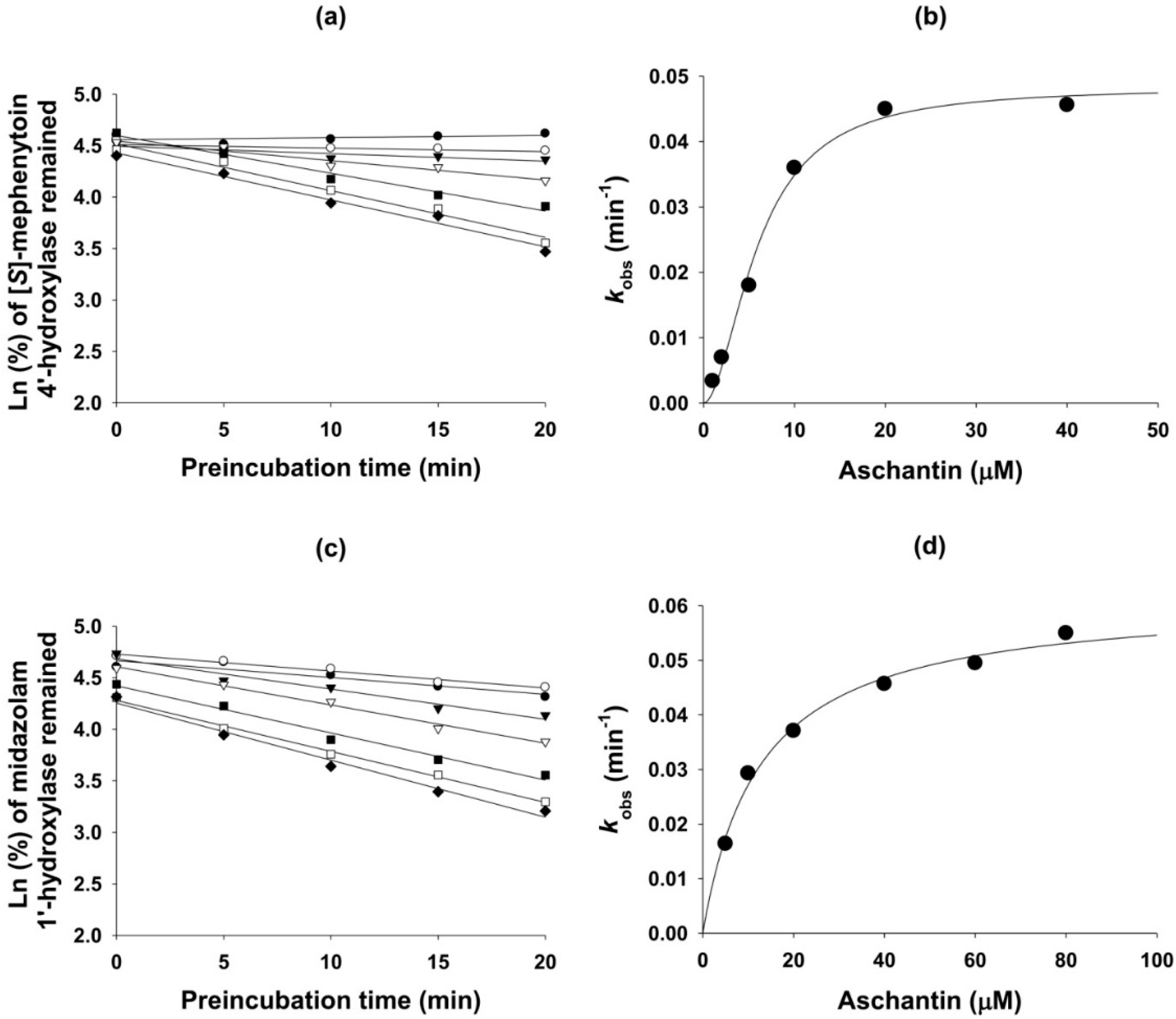
3.5. Mechanism-Based Inhibition of CYP Activities by Aschantin
3.6. Data analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, X.; Chen, C.; Ye, X.; Song, F.; Fan, G.; Wu, F. Analysis of lignans in Magnoliae Flos by turbulent flow chromatography with online solid-phase extraction and high-performance liquid chromatography with tandem mass spectrometry. J. Sep. Sci. 2016, 39, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Jang, J.H.; Lee, J.Y.; Lee, M.H.; Li, Y.; Ryu, H.W.; Choi, K.I.; Dong, Z.; Lee, H.S.; Oh, S.R.; et al. Aschantin targeting on the kinase domain of mammalian target of rapamycin suppresses epidermal growth factor-induced neoplastic cell transformation. Carcinogenesis 2015, 36, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.Y.; Chen, I.S.; Yeh, J.L.; Chen, J.J.; Ko, Y.C.; Cheng, J.S.; Liu, C.P.; Lo, Y.K.; Su, W.; Chou, K.J.; et al. Novel action of lignans isolated from Hernandia nymphaeifolia on Ca2+ signaling in renal tubular cells. Eur. J. Pharm. 2002, 443, 31–38. [Google Scholar] [CrossRef]
- Chao, Y.Y.; Jan, C.R.; Ko, Y.C.; Chen, J.J.; Jiann, B.P.; Lu, Y.C.; Chen, W.C.; Su, W.; Chen, I.S. Effect of lignans isolated from Hernandia nymphaeifolia on estrogenic compounds-induced calcium mobilization in human neutrophils. Life Sci. 2002, 70, 3109–3121. [Google Scholar] [CrossRef]
- Chao, Y.Y.; Su, W.; Jan, C.R.; Ko, Y.C.; Chen, J.J.; Cheng, J.S.; Liu, C.P.; Lo, Y.K.; Chou, K.J.; Lee, K.C.; et al. Novel action of lignans isolated from Hernandia nymphaeifolia on Ca2+ signaling in human neutrophils. Arch. Toxicol. 2002, 75, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, J.Y.; Lee, H.J.; Lim, H.J.; Lee da, Y.; Kim do, H.; Ryu, J.H. Suppression of inducible nitric oxide synthase expression by furfuran lignans from flower buds of Magnolia fargesii in BV-2 microglial cells. Phytother. Res. 2010, 24, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Ortet, R.; Prado, S.; Regalado, E.L.; Valeriote, F.A.; Media, J.; Mendiola, J.; Thomas, O.P. Furfuran lignans and a flavone from Artemisia gorgonum Webb and their in vitro activity against Plasmodium falciparum. J. Ethnopharm. 2011, 138, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Na, D.H.; Ji, H.Y.; Park, E.J.; Kim, M.S.; Liu, K.H.; Lee, H.S. Evaluation of metabolism-mediated herb-drug interactions. Arch. Pharm. Res. 2011, 34, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.Z.; Zhang, Y.L.; Zeng, M.Z.; He, F.Z.; Luo, Z.Y.; Luo, J.Q.; Wen, J.G.; Chen, X.P.; Zhou, H.H.; Zhang, W. Pharmacogenomics and herb-drug interactions: Merge of future and tradition. Evid. based Complement. Altern. Med. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.L.; Ma, Y.M. Pharmacokinetic herb-drug interactions with traditional Chinese medicine: Progress, causes of conflicting results and suggestions for future research. Drug Metab. Rev. 2016, 48, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Liu, K. Pharmacokinetic interactions between herbal medicines and prescribed drugs: Focus on drug metabolic enzymes and transporters. Curr. Drug Metab. 2014, 15, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Yoon, I.S. Pharmacokinetic interactions of herbs with cytochrome p450 and p-glycoprotein. Evid. Based Complement. Altern. Med. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Huang, M. Herb-drug interactions: Systematic review, mechanisms, and therapies. Evid. Based Complement. Altern. Med. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Haefeli, W.E.; Carls, A. Drug interactions with phytotherapeutics in oncology. Expert Opin. Drug Metab. Toxicol. 2014, 10, 359–377. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Scicchitano, F.; Whalley, B.J.; Mazzitello, C.; Ciriaco, M.; Esposito, S.; Patane, M.; Upton, R.; Pugliese, M.; Chimirri, S.; et al. Hypericum perforatum: Pharmacokinetic, mechanism of action, tolerability, and clinical drug-drug interactions. Phytother. Res. 2014, 28, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Unger, M. Pharmacokinetic drug interactions involving Ginkgo biloba. Drug Metab. Rev. 2013, 45, 353–385. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, L.; Shan, L.; Fan, G.; Gao, X. Liquorice, a unique “guide drug” of traditional Chinese medicine: A review of its role in drug interactions. J. Ethnopharm. 2013, 150, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.U.; Kong, T.Y.; Kwon, S.S.; Hong, S.W.; Yeon, S.H.; Choi, J.H.; Lee, J.Y.; Cho, Y.Y.; Lee, H.S. Effect of honokiol on cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2013, 18, 10681–10693. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; You, J.; Choi, H.G.; Kim, J.A.; Jee, J.G.; Lee, S. Selective inhibitory effects of machilin A isolated from Machilus thunbergii on human cytochrome P450 1A and 2B6. Phytomedicine 2015, 22, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Kang, H.E.; Cho, H.J.; Kim, Y.S.; Chung, S.J.; Yoon, I.S.; Kim, D.D. Metabolic interactions of magnolol with cytochrome P450 enzymes: Uncompetitive inhibition of CYP1A and competitive inhibition of CYP2C. Drug Dev. Ind. Pharm. 2016, 42, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Fan, X.; Wang, Y.; Tan, H.; Chen, P.; Zeng, H.; Huang, M.; Bi, H. Hepato-protective effects of six schisandra lignans on acetaminophen-induced liver injury are partially associated with the inhibition of CYP-mediated bioactivation. Chem. Biol. Interact. 2015, 231, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Fan, X.; Wang, Y.; Chen, P.; Zeng, H.; Tan, H.; Gonzalez, F.J.; Huang, M.; Bi, H. Schisandrol B protects against acetaminophen-induced hepatotoxicity by inhibition of CYP-mediated bioactivation and regulation of liver regeneration. Toxicol. Sci. 2015, 143, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Taesotikul, T.; Dumrongsakulchai, W.; Wattanachai, N.; Navinpipat, V.; Somanabandhu, A.; Tassaneeyakul, W.; Tassaneeyakul, W. Inhibitory effects of Phyllanthus amarus and its major lignans on human microsomal cytochrome P450 activities: Evidence for CYP3A4 mechanism-based inhibition. Drug Metab. Pharm. 2011, 26, 154–161. [Google Scholar] [CrossRef]
- Song, J.H.; Sun, D.X.; Chen, B.; Ji, D.H.; Pu, J.; Xu, J.; Tian, F.D.; Guo, L. Inhibition of CYP3A4 and CYP2C9 by podophyllotoxin: Implication for clinical drug-drug interactions. J. Biosci. 2011, 36, 879–885. [Google Scholar] [CrossRef] [PubMed]
- P450 Drug Interaction Table: Abbreviated “Clinically Relevant” Table. Available online: http://medicine.iupui.edu/clinpharm/ddis/table.asp (accessed on 26 April 2016).
- Lai, X.S.; Yang, L.P.; Li, X.T.; Liu, J.P.; Zhou, Z.W.; Zhou, S.F. Human CYP2C8: Structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr. Drug Metab. 2009, 10, 1009–1047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Zhou, Z.W.; Yang, L.P.; Cai, J.P. Substrates, inducers, inhibitors and structure-activity relationships of human Cytochrome P450 2C9 and implications in drug development. Curr. Med. Chem. 2009, 16, 3480–3675. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Jung, K.Y.; Ahn, K.S.; Oh, S.R.; Lee, I.S.; Park, S.H.; Kim, J.H.; Jang, H.W.; Hong, N.D.; (inventors); Korea Institute of Science & Technology; (assignee). New Lignans Isolated from Magnolia Flos with Leukotriene Synthesis Inhibitory Activity. Korea Patent 10-0321313-0000, 7 January 2002. [Google Scholar]
- Joo, J.; Lee, B.; Lee, T.; Liu, K.H. Screening of six UGT enzyme activities in human liver microsomes using liquid chromatography/triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYP | Enzyme | IC50 (µM) | kinact (min−1) | Ki (µM) | |
|---|---|---|---|---|---|
| No Pre-incubation | With Pre-incubation * | ||||
| 1A2 | Phenacetin O-de-ethylase | >100 | >100 | - | - |
| 2A6 | Coumarin 7-hydroxylase | >100 | >100 | - | - |
| 2B6 | Bupropion hydroxylase | >100 | 87.9 | - | - |
| 2C8 | Amodiaquine N-de-ethylase | 27.8 | 5.3 | 0.056 | 10.2 |
| 2C9 | Diclofenac 4′-hydroxylase | 40.5 | 14.1 | 0.044 | 3.7 |
| 2C19 | (S)-Mephenytoin 4′-hydroxylase | 22.7 | 6.1 | 0.048 | 5.8 |
| 2D6 | Bufuralol 1′-hydroxylase | >100 | >100 | - | - |
| 3A4 | Midazolam 1′-hydroxylase | 57.5 | 14.4 | 0.062 | 12.6 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, S.-S.; Kim, J.-H.; Jeong, H.-U.; Cho, Y.Y.; Oh, S.-R.; Lee, H.S. Inhibitory Effects of Aschantin on Cytochrome P450 and Uridine 5′-diphospho-glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules 2016, 21, 554. https://doi.org/10.3390/molecules21050554
Kwon S-S, Kim J-H, Jeong H-U, Cho YY, Oh S-R, Lee HS. Inhibitory Effects of Aschantin on Cytochrome P450 and Uridine 5′-diphospho-glucuronosyltransferase Enzyme Activities in Human Liver Microsomes. Molecules. 2016; 21(5):554. https://doi.org/10.3390/molecules21050554
Chicago/Turabian StyleKwon, Soon-Sang, Ju-Hyun Kim, Hyeon-Uk Jeong, Yong Yeon Cho, Sei-Ryang Oh, and Hye Suk Lee. 2016. "Inhibitory Effects of Aschantin on Cytochrome P450 and Uridine 5′-diphospho-glucuronosyltransferase Enzyme Activities in Human Liver Microsomes" Molecules 21, no. 5: 554. https://doi.org/10.3390/molecules21050554