
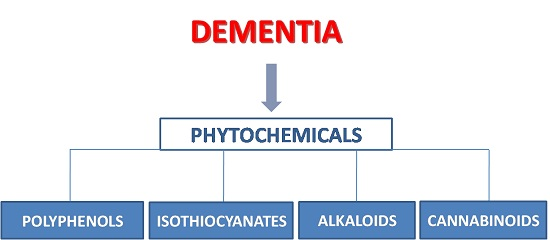
Natural Phytochemicals in the Treatment and Prevention of Dementia: An Overview


Abstract
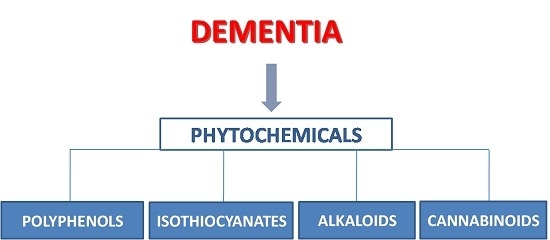
:
1. Introduction
The Etiopathogenesis of Dementia
2. Polyphenols
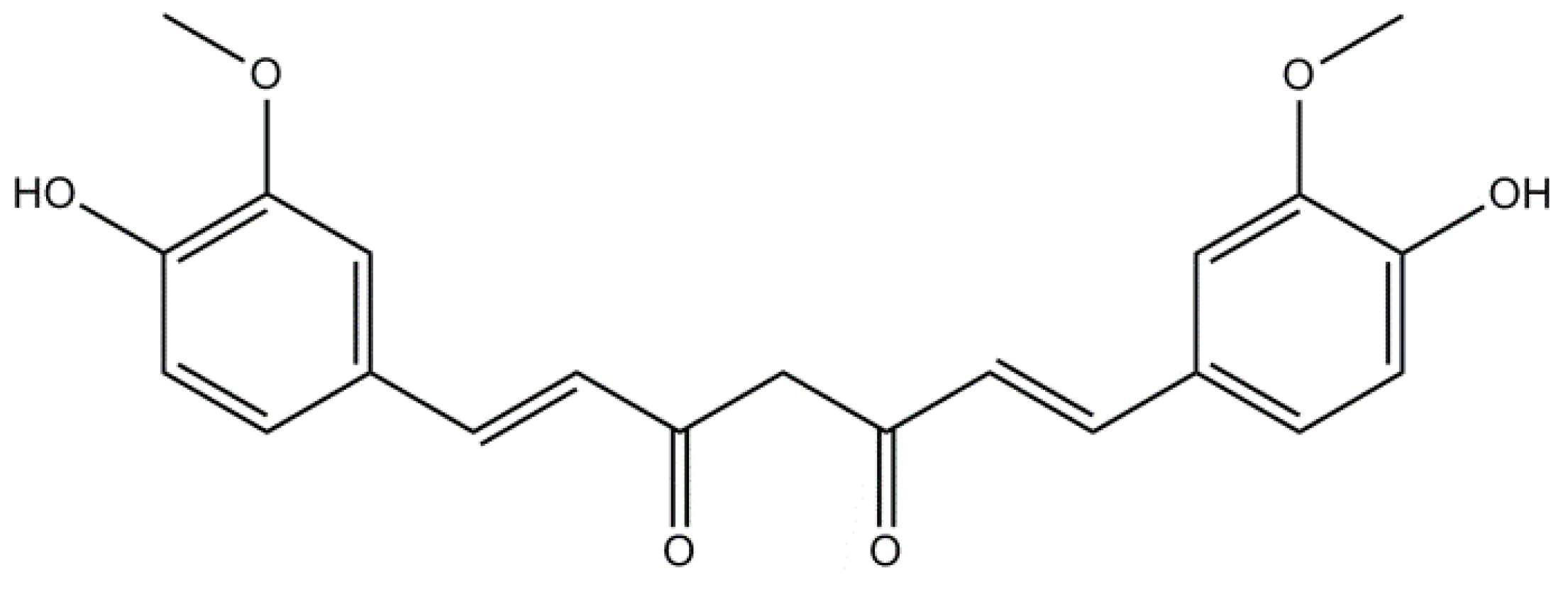
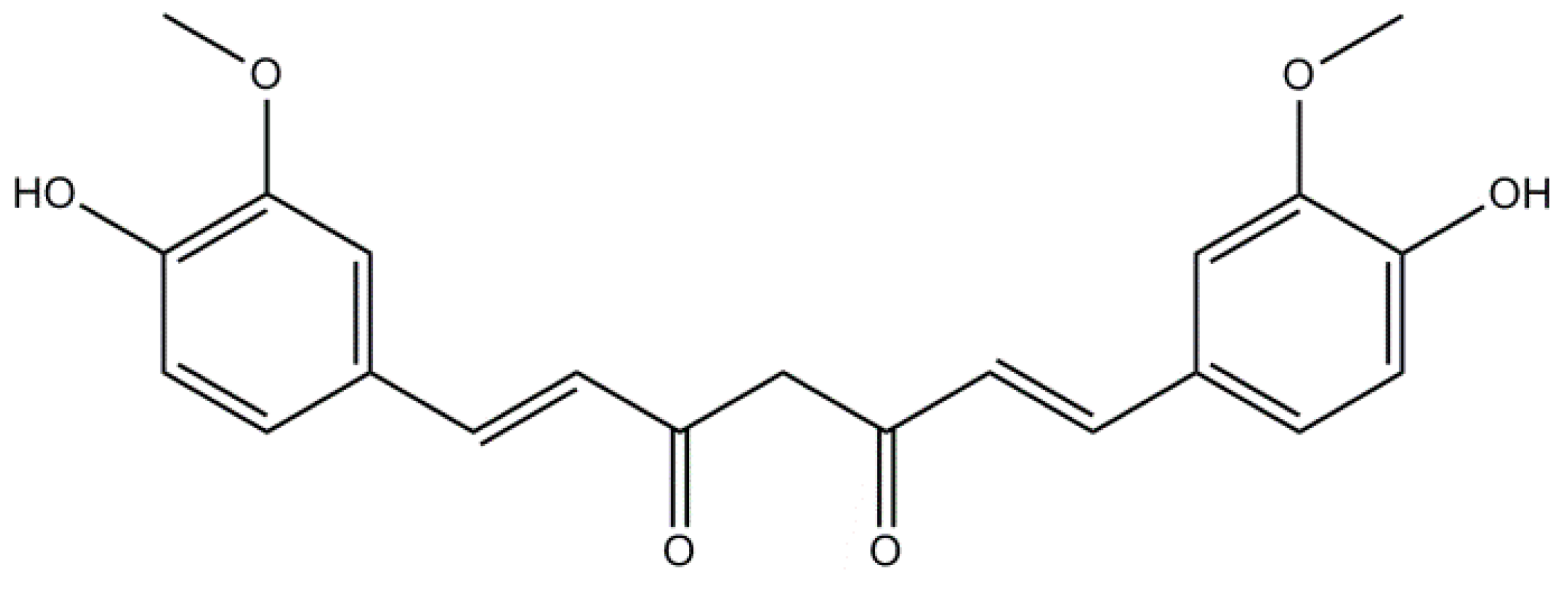
2.1. Curcumin: A Non-Flavonoid
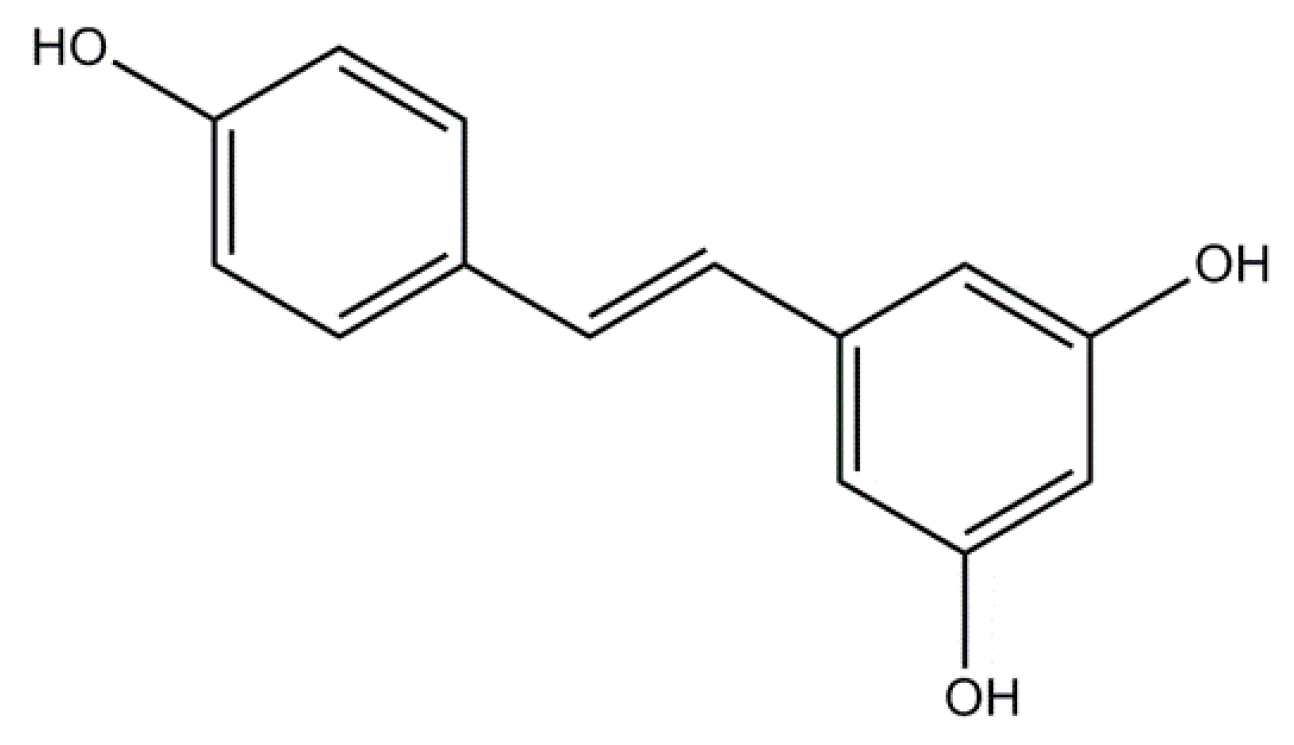
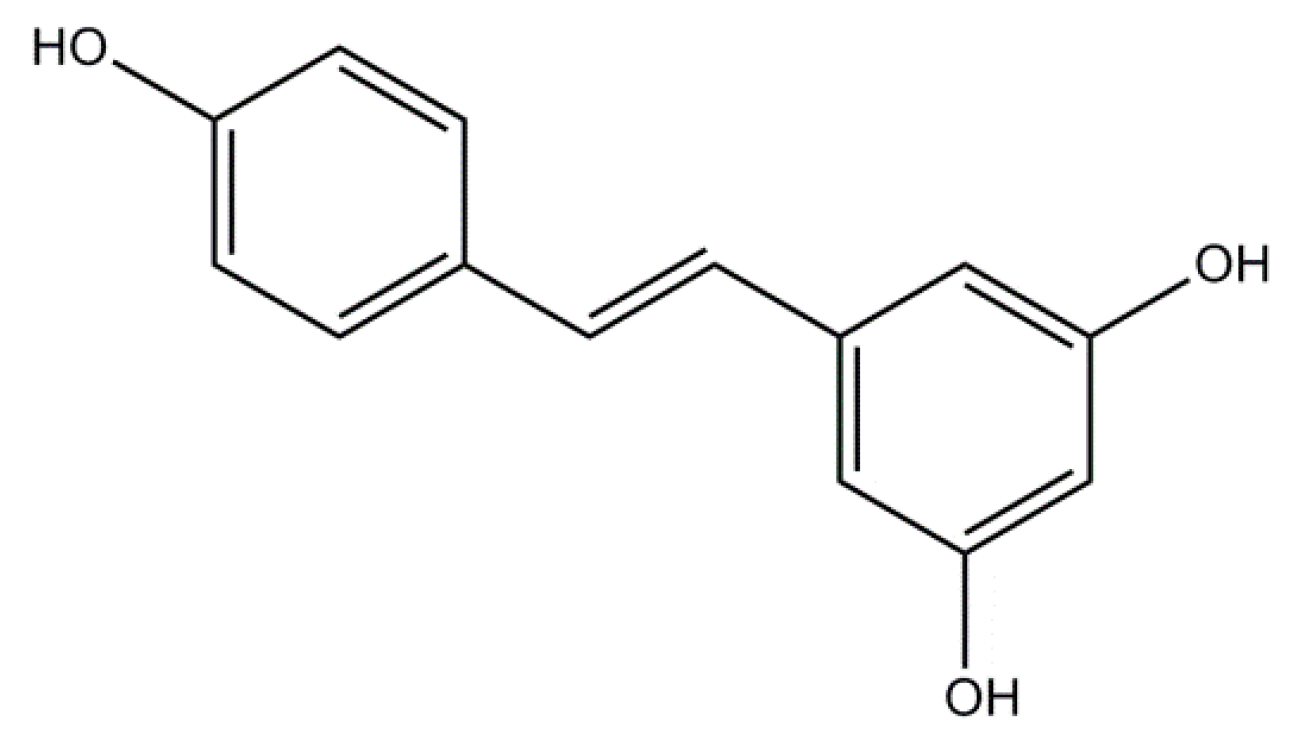
2.2. Resveratrol: A Non-Flavonoid
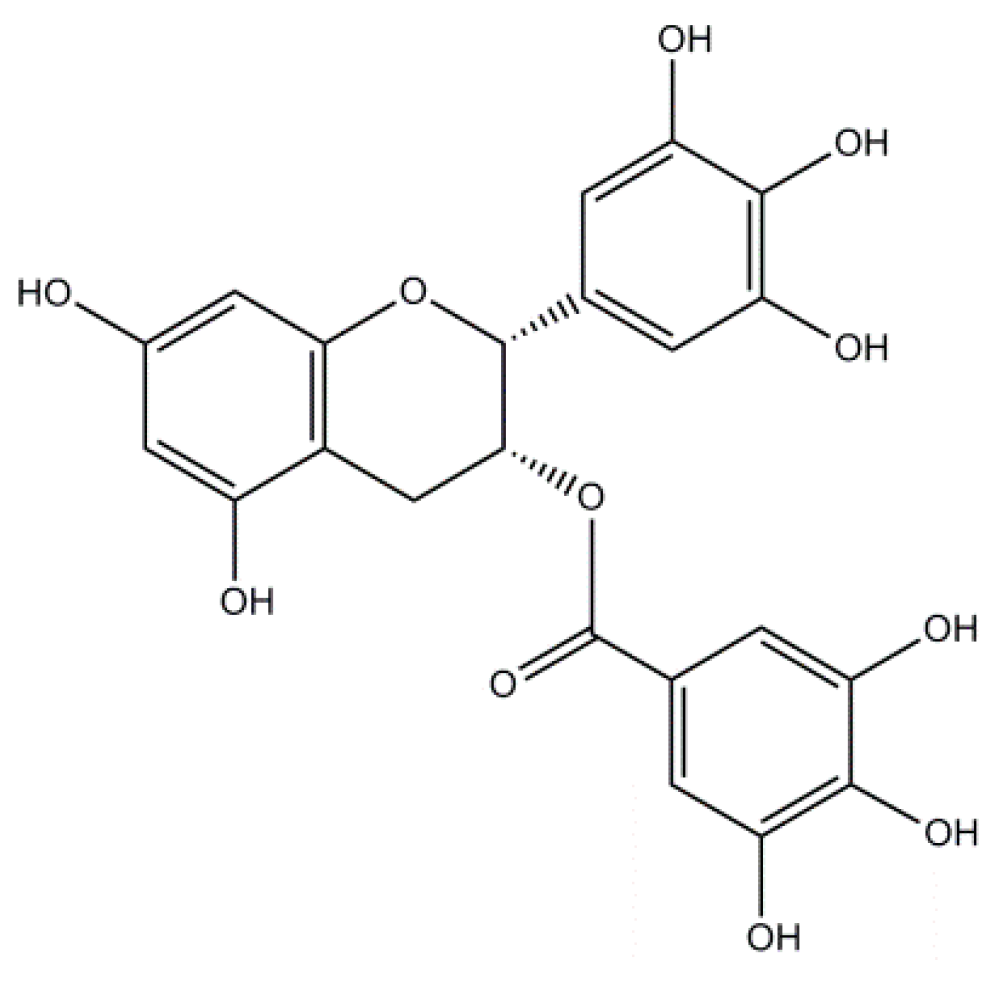
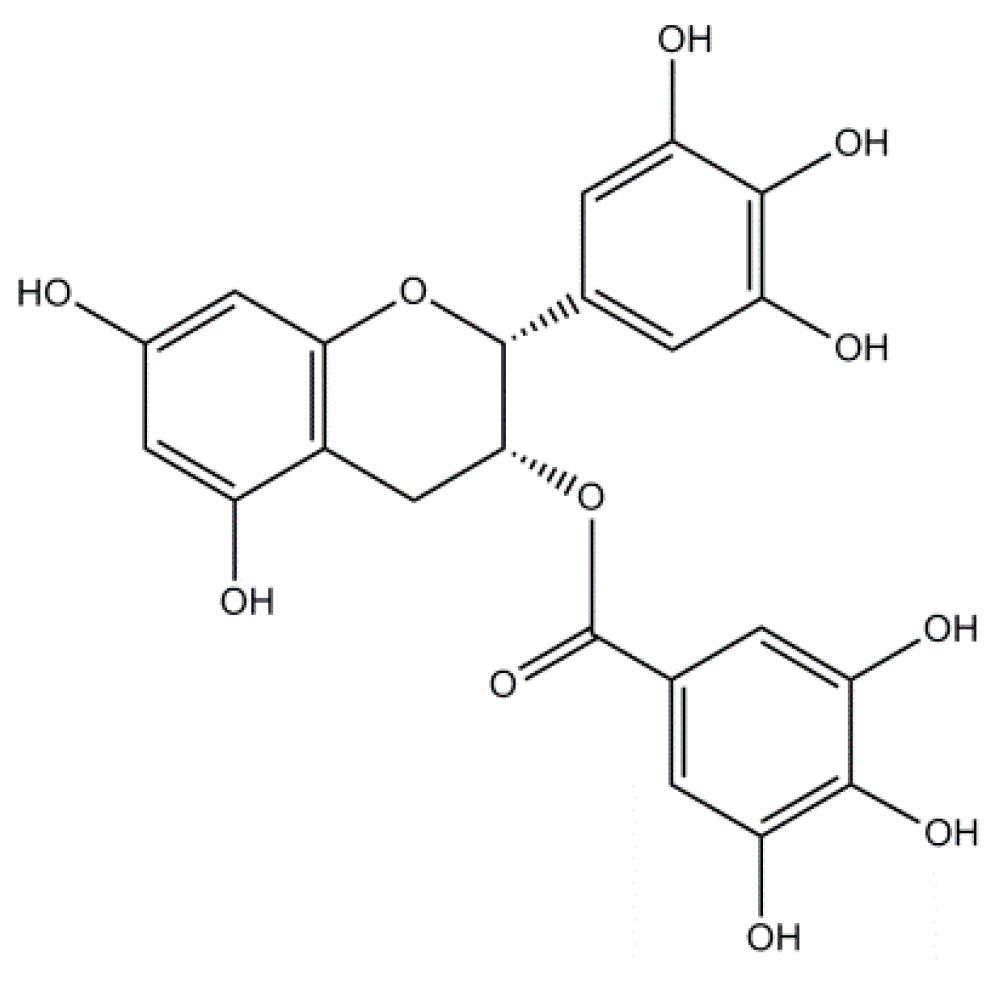
2.3. Epigallocatechin-3-Gallate: A Flavonoid
3. Isothiocyanates
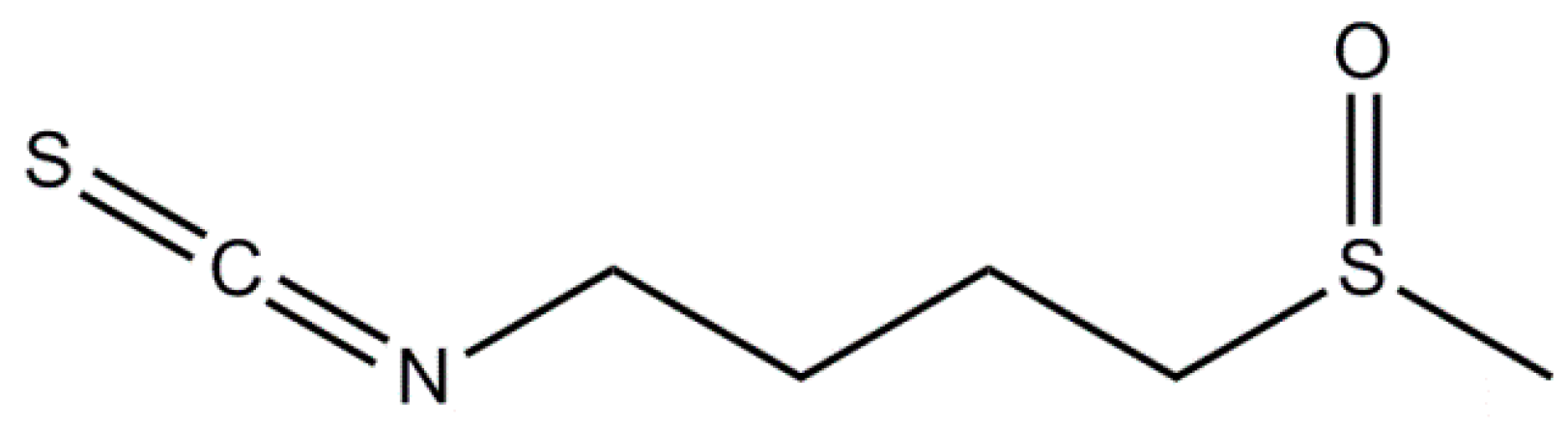
3.1. Sulforaphane
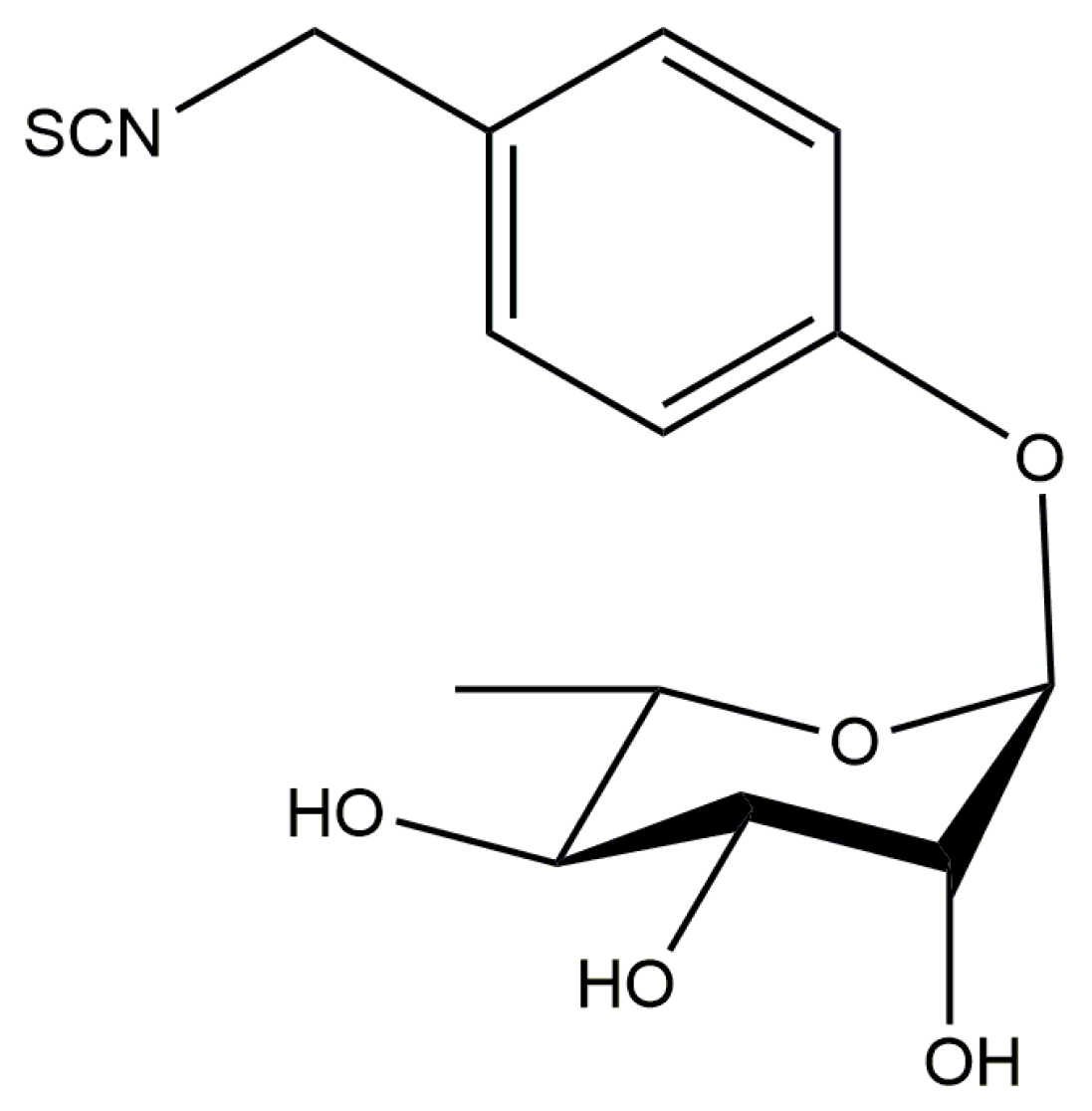
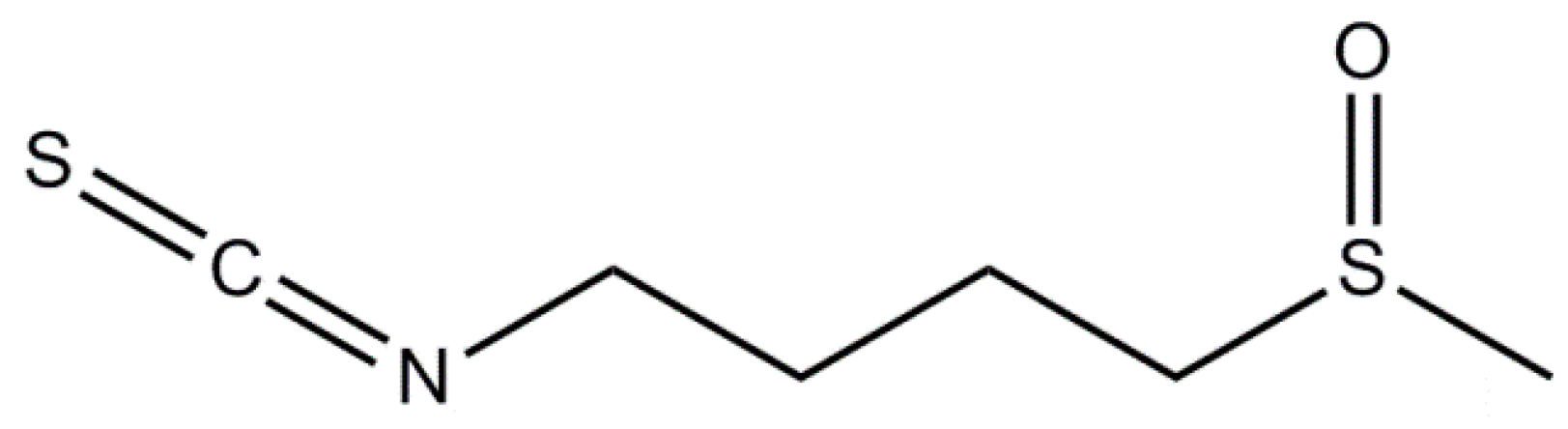
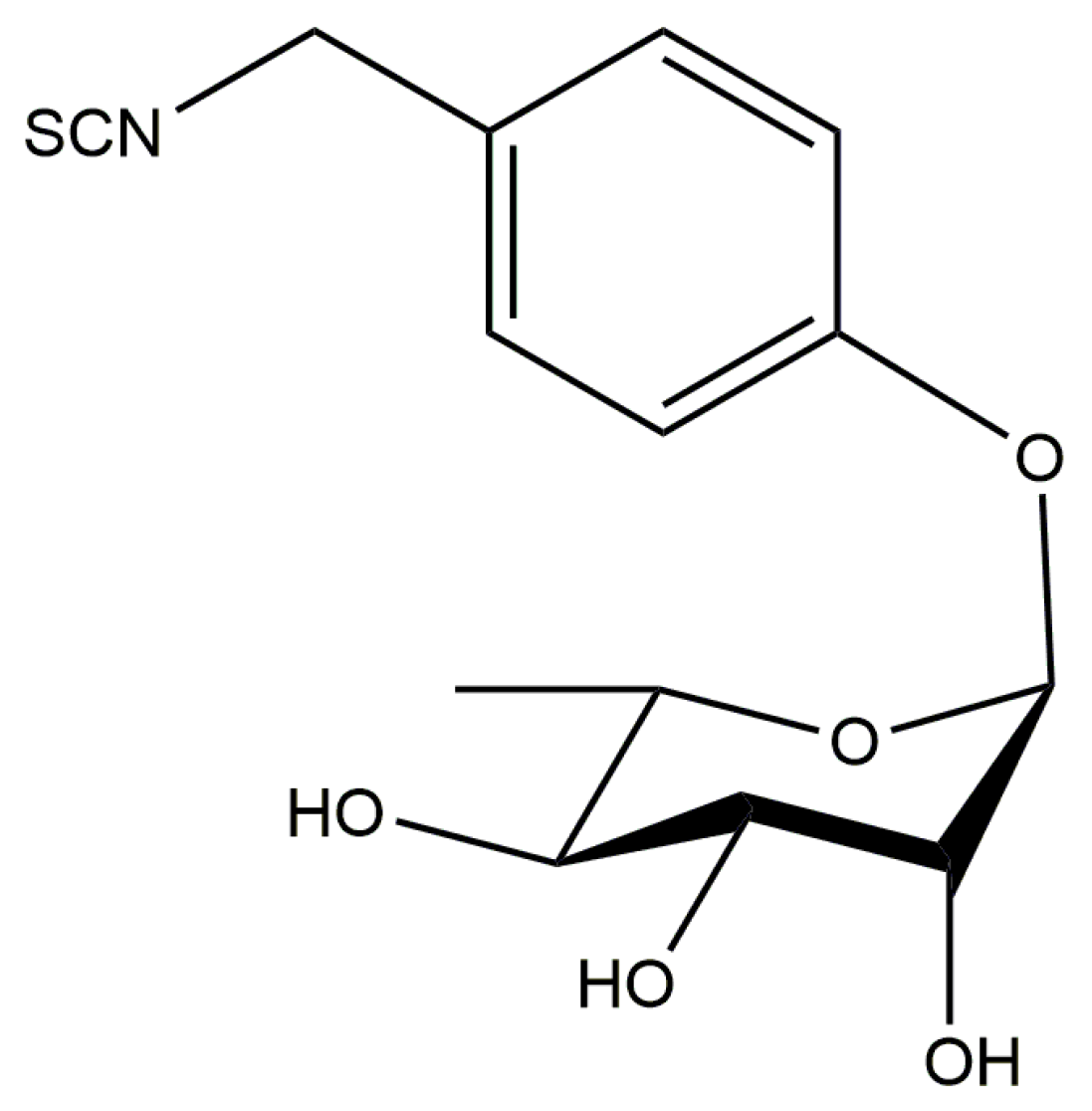
3.2. Moringin
4. Alkaloids
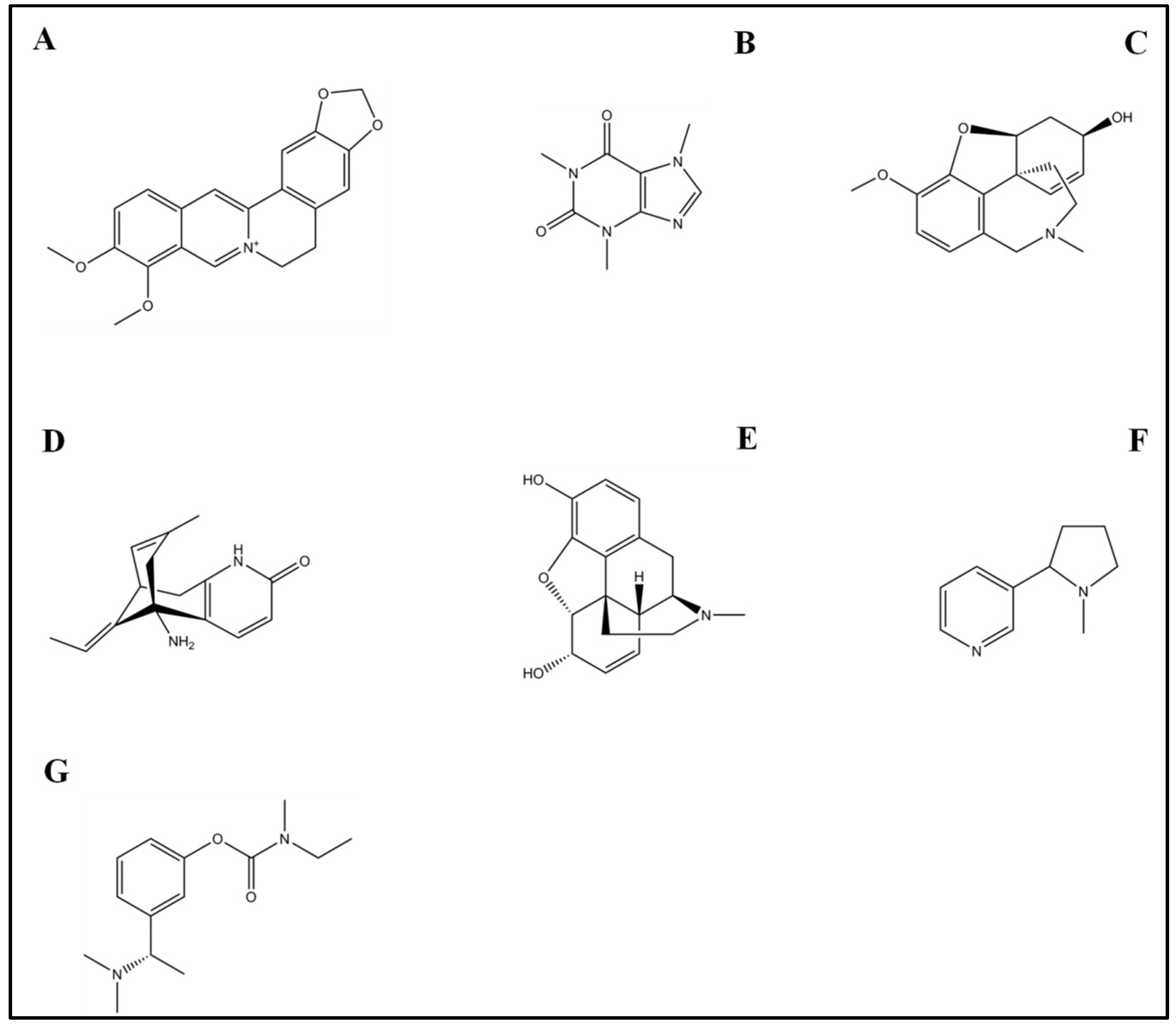
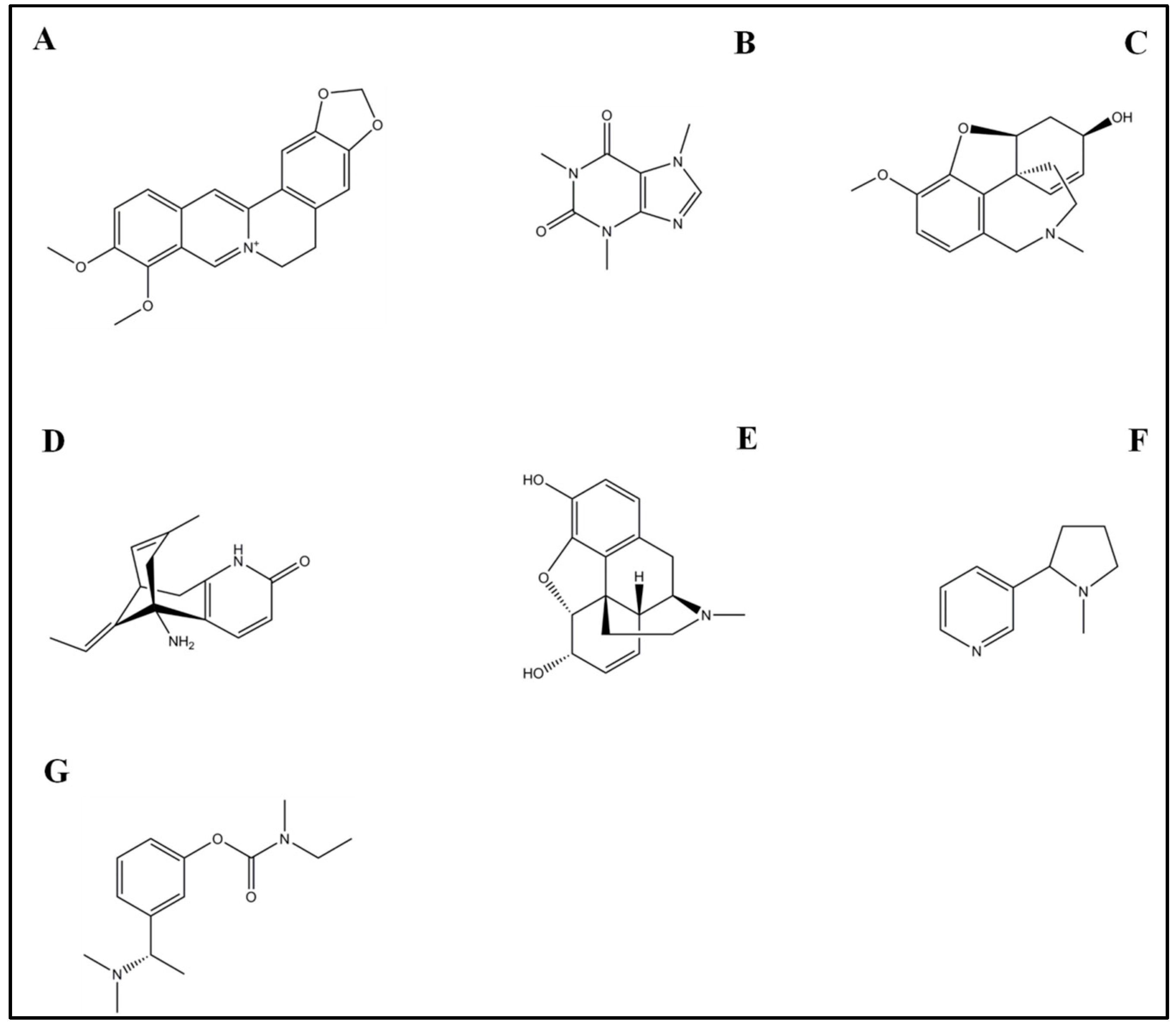
4.1. Rivastigmine
4.2. Galantamine
4.3. Morphine
4.4. Caffeine
4.5. Nicotine
4.6. Huperzine A
4.7. Berberine
5. Phytocannabinoids
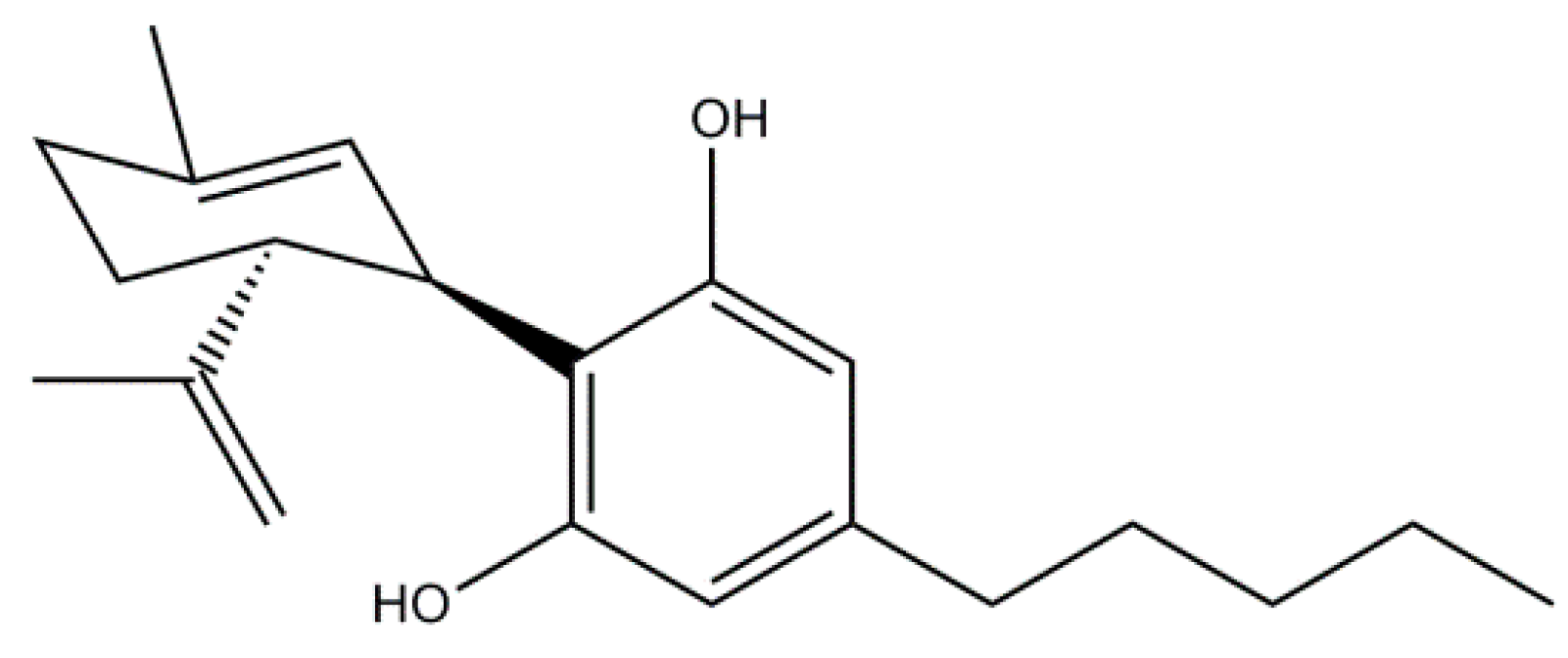
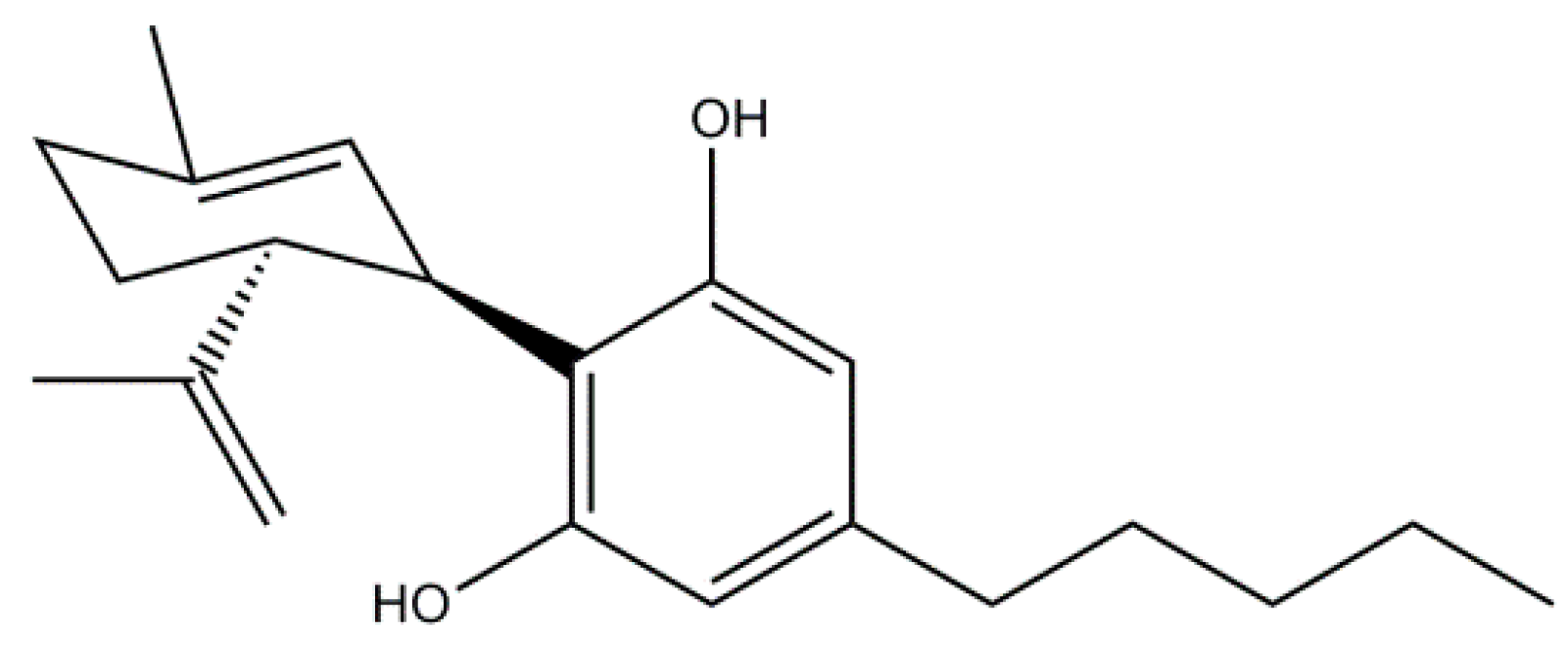
Cannabidiol
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| α-Syn | α-Synuclein |
| ACh | Acetylcholine |
| AChE | Acetylcholinesterase |
| AChEIs | Acetylcholinesterase inhibitors |
| AD | Alzheimer’s disease |
| Akt | protein kinase B |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AMPK | 5′ adenosine monophosphate-activated protein kinase activated protein kinase |
| APP | Amyloid Precursor Protein |
| APPswe | Swedish mutant of the Amyloid Precursor Protein |
| ARE | Antioxidant Responsive Element |
| Aβ | Amyloid beta |
| BACE-1 | Beta-site amyloid precursor protein cleaving enzyme 1 |
| BBB | Blood brain barrier |
| Bax | Bcl-2-associated X protein |
| BChE | Butyryl-cholinesterase |
| BER | Berberine |
| CAF | Caffeine |
| CBD | Cannabidiol |
| CCH | Chronic cerebral hypoperfusion |
| ChAT | Choline acetyltransferase |
| COX-2 | Cyclooxygenase 2 |
| CREB | cAMP-responsive element-binding protein |
| CUR | Curcumin |
| DLB | Dementia with Lewy bodies |
| EGCG | Epigallocatechin 3-Gallate |
| ERK | Extracellular-signal-regulated kinases |
| FDA | Food and Drug Administration |
| FTD | Frontotemporal Dementia |
| GAL | Galantamine |
| GCLC | Glutamate cysteine ligase |
| GFAP | Glial fibrillary acidic protein |
| GPx | Glutathione peroxidase |
| GSH | Glutathione |
| GSK3β | Glycogen synthase kinase 3 β |
| GST | glutathione-S-transferase |
| HEK293swe | HEK293 cells trasnsfected with APPswe variant |
| 5-HT1A | 5-hydroxytriptamine1A |
| HO-1 | Heme oxygenase-1 |
| Hsp | Heat shock protein |
| HupA | Huperzine A |
| icv | intracerebroventricular |
| icv-STZ | Intracerebroventricular administration of streptozocin |
| IL-1β | Interleukin-1beta |
| IL-6 | Interleukin-6 |
| iNOS | Inducible nitric oxide synthase |
| ITCs | Isothiocyanates |
| JNK | c-Jun N-terminal kinases |
| LPS | Lipopolysaccharide |
| MCP-1 | Monocyte Chemoattractant Protein-1 |
| MG | Moringin |
| MnSOD | Manganese-dependent Superoxide Dismutase |
| mTOR | Mammalian target of rapamycin |
| N2a/APPswe | Neuro2a cells overexpressing mutant APPswe gene |
| NFκB | Nuclear Factor Kappa B |
| NFTs | Neurofibrillary tangles |
| NIC | Nicotine |
| NMDA | N-methyl-d-aspartate |
| NO | Nitric oxide |
| NQO1 | NAD(P)H:quinone oxidoreductase 1 |
| Nrf2 | Nuclear erythroid-2 related factor |
| OGD/R | Oxygen-Glucose Deprivation/Reoxygenation |
| OKA | Okadaic acid |
| pCBs | Phytocannabinoids |
| PDD | Parkinson’s disease dementia |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphatidylinositol 3-Kinase |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PPARγ | Proliferator-activated receptor gamma antagonist |
| PS1 | Presenilin-1 |
| PS2 | Presenilin 2 |
| RESV | Resveratrol |
| RIV | Rivastigmine |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| SIRT-1 | Sirtuin-1 |
| SOD | Superoxide dismutase |
| TNF-α | Tumor Necrosis Factor-alpha |
| TrkA | Tropomyosin receptor kinase A |
| TrkB | Tropomyosin receptor kinase B |
| VAChT | Vesicular ACh transporter |
| VaD | Vascular Dementia |
| Δ9-THC | Delta-9 tetrahydrocannabinol |
References
- World Health Organization. “WHO | Dementia”. Available online: http://www.who.int/mediacentre/factsheets/fs362/en (accessed on 14 April 2016).
- Raz, L.; Knoefel, J.; Bhaskar, K. The neuropathology and cerebrovascular mechanisms of dementia. J. Cereb. Blood Flow Metab. 2015. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C. Dementia. Medicine 2012, 40, 628–631. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Barage, S.H.; Sonawane, K.D. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer’s disease. Neuropeptides 2015, 52. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, W.J.; Feen, E.; Grossberg, G.T. The Use of Cholinesterase Inhibitors Across All Stages of Alzheimer’s Disease. Drugs Aging 2015, 32, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.W. Alzheimer’s disease: Early diagnosis and treatment. Hong Kong Med. J. 2012, 18, 228–237. [Google Scholar] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.J.; Kim, M.K.; Lee, E.J.; Kim, J.N.; Choi, B.-R.; Kim, S.Y.; Cho, K.S.; Han, J.-S.; Kim, H.Y.; Shin, C.Y.; et al. Effects of donepezil, an acetylcholinesterase inhibitor, on neurogenesis in a rat model of vascular dementia. J. Neurol. Sci. 2014, 347, 66–77. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar] [PubMed]
- Higuchi, S.; Arai, H.; Matsushita, S.; Matsui, T.; Kimpara, T.; Takeda, A.; Shirakura, K. Mutation in the alpha-synuclein gene and sporadic Parkinson’s disease, Alzheimer’s disease, and dementia with Lewy bodies. Exp. Neurol. 1998, 153, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Mao, P. Oxidative Stress and Its Clinical Applications in Dementia. J. Neurodegener. Dis. 2013. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Masliah, E. Combination therapies: The next logical Step for the treatment of synucleinopathies? Mov. Disord. 2016, 31, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Jicha, G.A. Medical management of frontotemporal dementias: The importance of the caregiver in symptom assessment and guidance of treatment strategies. J. Mol. Neurosci. 2011, 45, 713–723. [Google Scholar] [PubMed]
- Howes, L.G. Cardiovascular effects of drugs used to treat Alzheimer’s disease. Drug Saf. 2014, 37, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Kröger, E.; Mouls, M.; Wilchesky, M.; Berkers, M.; Carmichael, P.-H.; van Marum, R.; Souverein, P.; Egberts, T.; Laroche, M.-L. Adverse Drug Reactions Reported With Cholinesterase Inhibitors: An Analysis of 16 Years of Individual Case Safety Reports from VigiBase. Ann. Pharmacother. 2015, 49, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Scheepens, A. Vascular action of polyphenols. Mol. Nutr. Food Res. 2009, 53, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Tsao, R. Chemistry and biochemistry of dietary polyphenols. Nutrients 2010, 2, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D. Effect of flavonoids on learning, memory and neurocognitive performance: Relevance and potential implications for Alzheimer’s disease pathophysiology. J. Sci. Food Agric. 2014, 94, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Mumper, R.J. Plant phenolics: Extraction, analysis and their antioxidant and anticancer properties. Molecules 2010, 15, 7313–7352. [Google Scholar] [CrossRef] [PubMed]
- Baraboui, V.A.; Medovar, B.I. Anti-radiation and anti-oxidation properties of some polyphenols. Ukr. Biokhim. Zhurnal 1963, 35, 924–930. [Google Scholar]
- Hur, J.-M.; Hyun, M.-S.; Lim, S.-Y.; Lee, W.-Y.; Kim, D. The combination of berberine and irradiation enhances anti-cancer effects via activation of p38 MAPK pathway and ROS generation in human hepatoma cells. J. Cell. Biochem. 2009, 107, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, H.; Ahmad, N. Tea polyphenols: Prevention of cancer and optimizing health. Am. J. Clin. Nutr. 2000, 71, 1698S–1702S. [Google Scholar] [PubMed]
- Vita, J.A. Polyphenols and cardiovascular disease: Effects on endothelial and platelet function. Am. J. Clin. Nutr. 2005, 81, 292S–297S. [Google Scholar] [PubMed]
- Albarracin, S.L.; Stab, B.; Casas, Z.; Sutachan, J.J.; Samudio, I.; Gonzalez, J.; Gonzalo, L.; Capani, F.; Morales, L.; Barreto, G.E. Effects of natural antioxidants in neurodegenerative disease. Nutr. Neurosci. 2012, 15. [Google Scholar] [CrossRef] [PubMed]
- Ngoungoure, V.L.N.; Schluesener, J.; Moundipa, P.F.; Schluesener, H. Natural polyphenols binding to amyloid: A broad class of compounds to treat different human amyloid diseases. Mol. Nutr. Food Res. 2015, 59, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P.E. The neuroprotective potential of flavonoids: A multiplicity of effects. Genes Nutr. 2008, 3, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008, 65, 1631–1652. [Google Scholar] [CrossRef] [PubMed]
- Ganguli, M.; Chandra, V.; Kamboh, M.I.; Johnston, J.M.; Dodge, H.H.; Thelma, B.K.; Juyal, R.C.; Pandav, R.; Belle, S.H.; DeKosky, S.T. Apolipoprotein E polymorphism and Alzheimer disease: The Indo-US Cross-National Dementia Study. Arch. Neurol. 2000, 57, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, H.J.; Lee, K.W. Naturally occurring phytochemicals for the prevention of Alzheimer’s disease. J. Neurochem. 2010, 112, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Cole, G.M.; Teter, B.; Frautschy, S.A. Neuroprotective effects of curcumin. Adv. Exp. Med. Biol. 2007, 595, 197–212. [Google Scholar] [PubMed]
- Waseem, M.; Parvez, S. Neuroprotective activities of curcumin and quercetin with potential relevance to mitochondrial dysfunction induced by oxaliplatin. Protoplasma 2016, 253, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Priyadarsini, K.I. Chemical and structural features influencing the biological activity of curcumin. Curr. Pharm. Des. 2013, 19, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.; Huebbe, P.; Pallauf, K.; Rimbach, G. Neuroprotective properties of curcumin in Alzheimer’s disease—Merits and limitations. Curr. Med. Chem. 2013, 20, 3955–3985. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Ng, A. Curcumin interaction with copper and iron suggests one possible mechanism of action in Alzheimer’s disease animal models. J. Alzheimers Dis. 2004, 6, 367–377; discussion 443–449. [Google Scholar] [PubMed]
- Jin, C.-Y.; Lee, J.-D.; Park, C.; Choi, Y.-H.; Kim, G.-Y. Curcumin attenuates the release of pro-inflammatory cytokines in lipopolysaccharide-stimulated BV2 microglia. Acta Pharmacol. Sin. 2007, 28, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zheng, Z.; Li, J.; Xiao, Z.; Qi, W.; Zhang, A.; Wu, Q.; Fang, Y. Curcumin inhibits Aβ-induced microglial inflammatory responses in vitro: Involvement of ERK1/2 and p38 signaling pathways. Neurosci. Lett. 2015, 594, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The Curry Spice Curcumin Reduces Oxidative Damage and Amyloid Pathology in an Alzheimer Transgenic Mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [PubMed]
- Rinwa, P.; Kaur, B.; Jaggi, A.S.; Singh, N. Involvement of PPAR-gamma in curcumin-mediated beneficial effects in experimental dementia. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 381, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Landreth, G.; Jiang, Q.; Mandrekar, S.; Heneka, M. PPARγ agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics 2008, 5, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Kim, H.-S.; Cho, E.-K.; Kwon, B.-Y.; Phark, S.; Hwang, K.-W.; Sul, D. Curcumin protected PC12 cells against beta-amyloid-induced toxicity through the inhibition of oxidative damage and tau hyperphosphorylation. Food Chem. Toxicol. 2008, 46, 2881–2887. [Google Scholar] [CrossRef] [PubMed]
- Durairajan, S.S.K.; Liu, L.-F.; Lu, J.-H.; Chen, L.-L.; Yuan, Q.; Chung, S.K.; Huang, L.; Li, X.-S.; Huang, J.-D.; Li, M. Berberine ameliorates β-amyloid pathology, gliosis, and cognitive impairment in an Alzheimer’s disease transgenic mouse model. Neurobiol. Aging 2012, 33, 2903–2919. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Deng, Y.; Yu, D.; Cao, H.; Wang, L.; Liu, L.; Yu, C.; Zhang, Y.; Guo, X.; Yu, G. Histone acetyltransferase p300 mediates histone acetylation of PS1 and BACE1 in a cellular model of Alzheimer’s disease. PLoS ONE 2014, 9, e103067. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, D.; Ibrahim, N.F.; Taguchi, H.; Morikawa, S.; Hirao, K.; Shirai, N.; Sogabe, T.; Tooyama, I. Curcumin derivative with the substitution at C-4 position, but not curcumin, is effective against amyloid pathology in APP/PS1 mice. Neurobiol. Aging 2015, 36, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, X.; Teng, Z.; Zhang, T.; Li, Y. Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur. J. Pharmacol. 2014, 740, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Su, C.; Li, R.; Wang, H.; Ren, Y.; Sun, H.; Yang, J.; Sun, J.; Shi, J.; Tian, J.; Jiang, S. Mechanisms and effects of curcumin on spatial learning and memory improvement in APPswe/PS1dE9 mice. J. Neurosci. Res. 2014, 92, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Duron, E.; Hanon, O. Vascular risk factors, cognitive decline, and dementia. Vasc. Health Risk Manag. 2008, 4, 363–381. [Google Scholar] [PubMed]
- Tian, M.; Zhang, X.; Wang, L.; Li, Y. Curcumin induces ABCA1 expression and apolipoprotein A-I-mediated cholesterol transmembrane in the chronic cerebral hypoperfusion aging rats. Am. J. Chin. Med. 2013, 41, 1027–1042. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Z.; Wang, Y.; Zhang, Q. Hippocampal expression of synaptic structural proteins and phosphorylated cAMP response element-binding protein in a rat model of vascular dementia induced by chronic cerebral hypoperfusion. Neural Regen. Res. 2012, 7, 821–826. [Google Scholar] [PubMed]
- Li, H.; Wang, J.; Wang, P.; Rao, Y.; Chen, L. Resveratrol Reverses the Synaptic Plasticity Deficits in a Chronic Cerebral Hypoperfusion Rat Model. J. Stroke Cerebrovasc. Dis. 2015, 25, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Lam, C.W.K.; Cheung, S.K.-K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Frautschy, S.A.; Teng, E.; Begum, A.N.; Bardens, J.; Beigi, M.; Gylys, K.H.; Badmaev, V.; Heath, D.D.; Apostolova, L.G.; et al. Oral curcumin for Alzheimer’s disease: Tolerability and efficacy in a 24-week randomized, double blind, placebo-controlled study. Alzheimers Res. Ther. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, N.; Takahashi, Y.; Amakusa, Y.; Tanno, Y.; Tuji, Y.; Niwa, H.; Murakami, N.; Krishna, U.K. Effects of turmeric on Alzheimer’s disease with behavioral and psychological symptoms of dementia. Ayu 2012, 33, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Brondino, N.; Re, S.; Boldrini, A.; Cuccomarino, A.; Lanati, N.; Barale, F.; Politi, P. Curcumin as a Therapeutic Agent in Dementia: A Mini Systematic Review of Human Studies. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Maiti, P.; Ma, Q.; Zuo, X.; Jones, M.R.; Cole, G.M.; Frautschy, S.A. Clinical development of curcumin in neurodegenerative disease. Expert Rev. Neurother. 2015, 15, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Gong, Q.; Dong, H.; Shi, J. Resveratrol, a neuroprotective supplement for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, J.; Rottinghaus, G.E.; Simonyi, A.; Lubahn, D.; Sun, G.Y.; Sun, A.Y. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002, 958, 439–447. [Google Scholar] [CrossRef]
- Rege, S.D.; Geetha, T.; Griffin, G.D.; Broderick, T.L.; Babu, J.R. Neuroprotective effects of resveratrol in Alzheimer disease pathology. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Mokni, M.; Elkahoui, S.; Limam, F.; Amri, M.; Aouani, E. Effect of resveratrol on antioxidant enzyme activities in the brain of healthy rat. Neurochem. Res. 2007, 32, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.A.; Lim, S.-Y.; Rhee, S.-H.; Park, K.Y.; Kim, C.-H.; Choi, B.T.; Lee, S.J.; Park, Y.-M.; Choi, Y.H. Resveratrol inhibits inducible nitric oxide synthase and cyclooxygenase-2 expression in beta-amyloid-treated C6 glioma cells. Int. J. Mol. Med. 2006, 17, 1069–1075. [Google Scholar] [PubMed]
- Jang, J.-H.; Surh, Y.-J. Protective effect of resveratrol on beta-amyloid-induced oxidative PC12 cell death. Free Radic. Biol. Med. 2003, 34, 1100–1110. [Google Scholar] [CrossRef]
- Han, Y.-S.; Zheng, W.-H.; Bastianetto, S.; Chabot, J.-G.; Quirion, R. Neuroprotective effects of resveratrol against beta-amyloid-induced neurotoxicity in rat hippocampal neurons: Involvement of protein kinase C. Br. J. Pharmacol. 2004, 141, 997–1005. [Google Scholar] [CrossRef] [PubMed]
- Racchi, M.; Mazzucchelli, M.; Pascale, A.; Sironi, M.; Govoni, S. Role of protein kinase Calpha in the regulated secretion of the amyloid precursor protein. Mol. Psychiatry 2003, 8, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yan, L.-J.; Li, K.; Quazi, S.H.; Zhao, B. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromol. Med. 2012, 14. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Jayasena, T.; Poljak, A.; Sachdev, P.S. Sirtuins in cognitive ageing and Alzheimer’s disease. Curr. Opin. Psychiatry 2012, 25, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Porquet, D.; Casadesús, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegrí, C.; Sanfeliu, C.; Camins, A.; Pallàs, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age 2013, 35, 1851–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porquet, D.; Griñán-Ferré, C.; Ferrer, I.; Camins, A.; Sanfeliu, C.; del Valle, J.; Pallàs, M. Neuroprotective role of trans-resveratrol in a murine model of familial Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 1209–1220. [Google Scholar] [PubMed]
- Ma, X.; Sun, Z.; Liu, Y.; Jia, Y.; Zhang, B.; Zhang, J. Resveratrol improves cognition and reduces oxidative stress in rats with vascular dementia. Neural Regen. Res. 2013, 8, 2050–2059. [Google Scholar] [PubMed]
- Ozacmak, V.H.; Sayan-Ozacmak, H.; Barut, F. Chronic treatment with resveratrol, a natural polyphenol found in grapes, alleviates oxidative stress and apoptotic cell death in ovariectomized female rats subjected to chronic cerebral hypoperfusion. Nutr. Neurosci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-K.; Ma, X.-R.; Jia, Y.-J.; Liu, Y.-R.; Zhang, J.-W.; Zhang, B.-A. Effects of resveratrol on apoptosis in a rat model of vascular dementia. Exp. Ther. Med. 2014, 7, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Lonze, B.E.; Ginty, D.D. Function and Regulation of CREB Family Transcription Factors in the Nervous System. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef]
- Davinelli, S.; Sapere, N.; Zella, D.; Bracale, R.; Intrieri, M.; Scapagnini, G. Pleiotropic protective effects of phytochemicals in Alzheimer’s disease. Oxid. Med. Cell. Longev. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Na, H.-K.; Surh, Y.-J. Modulation of Nrf2-mediated antioxidant and detoxifying enzyme induction by the green tea polyphenol EGCG. Food Chem. Toxicol. 2008, 46, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Cheng-Chung Wei, J.; Huang, H.-C.; Chen, W.-J.; Huang, C.-N.; Peng, C.-H.; Lin, C.-L. Epigallocatechin gallate attenuates amyloid β-induced inflammation and neurotoxicity in EOC 13.31 microglia. Eur. J. Pharmacol. 2015, 770, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Jeong, H.-J.; Lee, K.-M.; Myung, N.-Y.; An, N.-H.; Yang, W.M.; Park, S.K.; Lee, H.-J.; Hong, S.-H.; Kim, H.-M.; et al. Epigallocatechin-3-gallate suppresses NF-kappaB activation and phosphorylation of p38 MAPK and JNK in human astrocytoma U373MG cells. J. Nutr. Biochem. 2007, 18, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Dragicevic, N.; Smith, A.; Lin, X.; Yuan, F.; Copes, N.; Delic, V.; Tan, J.; Cao, C.; Shytle, R.D.; Bradshaw, P.C. Green tea epigallocatechin-3-gallate (EGCG) and other flavonoids reduce Alzheimer’s amyloid-induced mitochondrial dysfunction. J. Alzheimers Dis. 2011, 26, 507–521. [Google Scholar] [PubMed]
- Biasibetti, R.; Tramontina, A.C.; Costa, A.P.; Dutra, M.F.; Quincozes-Santos, A.; Nardin, P.; Bernardi, C.L.; Wartchow, K.M.; Lunardi, P.S.; Gonçalves, C.-A. Green tea (−)epigallocatechin-3-gallate reverses oxidative stress and reduces acetylcholinesterase activity in a streptozotocin-induced model of dementia. Behav. Brain Res. 2013, 236, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Lee, Y.K.; Ban, J.O.; Ha, T.Y.; Yun, Y.P.; Han, S.B.; Oh, K.W.; Hong, J.T. Green tea (−)-epigallocatechin-3-gallate inhibits beta-amyloid-induced cognitive dysfunction through modification of secretase activity via inhibition of ERK and NF-kappaB pathways in mice. J. Nutr. 2009, 139, 1987–1993. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, F.; Sha, L.; Wang, S.; Tao, L.; Yao, L.; He, M.; Yao, Z.; Liu, H.; Zhu, Z.; et al. (−)-Epigallocatechin-3-gallate ameliorates learning and memory deficits by adjusting the balance of TrkA/p75NTR signaling in APP/PS1 transgenic mice. Mol. Neurobiol. 2014, 49, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D.; Kennett, M.J.; Sang, S.; Reuhl, K.R.; Ju, J.; Yang, C.S. Hepatotoxicity of high oral dose (−)-epigallocatechin-3-gallate in mice. Food Chem. Toxicol. 2010, 48, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Mazzanti, G.; Menniti-Ippolito, F.; Moro, P.A.; Cassetti, F.; Raschetti, R.; Santuccio, C.; Mastrangelo, S. Hepatotoxicity from green tea: A review of the literature and two unpublished cases. Eur. J. Clin. Pharmacol. 2009, 65, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Conaway, C.C.; Getahun, S.M.; Liebes, L.L.; Pusateri, D.J.; Topham, D.K.; Botero-Omary, M.; Chung, F.L. Disposition of glucosinolates and sulforaphane in humans after ingestion of steamed and fresh broccoli. Nutr. Cancer 2000, 38, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Thornalley, P.J. Effect of storage, processing and cooking on glucosinolate content of Brassica vegetables. Food Chem. Toxicol. 2007, 45, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Abdull Razis, A.F.; Bagatta, M.; de Nicola, G.R.; Iori, R.; Ioannides, C. Up-regulation of cytochrome P450 and phase II enzyme systems in rat precision-cut rat lung slices by the intact glucosinolates, glucoraphanin and glucoerucin. Lung Cancer 2011, 71, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Fimognari, C.; Nüsse, M.; Cesari, R.; Iori, R.; Cantelli-Forti, G.; Hrelia, P. Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis 2002, 23, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Hanschen, F.S.; Lamy, E.; Schreiner, M.; Rohn, S. Reactivity and stability of glucosinolates and their breakdown products in foods. Angew. Chem. Int. Ed. Engl. 2014, 53, 11430–11450. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef] [PubMed]
- De Nicola, G.R.; Rollin, P.; Mazzon, E.; Iori, R. Novel gram-scale production of enantiopure R-sulforaphane from Tuscan black kale seeds. Molecules 2014, 19, 6975–6986. [Google Scholar] [CrossRef] [PubMed]
- Vergara, F.; Wenzler, M.; Hansen, B.G.; Kliebenstein, D.J.; Halkier, B.A.; Gershenzon, J.; Schneider, B. Determination of the absolute configuration of the glucosinolate methyl sulfoxide group reveals a stereospecific biosynthesis of the side chain. Phytochemistry 2008, 69, 2737–2742. [Google Scholar] [CrossRef] [PubMed]
- De Figueiredo, S.M.; Binda, N.S.; Nogueira-Machado, J.A.; Vieira-Filho, S.A.; Caligiorne, R.B. The antioxidant properties of organosulfur compounds (sulforaphane). Recent Pat. Endocr. Metab. Immune Drug Discov. 2015, 9, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Galuppo, M.; Montaut, S.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. An overview on neuroprotective effects of isothiocyanates for the treatment of neurodegenerative diseases. Fitoterapia 2015, 106, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacoppo, S.; Galuppo, M.; Iori, R.; de Nicola, G.R.; Bramanti, P.; Mazzon, E. The protective effects of bioactive (RS)-glucoraphanin on the permeability of the mice blood-brain barrier following experimental autoimmune encephalomyelitis. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 194–204. [Google Scholar] [PubMed]
- Galuppo, M.; Giacoppo, S.; de Nicola, G.R.; Iori, R.; Mazzon, E.; Bramanti, P. RS-Glucoraphanin bioactivated with myrosinase treatment counteracts proinflammatory cascade and apoptosis associated to spinal cord injury in an experimental mouse model. J. Neurol. Sci. 2013, 334, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Galuppo, M.; Iori, R.; de Nicola, G.R.; Cassata, G.; Bramanti, P.; Mazzon, E. Protective role of (RS)-glucoraphanin bioactivated with myrosinase in an experimental model of multiple sclerosis. CNS Neurosci. Ther. 2013, 19, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Iori, R.; de Nicola, G.R.; Bramanti, P.; Mazzon, E. Anti-inflammatory and anti-apoptotic effects of (RS)-glucoraphanin bioactivated with myrosinase in murine sub-acute and acute MPTP-induced Parkinson’s disease. Bioorg. Med. Chem. 2013, 21, 5532–5547. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Park, G.H.; Lee, S.-R.; Jang, J.-H. Attenuation of β-amyloid-induced oxidative cell death by sulforaphane via activation of NF-E2-related factor 2. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-M.; Kim, J.-A.; Kwak, M.-K. Protection against amyloid beta cytotoxicity by sulforaphane: Role of the proteasome. Arch. Pharm. Res. 2009, 32, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Gan, N.; Wu, Y.-C.; Brunet, M.; Garrido, C.; Chung, F.-L.; Dai, C.; Mi, L. Sulforaphane activates heat shock response and enhances proteasome activity through up-regulation of Hsp27. J. Biol. Chem. 2010, 285, 35528–35536. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.Y.; Goldberg, A.L. Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 2001, 29, 15–32. [Google Scholar] [CrossRef]
- Kwak, M.-K.; Cho, J.-M.; Huang, B.; Shin, S.; Kensler, T.W. Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 2007, 43, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.-O.; Kipp, M.; Lucius, R.; Pufe, T.; Wruck, C.J. Sulforaphane suppresses LPS-induced inflammation in primary rat microglia. Inflamm. Res. 2010, 59, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, J.; Fang, L.; Li, X.; Zhao, Y.; Shi, W.; An, L. Neuroprotective effects of sulforaphane on cholinergic neurons in mice with Alzheimer’s disease-like lesions. Int. J. Mol. Sci. 2014, 15, 14396–14410. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Miao, Q.-W.; Zhu, C.-X.; Zhao, Y.; Liu, L.; Yang, J.; An, L. Sulforaphane ameliorates neurobehavioral deficits and protects the brain from amyloid β deposits and peroxidation in mice with Alzheimer-like lesions. Am. J. Alzheimers Dis. Other Demen. 2015, 30, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, J.; Seo, S.G.; Choi, B.-R.; Han, J.-S.; Lee, K.W.; Kim, J. Sulforaphane alleviates scopolamine-induced memory impairment in mice. Pharmacol. Res. 2014, 85, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Molchan, S.E.; Martinez, R.A.; Hill, J.L.; Weingartner, H.J.; Thompson, K.; Vitiello, B.; Sunderland, T. Increased cognitive sensitivity to scopolamine with age and a perspective on the scopolamine model. Brain Res. Brain Res. Rev. 17, 215–226.
- Dwivedi, S.; Rajasekar, N.; Hanif, K.; Nath, C.; Shukla, R. Sulforaphane Ameliorates Okadaic Acid-Induced Memory Impairment in Rats by Activating the Nrf2/HO-1 Antioxidant Pathway. Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Rai, S.; Swarnkar, S.; Shukla, R.; Nath, C. Molecular and cellular mechanism of okadaic acid (OKA)-induced neurotoxicity: A novel tool for Alzheimer’s disease therapeutic application. Mol. Neurobiol. 2014, 50, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Abdull Razis, A.F.; Ibrahim, M.D.; Kntayya, S.B. Health benefits of Moringa oleifera. Asian Pac. J. Cancer Prev. 2014, 15, 8571–8576. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.N.; Mellon, F.A.; Foidl, N.; Pratt, J.H.; Dupont, M.S.; Perkins, L.; Kroon, P.A. Profiling glucosinolates and phenolics in vegetative and reproductive tissues of the multi-purpose trees Moringa oleifera L. (horseradish tree) and Moringa stenopetala L. J. Agric. Food Chem. 2003, 51, 3546–3553. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Giacoppo, S.; de Nicola, G.R.; Iori, R.; Navarra, M.; Lombardo, G.E.; Bramanti, P.; Mazzon, E. Antiinflammatory activity of glucomoringin isothiocyanate in a mouse model of experimental autoimmune encephalomyelitis. Fitoterapia 2014, 95, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Giacoppo, S.; Iori, R.; de Nicola, G.R.; Milardi, D.; Bramanti, P.; Mazzon, E. 4(α-l-rhamnosyloxy)-benzyl isothiocyanate, a bioactive phytochemical that defends cerebral tissue and prevents severe damage induced by focal ischemia/reperfusion. J. Biol. Regul. Homeost. Agents 2015, 29, 343–356. [Google Scholar] [PubMed]
- Giacoppo, S.; Galuppo, M.; de Nicola, G.R.; Iori, R.; Bramanti, P.; Mazzon, E. 4(α-l-rhamnosyloxy)-benzyl isothiocyanate, a bioactive phytochemical that attenuates secondary damage in an experimental model of spinal cord injury. Bioorg. Med. Chem. 2015, 23, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Sutalangka, C.; Wattanathorn, J.; Muchimapura, S.; Thukham-Mee, W. Moringa oleifera mitigates memory impairment and neurodegeneration in animal model of age-related dementia. Oxid. Med. Cell. Longev. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, R.; Guha, D. Alteration of brain monoamines & EEG wave pattern in rat model of Alzheimer’s disease & protection by Moringa oleifera. Indian J. Med. Res. 2008, 128, 744–751. [Google Scholar] [PubMed]
- Ganguly, R.; Hazra, R.; Ray, K.; Guha, D. Effect of Moringa oleifera in Experimental Model of Alzheimer’s Disease: Role of Antioxidants. Ann. Neurosci. 2005, 12, 33–36. [Google Scholar] [CrossRef]
- Preininger, V.; Thakur, R.S.; Santavý, F. Isolation and chemistry of alkaloids from plants of the family Papaveraceae LXVII: Corydalis cava (L.) Sch. et K. (C. tuberosa DC). J. Pharm. Sci. 1976, 65, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Schläger, S.; Dräger, B. Exploiting plant alkaloids. Curr. Opin. Biotechnol. 2015, 37, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Satheeshkumar, N.; Venkatesh, P.; Venkatesh, M. Lead finding for acetyl cholinesterase inhibitors from natural origin: Structure activity relationship and scope. Mini Rev. Med. Chem. 2011, 11, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Konrath, E.L.; Passos, C.; Dos, S.; Klein, L.C.; Henriques, A.T. Alkaloids as a source of potential anticholinesterase inhibitors for the treatment of Alzheimer’s disease. J. Pharm. Pharmacol. 2013, 65, 1701–1725. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Adem, A.; Sabbagh, M. New acetylcholinesterase inhibitors for Alzheimer’s disease. Int. J. Alzheimers. Dis. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.; Farlow, M.; Lefèvre, G. Pharmacokinetics of a novel transdermal rivastigmine patch for the treatment of Alzheimer’s disease: A review. Int. J. Clin. Pract. 2009, 63, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.L. A critical review of cholinesterase inhibitors as a treatment modality in Alzheimer’s disease. Dialogues Clin. Neurosci. 2000, 2, 111–128. [Google Scholar]
- Cummings, J.; Winblad, B. A rivastigmine patch for the treatment of Alzheimer’s disease and Parkinson’s disease dementia. Expert Rev. Neurother. 2007, 7, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Boot, B.P. Comprehensive treatment of dementia with Lewy bodies. Alzheimers. Res. Ther. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.S.; Chong, L.Y.; Grimley Evans, J. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst. Rev. 2015, 9. [Google Scholar] [CrossRef]
- Matsuzono, K.; Sato, K.; Kono, S.; Hishikawa, N.; Ohta, Y.; Yamashita, T.; Deguchi, K.; Nakano, Y.; Abe, K. Clinical Benefits of Rivastigmine in the Real World Dementia Clinics of the Okayama Rivastigmine Study (ORS). J. Alzheimers. Dis. 2015, 48, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Ehret, M.J.; Chamberlin, K.W. Current Practices in the Treatment of Alzheimer Disease: Where is the Evidence After the Phase III Trials? Clin. Ther. 2015, 37, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Spalletta, G.; Gianni, W.; Giubilei, F.; Casini, A.R.; Sancesario, G.; Caltagirone, C.; Cravello, L. Rivastigmine patch ameliorates depression in mild AD: Preliminary evidence from a 6-month open-label observational study. Alzheimer Dis. Assoc. Disord. 2013, 27, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Servello, A.; Andreozzi, P.; Bechini, F.; de Angelis, R.; Pontecorvo, M.L.; Vulcano, A.; Cerra, E.; Vigliotta, M.T.; Artini, M.; Selan, L.; et al. Effect of AChE and BuChE inhibition by rivastigmin in a group of old-old elderly patients with cerebrovascular impairment (SIVD type). Minerva Med. 2014, 105, 167–174. [Google Scholar] [PubMed]
- Birks, J.; McGuinness, B.; Craig, D. Rivastigmine for vascular cognitive impairment. Cochrane Database Syst. Rev. 2013, 5. [Google Scholar] [CrossRef]
- Ringman, J.M.; Cummings, J.L. Current and emerging pharmacological treatment options for dementia. Behav. Neurol. 2006, 17, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Stinton, C.; McKeith, I.; Taylor, J.-P.; Lafortune, L.; Mioshi, E.; Mak, E.; Cambridge, V.; Mason, J.; Thomas, A.; O’Brien, J.T. Pharmacological Management of Lewy Body Dementia: A Systematic Review and Meta-Analysis. Am. J. Psychiatry 2015, 172, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.; Lee Teoh, H. Galanthamine from snowdrop—The development of a modern drug against Alzheimer’s disease from local Caucasian knowledge. J. Ethnopharmacol. 2004, 92, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Koola, M.M.; Buchanan, R.W.; Pillai, A.; Aitchison, K.J.; Weinberger, D.R.; Aaronson, S.T.; Dickerson, F.B. Potential role of the combination of galantamine and memantine to improve cognition in schizophrenia. Schizophr. Res. 2014, 157, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, G.K.; Lilienfeld, S.; Gaens, E. Efficacy and safety of galantamine in patients with mild to moderate Alzheimer’s disease: Multicentre randomised controlled trial. Galantamine International-1 Study Group. BMJ 2000, 321, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Mantua, V.; Mecocci, P.; Pani, L.; Winblad, B.; et al. Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef] [PubMed]
- Miranda, L.F.J.R.; Gomes, K.B.; Silveira, J.N.; Pianetti, G.A.; Byrro, R.M.D.; Peles, P.R.H.; Pereira, F.H.; Santos, T.R.; Assini, A.G.; Ribeiro, V.; et al. Predictive factors of clinical response to cholinesterase inhibitors in mild and moderate Alzheimer’s disease and mixed dementia: A one-year naturalistic study. J. Alzheimers Dis. 2015, 45, 609–620. [Google Scholar] [PubMed]
- Richarz, U.; Gaudig, M.; Rettig, K.; Schauble, B. Galantamine treatment in outpatients with mild Alzheimer’s disease. Acta Neurol. Scand. 2014, 129, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Naharci, M.I.; Ozturk, A.; Yasar, H.; Cintosun, U.; Kocak, N.; Bozoglu, E.; Tasci, I.; Doruk, H. Galantamine improves sleep quality in patients with dementia. Acta Neurol. Belg. 2015, 115, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Matharu, B.; Gibson, G.; Parsons, R.; Huckerby, T.N.; Moore, S.A.; Cooper, L.J.; Millichamp, R.; Allsop, D.; Austen, B. Galantamine inhibits beta-amyloid aggregation and cytotoxicity. J. Neurol. Sci. 2009, 280, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.B.; Sousa, C.; Garção, P.; Oliveira, C.R.; Agostinho, P. Galantamine protects against oxidative stress induced by amyloid-beta peptide in cortical neurons. Eur. J. Neurosci. 2009, 29, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkova, D.; Obreshkova, D.; Zheleva-Dimitrova, D.; Saso, L. Antioxidant activity of galantamine and some of its derivatives. Curr. Med. Chem. 2013, 20, 4595–4608. [Google Scholar] [CrossRef] [PubMed]
- Ezoulin, M.J.M.; Ombetta, J.-E.; Dutertre-Catella, H.; Warnet, J.-M.; Massicot, F. Antioxidative properties of galantamine on neuronal damage induced by hydrogen peroxide in SK-N-SH cells. Neurotoxicology 2008, 29, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.; Craig, D. Galantamine for vascular cognitive impairment. Cochrane Database Syst. Rev. 2013, 4. [Google Scholar] [CrossRef]
- Auchus, A.P.; Brashear, H.R.; Salloway, S.; Korczyn, A.D.; de Deyn, P.P.; Gassmann-Mayer, C. Galantamine treatment of vascular dementia: A randomized trial. Neurology 2007, 69, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.; Royall, D.; Hershey, L.; Lichter, D.; Hake, A.; Farlow, M.; Pasquier, F.; Johnson, S. Efficacy and safety of galantamine in patients with dementia with Lewy bodies: A 24-week open-label study. Dement. Geriatr. Cogn. Disord. 2007, 23, 401–405. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Burns, A. Clinical practice with anti-dementia drugs: A revised (second) consensus statement from the British Association for Psychopharmacology. J. Psychopharmacol. 2011, 25, 997–1019. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, A.; Morlog, D.; Light, M.; Blair, M.; Davidson, W.; Jesso, S.; Brashear, R. Galantamine in frontotemporal dementia and primary progressive aphasia. Dement. Geriatr. Cogn. Disord. 2008, 25, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-D.; Zhang, J.; Wang, Y.; Yuan, J.-L.; Hu, W.-L. Efficacy of Cholinesterase Inhibitors in Vascular Dementia: An Updated Meta-Analysis. Eur. Neurol. 2016, 75, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Schug, S.A.; Zech, D.; Dörr, U. Cancer pain management according to WHO analgesic guidelines. J. Pain Symptom Manag. 1990, 5, 27–32. [Google Scholar] [CrossRef]
- Cui, J.; Wang, Y.; Dong, Q.; Wu, S.; Xiao, X.; Hu, J.; Chai, Z.; Zhang, Y. Morphine protects against intracellular amyloid toxicity by inducing estradiol release and upregulation of Hsp70. J. Neurosci. 2011, 31, 16227–16240. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.-X.; Liu, T.; Law, P.-Y.; Loh, H.H.; Qiu, Y.; Chen, H.-Z. μ-Opioid receptor attenuates Aβ oligomers-induced neurotoxicity through mTOR signaling. CNS Neurosci. Ther. 2015, 21, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Swiech, L.; Perycz, M.; Malik, A.; Jaworski, J. Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta 2008, 1784, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Parsons, R.G.; Gafford, G.M.; Helmstetter, F.J. Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J. Neurosci. 2006, 26, 12977–12983. [Google Scholar] [CrossRef] [PubMed]
- Husebo, B.S.; Ballard, C.; Cohen-Mansfield, J.; Seifert, R.; Aarsland, D. The response of agitated behavior to pain management in persons with dementia. Am. J. Geriatr. Psychiatry 2014, 22, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Husebo, B.S.; Ballard, C.; Sandvik, R.; Nilsen, O.B.; Aarsland, D. Efficacy of treating pain to reduce behavioural disturbances in residents of nursing homes with dementia: Cluster randomised clinical trial. BMJ 2011, 343. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.; Rodriguez, C.; Moser, D.; Toma, S.; Hofmeister, J.; Sinanaj, I.; van de Ville, D.; Giannakopoulos, P.; Lovblad, K.-O. Acute caffeine administration impact on working memory-related brain activation and functional connectivity in the elderly: A BOLD and perfusion MRI study. Neuroscience 2013, 250, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Palacios, N.; Gao, X.; McCullough, M.L.; Schwarzschild, M.A.; Shah, R.; Gapstur, S.; Ascherio, A. Caffeine and risk of Parkinson’s disease in a large cohort of men and women. Mov. Disord. 2012, 27, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Zhang, S.M.; Hernán, M.A.; Kawachi, I.; Colditz, G.A.; Speizer, F.E.; Willett, W.C. Prospective study of caffeine consumption and risk of Parkinson’s disease in men and women. Ann. Neurol. 2001, 50, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.; de Mendonça, A. Does caffeine intake protect from Alzheimer’s disease? Eur. J. Neurol. 2002, 9, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, K.; Carrière, I.; de Mendonca, A.; Portet, F.; Dartigues, J.F.; Rouaud, O.; Barberger-Gateau, P.; Ancelin, M.L. The neuroprotective effects of caffeine: A prospective population study (the Three City Study). Neurology 2007, 69, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Loewenstein, D.A.; Lin, X.; Zhang, C.; Wang, L.; Duara, R.; Wu, Y.; Giannini, A.; Bai, G.; Cai, J.; et al. High Blood caffeine levels in MCI linked to lack of progression to dementia. J. Alzheimers Dis. 2012, 30, 559–572. [Google Scholar] [PubMed]
- Laurent, C.; Eddarkaoui, S.; Derisbourg, M.; Leboucher, A.; Demeyer, D.; Carrier, S.; Schneider, M.; Hamdane, M.; Müller, C.E.; Buée, L.; et al. Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 2014, 35, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Schleif, W.; Rezai-Zadeh, K.; Jackson, E.K.; Zacharia, L.C.; Cracchiolo, J.R.; Shippy, D.; Tan, J. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 2006, 142, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Arendash, G.W.; Mori, T.; Cao, C.; Mamcarz, M.; Runfeldt, M.; Dickson, A.; Rezai-Zadeh, K.; Tane, J.; Citron, B.A.; Lin, X.; et al. Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer’s disease mice. J. Alzheimers Dis. 2009, 17, 661–680. [Google Scholar] [PubMed]
- Han, K.; Jia, N.; Li, J.; Yang, L.; Min, L.-Q. Chronic caffeine treatment reverses memory impairment and the expression of brain BNDF and TrkB in the PS1/APP double transgenic mouse model of Alzheimer’s disease. Mol. Med. Rep. 2013, 8, 737–740. [Google Scholar] [PubMed]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Prasanthi, J.R.P.; Dasari, B.; Marwarha, G.; Larson, T.; Chen, X.; Geiger, J.D.; Ghribi, O. Caffeine protects against oxidative stress and Alzheimer’s disease-like pathology in rabbit hippocampus induced by cholesterol-enriched diet. Free Radic. Biol. Med. 2010, 49, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife coffee and tea drinking and the risk of late-life dementia: A population-based CAIDE study. J. Alzheimers Dis. 2009, 16, 85–91. [Google Scholar] [PubMed]
- Kim, Y.-S.; Kwak, S.M.; Myung, S.-K. Caffeine intake from coffee or tea and cognitive disorders: A meta-analysis of observational studies. Neuroepidemiology 2015, 44, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Gelber, R.P.; Petrovitch, H.; Masaki, K.H.; Ross, G.W.; White, L.R. Coffee intake in midlife and risk of dementia and its neuropathologic correlates. J. Alzheimers Dis. 2011, 23, 607–615. [Google Scholar] [PubMed]
- Picciotto, M.R.; Zoli, M. Nicotinic receptors in aging and dementia. J. Neurobiol. 2002, 53, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, V.; Yarkov, A.; Aliev, G. Positive modulators of the α7 nicotinic receptor against neuroinflammation and cognitive impairment in Alzheimer’s disease. Prog. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kihara, T.; Shimohama, S.; Sawada, H.; Kimura, J.; Kume, T.; Kochiyama, H.; Maeda, T.; Akaike, A. Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann. Neurol. 1997, 42, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hasegawa, K.; Yamada, M.; Naiki, H. Nicotine breaks down preformed Alzheimer’s beta-amyloid fibrils in vitro. Biol. Psychiatry 2002, 52, 880–886. [Google Scholar] [CrossRef]
- Buckingham, S.D.; Jones, A.K.; Brown, L.A.; Sattelle, D.B. Nicotinic acetylcholine receptor signalling: Roles in Alzheimer’s disease and amyloid neuroprotection. Pharmacol. Rev. 2009, 61, 39–61. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.A.; Huckerby, T.N.; Gibson, G.L.; Fullwood, N.J.; Turnbull, S.; Tabner, B.J.; El-Agnaf, O.M.A.; Allsop, D. Both the d-(+) and l-(−) enantiomers of nicotine inhibit Abeta aggregation and cytotoxicity. Biochemistry 2004, 43, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Srivareerat, M.; Tran, T.T.; Salim, S.; Aleisa, A.M.; Alkadhi, K.A. Chronic nicotine restores normal Aβ levels and prevents short-term memory and E-LTP impairment in Aβ rat model of Alzheimer’s disease. Neurobiol. Aging 2011, 32, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.B.; Lee, S.H.; Chae, K.R.; Kim, C.K.; Hwang, D.Y.; Kim, B.G.; Jee, S.W.; Lee, S.H.; Sin, J.S.; Bae, C.J.; et al. Nicotine leads to improvements in behavioral impairment and an increase in the nicotine acetylcholine receptor in transgenic mice. Neurochem. Res. 2008, 33, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.M.; Terry, A. V Repeated nicotine exposure in rats: Effects on memory function, cholinergic markers and nerve growth factor. Neuroscience 2005, 130, 997–1012. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Green, K.N.; Liang, K.; Tran, L.; Chen, Y.; Leslie, F.M.; LaFerla, F.M. Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3046–3051. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Shen, C.; Wang, Y.-J.; Zhang, M.; Li, J.; Xu, Z.-Q.; Gao, C.-Y.; Fang, C.-Q.; Zhou, H.-D. Nicotine exacerbates tau phosphorylation and cognitive impairment induced by amyloid-beta 25-35 in rats. Eur. J. Pharmacol. 2010, 637, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hirohata, M.; Yamada, M. Anti-fibrillogenic and fibril-destabilizing activity of nicotine in vitro: Implications for the prevention and therapeutics of Lewy body diseases. Exp. Neurol. 2007, 205, 414–424. [Google Scholar] [CrossRef] [PubMed]
- White, H.K.; Levin, E.D. Chronic transdermal nicotine patch treatment effects on cognitive performance in age-associated memory impairment. Psychopharmacology 2004, 171, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, P.; Kellar, K.; Aisen, P.; White, H.; Wesnes, K.; Coderre, E.; Pfaff, A.; Wilkins, H.; Howard, D.; Levin, E.D. Nicotine treatment of mild cognitive impairment: A 6-month double-blind pilot clinical trial. Neurology 2012, 78, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Tan, C.; Zhu, D.; Gang, D.R.; Xiao, P. Huperzine A from Huperzia species—An ethnopharmacolgical review. J. Ethnopharmacol. 2007, 113, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.-H.; Zhu, C.-X.; Zhang, R.; An, L. Huperzine a in the treatment of Alzheimer’s disease and vascular dementia: A meta-analysis. Evid. Based Complement. Altern. Med. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.-L.; Wang, R.; Tang, X.-C. Huperzine A protects SHSY5Y neuroblastoma cells against oxidative stress damage via nerve growth factor production. Eur. J. Pharmacol. 2005, 519, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.Q.; Wang, R.; Han, Y.F.; Tang, X.C. Protective effects of huperzine A on beta-amyloid(25-35) induced oxidative injury in rat pheochromocytoma cells. Neurosci. Lett. 2000, 286, 155–158. [Google Scholar] [CrossRef]
- Gao, X.; Tang, X.C. Huperzine A attenuates mitochondrial dysfunction in beta-amyloid-treated PC12 cells by reducing oxygen free radicals accumulation and improving mitochondrial energy metabolism. J. Neurosci. Res. 2006, 83, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.Q.; Zhang, H.Y.; Tang, X.C. Huperzine A attenuates amyloid beta-peptide fragment 25-35-induced apoptosis in rat cortical neurons via inhibiting reactive oxygen species formation and caspase-3 activation. J. Neurosci. Res. 2002, 67, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yan, H.; Tang, X. Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine. Acta Pharmacol. Sin. 2006, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tang, X.C. Anticholinesterase effects of huperzine A, E2020, and tacrine in rats. Zhongguo Yao Li Xue Bao 1998, 19, 27–30. [Google Scholar] [PubMed]
- Ma, T.; Gong, K.; Yan, Y.; Zhang, L.; Tang, P.; Zhang, X.; Gong, Y. Huperzine A promotes hippocampal neurogenesis in vitro and in vivo. Brain Res. 2013, 1506, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, H.Y.; Tang, X.C. Huperzine A attenuates cognitive dysfunction and neuronal degeneration caused by beta-amyloid protein-(1-40) in rat. Eur. J. Pharmacol. 2001, 421, 149–156. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Yan, H.; Tang, X.C. Huperzine A enhances the level of secretory amyloid precursor protein and protein kinase C-alpha in intracerebroventricular beta-amyloid-(1-40) infused rats and human embryonic kidney 293 Swedish mutant cells. Neurosci. Lett. 2004, 360, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wang, Y.; Tian, J.; Liu, J.-P. Huperzine A for Alzheimer’s disease: A systematic review and meta-analysis of randomized clinical trials. PLoS ONE 2013, 8, e74916. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Q.; Liang, X.-M.; Wu, J.; Zhang, Y.-F.; Zhu, C.-X.; Jiang, X.-J. Treatment with Huperzine A improves cognition in vascular dementia patients. Cell Biochem. Biophys. 2012, 62, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Liu, M.; Liu, Z.; Lv, D. Huperzine A for vascular dementia. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Racková, L.; Májeková, M.; Kost’álová, D.; Stefek, M. Antiradical and antioxidant activities of alkaloids isolated from Mahonia aquifolium. Structural aspects. Bioorg. Med. Chem. 2004, 12, 4709–4715. [Google Scholar] [CrossRef] [PubMed]
- Küpeli, E.; Koşar, M.; Yeşilada, E.; Hüsnü, K.; Başer, C. A comparative study on the anti-inflammatory, antinociceptive and antipyretic effects of isoquinoline alkaloids from the roots of Turkish Berberis species. Life Sci. 2002, 72, 645–657. [Google Scholar] [CrossRef]
- Kettmann, V.; Kosfálová, D.; Jantová, S.; Cernáková, M.; Drímal, J. In vitro cytotoxicity of berberine against HeLa and L1210 cancer cell lines. Pharmazie 2004, 59, 548–551. [Google Scholar] [PubMed]
- Tran, Q.L.; Tezuka, Y.; Ueda, J.; Nguyen, N.T.; Maruyama, Y.; Begum, K.; Kim, H.-S.; Wataya, Y.; Tran, Q.K.; Kadota, S. In vitro antiplasmodial activity of antimalarial medicinal plants used in Vietnamese traditional medicine. J. Ethnopharmacol. 2003, 86, 249–252. [Google Scholar] [CrossRef]
- Han, J.; Lin, H.; Huang, W. Modulating gut microbiota as an anti-diabetic mechanism of berberine. Med. Sci. Monit. 2011, 17, RA164–RA167. [Google Scholar] [CrossRef] [PubMed]
- Shvarev, I.F.; Tsetlin, A.L. Anti-blastic properties of berberine and its derivatives. Farmakol. Toksikol. 1972, 35, 73–75. [Google Scholar] [PubMed]
- Wang, X.; Wang, R.; Xing, D.; Su, H.; Ma, C.; Ding, Y.; Du, L. Kinetic difference of berberine between hippocampus and plasma in rat after intravenous administration of Coptidis rhizoma extract. Life Sci. 2005, 77, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.K.; Dhir, A. Berberine: A plant alkaloid with therapeutic potential for central nervous system disorders. Phytother. Res. 2010, 24, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Xie, S.; Wei, H.; Yan, J.; Huang, L.; Li, X. Synthesis and biological evaluation of berberine-thiophenyl hybrids as multi-functional agents: Inhibition of acetylcholinesterase, butyrylcholinesterase, and Aβ aggregation and antioxidant activity. Bioorg. Med. Chem. 2013, 21, 5830–5840. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Chen, S.; Liang, Y.; Guo, Y. The Role of Berberine in the Multi-Target Treatment of Senile Dementia. Curr. Top. Med. Chem. 2016, 16, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Kim, S.-H.; Yang, W.M. Mechanisms of action of phytochemicals from medicinal herbs in the treatment of Alzheimer’s disease. Planta Med. 2014, 80, 1249–1258. [Google Scholar] [PubMed]
- Asai, M.; Iwata, N.; Yoshikawa, A.; Aizaki, Y.; Ishiura, S.; Saido, T.C.; Maruyama, K. Berberine alters the processing of Alzheimer’s amyloid precursor protein to decrease Abeta secretion. Biochem. Biophys. Res. Commun. 2007, 352, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wu, F.; Ma, Y.; Liu, G.; Li, Z.; Sun, Y.; Pei, Z. Decrease in the production of β-amyloid by berberine inhibition of the expression of β-secretase in HEK293 cells. BMC Neurosci. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-H.; Huang, S.-M.; Tan, T.-W.; Lin, H.-Y.; Chen, P.-Y.; Yeh, W.-L.; Chou, S.-C.; Tsai, C.-F.; Wei, I.-H.; Lu, D.-Y. Berberine induces heme oxygenase-1 up-regulation through phosphatidylinositol 3-kinase/AKT and NF-E2-related factor-2 signaling pathway in astrocytes. Int. Immunopharmacol. 2012, 12, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Liu, J.; Song, Z.; Pan, X.; Chen, L.; Cui, X.; Wang, M. Berberine suppresses amyloid-beta-induced inflammatory response in microglia by inhibiting nuclear factor-kappaB and mitogen-activated protein kinase signalling pathways. J. Pharm. Pharmacol. 2012, 64, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.H.; Choi, H.S.; Shin, K.S.; Lee, B.K.; Lee, C.K.; Hwang, B.Y.; Lim, S.C.; Lee, M.K. Effects of berberine on 6-hydroxydopamine-induced neurotoxicity in PC12 cells and a rat model of Parkinson’s disease. Neurosci. Lett. 2010, 486, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xing, H.; Ye, J. Efficacy of berberine in patients with type 2 diabetes mellitus. Metabolism 2008, 57, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.-M.; Xia, M.-F.; Wang, Y.; Chang, X.-X.; Yao, X.-Z.; Rao, S.-X.; Zeng, M.-S.; Tu, Y.-F.; Feng, R.; Jia, W.-P.; et al. Efficacy of Berberine in Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0134172. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Tao, C.; Liu, Z.; Lu, M.; Pan, Q.; Zheng, L.; Li, Q.; Song, Z.; Fichna, J. A Randomized Clinical Trial of Berberine Hydrochloride in Patients with Diarrhea-Predominant Irritable Bowel Syndrome. Phytother. Res. 2015, 29, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Guy, G.W. A tale of two cannabinoids: The therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med. Hypotheses 2006, 66, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Mandolino, G.; Galuppo, M.; Bramanti, P.; Mazzon, E. Cannabinoids: New promising agents in the treatment of neurological diseases. Molecules 2014, 19, 18781–18816. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; de Filippis, D.; Carnuccio, R.; Izzo, A.A.; Iuvone, T. The marijuana component cannabidiol inhibits beta-amyloid-induced tau protein hyperphosphorylation through Wnt/beta-catenin pathway rescue in PC12 cells. J. Mol. Med. 2006, 84, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; de Filippis, D.; Maiuri, M.C.; de Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Iuvone, T.; Esposito, G.; Esposito, R.; Santamaria, R.; di Rosa, M.; Izzo, A.A. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem. 2004, 89, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; de Filippis, D.; Steardo, L.; Scuderi, C.; Savani, C.; Cuomo, V.; Iuvone, T. CB1 receptor selective activation inhibits beta-amyloid-induced iNOS protein expression in C6 cells and subsequently blunts tau protein hyperphosphorylation in co-cultured neurons. Neurosci. Lett. 2006, 404, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Hampson, A.J.; Grimaldi, M.; Lolic, M.; Wink, D.; Rosenthal, R.; Axelrod, J. Neuroprotective antioxidants from marijuana. Ann. N. Y. Acad. Sci. 2000, 899, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5YAPP+ cells through PPARγ involvement. Phytother. Res. 2014, 28, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; de Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L.; de Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Martín-Moreno, A.M.; Reigada, D.; Ramírez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Vaney, C.; Heinzel-Gutenbrunner, M.; Jobin, P.; Tschopp, F.; Gattlen, B.; Hagen, U.; Schnelle, M.; Reif, M. Efficacy, safety and tolerability of an orally administered cannabis extract in the treatment of spasticity in patients with multiple sclerosis: A randomized, double-blind, placebo-controlled, crossover study. Mult. Scler. 2004, 10, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Sánchez-Pla, A.; Vegas-Lozano, E.; Maldonado, R.; Ferrer, I. Cannabis-based medicine reduces multiple pathological processes in AβPP/PS1 mice. J. Alzheimers Dis. 2015, 43, 977–991. [Google Scholar] [PubMed]
- Iring, A.; Ruisanchez, É.; Leszl-Ishiguro, M.; Horváth, B.; Benkő, R.; Lacza, Z.; Járai, Z.; Sándor, P.; di Marzo, V.; Pacher, P.; et al. Role of endocannabinoids and cannabinoid-1 receptors in cerebrocortical blood flow regulation. PLoS ONE 2013, 8, e53390. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Járai, Z.; Bátkai, S.; Kunos, G. Hemodynamic effects of cannabinoids: Coronary and cerebral vasodilation mediated by cannabinoid CB(1) receptors. Eur. J. Pharmacol. 2001, 423, 203–210. [Google Scholar] [CrossRef]
- Walther, S.; Halpern, M. Cannabinoids and Dementia: A Review of Clinical and Preclinical Data. Pharmaceuticals 2010, 3, 2689–2708. [Google Scholar] [CrossRef]
- Mishima, K.; Hayakawa, K.; Abe, K.; Ikeda, T.; Egashira, N.; Iwasaki, K.; Fujiwara, M. Cannabidiol prevents cerebral infarction via a serotonergic 5-hydroxytryptamine1A receptor-dependent mechanism. Stroke 2005, 36, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Walther, S.; Mahlberg, R.; Eichmann, U.; Kunz, D. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology 2006, 185, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Volicer, L.; Stelly, M.; Morris, J.; McLaughlin, J.; Volicer, B.J. Effects of dronabinol on anorexia and disturbed behavior in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 1997, 12, 913–919. [Google Scholar] [CrossRef]
- Krishnan, S.; Cairns, R.; Howard, R. Cannabinoids for the treatment of dementia. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subclass | Polyphenols | Source |
|---|---|---|
| Non-Flavonoids | ||
| Phytochemical | ||
| Stilbenes | resveratrol | grapeskin, red wine, blueberries and blackberries |
| Lignans | secoisolariciresinol | linseed, cereals and grain |
| Flavonoids | ||
| Flavones | apigenin, luteolin | parsley and celery |
| Flavonols | kaempferol, quercetin | onions, leeks and broccoli |
| Flavanols | catechin, epicatechin, epigallocatechin and epigallocatechin gallate | green tea, red wine and chocolate |
| Flavanones | hesperetin, naringenin | citrus fruits and tomatoes |
| Isoflavones | daidzein, genistein, glycetin | soy and soy products |
| Anthocyanins | pelargonidin, cyanidin, malvidin | red wine and berry fruits |
| Model | CUR-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vitro | ||||
| LPS-stimulated rat BV2 microglia | antioxidative, anti-inflammatory | iNOS, NO, COX-2, PGE2, IL-1β, IL-6, TNF-α | ↓ | [37] |
| Aβ-induced murine primary microglia | anti-inflammatory, anti-amyloidogenic | IL-1β, IL-6, TNF-α, MAPK, ERK1/2 | ↓ | [38] |
| Aβ-induced rat PC12 cells | anti-amyloidogenic | intracellular calcium, Tau hyperphosphorylation | ↓ | [42] |
| Mutant APPswe over expression in SH-SY5Y | anti-amyloidogenic | GSK3β activity, APP and Tau hyperphosphorylation | ↓ | [43] |
| Mutant APPswe over expression in Neuro2A | anti-amyloidogenic | PS1, BACE-1, Aβ plaques | ↓ | [37] |
| In vivo | ||||
| Tg2576 mice expressing mutant APP | anti-inflammatory, anti-amyloidogenic | IL-1β, GFAP, amyloid plaques | ↓ | [39] |
| Icv-STZ mice model for AD | anti-inflammatory, antioxidative | AChE, oxidative stress, memory deficits PPARγ receptor activation | ↓ ↑ | [40] |
| APP/PS1 double transgenic AD mice | anti-amyloidogenic | Aβ deposits, cognitive deficit | ↓ | [38] |
| APP/PS1 double transgenic AD mice | anti-amyloidogenic | PI3K/Akt/mTOR pathway | ↓ | [46] |
| APP/PS1 double transgenic AD mice | anti-amyloidogenic | insulin-degrading enzymes and neprilysin | ↑ | [47] |
| CCH rats | anti-cholesterol | ATP-binding cassette transporter and Apolipoprotein A1 | ↑ | [50] |
| Model | RESV-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vitro | ||||
| Aβ-induced rat C6 glioma cells | anti-inflammatory | iNOS, NO, COX-2, PGE2 | ↓ | [62] |
| Aβ-induced rat PC12 cells | anti-apoptotic anti-inflammatory | ROS, Bax, JNK, NFκB | ↓ | [63] |
| Aβ-induced rat hippocampal cells | anti-apoptotic | PKCphosphorylation | ↑ | [64] |
| Mutant APPswe over expression in Neuro 2A and in HEK293 cells | anti-amyloidogenic | AMPK | ↑ | [66] |
| In vivo | ||||
| Healthy rats | antioxidative | SOD, CAT MDA | ↑ ↓ | [61] |
| SAMP8 mice | anti-amyloidogenic antioxidative | AMPK, SIRT-1 | ↑ | [68] |
| APP/PS1 double transgenic AD mice | anti-amyloidogenic antioxidative | AMPK, SIRT-1 | ↑ | [69] |
| CCH rats | antioxidative | MDA GSH, SOD, GST | ↓ ↑ | [71] [72] |
| CCH rats | anti-apoptotic | Bax, PARP | ↓ | [73] |
| CCH rats | spatial learning and memory improvement | PKA, CREB phosphorylation | ↑ | [74] |
| Model | EGCG-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vitro | ||||
| EOC 13.31 | anti-inflammatory | TNF-α, IL-1β, IL-6, iNOS. | ↓ | [77] |
| Neuro2a | antioxidative | Nrf2, HO-1 | ↑ | [77] |
| IL-1β/Aβ exposed U373MG cells | anti-inflammatory | IL-6, IL-8, VEGF, PGE, COX2. NFκB, MAPK, JNK | ↓ | [78] |
| In vivo | ||||
| APP/PS1 double transgenic AD mice | antioxidative anti-amyloidogenic | ROS ATP | ↓ ↑ | [79] |
| icv-STZ rats | anti-amyloidogenic anti-oxidative | ROS, AChE | ↓ | [80] |
| AD (PS2-mutant) transgenic mice; Aβ-treated mice | anti-amyloidogenic | ERK/NFκB, γ-secretases, β-secretases | ↓ | [81] |
| APP/PS1 double transgenic AD mice | neurogenesis anti-amyloidogenic anti-apoptotic | NGF, TrKa p75NTR, JNK/cleaved-caspase 3 | ↑ ↓ | [82] |
| Model | SFN-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vitro | ||||
| Aβ-exposed SHSY5Y cells | anti-apoptotic antioxidative | JNK Nrf2 | ↓ ↑ | [102] |
| Neuro 2A cells N1E115 cells | anti-amyloidogenic antioxidative | Nrf2 | ↑ | [103] |
| Hela and COS-1 cells | antioxidative anti-amyloidogenic | Hsp27 | ↑ | [104] |
| BV2 microglia cells | anti-inflammatory anti-apoptotic | NFκB, ERK1/2, JNK | ↓ | [107] |
| In vivo | ||||
| Scopolamine-infused mice | improve scopolamine-induced memory impairment | ACh | ↑ | [110] |
| Rats treated with OKA | antioxidative anti-inflammatory | Nrf2 | ↑ | [112] |
| Model | MG-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vivo | ||||
| AF64A rats | antioxidative | SOD, CAT MDA, AChE | ↑ ↓ | [119] |
| Rats infused with colchicine | ameliorating cognitive functions | SOD, CAT | ↑ | [120] |
| Model | MOR-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vitro | ||||
| Aβ-exposed rat primary neurons | anti-amyloidogenic | Hsp70 | ↑ | [157] |
| Aβ-primary cortical neurons | anti-amyloidogenic | mTOR | ↓ | [158] |
| In vivo | ||||
| APP/PS1 double transgenic AD mice | anti-amyloidogenic | Hsp70 | ↑ | [159] |
| Model | CAF-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vivo | ||||
| THY-Tau22 Transgenic mouse | anti-inflammatory antioxidative | TNF-α, GFAP, MAPK, Nrf2, MnSOD | ↓ ↑ | [170] |
| AD transgenic mouse model (Tg APPswe) | anti-amyloidogenic | PS1, BACE-1 | ↓ | [171] |
| APP/PS1 double transgenic AD mice | anti-amyloidogenic | BDNF, TrkB | ↑ | [173] |
| Model | NIC-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vivo | ||||
| AD rat model | anti-amyloidogenic | BACE-1 | ↓ | [185] |
| AD transgenic mouse model (Tg APPswe) | anti-amyloidogenic | nAchRα7 | ↑ | [186] |
| Male Wistar rats | improved memory performance | ChAT, VAChT NGF, TrkA | ↑ | [187] |
| Model | HupA-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vitro | ||||
| SHSY5Y exposed to H2O2 | antioxidative | NGF, P75NTR, MAPK/ERK | ↑ | [195] |
| Aβ-exposed cell lines | antioxidative anti-amyloidogenic anti-apoptotic | GPx, CAT, ATP ROS Cleaved-caspase 3 | ↑ ↓ | [196,197] [198] |
| Mutant APPswe over expression in HEK293 cells | anti-amyloidogenic | PKC | ↑ | [203] |
| In vivo | ||||
| Aβ-infused rats | anti-amyloidogenic | PKC | ↑ | [203] |
| Aβ-infused rats | neurogenesis | MAPK/ERK | ↑ | [201] |
| Aβ-infused rats | improved memory performance anti-apoptotic | Bax, p53 | ↓ | [202] |
| Model | BER-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| In vitro | ||||
| Mutant APPswe over expression in H4 | anti-amyloidogenic | β-secretase | ↓ | [218] |
| Mutant APPswe over expression in HEK293 cells | anti-amyloidogenic | β-secretase ERK1/2 | ↓ ↑ | [219] |
| Mutant APPswe over expression in Neuro 2A | anti-amyloidogenic | GSK3β | ↓ | [43] |
| rat primary astrocytes | antioxidative | PI3-kinase/Akt, HO-1 | ↑ | [220] |
| Aβ-exposed microglia BV2 cells | anti-inflammatory | MAPK, NF-kB | ↓ | [220] |
| In vivo | ||||
| AD transgenic mouse model (TgCRND8) | improved learning deficits and long-term spatial memory | [43] |
| Model | CBD-Mediated Protective Effects | Proposed Mechanisms Involved | Up/Down | References |
|---|---|---|---|---|
| Aβ-stimulated PC12 neuronal cells | anti-amyloidogenic | GSK3β Wnt/β-catenin | ↓ ↑ | [228] |
| Aβ-stimulated PC12 neuronal cells | antioxidative anti-apoptotic | ROS, iNOS, NO, Casp3 | ↓ | [230,231] |
| rat cortical neurons exposed to toxic glutamate | antioxidative anti-apoptotic | NMDA, AMPA and kainate receptor toxicity | ↓ | [232] |
| SHSY5Y overexpressing APPswe | anti-amyloidogenic | PPARγ | ↑ | [233] |
| In vivo | ||||
| Aβ-infused mice | anti-amyloidogenic antioxidative anti-inflammatory | iNOS, NO, MAPK, NFκB, IL-1β | ↓ | [235] |
| Aβ-injected rats | anti-inflammatory | PPARγ | ↑ | [234] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libro, R.; Giacoppo, S.; Soundara Rajan, T.; Bramanti, P.; Mazzon, E. Natural Phytochemicals in the Treatment and Prevention of Dementia: An Overview. Molecules 2016, 21, 518. https://doi.org/10.3390/molecules21040518
Libro R, Giacoppo S, Soundara Rajan T, Bramanti P, Mazzon E. Natural Phytochemicals in the Treatment and Prevention of Dementia: An Overview. Molecules. 2016; 21(4):518. https://doi.org/10.3390/molecules21040518
Chicago/Turabian StyleLibro, Rosaliana, Sabrina Giacoppo, Thangavelu Soundara Rajan, Placido Bramanti, and Emanuela Mazzon. 2016. "Natural Phytochemicals in the Treatment and Prevention of Dementia: An Overview" Molecules 21, no. 4: 518. https://doi.org/10.3390/molecules21040518
APA StyleLibro, R., Giacoppo, S., Soundara Rajan, T., Bramanti, P., & Mazzon, E. (2016). Natural Phytochemicals in the Treatment and Prevention of Dementia: An Overview. Molecules, 21(4), 518. https://doi.org/10.3390/molecules21040518