
Calculation of Relative Binding Free Energy in the Water-Filled Active Site of Oligopeptide-Binding Protein A


Abstract
:
1. Introduction
1.1. Ligand Binding and Promiscuity
1.2. Crystal Structure and Water
1.3. Thermodynamic Contributions to Binding
1.4. Scope
2. Materials and Methods
2.1. Simulation Settings
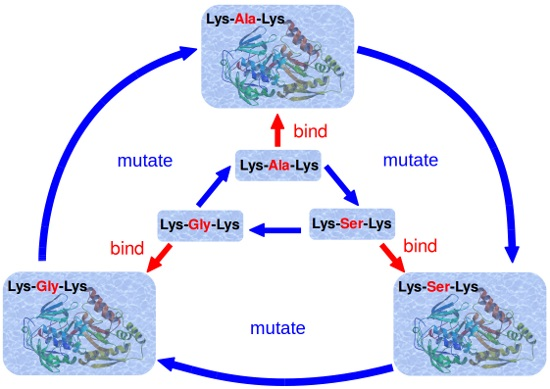
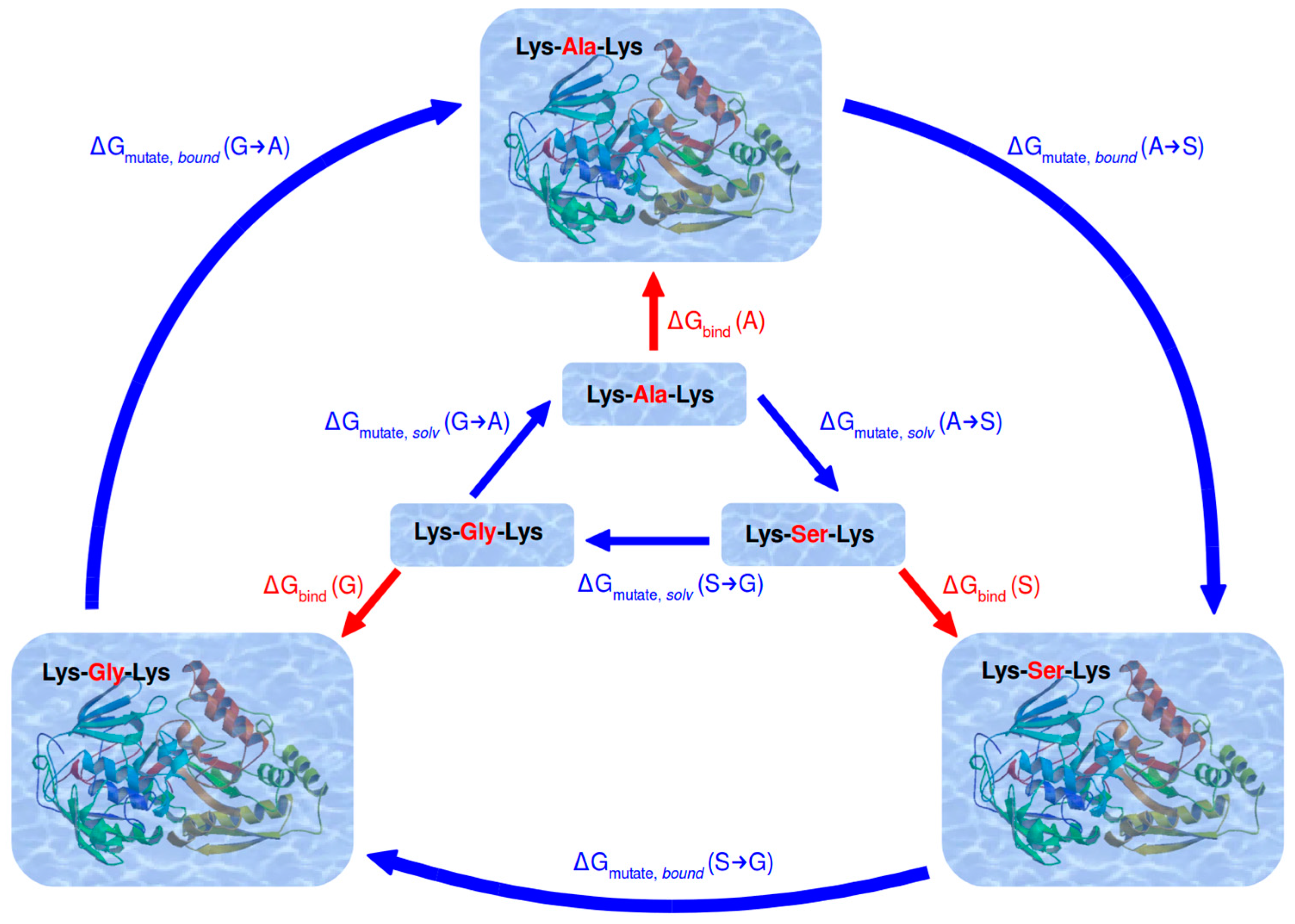
2.2. Simulation Scheme
2.3. Calculation of Relative Binding Free Energy
3. Results and Discussion
3.1. TI of Ligands Free in Solvent
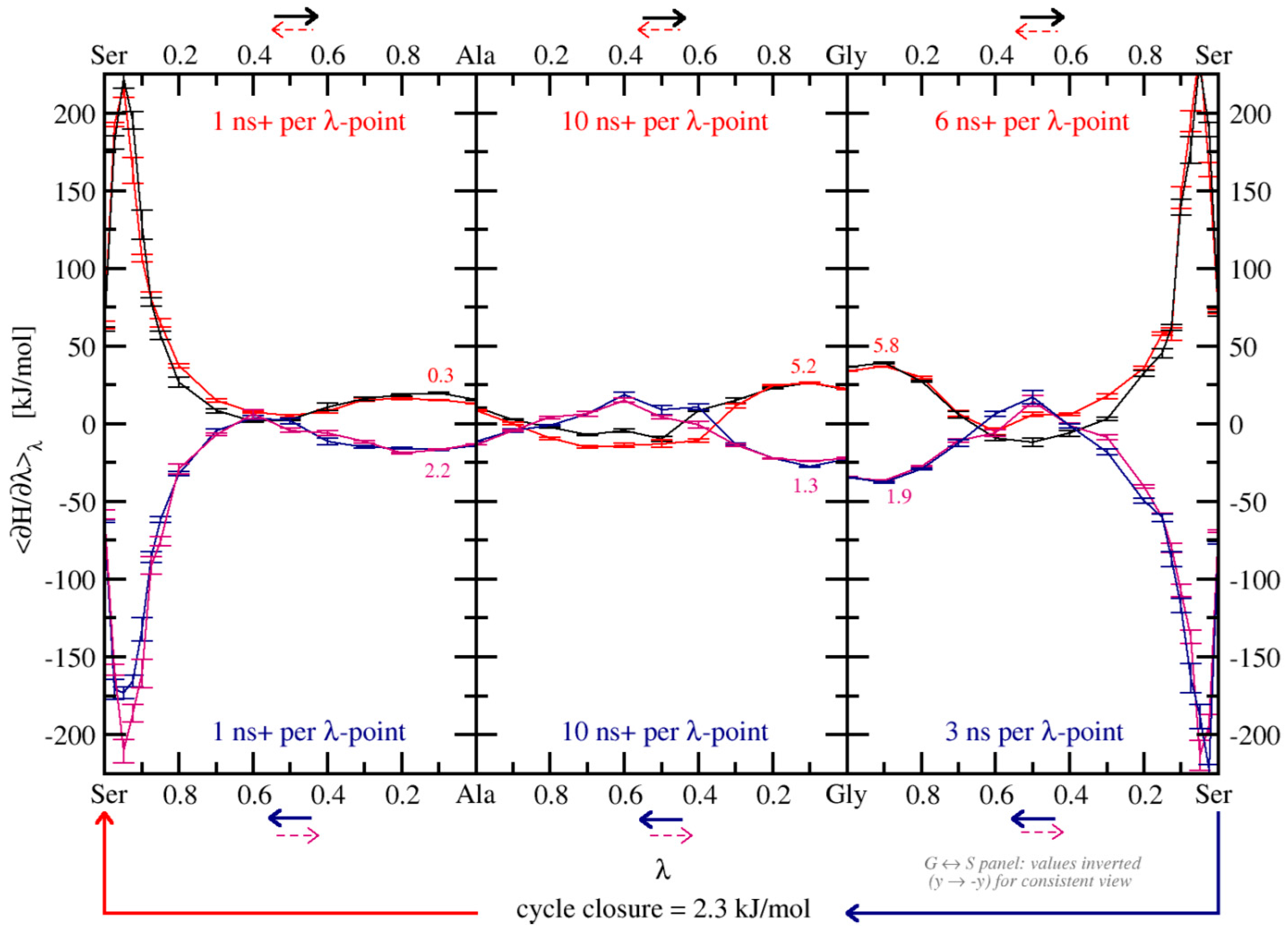
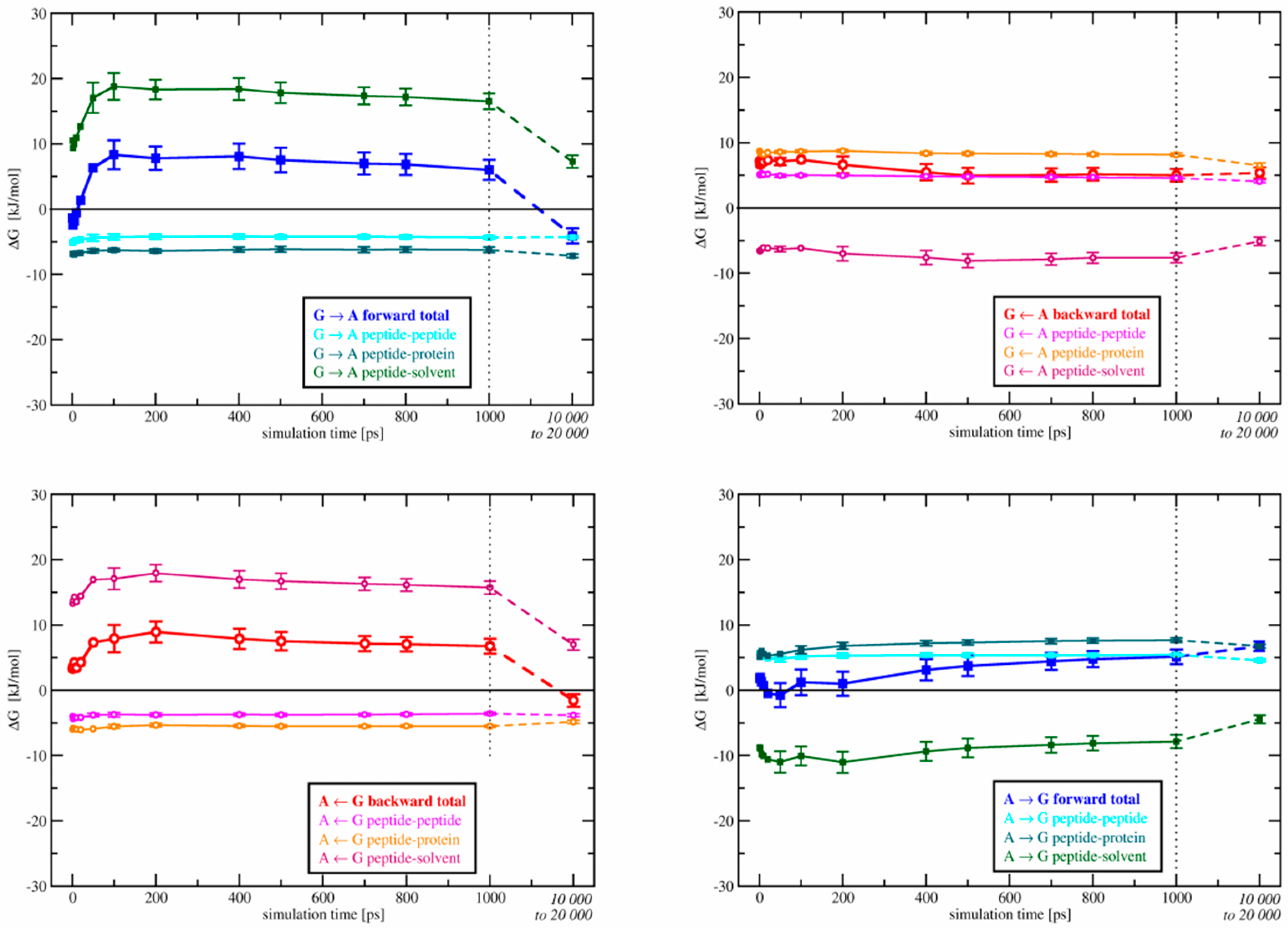
3.1.1. Free-Energy Convergence
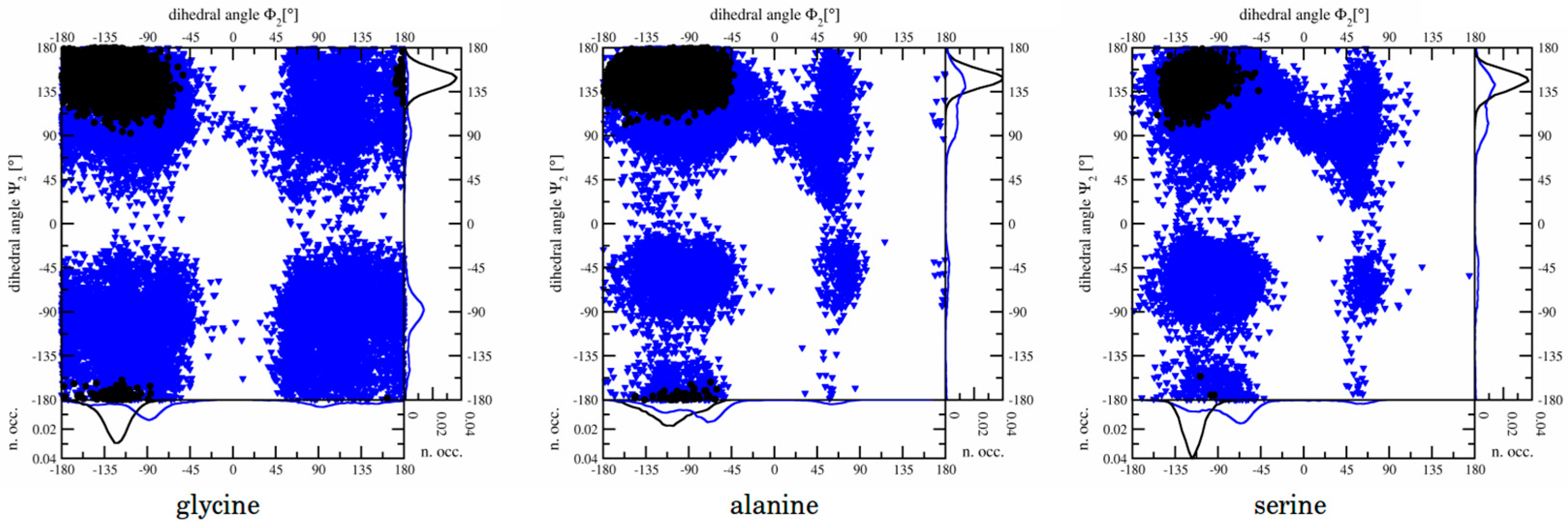
3.1.2. Dihedral Angles
3.2. TI of Ligands Bound to the Protein
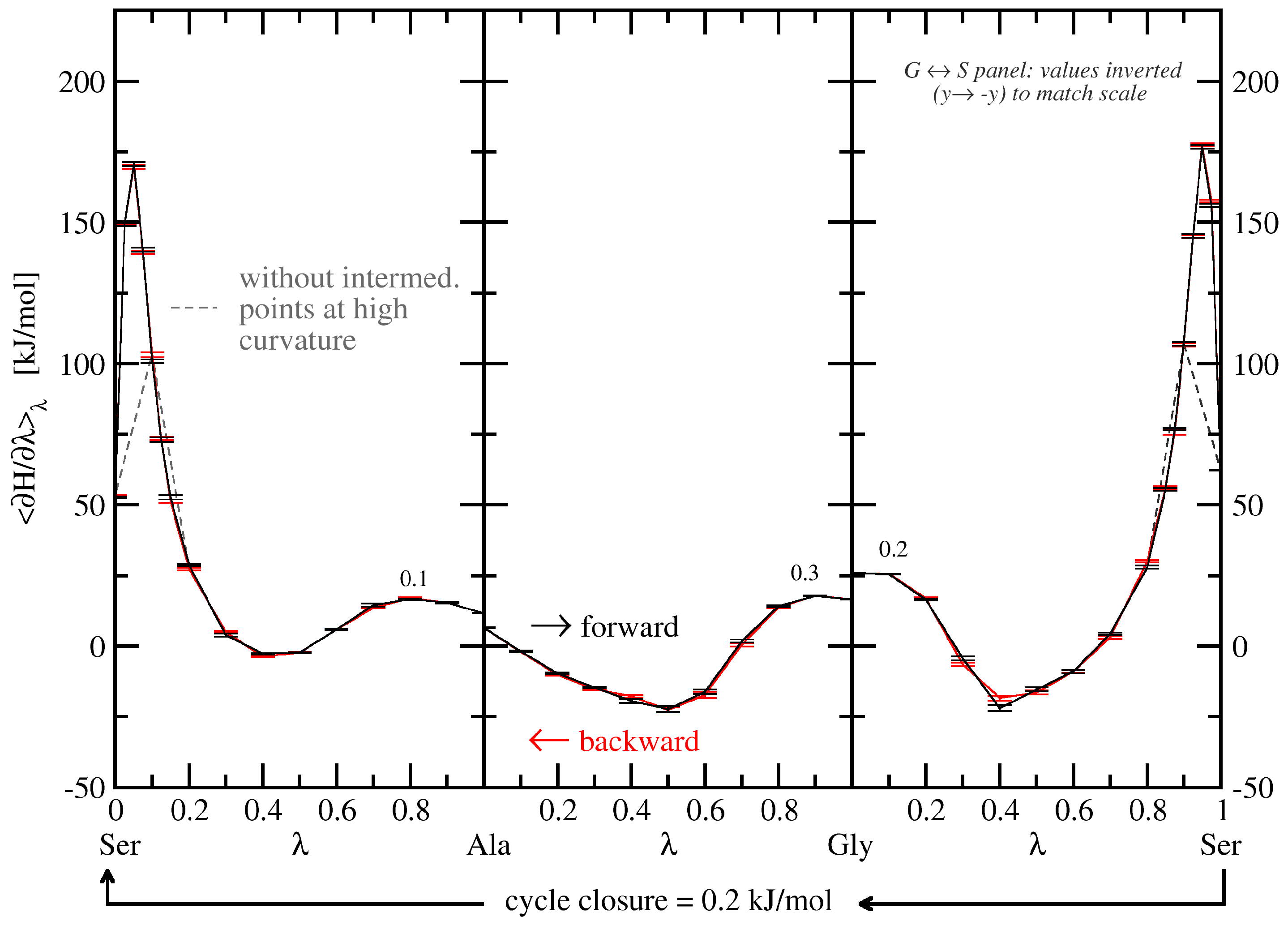
3.2.1. Free-Energy Convergence
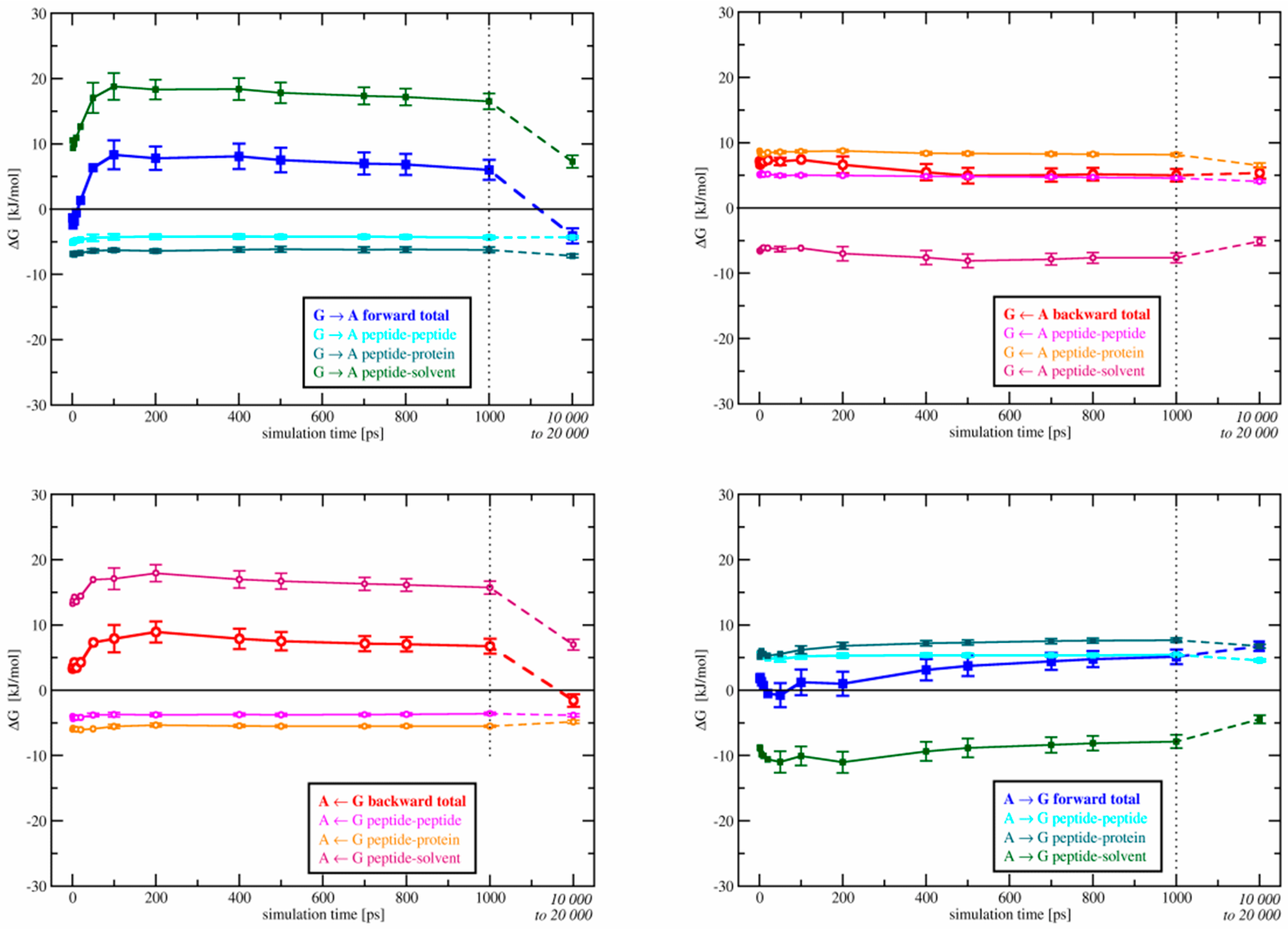
3.2.2. Energy Contribution Analysis
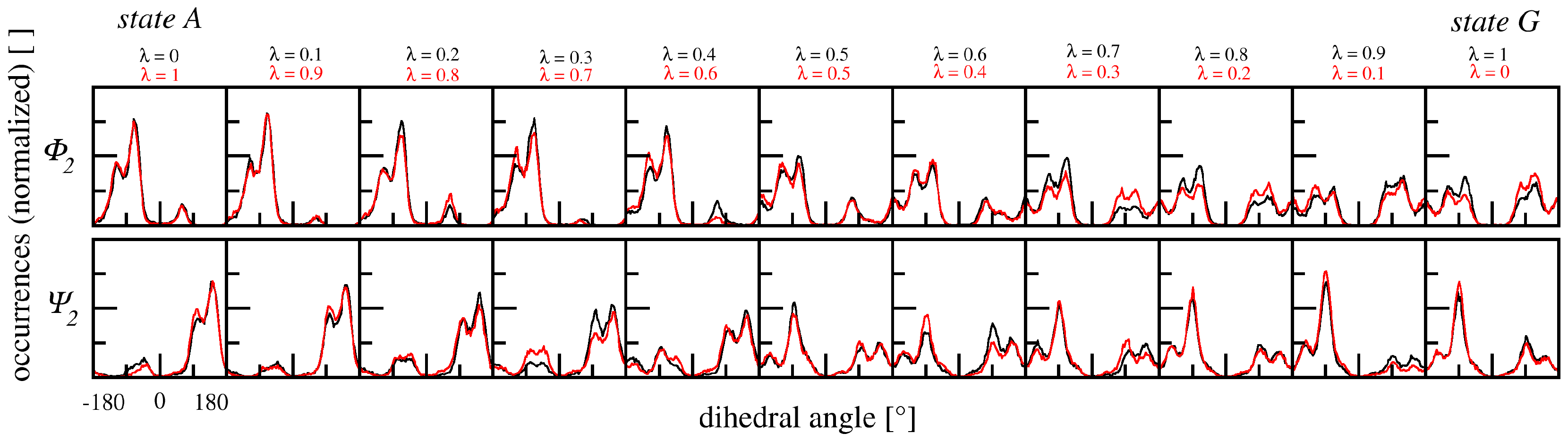
3.2.3. Dihedral Angles
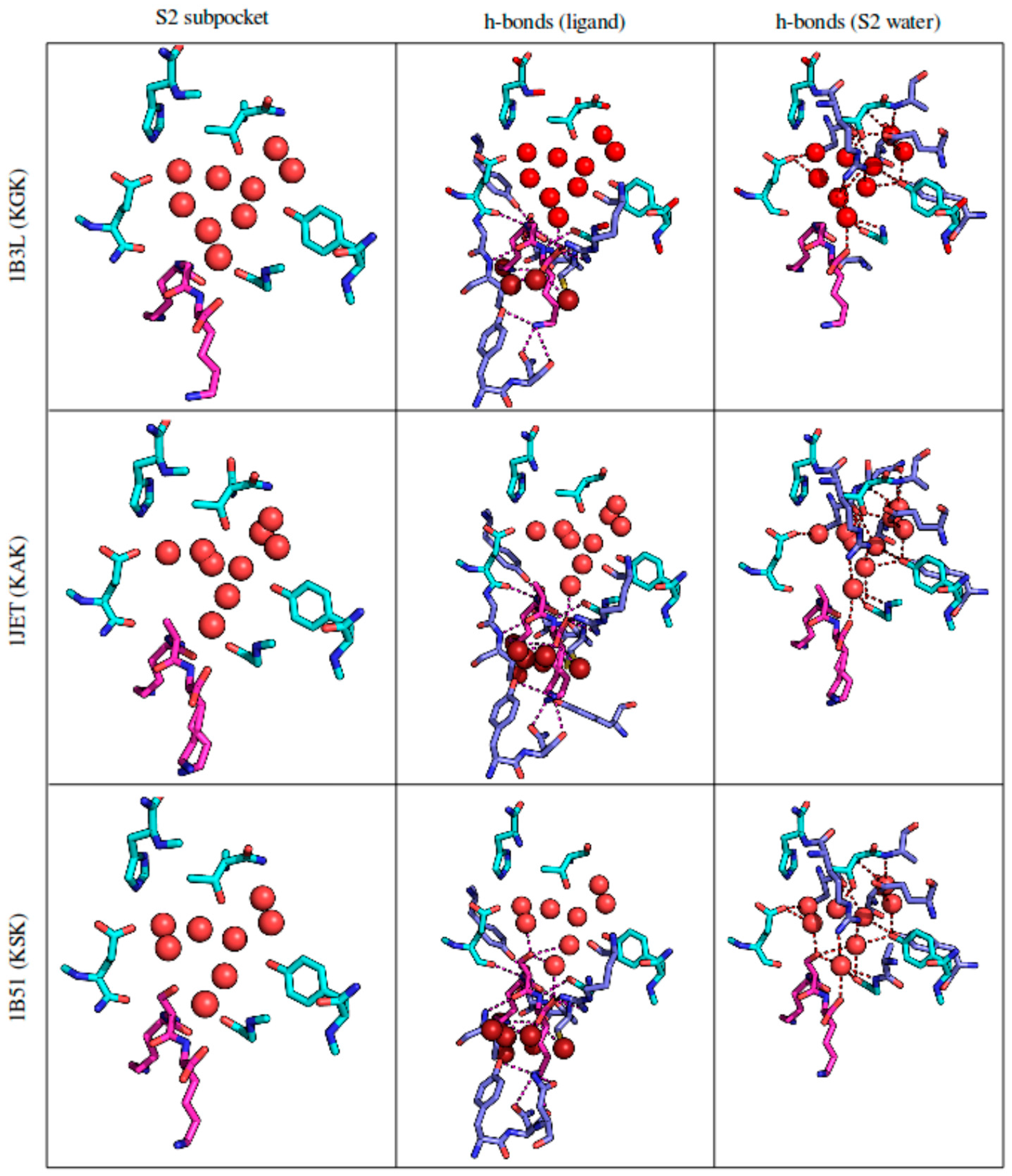
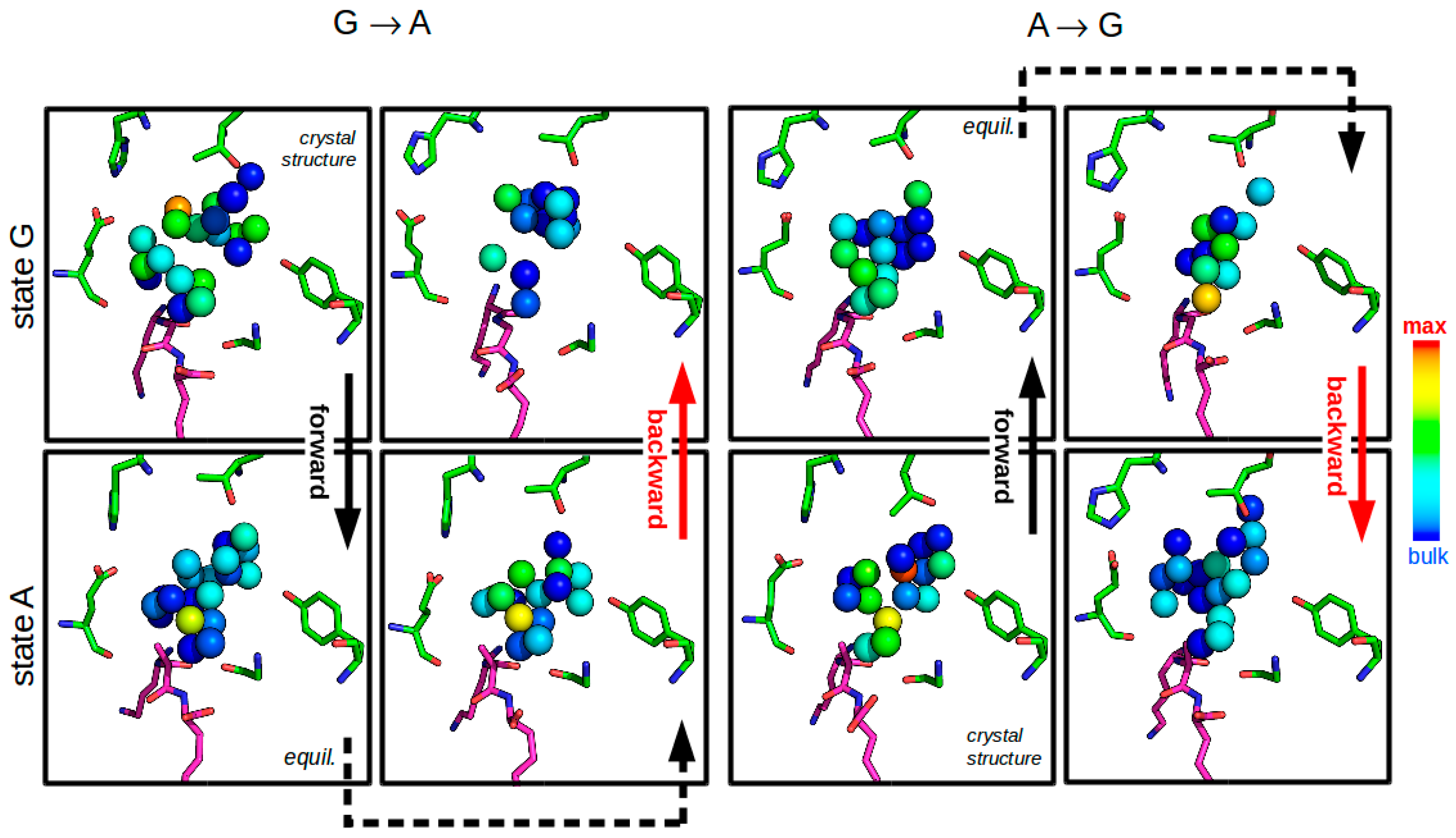
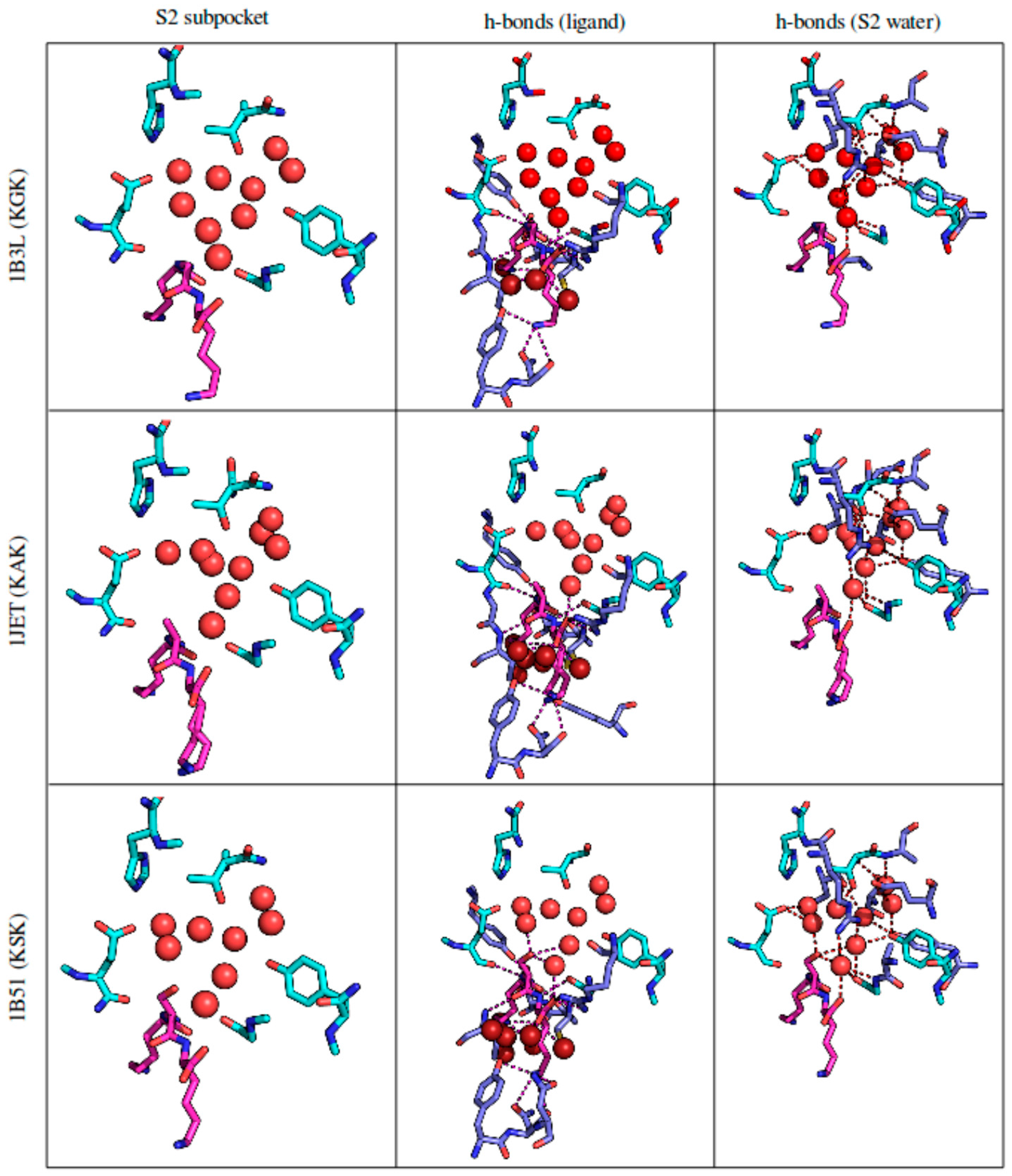
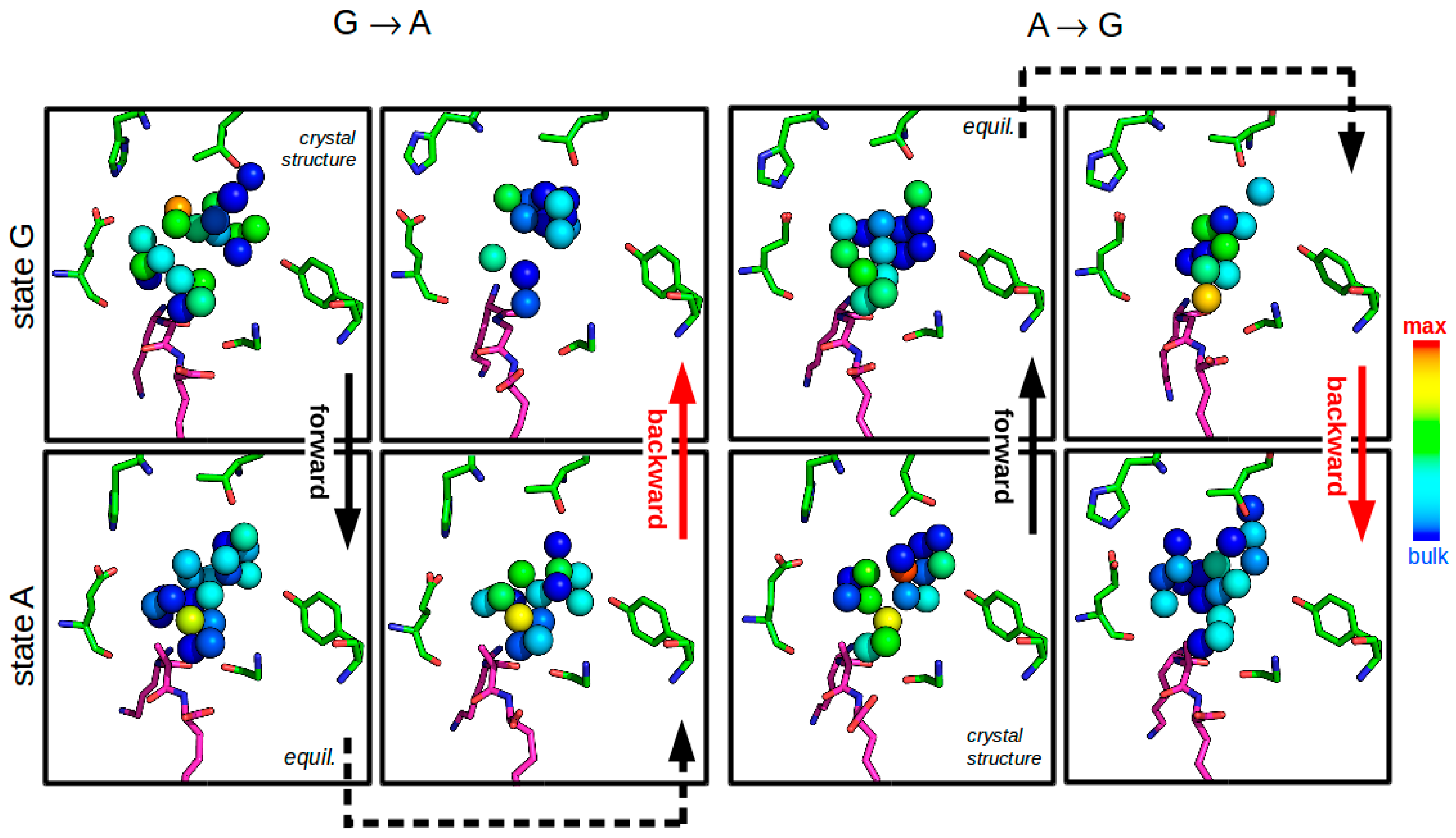
3.2.4. Solvent Network Rearrangement
3.3. Relative Binding Free Energies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MD | molecular dynamics |
| n.occ. | number of occurrences |
| OppA | oligopeptide-binding protein A |
| QSAR | quantitative structure-activity-relationship |
| TI | thermodynamic integration |
References
- Levitt, M.; Park, B.H. Water: Now you see it, now you don’t. Structure 1993, 1, 223–226. [Google Scholar] [CrossRef]
- Li, Z.; Lazaridis, T. The effect of water displacement on binding thermodynamics: Concanavalin A. J. Phys. Chem. B 2005, 109, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lazaridis, T. Water at biomolecular binding interfaces. Phys. Chem. Chem. Phys. 2007, 9, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F.; Hardie, M.M. Periplasmic protein associated with the oligopeptide permeases of Salmonella typhimurium and Escherichia coli. J. Bacteriol. 1983, 155, 1434–1438. [Google Scholar] [PubMed]
- Monnet, V. Bacterial oligopeptide-binding proteins. Cell. Mol. Life Sci. CMLS 2003, 60, 2100–2114. [Google Scholar] [CrossRef] [PubMed]
- Kornacki, J.A.; Oliver, D.B. Lyme disease-causing Borrelia species encode multiple lipoproteins homologous to peptide-binding proteins of ABC-type transporters. Infect. Immun. 1998, 66, 4115–4122. [Google Scholar] [PubMed]
- Hammond, S.M.; Claesson, A.; Jansson, A.M.; Larsson, L.G.; Pring, B.G.; Town, C.M.; Ekström, B. A new class of synthetic antibacterials acting on lipopolysaccharide biosynthesis. Nature 1987, 327, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Goodell, E.W.; Higgins, C.F. Uptake of cell wall peptides by Salmonella typhimurium and Escherichia coli. J. Bacteriol. 1987, 169, 3861–3865. [Google Scholar] [PubMed]
- Staskawicz, B.J.; Panopoulos, N.J. Phaseolotoxin transport in Escherichia coli and Salmonella typhimurium via the oligopeptide permease. J. Bacteriol. 1980, 142, 474–479. [Google Scholar] [PubMed]
- Tame, J.R.; Sleigh, S.H.; Wilkinson, A.J.; Ladbury, J.E. The role of water in sequence-independent ligand binding by an oligopeptide transporter protein. Nat. Struct. Biol. 1996, 3, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, M.M.; Kovermann, M.; Löw, C.; Balbach, J.; Permentier, H.P.; Fusetti, F.; de Gier, J.W.; Slotboom, D.J.; Berntsson, R.P.-A. Escherichia coli peptide binding protein OppA has a preference for positively charged peptides. J. Mol. Biol. 2011, 414, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, S.H.; Tame, J.R.H.; Dodson, E.J.; Wilkinson, A.J. Peptide binding in OppA, the crystal structures of the periplasmic oligopeptide binding protein in the unliganded form and in complex with lysyllysine. Biochemistry 1997, 36, 9747–9758. [Google Scholar] [CrossRef] [PubMed]
- Tame, J.R.; Dodson, E.J.; Murshudov, G.; Higgins, C.F.; Wilkinson, A.J. The crystal structures of the oligopeptide-binding protein OppA complexed with tripeptide and tetrapeptide ligands. Structure 1995, 3, 1395–1406. [Google Scholar] [CrossRef]
- Davies, T.G.; Hubbard, R.E.; Tame, J.R. Relating structure to thermodynamics: The crystal structures and binding affinity of eight OppA-peptide complexes. Protein Sci. Publ. Protein Soc. 1999, 8, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, S.H.; Seavers, P.R.; Wilkinson, A.J.; Ladbury, J.E.; Tame, J.R.H. Crystallographic and calorimetric analysis of peptide binding to OppA protein. J. Mol. Biol. 1999, 291, 393–415. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Yang, L.; Lv, F.; Luo, X.; Pan, Y. Why OppA protein can bind sequence-independent peptides? A combination of QM/MM, PB/SA, and structure-based QSAR analyses. Amino Acids 2010, 40, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, R.P.-A.; Doeven, M.K.; Fusetti, F.; Duurkens, R.H.; Sengupta, D.; Marrink, S.-J.; Thunnissen, A.-M.W.H.; Poolman, B.; Slotboom, D.-J. The structural basis for peptide selection by the transport receptor OppA. EMBO J. 2009, 28, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wade, R.C. Comparative binding energy (COMBINE) analysis of OppA-peptide complexes to relate structure to binding thermodynamics. J. Med. Chem. 2002, 45, 4828–4837. [Google Scholar] [CrossRef] [PubMed]
- De Beer, S.; Vermeulen, N.; Oostenbrink, C. The role of water molecules in computational drug design. Curr. Top. Med. Chem. 2010, 10, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Christ, C.D.; Christen, M.; Eichenberger, A.P.; van Gunsteren, W.F. Architecture, implementation and parallelisation of the GROMOS software for biomolecular simulation. Comput. Phys. Commun. 2012, 183, 890–903. [Google Scholar] [CrossRef]
- Schuler, L.D.; Daura, X.; van Gunsteren, W.F. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 2001, 22, 1205–1218. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces; Pullman, B., Ed.; The Jerusalem Symposia on Quantum Chemistry and Biochemistry; Reidel: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Tironi, I.G.; Sperb, R.; Smith, P.E.; van Gunsteren, W.F. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys. 1995, 102, 5451–5459. [Google Scholar] [CrossRef]
- Heinz, T.N.; van Gunsteren, W.F.; Hünenberger, P.H. Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J. Chem. Phys. 2001, 115, 1125–1136. [Google Scholar] [CrossRef]
- Beutler, T.C.; Mark, A.E.; van Schaik, R.C.; Gerber, P.R.; van Gunsteren, W.F. Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem. Phys. Lett. 1994, 222, 529–539. [Google Scholar] [CrossRef]
- De Ruiter, A.; Boresch, S.; Oostenbrink, C. Comparison of thermodynamic integration and Bennett acceptance ratio for calculating relative protein-ligand binding free energies. J. Comput. Chem. 2013, 34, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.T.; Shirts, M.R. Optimal pairwise and non-pairwise alchemical pathways for free energy calculations of molecular transformation in solution phase. J. Chem. Phys. 2012, 136, 124120. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Riniker, S.; Christ, C.D.; Hansen, H.S.; Hünenberger, P.H.; Oostenbrink, C.; Steiner, D.; van Gunsteren, W.F. Calculation of relative free energies for ligand-protein binding, solvation, and conformational transitions using the GROMOS software. J. Phys. Chem. B 2011, 115, 13570–13577. [Google Scholar] [CrossRef] [PubMed]
- Lawrenz, M.; Baron, R.; Wang, Y.; McCammon, J.A. Independent-trajectory thermodynamic integration: A practical guide to protein-drug binding free energy calculations using distributed computing. Methods Mol. Biol. 2012, 819, 469–486. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alchemical Mutation | ΔGmutate,solv | ΔGmutate,bound | ΔΔGbind,calc | ΔΔGbind,exp |
|---|---|---|---|---|
| A → G | −3.9 ± 0.4 | 6.8 ± 0.7 | ||
| A ← G | 4.2 ± 0.4 | −1.6 ± 1.0 | ||
| G → A | −4.1 ± 1.1 | |||
| G ← A | 5.4 ± 0.9 | |||
| average A → G | −4.0 ± 0.4 | 4.4 ± 0.9 | 8.5 ± 1.0 | 7.5 ± 2.4 |
| G → S | −22.5 ± 0.5 | −31.6 ± 1.9 | ||
| G ← S | 22.4 ± 0.5 | 37.4 ± 1.7 | ||
| S → G | −35.7 ± 2.2 | |||
| S ← G | −33.8 ± 2.0 | |||
| average G → S | −22.4 ± 0.5 | −34.6 ± 2.0 | −12.2 ± 2.0 | −8.4 ± 0.9 |
| A → S | −26.2 ± 0.4 | −30.2 ± 1.7 | ||
| A ← S | 26.3 ± 0.4 | 32.4 ± 2.0 | ||
| S → A | 33.5 ± 1.9 | |||
| S ← A | −33.9 ± 1.4 | |||
| average A → S | −26.3 ± 0.4 | −32.5 ± 1.7 | −6.3 ± 1.8 | −0.9 ± 2.2 |
| cycle | −0.2 ± 0.8 | 2.3 ± 2.8 | 2.5 ± 2.9 | 0 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maurer, M.; De Beer, S.B.A.; Oostenbrink, C. Calculation of Relative Binding Free Energy in the Water-Filled Active Site of Oligopeptide-Binding Protein A. Molecules 2016, 21, 499. https://doi.org/10.3390/molecules21040499
Maurer M, De Beer SBA, Oostenbrink C. Calculation of Relative Binding Free Energy in the Water-Filled Active Site of Oligopeptide-Binding Protein A. Molecules. 2016; 21(4):499. https://doi.org/10.3390/molecules21040499
Chicago/Turabian StyleMaurer, Manuela, Stephanie B. A. De Beer, and Chris Oostenbrink. 2016. "Calculation of Relative Binding Free Energy in the Water-Filled Active Site of Oligopeptide-Binding Protein A" Molecules 21, no. 4: 499. https://doi.org/10.3390/molecules21040499