
Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease


Abstract
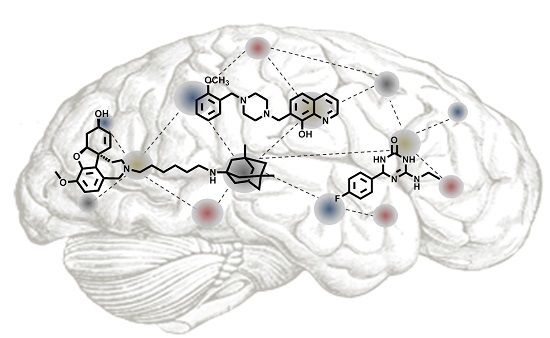
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
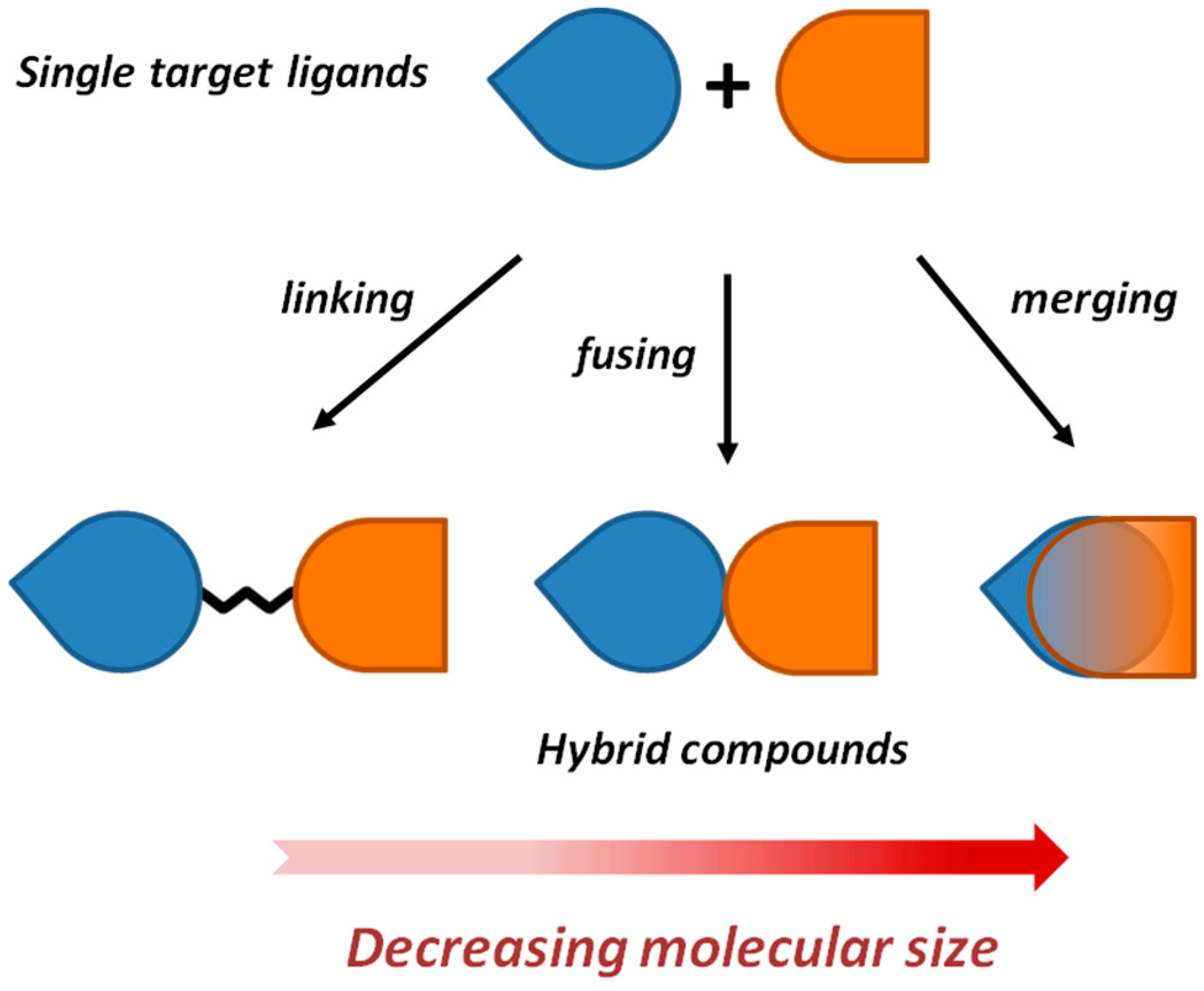
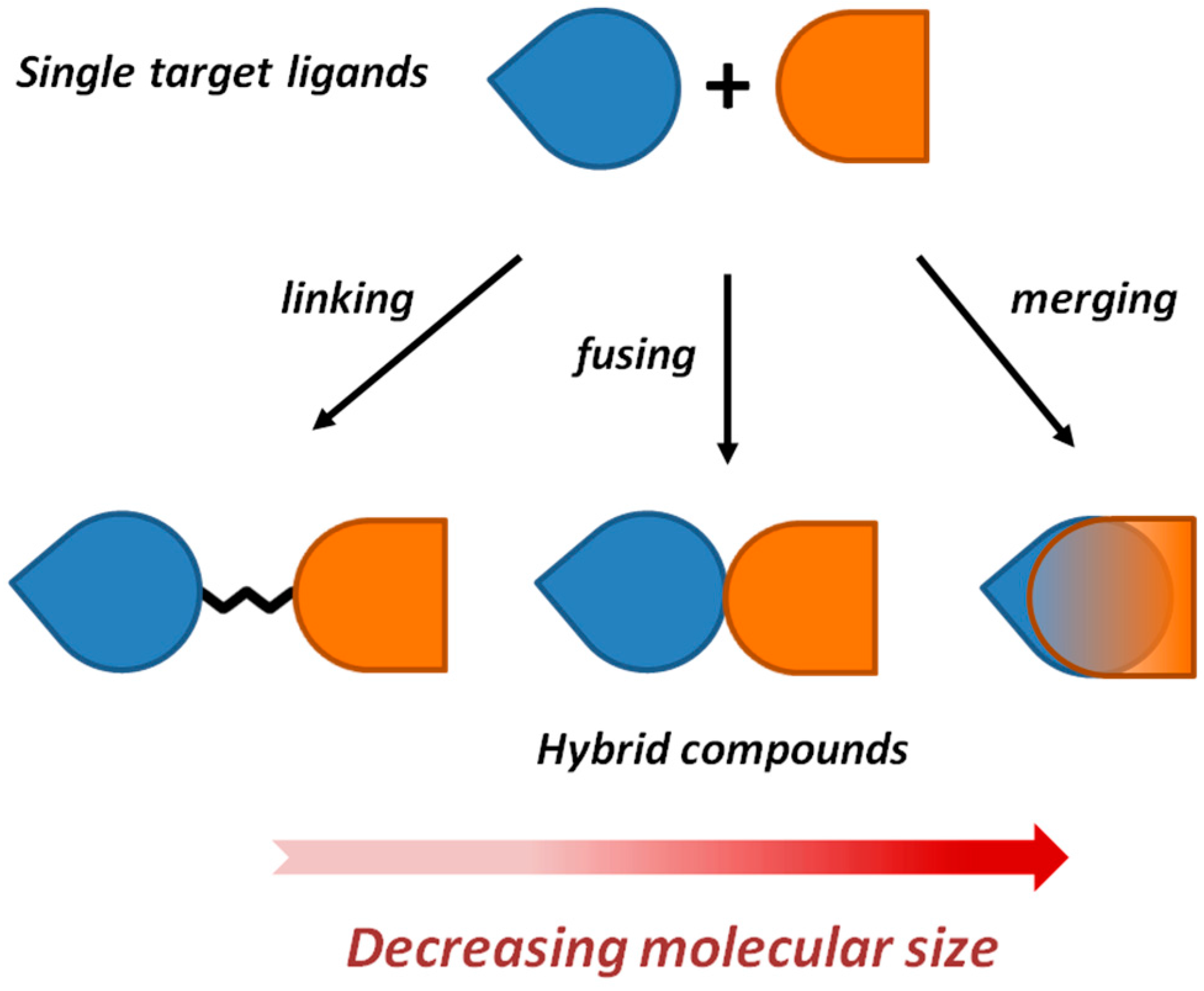
2. Framework Combination Strategies Applied to the Discovery of Hybrid Anti-AD Lead Candidates
2.1. Development of Memantine-Galantamine Hybrids via a Linking Strategy
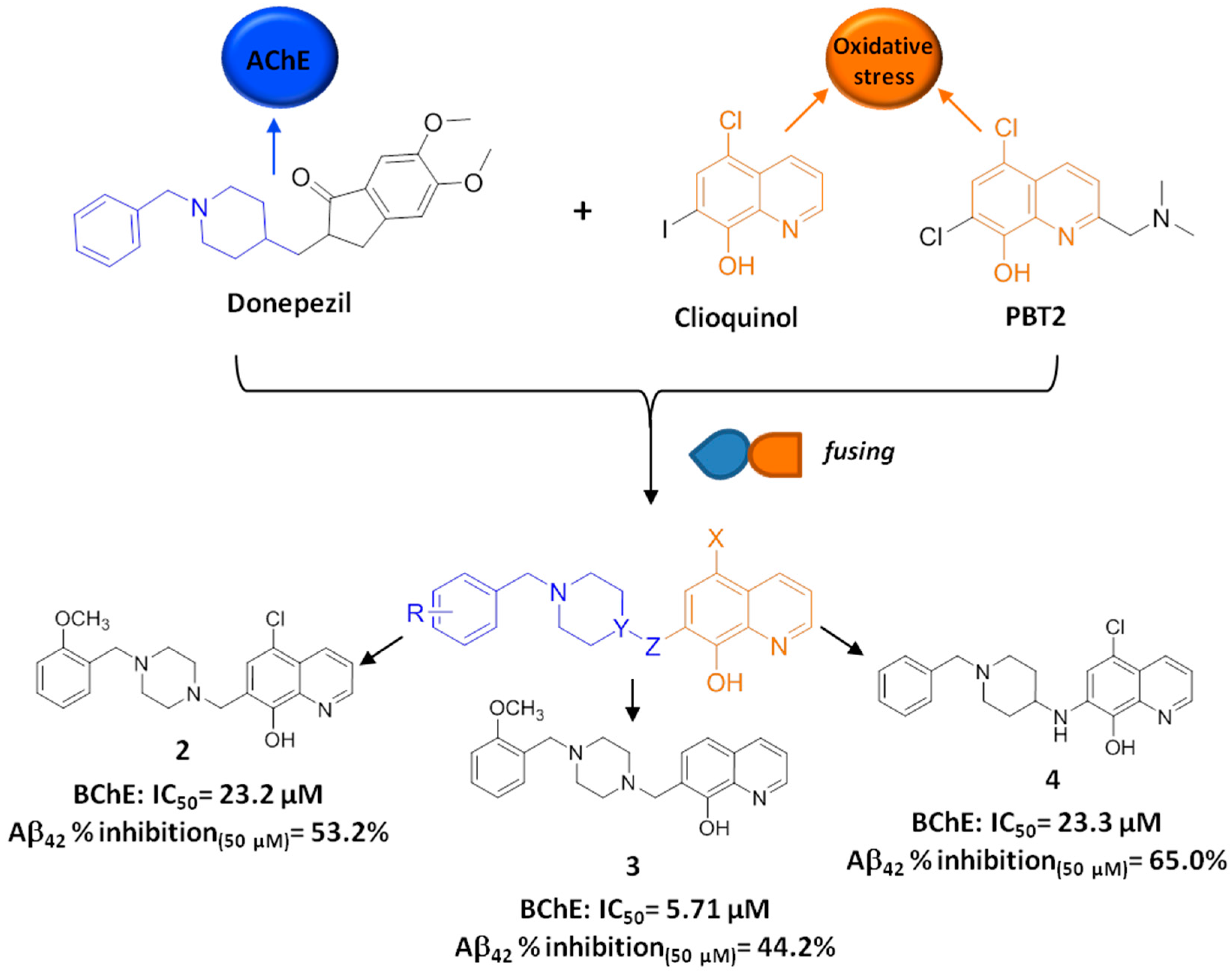
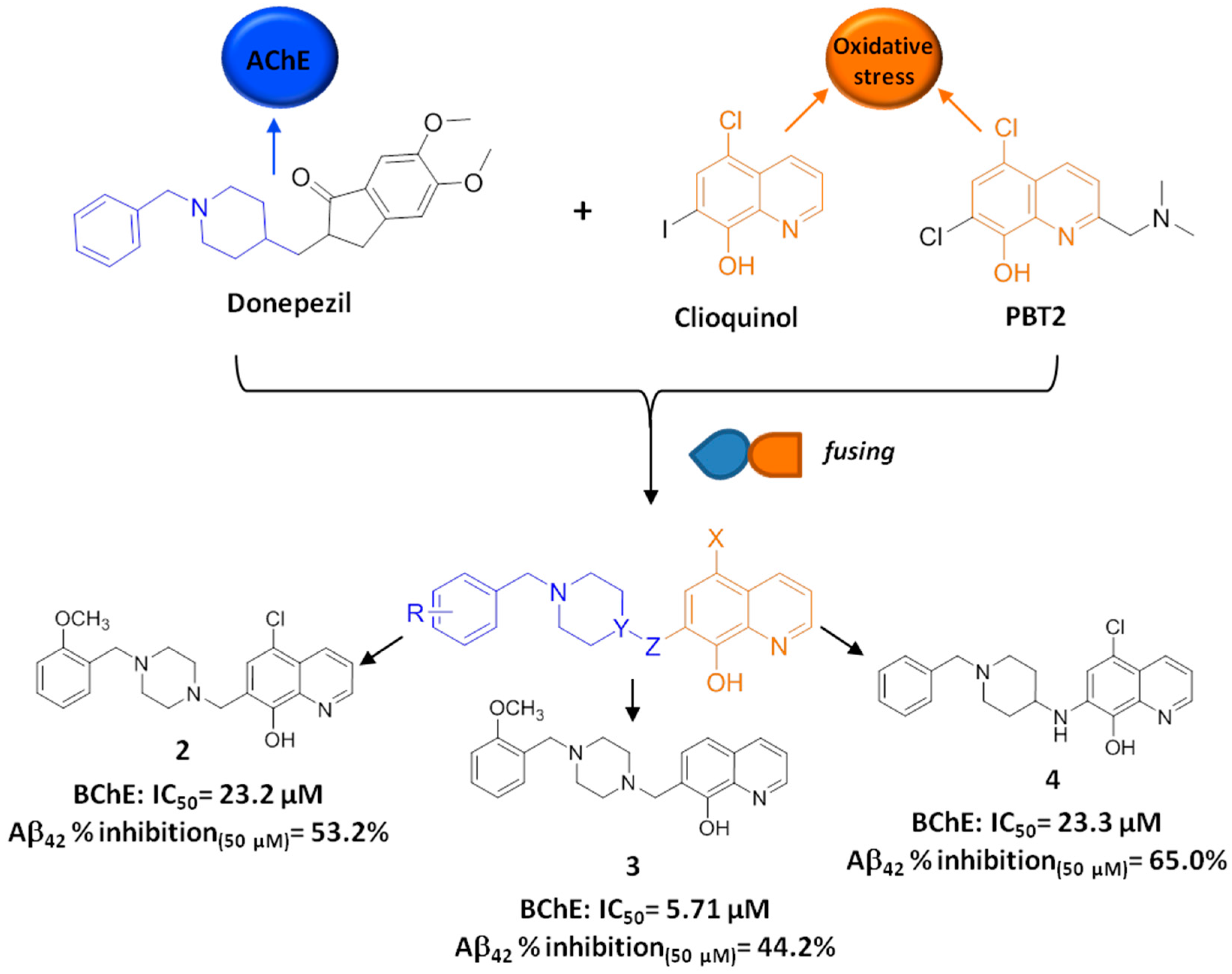
2.2. Development of Clioquinol-Donepezil Hybrids via a Fusing Strategy
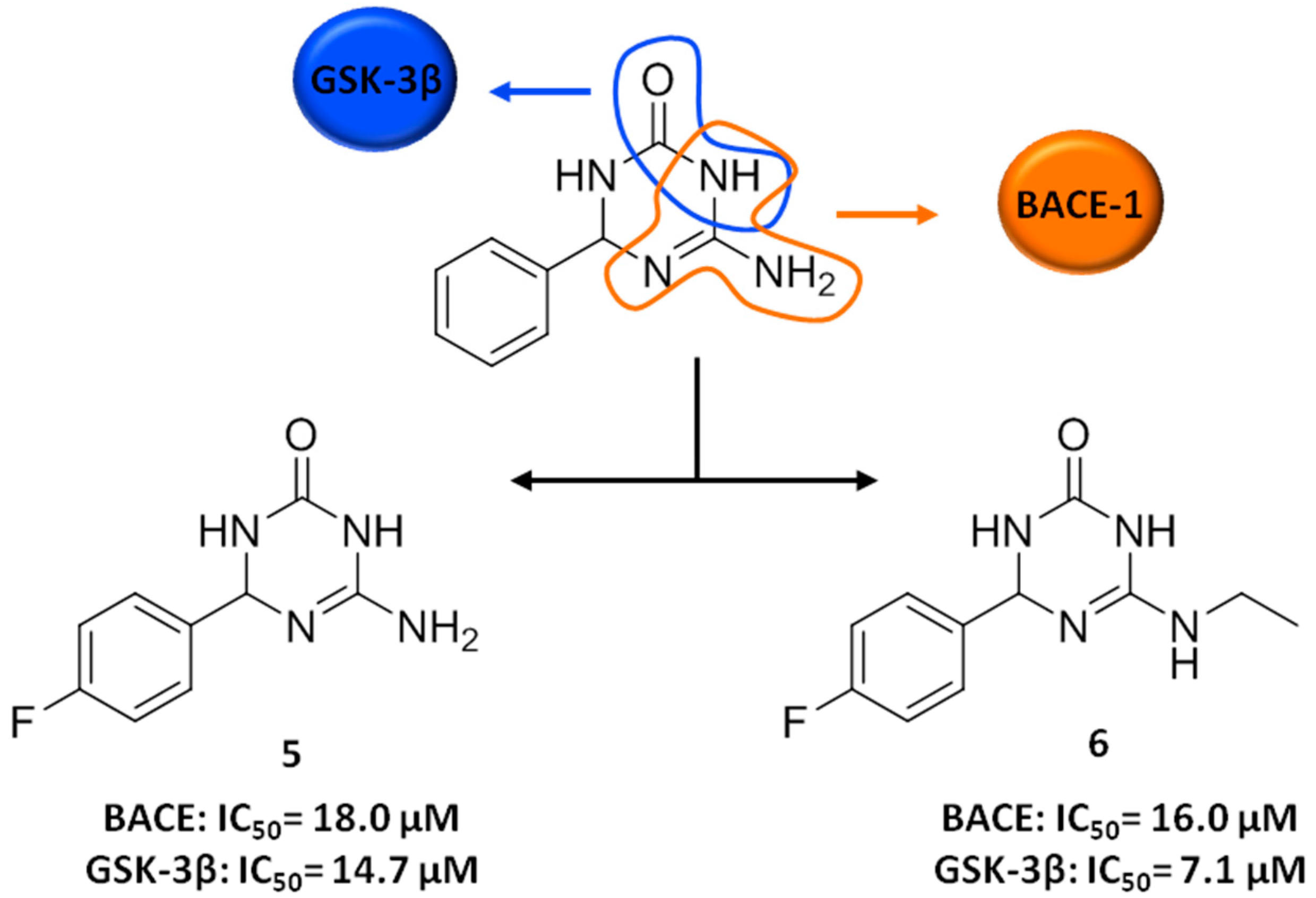
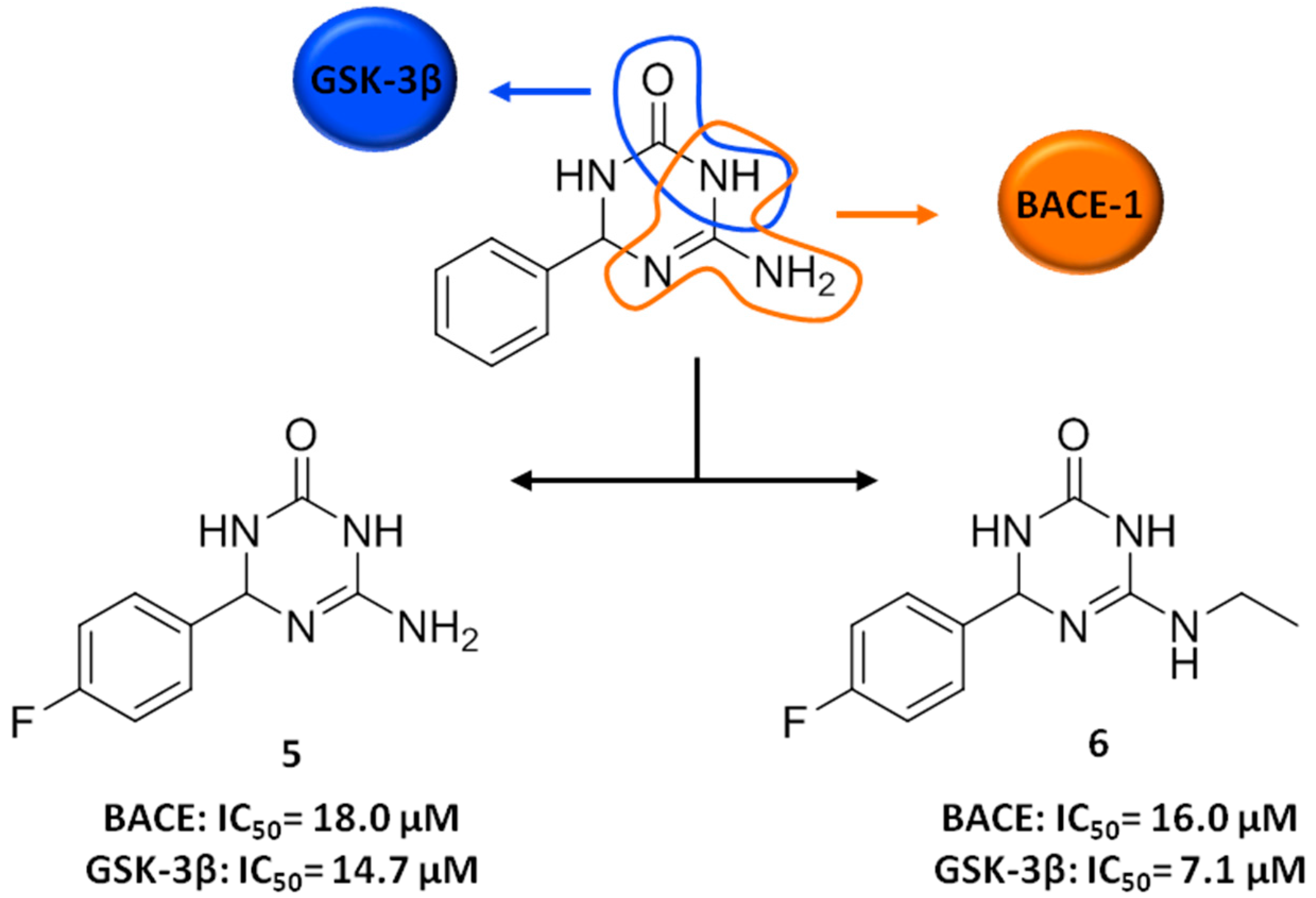
3. FBDD Applied to the Discovery of Anti-AD Lead Candidates
4. Conclusions
- From an academic medicinal chemistry perspective, we need a focused effort to promote a deeper understanding of the mechanism of multitarget action and the use of a rigorous proof of concept. Robust pharmacokinetic/pharmacodynamic data should guide the on/off decision to further develop a hit candidate that truly possesses a multitarget profile, i.e., (i) effectively reaches the brain; (ii) simultaneously modulates multiple targets and (iii) is more effective and safer than a single-targeted reference drug.
- In terms of target selection, we need to move from targets, such as AChE and amyloid, which have been over searched in favour of other ones with more credential for a disease-modifying effect [57]. As a recent Nature article bluntly put it, “we are over-reliant on amyloid” and “the time has come to face our fears and reject the amyloid cascade hypothesis” [58]. Indeed, potentially more promising targets, such as tau, have remained largely under-explored so far.
- Even more important is that the combination of targets to be addressed is made by using integrated network modelling and systems biology strategies. Multitarget approaches based on molecular network analysis may provide a fine-tuned modulation of the selected pathways instead of their complete blockade [51]. This appears to be critical for an effective therapeutic outcome.
- Generally, turning a small molecule hit into a lead is a formidable challenge, as numerous hurdles beyond activity have to be overcome. This is even truer when dealing with multitarget drug discovery, given the added complexity of optimizing against multiple targets. Thus, it is vital to identify a high-quality starting hit for lead optimization, as this is crucial in determining the later potential for success [17].
- Separate, although clearly related to this, is to push forward the use of cellular models as early as possible in the discovery phase. Different from isolated proteins, cell-screening systems maintain a reasonable experimental efficiency while preserving molecular-pathway interactions. Undoubtedly, this is indispensable in a network perspective. In particular, derivation of specific neural cells from patients’ induced pluripotent stem cells has made it possible to create in vitro models that recapitulate AD disease phenotypes. From a translation perspective, they represent unique platforms for AD (multitarget) drug discovery [59]. In this respect, treatment with approved or already validated combinations may provide clues or surrogate endpoints to track in vitro.
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 8HQ | 8-hydroxyquinoline |
| AChE | acetylcholinesterase |
| AChEI | acetylcholinesterase inhibitor |
| AD | Alzheimer’s disease |
| Aβ | β-amyloid |
| BACE-1 | beta-secretasie-1 |
| BBB | blood-brain barrier |
| CAS | catalytic anionic site |
| ChE | cholinesterase |
| CLQ | clioquinol |
| CNS | central nervous system |
| FBDD | fragment-based drug discovery |
| GSK-3β | glycogen synthase kinase-3 |
| hBChE | human butyrylcholinesterase |
| HTS | high throughput screening |
| JNK3 | c-Jun N-terminal kinase 3 |
| LE | ligand efficiency |
| MAO | monoamine oxidase |
| MPACs | metal-protein attenuation compounds |
| MTDL | multitarget-directed ligand |
| MW | molecular weight |
| NMDAR | N-methyl-d-aspartate receptor |
| PAMPA | parallel artificial membrane permeability assay |
| PAS | peripheral anionic site |
| PPAR | peroxisome proliferator-activated receptor |
| ROS | reactive oxygen species |
| SAR | structure-activity relationships |
References
- Csermely, P.; Agoston, V.; Pongor, S. The efficiency of multi-target drugs: The network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Fridkin, M.; Youdim, M. New approaches to treating Alzheimer’s disease. Perspect. Med. Chem. 2015, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Uliassi, E.; Bolognesi, M.L. Two diseases, one approach: Multitarget drug discovery in Alzheimer’s and neglected tropical diseases. MedChemComm 2014, 5, 853–861. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, A.; Bolognesi, M.L.; Capsoni, S.; Andrisano, V.; Bartolini, M.; Margotti, E.; Cattaneo, A.; Recanatini, M.; Melchiorre, C. A small molecule targeting the multifactorial nature of Alzheimer’s disease. Angew. Chem. Int. Ed. 2007, 46, 3689–3692. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Mason, J.S.; Overington, J.P. Can we rationally design promiscuous drugs? Curr. Opin. Struct. Biol. 2006, 16, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Designing multiple ligands—Medicinal chemistry strategies and challenges. Curr. Pharm. Des. 2009, 15, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. The physicochemical challenges of designing multiple ligands. J. Med. Chem. 2006, 49, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Favia, A.D.; Recanatini, M.; Cavalli, A. The role of fragment-based and computational methods in polypharmacology. Drug Discov. Today 2012, 17, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R. The influence of target family and functional activity on the physicochemical properties of pre-clinical compounds. J. Med. Chem. 2006, 49, 2969–2978. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. Fragments, network biology and designing multiple ligands. Drug Discov. Today 2007, 12, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Morphy, J.R.; Harris, C.J. Designing Multi-target Drugs; Royal Society of Chemistry: Cambridge, UK, 2012; Volume 21. [Google Scholar]
- Decker, M. Recent advances in the development of hybrid molecules/designed multiple compounds with antiamnesic properties. Mini Rev. Med. Chem. 2007, 7, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Viayna, E.; Sabate, R.; Munoz-Torrero, D. Dual inhibitors of beta-amyloid aggregation and acetylcholinesterase as multi-target anti-Alzheimer drug candidates. Curr. Top. Med. Chem. 2013, 13, 1820–1842. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; van der Schyf, C.J. Designing drugs with multi-target activity: The next step in the treatment of neurodegenerative disorders. Expert Opin. Drug Discov. 2012, 8, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Leon, R.; Garcia, A.G.; Marco-Contelles, J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med. Res. Rev. 2013, 33, 139–189. [Google Scholar] [CrossRef] [PubMed]
- Agis-Torres, A.; Solhuber, M.; Fernandez, M.; Sanchez-Montero, J.M. Multi-target-directed ligands and other therapeutic strategies in the search of a real solution for Alzheimer’s disease. Curr. Neuropharmacol. 2014, 12, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Guzior, N.; Wieckowska, A.; Panek, D.; Malawska, B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 2015, 22, 373–404. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.I.; Martinez, A.; Gil, C.; Campillo, N.E. From bitopic inhibitors to multitarget drugs for the future treatment of Alzheimer’s disease. Curr. Med. Chem. 2015, 22, 3789–3806. [Google Scholar] [CrossRef] [PubMed]
- Simoni, E.; Daniele, S.; Bottegoni, G.; Pizzirani, D.; Trincavelli, M.L.; Goldoni, L.; Tarozzo, G.; Reggiani, A.; Martini, C.; Piomelli, D.; et al. Combining galantamine and memantine in multitargeted, new chemical entities potentially useful in Alzheimer’s disease. J. Med. Chem. 2012, 55, 9708–9721. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Grossberg, G.T. Combination therapy for Alzheimer’s disease. Drugs Aging 2011, 28, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Parsons, C.G.; Danysz, W.; Dekundy, A.; Pulte, I. Memantine and cholinesterase inhibitors: Complementary mechanisms in the treatment of Alzheimer’s disease. Neurotox. Res. 2013, 24, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Bartolini, M.; Cavalli, A.; Ceccarini, L.; Pascu, N.; McClymont, D.W.; Tarozzi, A.; Bolognesi, M.L.; Minarini, A.; et al. Inhibition of acetylcholinesterase, beta-amyloid aggregation, and nmda receptors in Alzheimer’s disease: A promising direction for the multi-target-directed ligands gold rush. J. Med. Chem. 2008, 51, 4381–4384. [Google Scholar] [CrossRef] [PubMed]
- Rook, Y.; Schmidtke, K.U.; Gaube, F.; Schepmann, D.; Wunsch, B.; Heilmann, J.; Lehmann, J.; Winckler, T. Bivalent beta-carbolines as potential multitarget anti-Alzheimer agents. J. Med. Chem. 2010, 53, 3611–3617. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lv, C.; Zhao, W.; Song, Y.; Pei, D.; Xu, T. Nr2b-containing nmda receptors expression and their relationship to apoptosis in hippocampus of Alzheimer’s disease-like rats. Neurochem. Res. 2012, 37, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Mony, L.; Kew, J.N.; Gunthorpe, M.J.; Paoletti, P. Allosteric modulators of Nr2b-containing nmda receptors: Molecular mechanisms and therapeutic potential. Br. J. Pharmacol. 2009, 157, 1301–1317. [Google Scholar] [CrossRef] [PubMed]
- Chazot, P.L. The NMDA receptor Nr2b subunit: A valid therapeutic target for multiple CNS pathologies. Curr. Med. Chem. 2004, 11, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Minarini, A.; Melchiorre, C. Multi-target design strategies in the context of Alzheimer’s disease: Acetylcholinesterase inhibition and NMDA receptor antagonism as the driving forces. Neurochem. Res. 2014, 39, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.P.; Tarozzo, G.; Reggiani, A.; Piomelli, D.; Cavalli, A. Galantamine potentiates the neuroprotective effect of memantine against NMDA-induced excitotoxicity. Brain Behav. 2013, 3, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Takada-Takatori, Y.; Kume, T.; Sugimoto, M.; Katsuki, H.; Sugimoto, H.; Akaike, A. Acetylcholinesterase inhibitors used in treatment of Alzheimer’s disease prevent glutamate neurotoxicity via nicotinic acetylcholine receptors and phosphatidylinositol 3-kinase cascade. Neuropharmacology 2006, 51, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Bergamini, C.; Fato, R.; Soukup, O.; Korabecny, J.; Andrisano, V.; Bartolini, M.; Bolognesi, M.L. Novel 8-hydroxyquinoline derivatives as multitarget compounds for the treatment of Alzheimer’s disease. ChemMedChem 2016. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Suh, S.W.; Jensen, K.B.; Jensen, M.S.; Silva, D.S.; Kesslak, P.J.; Danscher, G.; Frederickson, C.J. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 2000, 852, 274–278. [Google Scholar] [CrossRef]
- Curtain, C.C.; Ali, F.; Volitakis, I.; Cherny, R.A.; Norton, R.S.; Beyreuther, K.; Barrow, C.J.; Masters, C.L.; Bush, A.I.; Barnham, K.J. Alzheimer’s disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 2001, 276, 20466–20473. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim. Biophys. Acta 2007, 1768, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Kim, Y.K.; Chang, Y.T. Investigations of the molecular mechanism of metal-induced abeta (1–40) amyloidogenesis. Biochemistry 2007, 46, 13523–13532. [Google Scholar] [CrossRef] [PubMed]
- Cuajungco, M.P.; Goldstein, L.E.; Nunomura, A.; Smith, M.A.; Lim, J.T.; Atwood, C.S.; Huang, X.; Farrag, Y.W.; Perry, G.; Bush, A.I. Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of abeta by zinc. J. Biol. Chem. 2000, 275, 19439–19442. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial abeta. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Kaur, D.; Yantiri, F.; Rajagopalan, S.; Kumar, J.; Mo, J.Q.; Boonplueang, R.; Viswanath, V.; Jacobs, R.; Yang, L.; Beal, M.F.; et al. Genetic or pharmacological iron chelation prevents mptp-induced neurotoxicity in vivo: A novel therapy for Parkinson’s disease. Neuron 2003, 37, 899–909. [Google Scholar] [CrossRef]
- Nguyen, T.; Hamby, A.; Massa, S.M. Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 11840–11845. [Google Scholar] [CrossRef] [PubMed]
- Zatta, P.; Drago, D.; Bolognin, S.; Sensi, S.L. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol. Sci. 2009, 30, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S. 8-hydroxyquinolines: A review of their metal chelating properties and medicinal applications. Drug Des. Devel. Ther. 2013, 7, 1157–1178. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Bachiller, M.I.; Perez, C.; Gonzalez-Munoz, G.C.; Conde, S.; Lopez, M.G.; Villarroya, M.; Garcia, A.G.; Rodriguez-Franco, M.I. Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Fridkin, M.; Youdim, M. From single target to multitarget/network therapeutics in Alzheimer’s therapy. Pharmaceuticals 2014, 7, 113–135. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; de Simone, A.; Bisignano, P.; Armirotti, A.; Summa, M.; Pizzirani, D.; Scarpelli, R.; Perez, D.I.; Andrisano, V.; Perez-Castillo, A.; et al. Multitarget drug discovery for Alzheimer’s disease: Triazinones as bace-1 and gsk-3beta inhibitors. Angew. Chem. Int. Ed. 2015, 54, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Venkatraman, S.; Zheng, Y.; McKeever, B.M.; Dillard, L.W.; Singh, S.B. Structure-based design of beta-site app cleaving enzyme 1 (bace1) inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2013, 56, 4156–4180. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Osswald, H.L. Bace1 (beta-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Schmidt, B.; Lo Monte, F. Small-molecule inhibitors of gsk-3: Structural insights and their application to Alzheimer’s disease models. Int. J. Alzheimers Dis. 2012, 2012, 381029. [Google Scholar] [CrossRef] [PubMed]
- Pena-Altamira, E.; Prati, F.; Massenzio, F.; Virgili, M.; Contestabile, A.; Bolognesi, M.L.; Monti, B. Changing paradigm to target microglia in neurodegenerative diseases: From anti-inflammatory strategy to active immunomodulation. Expert Opin. Ther. Targets 2016, 20, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; de Simone, A.; Armirotti, A.; Summa, M.; Pizzirani, D.; Scarpelli, R.; Bertozzi, S.M.; Perez, D.I.; Andrisano, V.; Perez-Castillo, A.; et al. 3,4-Dihydro-1,3,5-triazin-2(1h)-ones as the first dual bace-1/gsk-3beta fragment hits against Alzheimer’s disease. ACS Chem. Neurosci. 2015, 6, 1665–1682. [Google Scholar] [CrossRef] [PubMed]
- Banik, A.; Brown, R.E.; Bamburg, J.; Lahiri, D.K.; Khurana, D.; Friedland, R.P.; Chen, W.; Ding, Y.; Mudher, A.; Padjen, A.L.; et al. Translation of pre-clinical studies into successful clinical trials for Alzheimer’s disease: What are the roadblocks and how can they be overcome? J. Alzheimers Dis. 2015, 47, 815–843. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Xie, M.; Cao, N.; Ding, S. Patient-specific induced pluripotent stem cells for disease modeling and phenotypic drug discovery. J. Med. Chem. 2016, 59, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prati, F.; Cavalli, A.; Bolognesi, M.L. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules 2016, 21, 466. https://doi.org/10.3390/molecules21040466
Prati F, Cavalli A, Bolognesi ML. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules. 2016; 21(4):466. https://doi.org/10.3390/molecules21040466
Chicago/Turabian StylePrati, Federica, Andrea Cavalli, and Maria Laura Bolognesi. 2016. "Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease" Molecules 21, no. 4: 466. https://doi.org/10.3390/molecules21040466