
New Look on 3-Hydroxyiminoflavanone and Its Palladium(II) Complex: Crystallographic and Spectroscopic Studies, Theoretical Calculations and Cytotoxic Activity

 and
and
Abstract
:1. Introduction
2. Results and Discussion
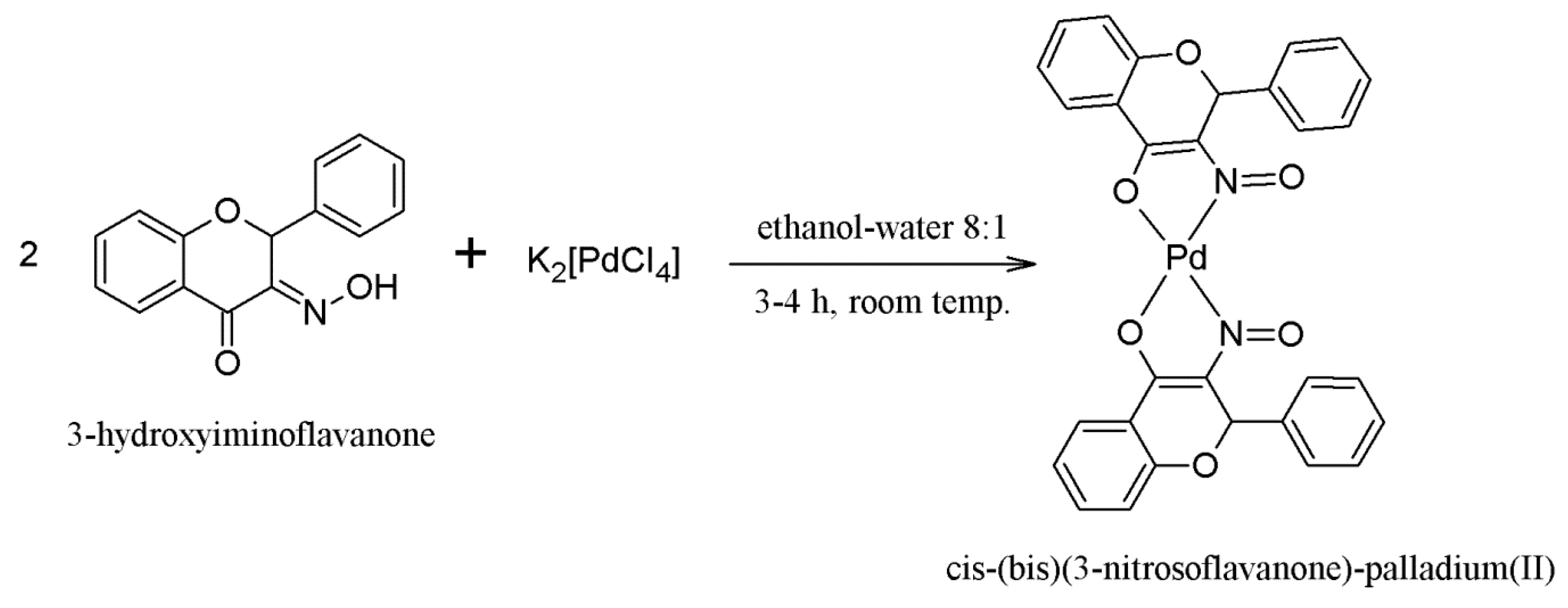
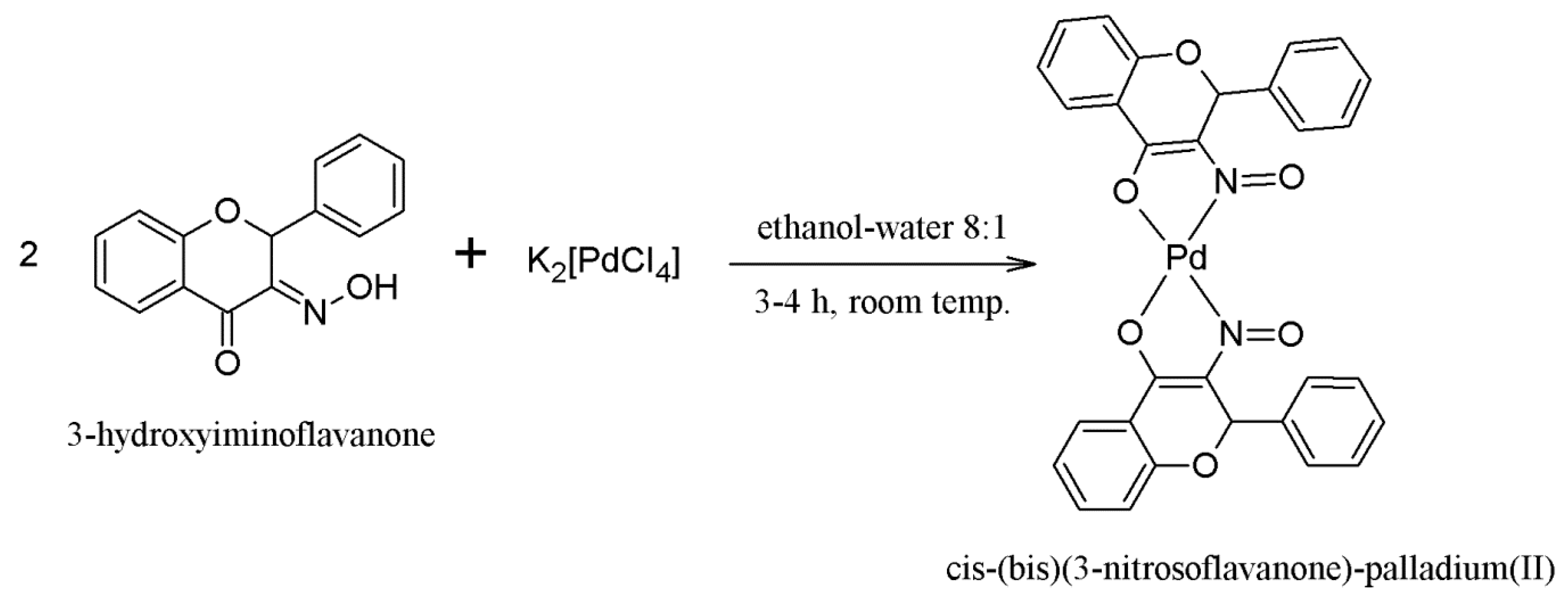
2.1. Synthesis of the Compounds 1 and 2
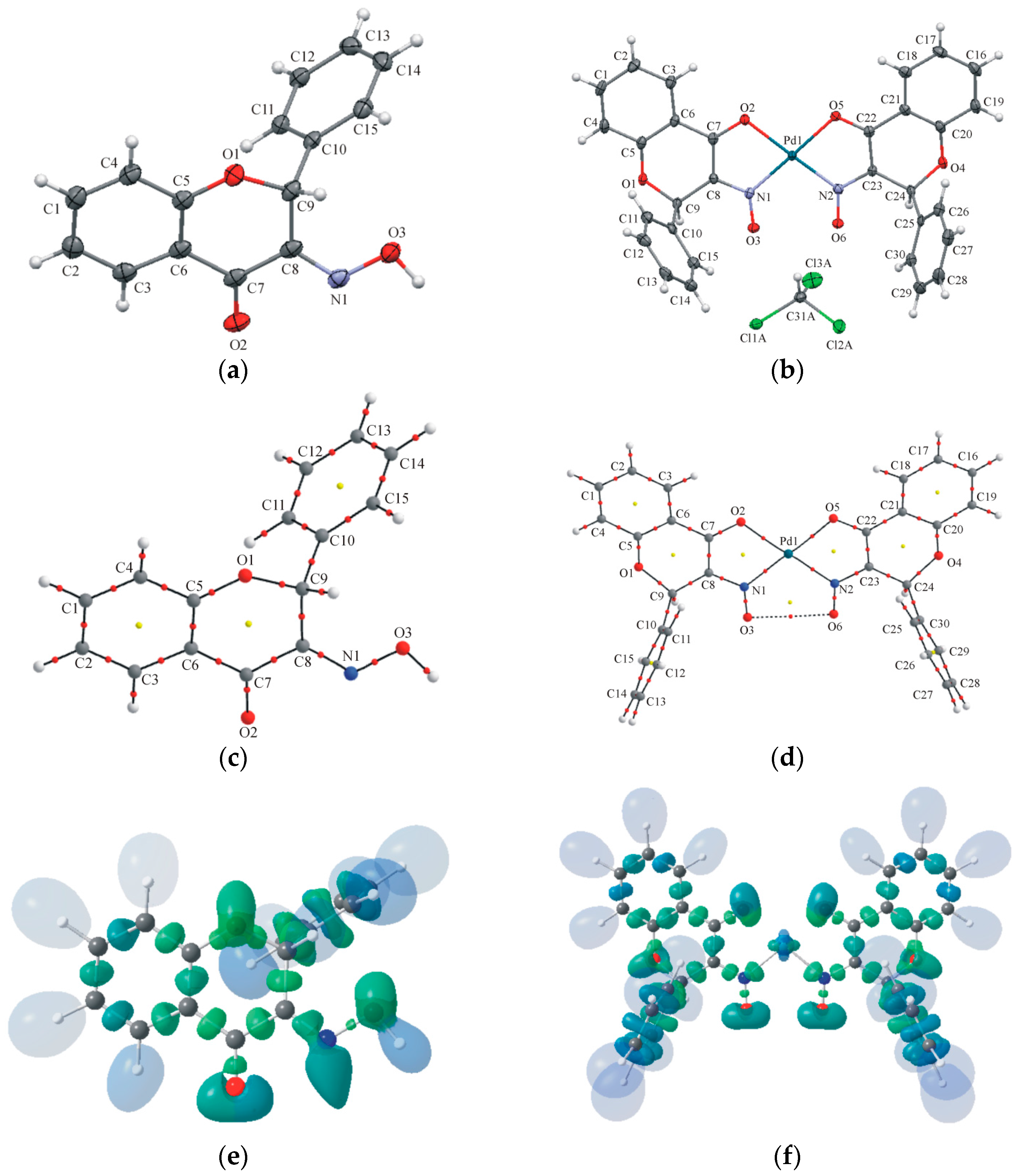
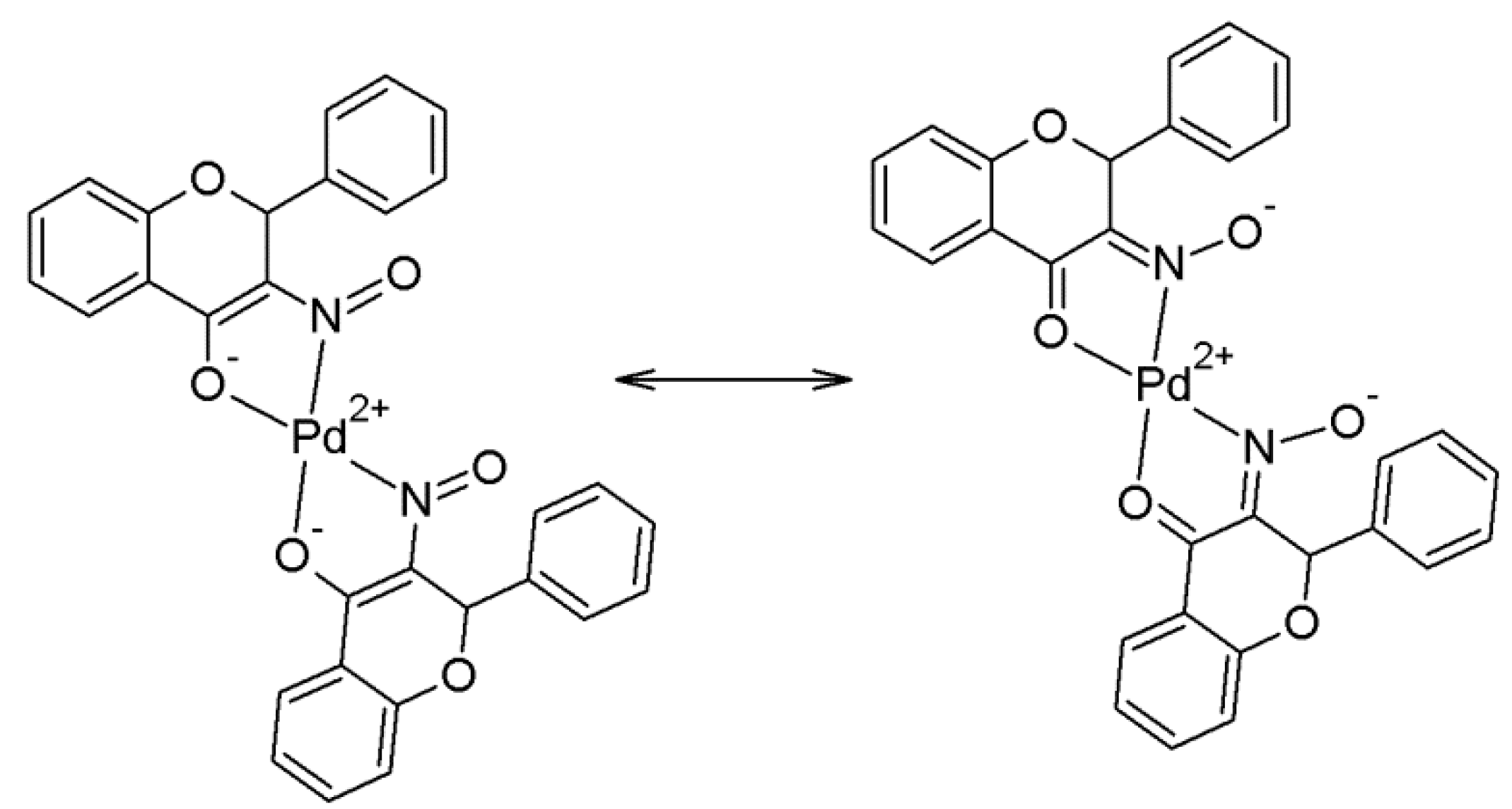
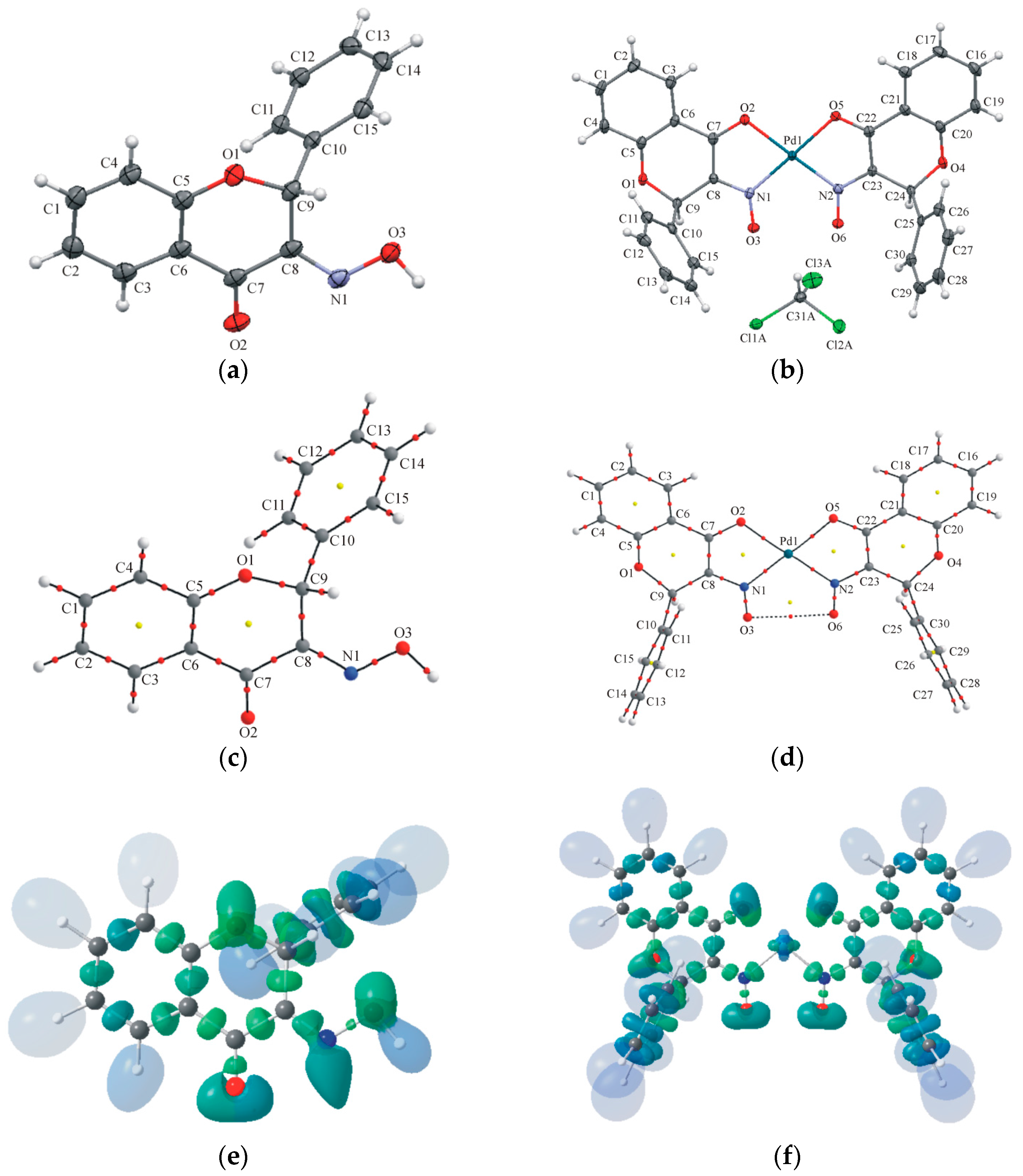
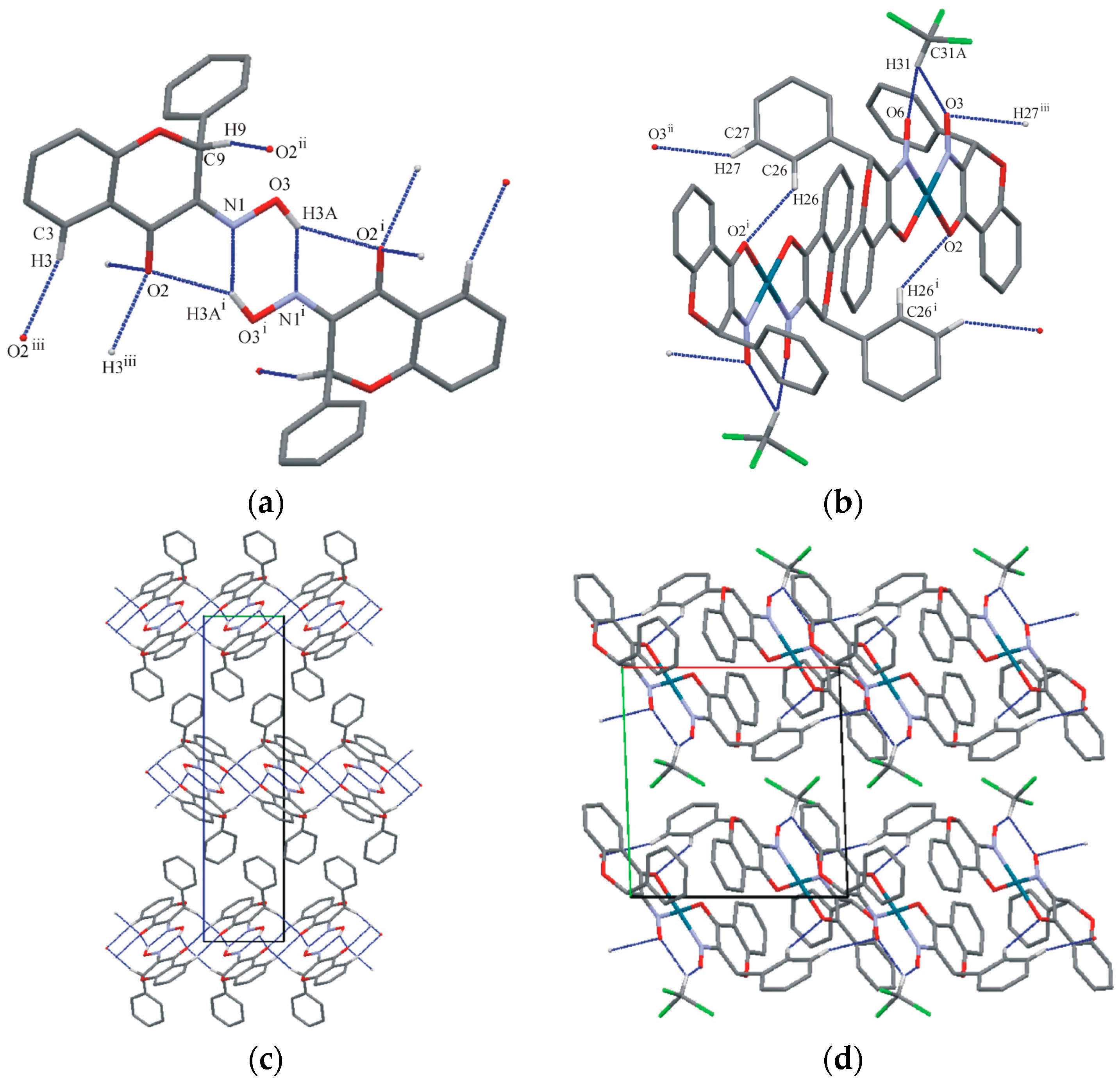
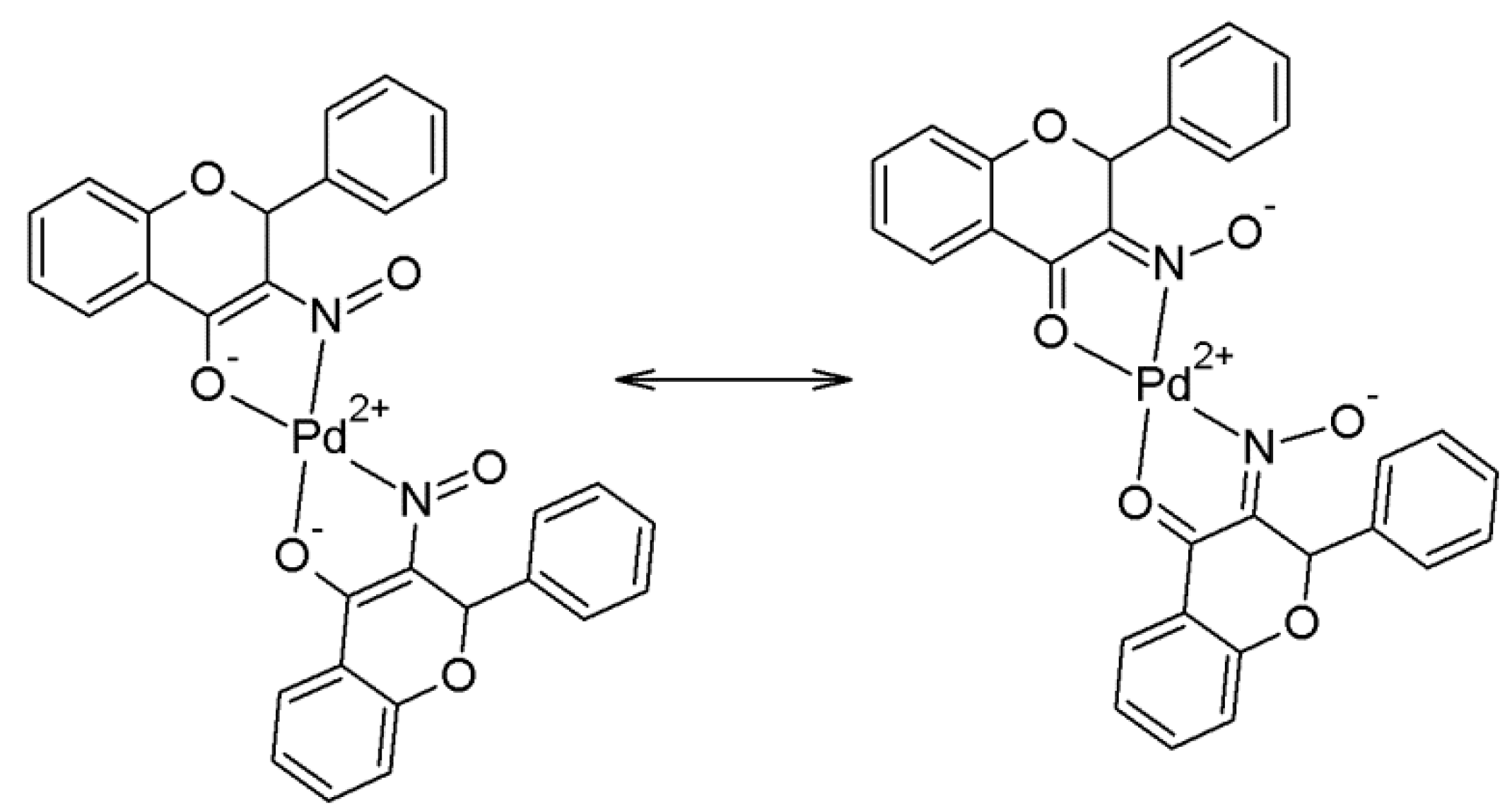
2.2. Crystallography and Theoretical Calculations
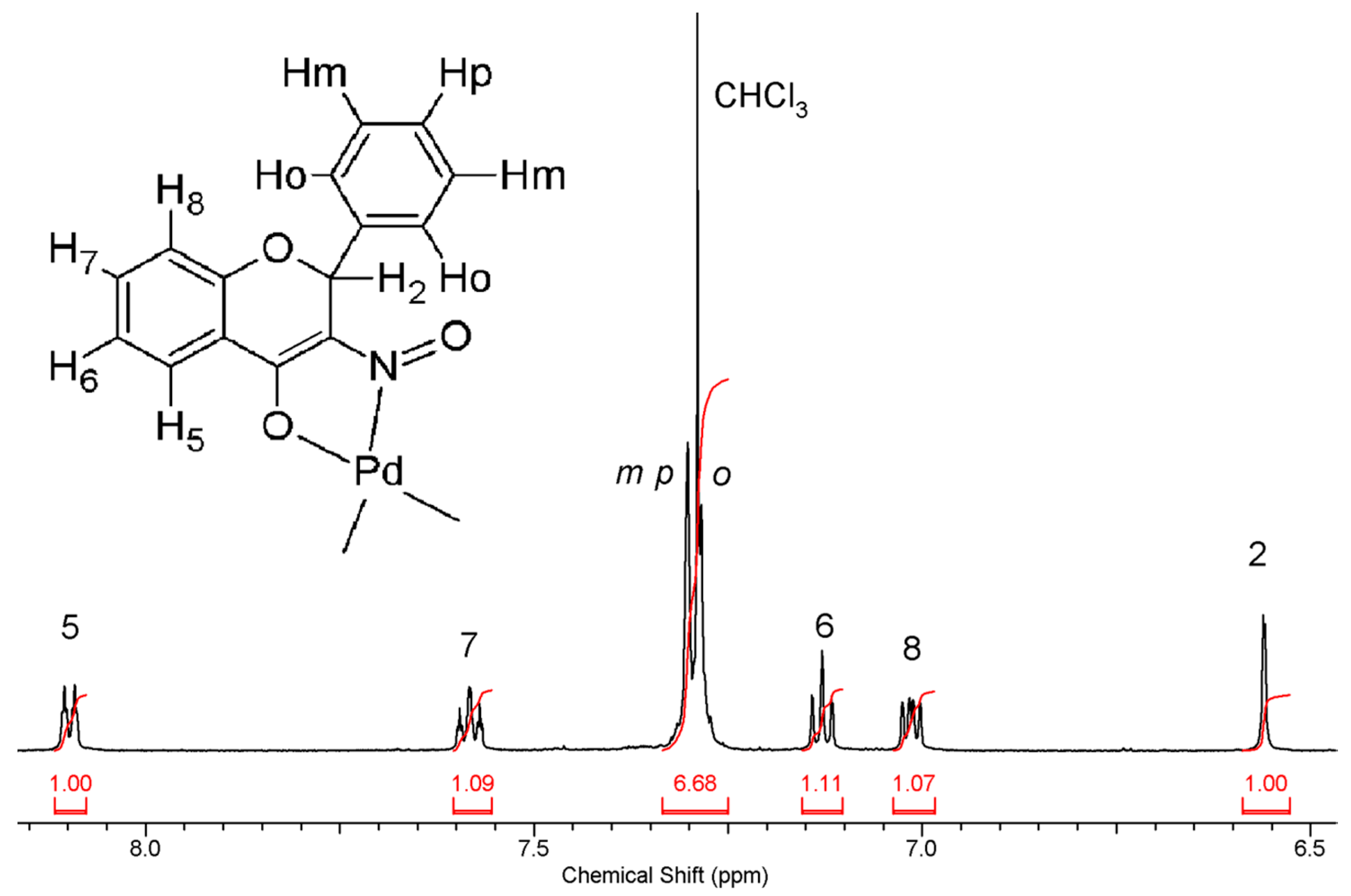
2.3. NMR Spectroscopic Analysis and Solution Stability Studies
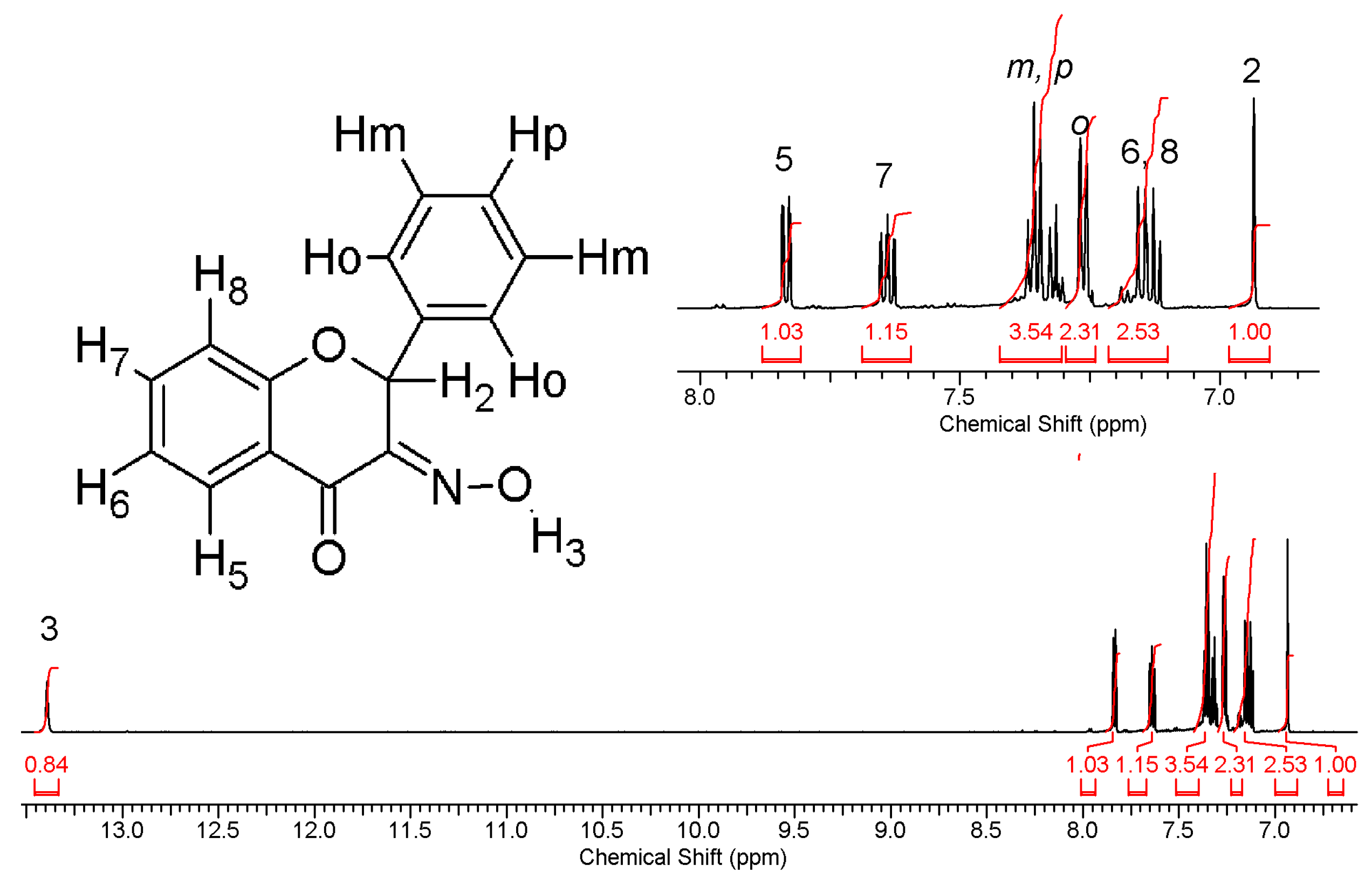
2.3.1. Compound 1
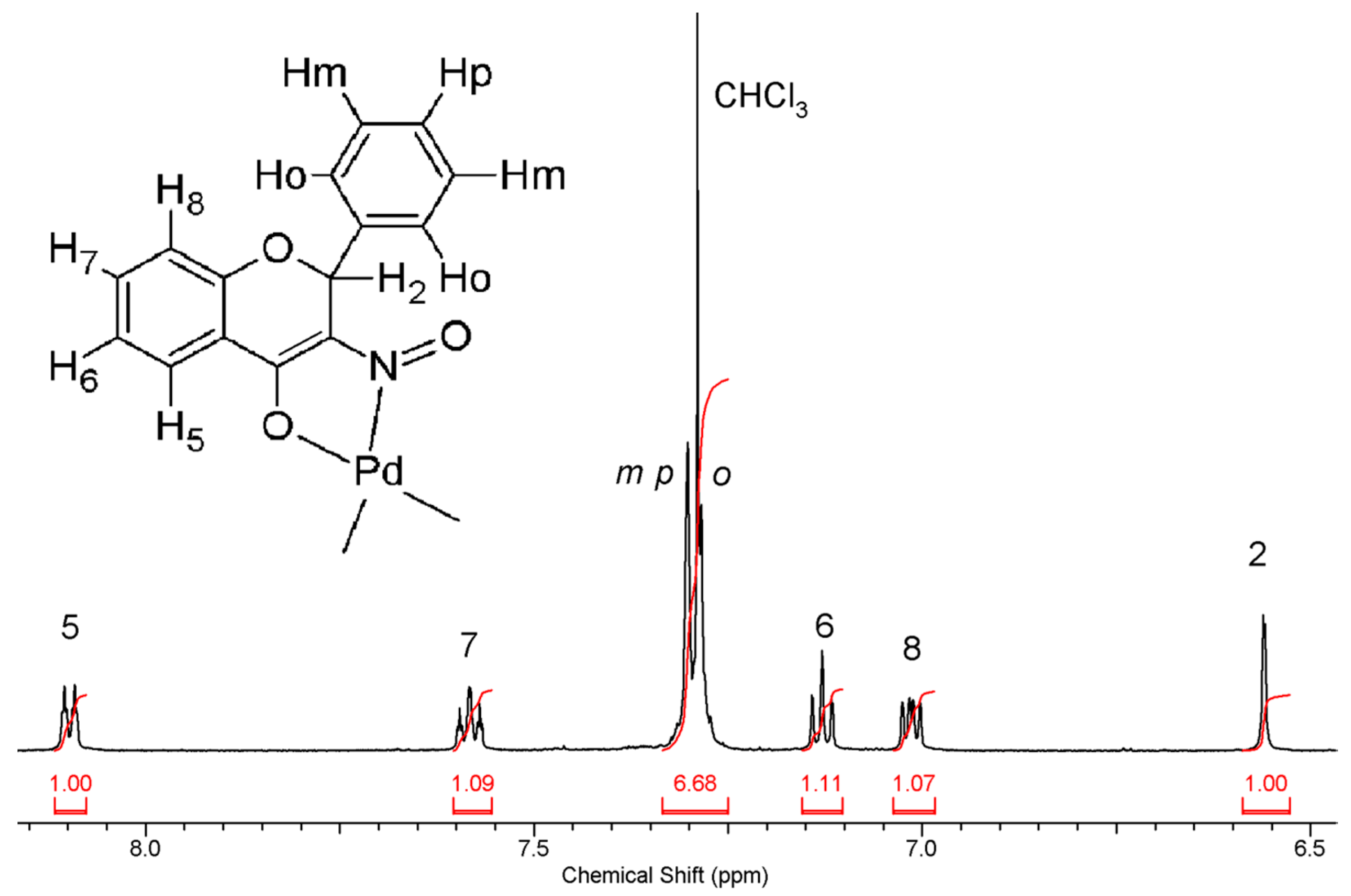
2.3.2. Compound 2
2.4. Cytotoxicity Assay (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide or MTT Assay)
3. Materials and Methods
3.1. Synthetic Procedures
3.1.1. Synthesis of Compound 1—(3-Hydroxyminoflavanone or 3-HIF)
3.1.2. Synthesis of Compound 2—[cis-(bis)(3-Nitrosoflavanone)-palladium(II)]
3.2. Crystallography and Theoretical Calculations
3.2.1. X-ray Measurements and Spherical Structure-Refinements (IAM-Independent-Atom Models)
3.2.2. X-ray Wavefunction Refinement (XWR)
3.2.3. Theoretical Calculations
3.2.4. QTAIM (Quantum Theory of Atoms in Molecules) and ELI-D (Electron Localizability Indicator) Analyses
3.3. NMR Spectroscopic Analysis
3.4. Solution Stability Studies: UV-Vis Spectroscopy
3.5. Cell Culture and Cytotoxicity Assay
3.5.1. Cell Culture
3.5.2. Cytotoxicity Assay (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide or MTT Assay)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 1 | 3-hydroxyiminoflavanone |
| 2 | cis-(bis)(3-nitrosoflavanone)palladium(II) |
| CDDP | cis-diaminnadichloridoplatinum(II) or cisplatin |
| DMF | N′-N′-dimethylformamide |
| DMSO | dimethylsulfoxide |
| ELI-D | Electron Localizability Indicator |
| HAR | Hirshfeld Atom Refinement |
| IAM | Independent Atom Model |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NAMI-A | New Anti-tumor Metastasis Inhibitor or imidazolium trans-DMSO-imidazole-tetrachlororuthenate(III) |
| NMR | Nuclear Magnetic Resonance |
| QTAIM | Quantum Theory of Atoms in Molecules |
| XWR | X-ray wavefunction refinement |
References
- Heffeter, P.; Jungwirth, U.; Jakupec, M.; Hartinger, C.; Galanski, M.; Elbling, L.; Micksche, M.; Keppler, B.; Berger, W. Resistance against novel anticancer metal compounds: Differences and similarities. Drug Resist. Updates 2008, 11. [Google Scholar] [CrossRef] [PubMed]
- Kostova, I. Platinum complexes as anticancer agents. Recent Pat. Anti-Cancer Drug Discov. 2006, 1. [Google Scholar] [CrossRef]
- Zhang, C.X.; Lippard, S.J. New metal complexes as potential therapeutics. Curr. Opin. Chem. Biol. 2003, 7, 481–489. [Google Scholar] [CrossRef]
- Garoufis, A.; Hadjikakou, S.K.; Hadjiliadis, N. Palladium coordination compounds as anti-viral, anti-fungal, anti-microbial and anti-tumor agents. Coord. Chem. Rev. 2009, 253, 1384–1397. [Google Scholar] [CrossRef]
- Iakovidou, Z.; Papageorgiou, A.; Demertzis, M.A.; Mioglou, E.; Mourelatos, D.; Kotsis, A.; Yadav, P.N.; Kovala-Demertzi, D. Platinum(II) and palladium(II) complexes with 2-acetylpyridine thiosemicarbazone: Cytogenetic and antineoplastic effects. Anti-Cancer Drugs 2001, 12, 65–70. [Google Scholar] [CrossRef]
- Zhao, G.H.; Lin, H.K.; Yu, P.; Sun, H.W.; Zhu, S.R.; Su, X.C.; Chen, Y.T. Ethylenediamine-palladium(II) complexes with pyridine and its derivatives: Synthesis, molecular structure and initial antitumor studies. J. Inorg. Biochem. 1999, 73, 145–149. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Budzisz, E.; Keppler, B.K.; Giester, G.; Wozniczka, M.; Kufelnicki, A.; Nawrot, B. Synthesis, crystal structure and biological characterization of a novel palladium(II) complex with a coumarin-derived ligand. Eur. J. Inorg. Chem. 2004, 2004, 4412–4419. [Google Scholar] [CrossRef]
- Kasprzak, M.M.; Erxleben, A.; Ochocki, J. Properties and applications of flavonoid metal complexes. RSC Adv. 2015, 5, 45853–45877. [Google Scholar] [CrossRef]
- Kosmider, B.; Osiecka, R.; Ciesielska, E.; Szmigiero, L.; Zyner, E.; Ochocki, J. Induction of apoptosis and necrosis in lymphocytes by the cis-Pt(II) complex of 3-aminoflavone in comparison with cis-DDP. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2004, 558, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Jakupec, M.A.; Galanski, M.; Keppler, B.K. Tumour-inhibiting platinum complexes—State of the art and future perspectives. Rev. Physiol., Biochem. Pharmacol. 2003, 146, 1–53. [Google Scholar]
- Zyner, E.; Ochocki, J. Platinum(II) and palladium(II) N,O-chelates with substituted flavanone containing ligands. Acta Pol. Pharm. Drug Res. 1999, 56, 159–167. [Google Scholar]
- Kostka, K.; Zyner, E. Structure and protolytic equilibria of 3-hydroxyiminoflavanone. Chem. Anal. 1987, 32, 253–261. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Dittrich, B. X-ray structure refinement using aspherical atomic density functions obtained from quantum-mechanical calculations. Acta Crystallogr. Sect. A 2008, 64, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Capelli, S.C.; Bürgi, H.-B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld atom refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Ha, K. A second monoclinic polymorph of (pyridine-2-carboxaldehyde oximato-κ2N,N′)(pyridine-2-carboxaldehyde oxime-κ2N,N′)palladium(II) chloride. Acta Crystallogr. Sect. E 2012, 68, m176–m177. [Google Scholar] [CrossRef] [PubMed]
- Torabi, A.A.; Soudozi, A.; Welter, R. Crystal structure of (pyridine-2-aldoxime-N,N′)palladium(II) chloride, [Pd(C12H11ClN4O2)]Cl. Z. Kristallogr. NCS 2007, 222, 197–198. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Duax, L.; Norton, D.A. Atlas of Steroid Structure; IFI/Plenum: New York, NY, USA, 1975; Volume 1, pp. 16–22. [Google Scholar]
- Bader, R.F.W. Atoms in molecules. In A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Grabowsky, S.; Luger, P.; Buschmann, J.; Schneider, T.; Schirmeister, T.; Sobolev, A.N.; Jayatilaka, D. The significance of ionic bonding in sulfur dioxide: Bond orders from X-ray diffraction data. Angew. Chem. Int. Ed. 2012, 51, 6776–6779. [Google Scholar] [CrossRef] [PubMed]
- Kohout, M. A measure of electron localizability. Int. J. Quantum Chem. 2004, 97, 651–658. [Google Scholar] [CrossRef]
- Kohout, M.; Wagner, F.R.; Grin, Y. Electron localizability indicator for correlated wavefunctions. III: Singlet and triplet pairs. Theor. Chem. Acc. 2008, 119, 413–420. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Slee, T.S.; Cremer, D.; Kraka, E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5061–5068. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Stephens, M.E. Spatial localization of the electronic pair and number distributions in molecules. J. Am. Chem. Soc. 1975, 97, 7391–7399. [Google Scholar] [CrossRef]
- Fradera, X.; Austen, M.A.; Bader, R.F.W. The Lewis model and beyond. J. Phys. Chem. A 1999, 103, 304–314. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Raub, S.; Jansen, G. A quantitative measure of bond polarity from the electron localization function and the theory of atoms in molecules. Theor. Chem. Acc. 2001, 106, 223–232. [Google Scholar] [CrossRef]
- Vidal, I.; Melchor, S.; Dobado, J.A. On the nature of metal-carbon bonding: AIM and ELF analyses of MCHn (n = 1–3) compounds containing early transition metals. J. Phys. Chem. A 2005, 109, 7500–7508. [Google Scholar] [CrossRef] [PubMed]
- Gonewar, N.R.; Jadhav, V.B.; Jadhav, K.D.; Sarawadekar, R.G. Theoretical calculations of infrared, NMR and electronic spectra of 2-nitroso-1, naphthol or 1–2 naphthoquinine-2 oxime and comparison with experimental data. Res. Pharm. 2012, 2, 18–25. [Google Scholar]
- Belmar, J.; Quezada, J.; Jimenez, C.A.; Diaz-Gallifa, P.; Pasan, J.; Ruiz-Perez, C. Synthesis, crystal structures and tautomerism in novel oximes based on hydroxyalkylpyrazolones. New J. Chem. 2013, 37, 2002–2010. [Google Scholar] [CrossRef]
- Enchev, V.; Angelova, S. Does tautomeric equilibrium exist in 4-nitroso-5-pyrazolones? J. Mol. Struct. Theochem. 2009, 897, 55–60. [Google Scholar] [CrossRef]
- Enchev, V.; Ivanova, G.; Stoyanov, N. Tautomeric and conformational equilibrium of 2-nitrosophenol and 9,10-phenanthrenequinonemonooxime: Ab initio and NMR study. J. Mol. Struct. Theochem. 2003, 640, 149–162. [Google Scholar] [CrossRef]
- Enchev, V.; Ivanova, G.; Ugrinov, A.; Neykov, G.D. Tautomeric and conformational equilibrium of acenaphthenequinonemonooxime. J. Mol. Struct. 1999, 508, 149–161. [Google Scholar] [CrossRef]
- Fabijanska, M.; Studzian, K.; Szmigiero, L.; Rybarczyk-Pirek, A.J.; Pfitzner, A.; Cebula-Obrzut, B.; Smolewski, P.; Zyner, E.; Ochocki, J. Trans-platinum(II) complex of 3-aminoflavone—Synthesis, X-ray crystal structure and biological activities in vitro. Dalton Trans. 2015, 44, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska-Lis, U.; Ochocki, J.; Matlawska-Wasowska, K. Trans geometry in platinum antitumor complexes. Coord. Chem. Rev. 2008, 252, 1328–1345. [Google Scholar] [CrossRef]
- Rademaker-Lakhai, J.M.; van den Bongard, D.; Pluim, D.; Beijnen, J.H.; Schellens, J.H. A Phase I and pharmacological study with imidazolium-trans-DMSO-imidazole-tetrachlororuthenate, a novel ruthenium anticancer agent. Clin. Cancer Res. 2004, 10, 3717–3727. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Gasparoli, L.; Stefanini, M.; Ristori, M.; D’Amico, M.; Alessio, E.; Scaletti, F.; Becchetti, A.; Arcangeli, A.; Messori, L. NAMI-A is highly cytotoxic toward leukaemia cell lines: Evidence of inhibition of KCa 3.1 channels. Dalton Trans. 2014, 43, 12150–12155. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis PRO; Ver. 1.171.36.32; Agilent Technologies: Yarnton, UK, 2013.
- CrysAlis RED; Ver. 1.171.32.29; Oxford Diffraction: Abingdon, UK, 2008.
- Sheldrick, G.M. A short history of shelx. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic-behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Grimwood, D.J. Tonto: A fortran based object-oriented system for quantum chemistry and crystallography. In Computational Science—ICCS 2003; Part 4; Springer: New York, 2003; Volume 2660, pp. 142–151. [Google Scholar]
- Peterson, K.A.; Figgen, D.; Dolg, M.; Stoll, H. Energy-consistent relativistic pseudopotentials and correlation consistent basis sets for the 4d elements Y-Pd. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Rev. D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Keith, T.A. AIMAll (Ver. 14.11.23); TK Gristmill Software: Overland Park, KS, USA, 2014. [Google Scholar]
- Kohout, M. Dgrid-4.6. Available online: http://www.cpfs.mpg.de/~kohout/dgrid.html (accessed on 15 May 2015).
- Hübschle, C.B.; Luger, P. Moliso—A program for colour-mapped iso-surfaces. J. Appl. Crystallogr. 2006, 39, 901–904. [Google Scholar] [CrossRef]
- Fischer, S.J.; Benson, L.M.; Fauq, A.; Naylor, S.; Windebank, A.J. Cisplatin and dimethyl sulfoxide react to form an adducted compound with reduced cytotoxicity and neurotoxicity. Neurotoxicology 2008, 29, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Ahmed, K.J.; Hollis, L.S.; Lippard, S.J. Solvolysis reactions of cis- and trans-diamminedichloroplatinum(ii) in dimethyl sulfoxide. Structural characterization and DNA binding of trans-bis(ammine)chloro(DMSO)platinum(1+). Inorg. Chem. 1987, 26, 1524–1528. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1 and 2 are available from the authors.
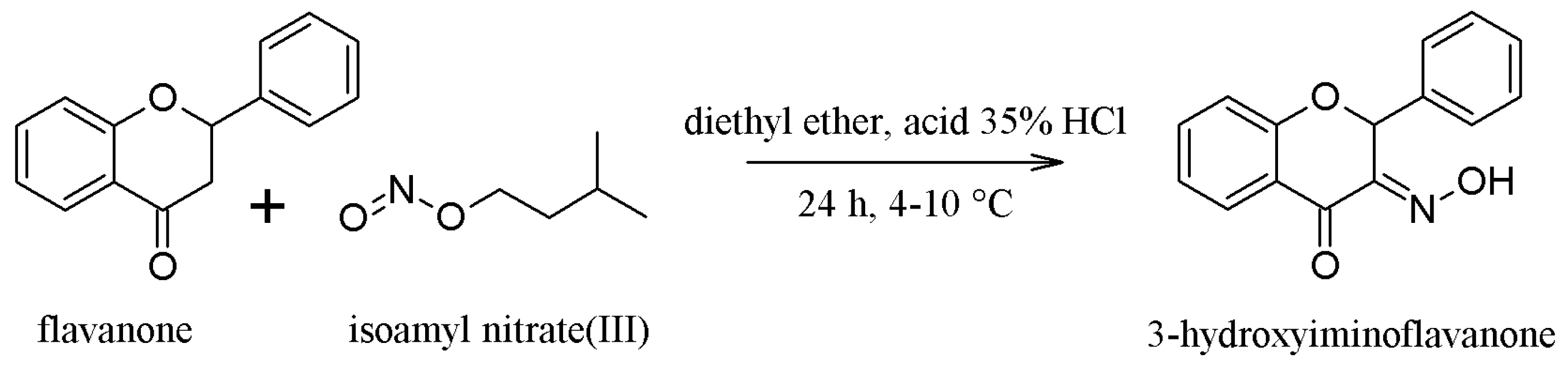



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
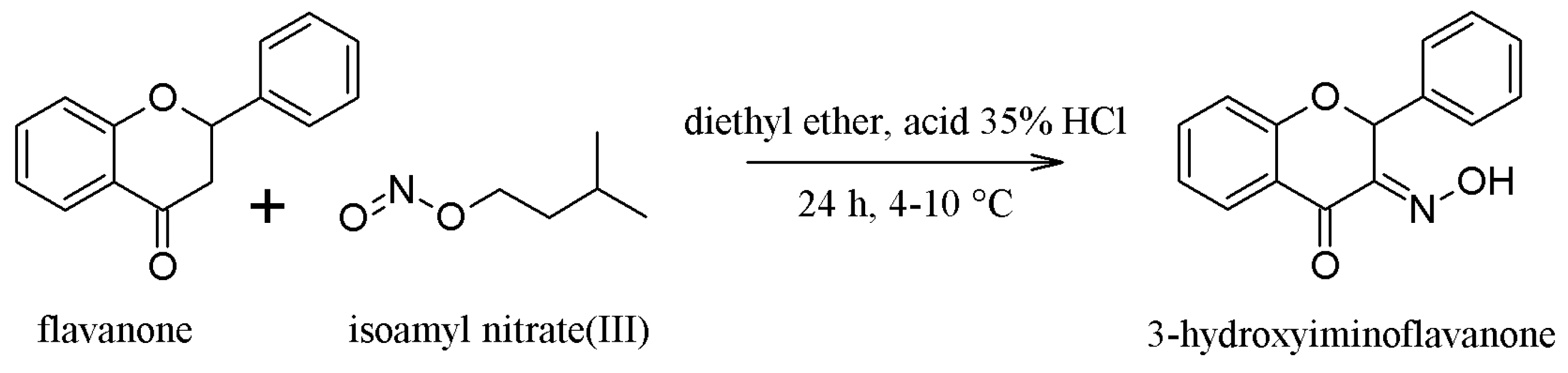{kind=link}
{kind=link}
| 1 | 2 | |
|---|---|---|
| Empirical formula | C15H11NO3 | C30H20N2O6Pd·CHCl3 |
| Formula weight | 253.25 | 730.25 |
| Crystal system, space group | Monoclinic, P21/n | Triclinic, P |
| a, b, c (Å) | 8.6182(17), 5.7715(12), 23.468(5) | 10.3894(7), 10.9265(8), 13.1736(8) |
| α, β, γ (°) | 90.00, 94.06(3), 90.00 | 83.848(6), 80.996(5), 86.933(6) |
| V (Å3) | 1164.4(4) | 1467.50(17) |
| Z | 4 | 2 |
| Dx (Mg/m3) | 1.445 | 1.653 |
| Crystal size (mm) | 0.25 × 0.06 × 0.03 | 0.24 × 0.13 × 0.07 |
| Crystal habit and color | Plate, colorless | Plate, red |
| λ (Å) | 0.71073 (MoKα) | 0.71073 (MoKα) |
| μ (mm−1) | 0.10 | 0.95 |
| T (K) | 100(2) | 100(2) |
| Measured/unique reflections | 18731/3055 | 12395/6905 |
| Rint | 0.021 | 0.023 |
| θ range (°) | 2.5–29.0 | 3.1–28.0 |
| Completeness to θmax (%) | 98.9 | 97.2 |
| IAM model (spherical) | ||
| Observed reflections [I > 2σ(I)] | 2741 | 5695 |
| Data/restraints/parameters | 3055/0/176 | 6905/0/429 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| S | 1.112 | 0.927 |
| R [I > 2σ(I)], wR [I > 2σ(I)] | 0.0452, 0.1009 | 0.0257, 0.0517 |
| R (all data), wR (all data) | 0.0497, 0.1034 | 0.0351, 0.0527 |
| (Δ/σ)max | <0.001 | 0.001 |
| Δρmax (eÅ−3), Δρmin (eÅ−3) | 0.40, −0.19 | 0.50, −0.75 |
| XWR: HAR model (aspherical) | ||
| Refinement method | on F with weight = 1/σ(|F|) | |
| Functional/basis-set | blyp/cc-pVTZ | |
| Data/restraints/parameters | 2974/0/216 | |
| χ2 | 10.17 | |
| S | 3.19 | |
| R (F), wR (F) | 0.0340, 0.0233 | |
| XWR: XCW (fitting procedure) | ||
| Refinement method | on F with weight = 1/σ(|F|) | |
| Functional/basis-set | blyp/cc-pVTZ | |
| Data/restraints/parameters | 2974/0/1 | |
| χ2 (λ = 0.10) | 6.42 | |
| S | 2.53 | |
| R (F), wR (F) | 0.0303, 0.0192 | |
| Interaction | D-H | H···A | D···A | D-H···A |
|---|---|---|---|---|
| 1 | ||||
| O3-H3A···O2 i | 0.96(2) | 2.49(2) | 3.3359(18) | 146(2) |
| O3-H3A···N1 i | 0.96(2) | 1.98(2) | 2.7996(18) | 142(2) |
| C9-H9···O2 ii | 0.98 | 2.41 | 3.283(2) | 147 |
| C3-H3···O2 iii | 0.93 | 2.46 | 3.297(2) | 149 |
| C1-H1···Cg ii,b | 0.93 | 2.79 | 3.628(2) | 150 |
| 2 | ||||
| C31-H31···O3 | 0.99(2) | 2.26(2) | 2.990(4) | 130(2) |
| C31-H31···O6 | 0.99(2) | 1.29(3) | 2.964(4) | 124(2) |
| C26-H26···O2 i | 0.93 | 2.51 | 3.259(2) | 137 |
| C27-H27···O3 ii | 0.93 | 2.63 | 3.395(2) | 140 |
| Bond | d | ρbcp | ∇2ρbcp | ε | G/ρbcp | H/ρbcp | δ | VELI | ELIpop | Ymax | ΔELI | RJI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (1-XWR) | ||||||||||||
| O1-C5 | 1.3520(6) | 1.94 | −7.6 | 0.04 | 1.15 | −1.43 | 0.96 | 1.16 | 1.39 | 1.56 | 0.06 | 78.4 |
| O1-C9 | 1.4405(6) | 1.61 | −9.6 | 0.07 | 0.87 | −1.29 | 0.80 | 1.21 | 1.40 | 1.58 | 0.06 | 82.3 |
| O2-C7 | 1.2218(5) | 2.75 | −10.9 | 0.16 | 1.42 | −1.70 | 1.37 | 4.28 | 2.29 | 1.54 | 0.04 | 73.2 |
| O3-N1 | 1.3641(5) | 2.24 | −7.6 | 0.03 | 0.67 | −0.90 | 1.34 | 0.56 | 0.99 | 1.46 | 0.02 | 55.0 |
| N1-C8 | 1.2856(6) | 2.57 | −22.8 | 0.39 | 0.92 | −1.54 | 1.50 | 6.88 | 3.08 | 1.71 | 0.17 | 69.2 |
| C5-C6 | 1.4026(6) | 2.09 | −23.3 | 0.25 | 0.36 | −1.14 | 1.25 | 7.92 | 2.87 | 1.82 | 0.03 | 54.2 |
| C6-C7 | 1.4614(7) | 1.96 | −21.7 | 0.12 | 0.27 | −1.05 | 1.04 | 4.84 | 2.38 | 1.95 | 0.01 | 50.8 |
| C7-C8 | 1.4839(7) | 1.89 | −20.2 | 0.15 | 0.26 | −1.01 | 0.95 | 5.22 | 2.40 | 1.97 | 0.03 | 52.1 |
| C8-C9 | 1.5015(7) | 1.79 | −17.9 | 0.15 | 0.26 | −0.96 | 0.94 | 3.82 | 2.21 | 1.95 | 0.05 | 55.9 |
| (2-OPT) | ||||||||||||
| N1-Pd1 | 2.032 | 0.80 | 8.2 | 0.07 | 1.07 | −0.36 | 0.76 | 6.42 | 2.53 | 1.77 | 0.09 | 94.5 |
| O2-Pd1 | 2.112 | 0.57 | 8.6 | 0.04 | 1.27 | −0.22 | 0.56 | 2.80 | 2.02 | 1.61 | 0.03 | 97.1 |
| O1-C5 | 1.364 | 1.97 | −14.1 | 0.05 | 0.95 | −1.45 | 0.97 | 1.23 | 1.47 | 1.59 | 0.05 | 76.1 |
| O1-C9 | 1.485 | 1.51 | −11.3 | 0.03 | 0.61 | −1.14 | 0.83 | 1.10 | 1.24 | 1.56 | 0.06 | 76.4 |
| O2-C7 | 1.286 | 2.40 | −15.5 | 0.07 | 1.14 | −1.60 | 1.19 | 1.94 | 1.82 | 1.57 | 0.01 | 73.7 |
| N1-O3 | 1.249 | 3.15 | −21.7 | 0.06 | 0.74 | −1.22 | 1.66 | 0.90 | 1.50 | 1.50 | 0.02 | 56.8 |
| N1-C8 | 1.359 | 2.18 | −19.6 | 0.34 | 0.77 | −1.40 | 1.22 | 3.86 | 2.51 | 1.72 | 0.05 | 75.1 |
| C5-C6 | 1.420 | 2.05 | −22.2 | 0.20 | 0.32 | −1.08 | 1.23 | 7.68 | 2.79 | 1.87 | 0.04 | 53.5 |
| C7-C6 | 1.458 | 1.92 | −19.8 | 0.14 | 0.27 | −0.99 | 1.07 | 5.57 | 2.46 | 1.95 | 0.03 | 53.2 |
| C8-C7 | 1.423 | 2.07 | −22.6 | 0.22 | 0.31 | −1.07 | 1.14 | 10.21 | 3.16 | 1.90 | 0.01 | 56.7 |
| C8-C9 | 1.508 | 1.74 | −16.0 | 0.09 | 0.24 | −0.88 | 0.95 | 3.51 | 2.16 | 1.98 | 0.02 | 52.4 |
| Cell Line | 1 IC50 (µM) | 1 15 µM (%) a | 2 15 µM (%) a | CDDP 15 µM (%) a | CDDP IC50 (µM) |
|---|---|---|---|---|---|
| A2780 | 57 ± 3 | ni | 54 ± 9 b | <10 c | 0.6 ± 0.03 |
| A2780cis | 50 ± 6 | ni | 65 ± 3 | 65 ± 1 | 20.2 ± 0.7 |
| A549 | 79 ± 13 | ni | 71 ± 13 b | 21 ± 1.5 | 3.9 ± 0.5 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasprzak, M.; Fabijańska, M.; Chęcińska, L.; Szmigiero, L.; Ochocki, J. New Look on 3-Hydroxyiminoflavanone and Its Palladium(II) Complex: Crystallographic and Spectroscopic Studies, Theoretical Calculations and Cytotoxic Activity. Molecules 2016, 21, 455. https://doi.org/10.3390/molecules21040455
Kasprzak M, Fabijańska M, Chęcińska L, Szmigiero L, Ochocki J. New Look on 3-Hydroxyiminoflavanone and Its Palladium(II) Complex: Crystallographic and Spectroscopic Studies, Theoretical Calculations and Cytotoxic Activity. Molecules. 2016; 21(4):455. https://doi.org/10.3390/molecules21040455
Chicago/Turabian StyleKasprzak, Maria, Małgorzata Fabijańska, Lilianna Chęcińska, Leszek Szmigiero, and Justyn Ochocki. 2016. "New Look on 3-Hydroxyiminoflavanone and Its Palladium(II) Complex: Crystallographic and Spectroscopic Studies, Theoretical Calculations and Cytotoxic Activity" Molecules 21, no. 4: 455. https://doi.org/10.3390/molecules21040455
APA StyleKasprzak, M., Fabijańska, M., Chęcińska, L., Szmigiero, L., & Ochocki, J. (2016). New Look on 3-Hydroxyiminoflavanone and Its Palladium(II) Complex: Crystallographic and Spectroscopic Studies, Theoretical Calculations and Cytotoxic Activity. Molecules, 21(4), 455. https://doi.org/10.3390/molecules21040455