
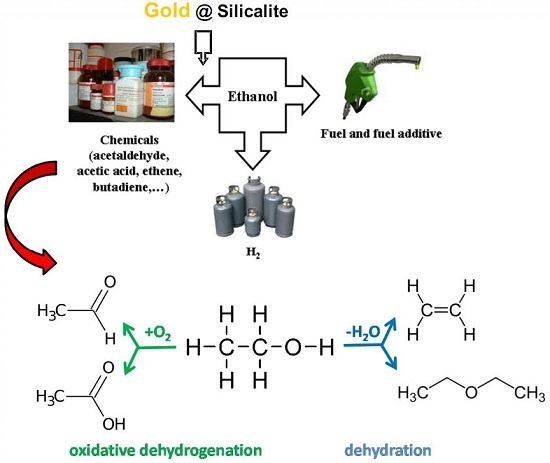
Clean Transformation of Ethanol to Useful Chemicals. The Behavior of a Gold-Modified Silicalite Catalyst



Abstract
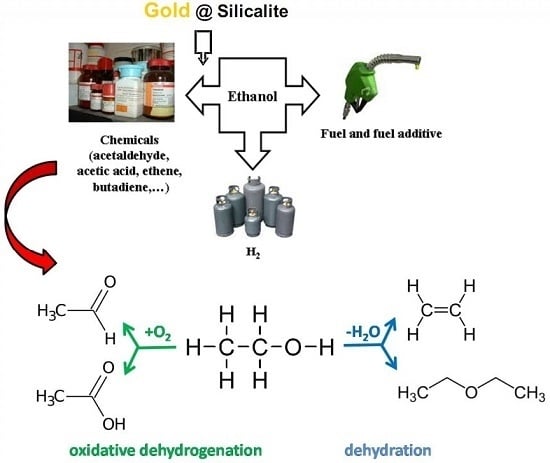
:
1. Introduction
2. Results and Discussion
2.1. Catalytic Tests
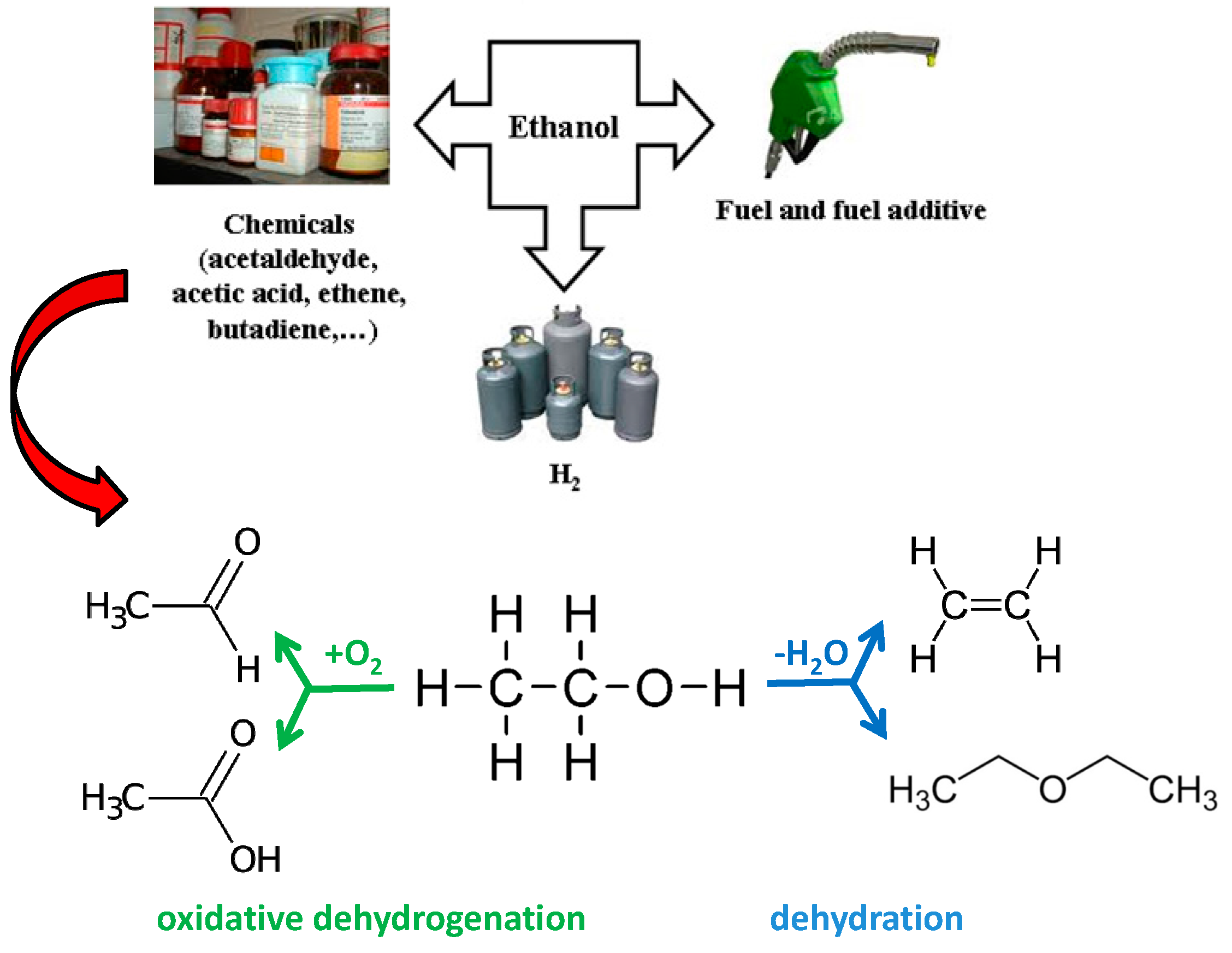
2.1.1. Ethanol Dehydration to Ethene and Diethyl Ether
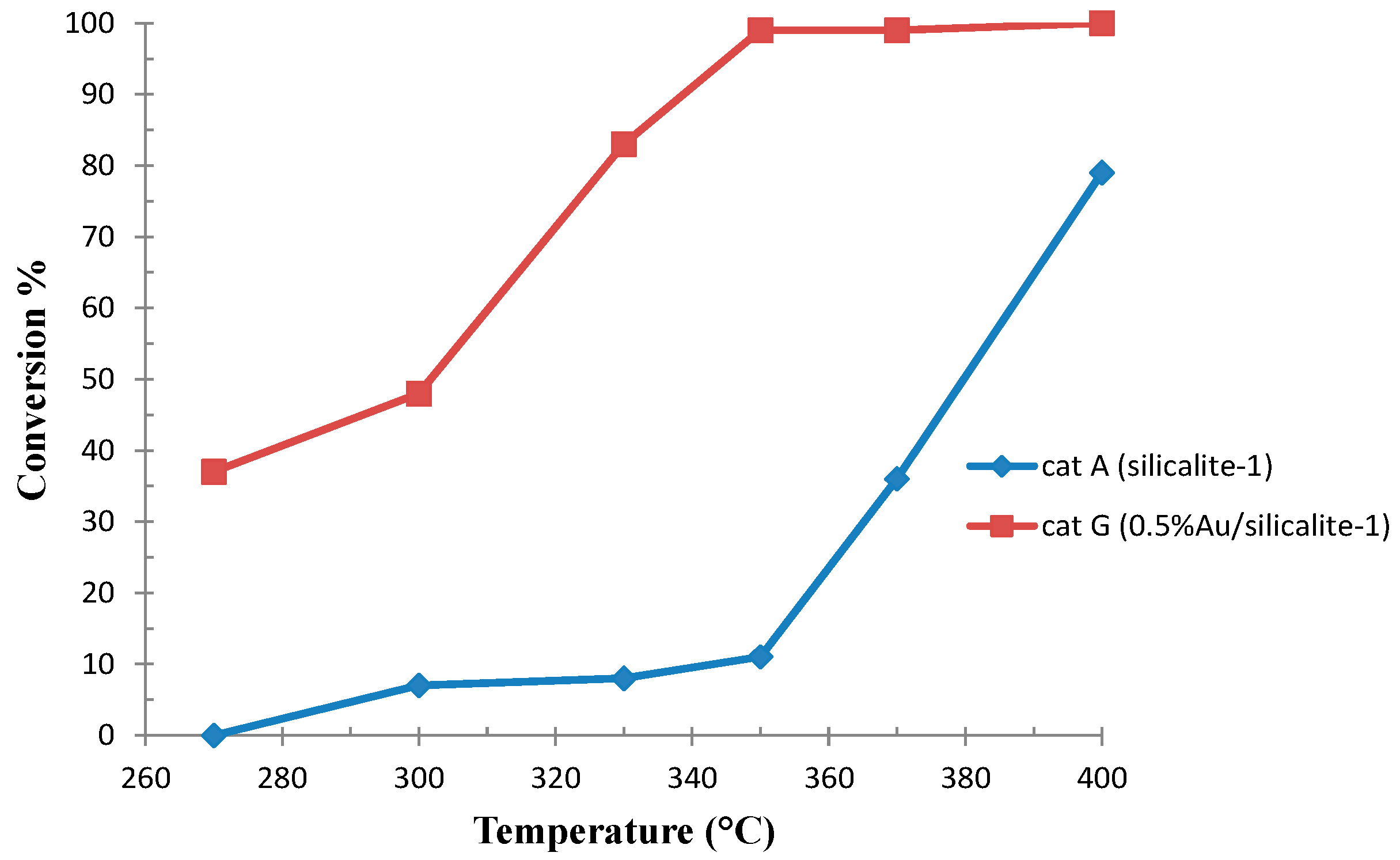
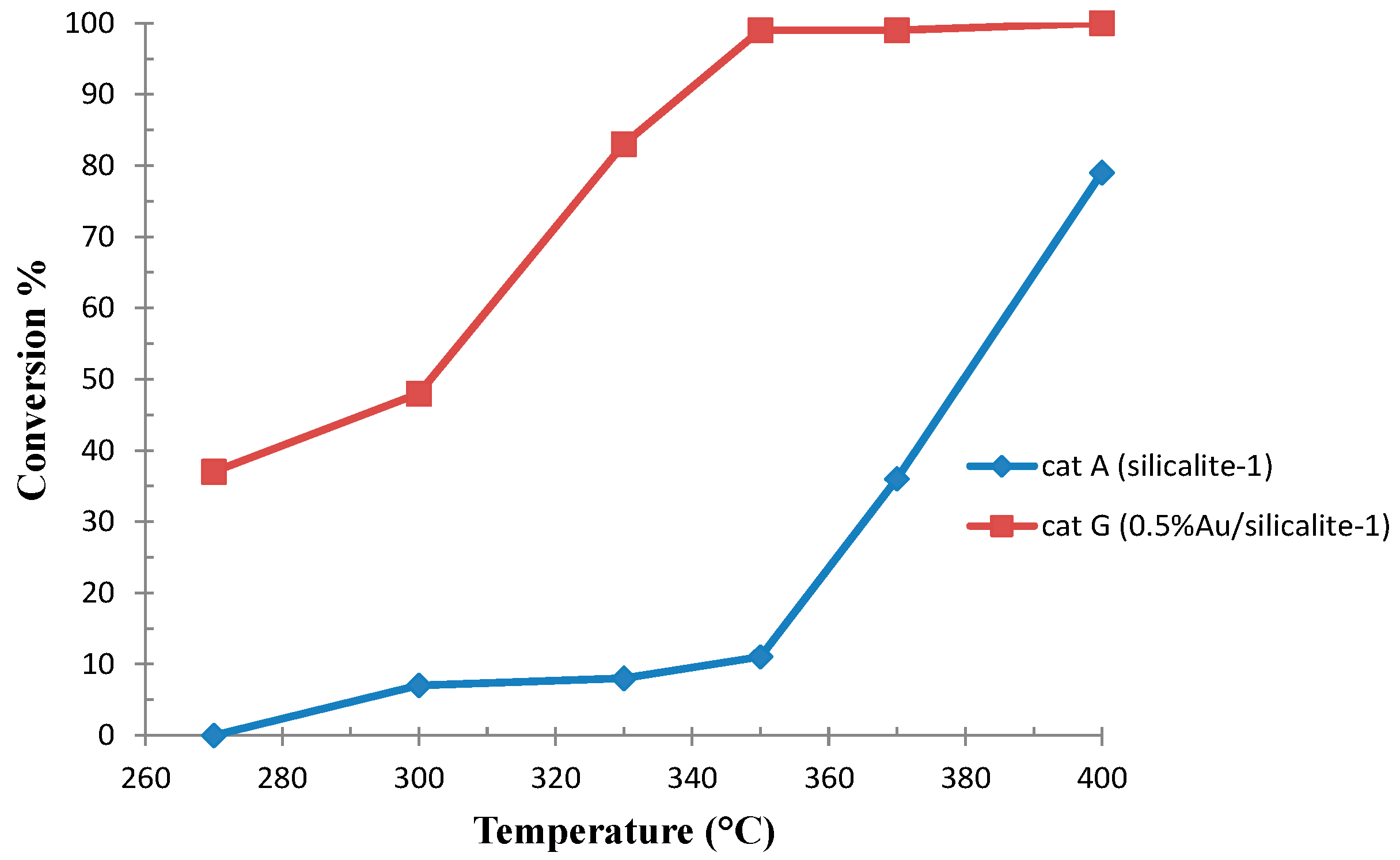
2.1.2. Ethanol Oxidative Dehydrogenation to Acetaldehyde and Oxidation to Acetic Acid
2.2. Catalyst Characterization
3. Materials and Methods
3.1. Reagents and Instruments
3.2. Catalyst Preparation
- cat A: 3 g silicalite-1 was heated under air at 350 °C for 4 h;
- cat B: as cat A but impregnated with diluted HCl (0.03 mmol/g);
- cat C: as cat A but impregnated with concentrated HCl (3 mmol/g);
- cat D: as cat A but impregnated with CH3COOK·3H2O (0.3 mmol/g);
- cat E: cat B after 2 h on stream at 300 °C, thereafter impregnated with CH3COOK·3H2O (0.3 mol/g);
- cat F: cat E after 2 h on stream at 300 °C, thereafter impregnated with HCl (0.3 mmol/g);
- cat G: 3 g of cat A were impregnated with 1 mL HAuCl4 solution (Au = 15 mg/mL) thereafter heated under air at 350 °C for 2 h to produce 0.5% Au/silicalite-1;
- cat H: as cat G but with the addition of 228 mg CH3COOK·3H2O into HAuCl4 solution (CH3COOK)/Au = 10, molar ratio);
- cat I: as cat G but with the addition of 517 mg CH3COOK·3H2O into HAuCl4 solution (CH3COOK)/Au = 20, molar ratio);
- cat L: as cat G but with the addition of 1293 mg CH3COOK·3H2O into HAuCl4 solution;
- cat M: as cat I but impregnated with 1 mL HAuCl4 solution (Au = 490 mg/mL) to produce 16.3% Au/silicalite-1.
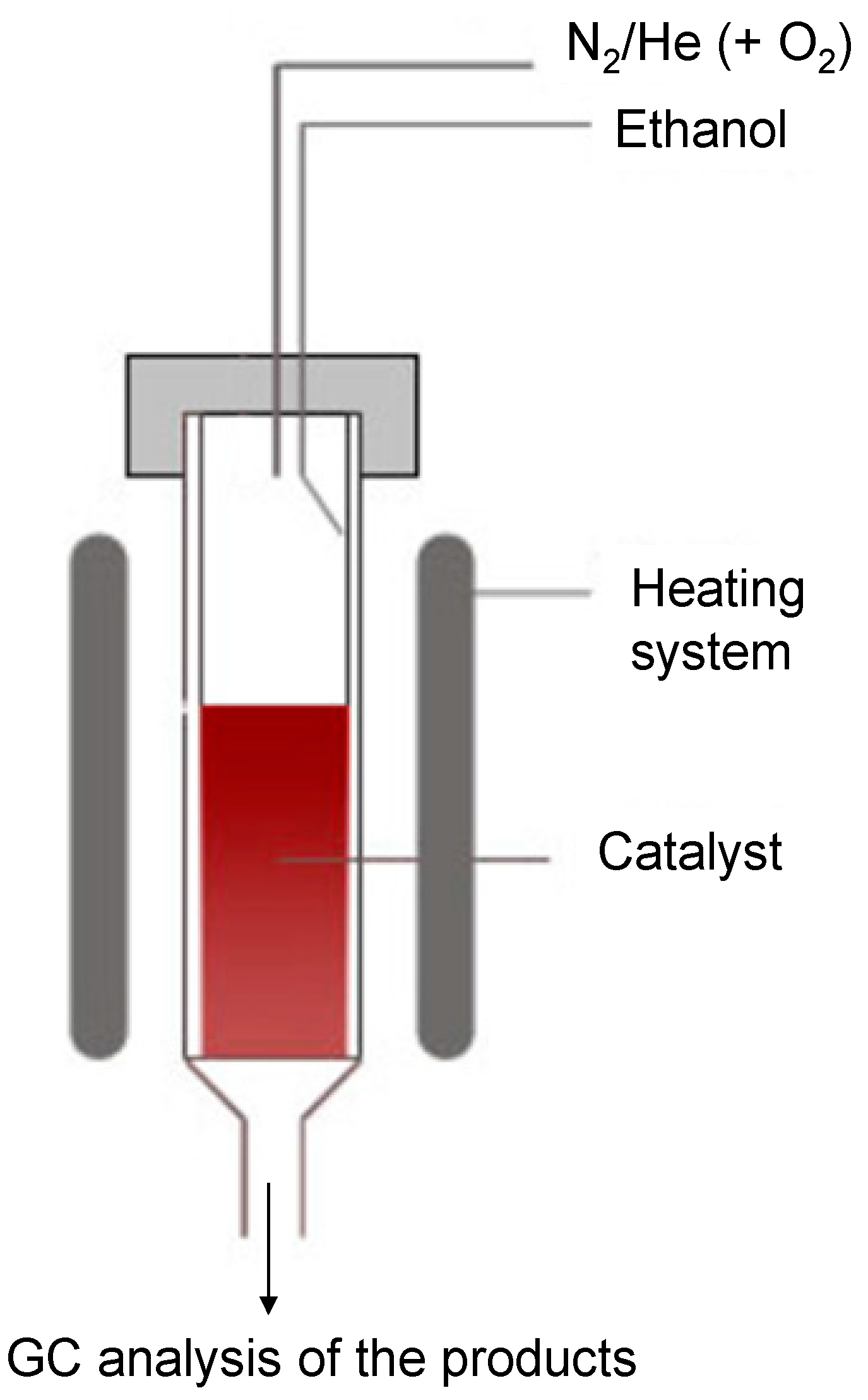
3.3. Catalytic Test Apparatus
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- IEA (International Energy Agency). Biofuels for Transport-An International Perspective; International Energy Agency: Paris, France, 2004. [Google Scholar]
- Jensen, K.H.; Thyø, K.A. 2nd Generation Bioethanol for Transport: The IBUS Concept. Master’s Thesis, Technical University of Denmark, Kongens Lyngby, Copenhagen, Denmark, 2007. [Google Scholar]
- Wang, M. An Update of Energy and Greenhouse Emission Impacts of Fuel Ethanol. In Proceedings of the 10th Annual National Ethanol Conference, Scottsdale, AZ, USA, 8 February 2005.
- Larsen, U.; Johansen, T.; Schramm, J. Ethanol as a Fuel for Road Transportation; IEA (International Energy Agency)-AMF (Advanced Motor Fuels); 2009; Available online: http://www.iea-amf.org/app/.../AMF_Annex_35-1.pdf (accessed on May 2009).
- Teles, J.H.; Hermans, I.; Franz, G.; Sheldon, R.A. Oxidation. In Ulmann’s Encyclopedia of Industrial Chemistry, 7th ed.; Barbara Elvers Editor-in Chief; Wiley-VCH: Weiheim, Germany, 2015; pp. 29–30. [Google Scholar]
- Hromatka, O.; Ebner, H. Vinegar by submerged oxidative fermentation. Ind. Eng. Chem. 1959, 51, 1279–1280. [Google Scholar] [CrossRef]
- Partridge, E.P. Acetic Acid and Cellulose Acetate in the United States: A General Survey of Economic and Technical Developments. Ind. Eng. Chem. 1931, 23, 482–498. [Google Scholar] [CrossRef]
- Hromatka, O.; Ebner, H. Investigations on vinegar fermentation: generator for vinegar fermentation and aeration procedures. Enzymologia 1949, 13, 369–387. [Google Scholar]
- Haynes, A. Catalytic methanol carbonylation. Adv. Catal. 2010, 53, 1–45. [Google Scholar]
- Elliott, P.I.P.; Haak, S.; Meijer, A.J.; Meijer, H.M.; Sunley, G.J.; Haynes, A. Reactivity of Ir(III) carbonyl complexes with water: alternative by-product formation pathways in catalytic methanol carbonylation. Dalton Trans. 2013, 42, 16538–16546. [Google Scholar] [CrossRef] [PubMed]
- Sunley, G.J.; Watson, D.J. High productivity methanol carbonylation catalysis using iridium-The CativaTM process for the manufacture of acetic acid. Catal. Today 2000, 58, 293–307. [Google Scholar] [CrossRef]
- Thayer, A.M. Biocatalysis. In C&EN: Cover Story; American Chemical Society: Washington, DC, USA, 2001; Volume 79, pp. 27–34. [Google Scholar]
- Serrano, D.P.; Calleja, G.; Botas, J.A.; Gutierrez, F.J. Characterization of adsorptive and hydrophobic properties of silicalite-1, ZSM-5, TS-1 and Beta zeolites by TPD techniques. Separ. Purif. Technol. 2007, 54, 1–9. [Google Scholar] [CrossRef]
- Yoshitake, H.; Aoki, Y.; Hemmi, S. Mesoporous titania supported-molybdenum catalysts: The formation of a new mesophase and use in ethanol-oxygen catalytic reactions. Micropor. Mesopor. Mat. 2006, 93, 294–303. [Google Scholar] [CrossRef]
- Srihari, V.; Viswanath, D.S. Vapor phase oxidation of ethanol over thorium molybdate catalyst. J. Catal. 1976, 43, 43–52. [Google Scholar] [CrossRef]
- Velu, S.; Wang, L.; Okazaki, M.; Suzuki, K.; Tomura, S. Characterization of MCM-41 mesoporous molecular sieves containing copper and zinc and their catalytic performance in the selective oxidation of alcohols to aldehydes. Micropor. Mesopor. Mat. 2002, 54, 113–126. [Google Scholar] [CrossRef]
- Castro, P.F.; Viola, M.C.; Pedregosa, J.C.; Gomez, M.F.; Abello, M.C. Oxidative dehydrogenation of ethanol over MgCrO catalysts. J. Argentine Chem. Soc. 2009, 97, 242–249. [Google Scholar]
- Prasad, R. Highly active copper chromite catalyst produced by thermal decomposition of ammoniac copper oxalate chromate. Mat. Lett. 2005, 59, 3945–3949. [Google Scholar] [CrossRef]
- Ten Brink, G.-J.; Arends, I.W.C.E.; Sheldon, R.A. Green, catalytic oxidation of alcohols in water. Science 2000, 287, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on platinum metal catalysts in aqueous solutions. Catal. Today 1994, 19, 247–283. [Google Scholar] [CrossRef]
- Ebitani, K.; Fujie, Y.; Kaneda, K. Immobilization of a ligand-preserved giant palladium cluster on a metal oxide surface and its nobel heterogeneous catalysis for oxidation of allylic alcohols in the presence of molecular oxygen. Langmuir 1999, 15, 3557–3562. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Mizuno, N. Supported ruthenium catalyst for the heterogeneous oxidation of alcohols with molecular oxygen. Angew Chem. Int. Ed. 2002, 41, 4538–4542. [Google Scholar] [CrossRef]
- Xu, C.; Shen, P.K.; Liu, Y. Ethanol electrooxidation on Pt/C and Pd/C catalysts promoted with oxide. J. Power Sources 2007, 164, 527–553. [Google Scholar] [CrossRef]
- Prati, L.; Rossi, M. Gold on carbon as a new catalyst for selective liquid phase oxidation of diols. J. Catal. 1998, 176, 552–560. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Catalysis by gold. Catal. Rev. Sci. Eng. 1999, 41, 319–388. [Google Scholar] [CrossRef]
- Biella, S.; Rossi, M. Gas phase oxidation of alcohols to aldehydes or ketones catalysed by supported gold. Chem. Commun. 2003, 378–379. [Google Scholar] [CrossRef]
- Comotti, M.; Della Pina, C.; Matarrese, R.; Rossi, M. The catalytic activity of “naked” gold particles. Angew Chem. Int. Ed. 2004, 43, 5812–5815. [Google Scholar] [CrossRef] [PubMed]
- Sheng, P.-Y.; Bowmaker, G.A.; Idriss, H. The reactions of ethanol over Au/CeO2. Appl. Catal. A 2004, 261, 171–181. [Google Scholar] [CrossRef]
- Hughes, M.D.; Xu, Y.-J.; Jenkins, P.; McMorn, P.; Landon, P.; Enache, D.I.; Carley, A.F.; Attard, G.-A.; Hutchings, G.J.; King, F.; et al. Tunable gold catalysts for selective hydrocarbon oxidation under mild conditions. Nature 2005, 437, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Hutchings, G.J. Gold catalysis. Angew Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Christensen, C.H.; Jørgersen, B.; Rass-Hassen, J.; Egeblad, K.; Madsen, R.; Klitgaard, S.K.; Hansen, S.M.; Hansen, M.R.; Andersen, H.C.; Riisager, A. Formation of acetic acid by aqueous-phase oxidation of ethanol with air in the presence of a heterogeneous gold catalyst. Angew. Chem. Int. Ed. 2006, 45, 4648–4651. [Google Scholar] [CrossRef] [PubMed]
- Milone, C.; Crisafulli, C.; Ingoglia, R.; Schipilliti, L.; Galvagno, S. A comparative study on the selective hydrogenation of α,β unsaturated aldehyde and ketone to unsaturated alcohols on Au supported catalysts. Catal. Today 2007, 122, 341–351. [Google Scholar] [CrossRef]
- Jørgensen, B.; Christensen, S.E.; Thomsen, M.L.D.; Christensen, C.H. Aerobic oxidation of aqueous ethanol using heterogeneous gold catalysts: Efficient routes to acetic acid and ethyl acetate. J. Catal. 2007, 251, 332–337. [Google Scholar] [CrossRef]
- Della Pina, C.; Falletta, E.; Prati, L.; Rossi, M. Selective oxidation using gold. Chem. Soc. Rev. 2008, 37, 2077–2095. [Google Scholar] [CrossRef] [PubMed]
- Della Pina, C.; Falletta, E.; Rossi, M. Highly selective oxidation of benzyl alcohol to benzaldehyde catalyzed by bimetallic gold-copper catalyst. J. Catal. 2008, 260, 384–386. [Google Scholar] [CrossRef]
- Chen, Z.; Della Pina, C.; Falletta, E.; Lo Faro, M.; Pasta, M.; Rossi, M.; Santo, N. Facile synthesis of polyaniline using gold catalyst. J. Catal. 2008, 259, 1–4. [Google Scholar] [CrossRef]
- Corma, A.; Garcia, H. Supported gold nanoparticles as catalysts for organic reactions. Chem. Soc. Rev. 2008, 37, 2096–2126. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.-Q.; Luo, S.-W.; Xu, N.; Xu, B.-Q. Gold nanosize effect in Au/SiO2 for selective ethanol oxidation in aqueous solution. Catal. Lett. 2008, 124, 238–242. [Google Scholar] [CrossRef]
- Zhu, B.; Lazar, M.; Trewyn, B.G.; Angelici, R.J. Aerobic oxidation of amines to imines catalyzed by bulk gold powder and by alumina-supported gold. J. Catal. 2008, 260, 1–6. [Google Scholar] [CrossRef]
- Della Pina, C.; Falletta, E.; Lo Faro, M.; Pasta, M.; Rossi, M. Gold-catalysed synthesis of polypyrrole. Gold Bull. 2009, 42, 27–33. [Google Scholar] [CrossRef]
- Guan, Y.; Hensen, E.J.M. Ethanol dehydrogenation by gold catalysts: The effect of the gold particle size and the presence of oxygen. Appl. Catal. A 2009, 361, 49–56. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Homogeneous gold catalysis beyond assumptions and proposals-characterized intermediates. Angew Chem. Int. Ed. 2010, 49, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Lothschtz, C.; Ackermann, M.; Doepp, R.; Anantharaman, S.; Marchetti, B.; Bertagnolli, H.; Rominger, F. Gold catalysis: In situ EXAFS study of homogeneous oxidative esterification. Chem. Eur. J. 2010, 16, 8012–8019. [Google Scholar] [CrossRef] [PubMed]
- Della Pina, C.; Falletta, E.; Gervasini, A.; Teles, J.H.; Rossi, M. Catalytic transformation of ethanol with silicalite-1: Influence of pretreatments and conditions on activity and selectivity. Chem. Cat. Chem. 2010, 2, 1587–1593. [Google Scholar] [CrossRef]
- Zhou, Y.; Angelici, R.J.; Woo, L.K. Bulk gold-catalyzed reactions of diazoalkanes with amines and O2 to give enamines. Catal. Lett. 2010, 137, 8–15. [Google Scholar] [CrossRef]
- Han, D.; Xu, T.; Xu, X.; Ding, Y. Gas-phase selective oxidation of benzyl alcohol to benzaldehyde with molecular oxygen over unsupported nanoporous gold. Chem. Cat. Chem. 2010, 2, 383–386. [Google Scholar] [CrossRef]
- Della Pina, C.; Falletta, E.; Rossi, M. Update on selective oxidation using gold. Chem. Soc. Rev. 2012, 41, 350–369. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, M.P.; Carabineiro, S. The best of two worlds from the gold catalysis universe: Making homogeneous heterogeneous. Chem. Cat. Chem. 2012, 4, 18–29. [Google Scholar] [CrossRef]
- Liu, P.; Hensen, E.J.M. Highly efficient and robust Au/MgCuCr2O4 catalyst for gas-phase oxidation of ethanol to acetaldehyde. J. Am. Chem. Soc. 2013, 135, 14032–14035. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Dual gold catalysis. Acc. Chem. Res. 2014, 47, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Wang, D.; Su, D.S.; Prati, L. New challenges in gold catalysis: Bimetallic systems. Catal. Sci. Technol. 2015, 5, 55–68. [Google Scholar] [CrossRef]
- Rossi, M.; Della Pina, C.; Falletta, E. Gold nanomaterials: From preparation to pharmaceutical design and application. Curr. Pharm. Des. 2016, 22, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Herron, N.; Schwarz, S.; Druliner, J.D. Gold Catalyst for Selective Oxidation. WO 2002016298 A1, 23 February 2002. [Google Scholar]
- Baker, M.J.; Johnston, P.; Murphy, D. Process for the Preparation of a Metal-Impregnated Microspheroidal Catalyst. WO 2003061829 A1, 31 July 2003. [Google Scholar]
- Wang, T.; Wade, L.E.; Wong, V.; Sokolovskii, V. Layered Support Material for Catalysts for Manufacture of Alkenyl Alkanoates. WO 2005065821 A1, 21 July 2005. [Google Scholar]
- Rossi, M.; Della Pina, C.; Falletta, E. Process for the Preparation of 3-Hydroxypropionic Acid and Related Salts. Italian Patent Application N. 98 MI2006A001326–UNIMI, 7 July 2006. [Google Scholar]
- Carter, M.K. Catalytic Conversion of Alcohols to Aldehydes and Ketones. U.S. Patent 20140046098 A1, 13 February 2014. [Google Scholar]
- Sample Availability: No sample of catalysts are available.
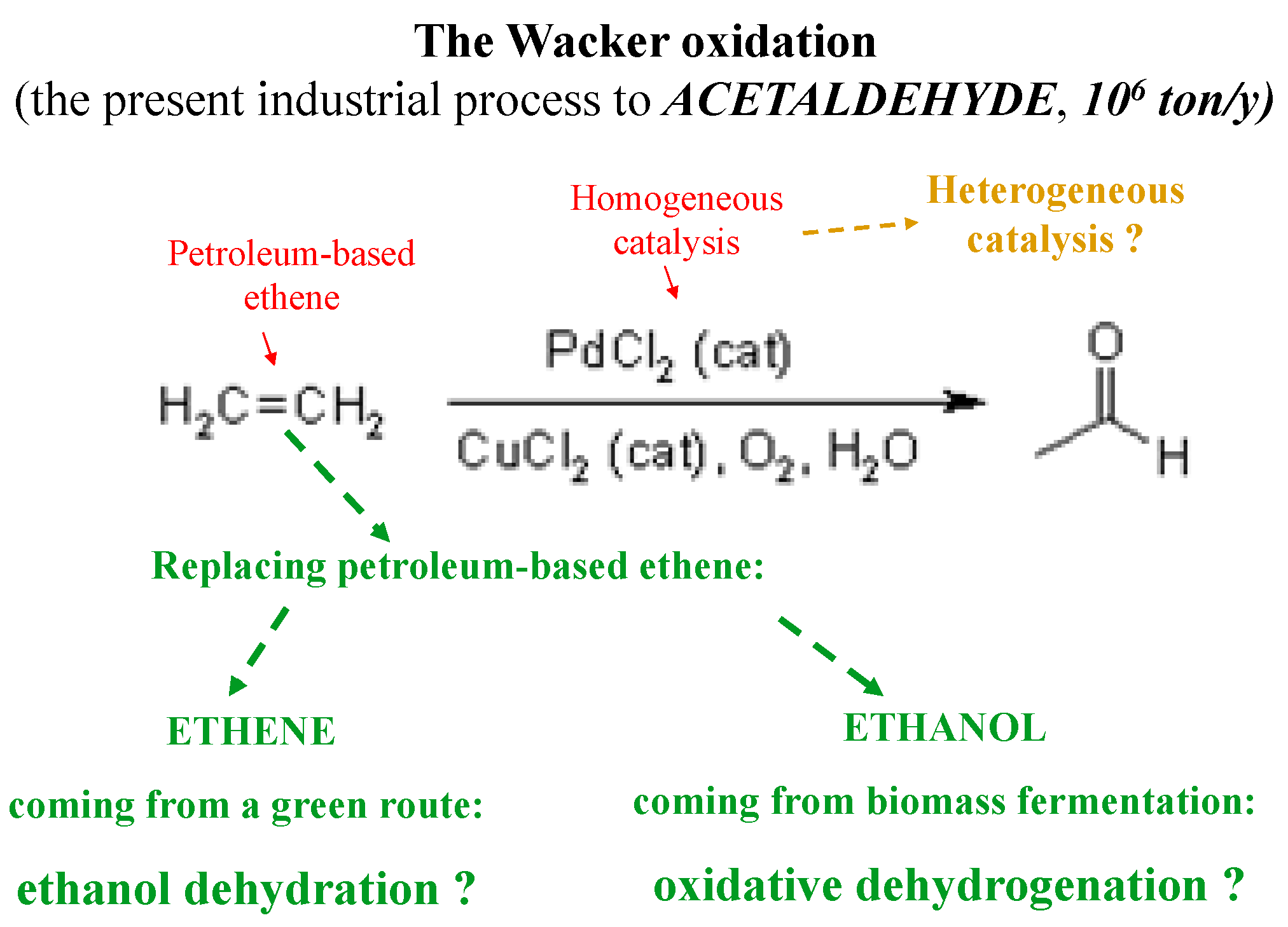



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Cat A | Cat B | Cat C | Cat D | Cat E | Cat F | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T (°C) | 300 | 400 | 240 | 300 | 240 | 300 | 300 | 400 | 300 | 400 | 240 | 400 |
| Conv. % | 7 | 79 | 39 | 100 | 58 | 100 | 0 | 10 | 5 | 42 | 36 | 100 |
| Sel Et2O % | 82 | 0 | 98 | 0 | 82 | 0 | 0 | 0 | 0 | 0 | 96 | 0 |
| Sel C2H4 % | 18 | 98 | 2 | 100 | 18 | 100 | 0 | 56 | 100 | 68 | 4 | 100 |
| Sel CH3CHO % | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 42 | 0 | 32 | 0 | 0 |
| Exp | O2/EtOH (mol ratio) | T (°C) | Conv. (%) | Selectivity (%) | ||||
|---|---|---|---|---|---|---|---|---|
| CH3CHO | CH3COOH | CO2 | Et2O | C2H4 | ||||
| 1 | 0 | 270 | 37 | 6 | 0 | 0 | 90 | 2 |
| 2 | 0.3 | 300 | 48 | 2 | 0 | 0 | 77 | 8 |
| 3 | 330 | 83 | 2 | 0 | 0 | 7 | 91 | |
| 4 | 350 | 99 | 2 | 0 | 0 | 0 | 98 | |
| 5 | 370 | 99 | 3 | 0 | 0 | 0 | 97 | |
| 6 | 0 | 400 | 100 | 4 | 0 | 0 | 0 | 96 |
| Exp | Catalysts | O2/EtOH (Mol Ratio) | T (°C) | Conv. (%) | Selectivity (%) | ||
|---|---|---|---|---|---|---|---|
| CH3CHO | CH3COOH | CO2 | |||||
| 7 | H | 0 | 270 | 40 | 100 | 0 | 0 |
| 8 | 0.3 | 68 | 86 | 13 | 1 | ||
| 9 | I | 0 | 19 | 100 | 0 | 0 | |
| 10 | 0.3 | 71 | 97 | 2 | 1 | ||
| 11 | L | 0 | 4 | 100 | 0 | 0 | |
| 12 | 0.3 | 46 | 72 | 21 | 7 | ||
| 13 | I | 0 | 300 | 41 | 100 | 0 | 0 |
| 14 | 0.3 | 75 | 86 | 12 | 2 | ||
| 15 | 0 | 350 | 54 | 100 | 0 | 0 | |
| O2/EtOH | T (°C) | Conversion % | Selectivity % | |||
|---|---|---|---|---|---|---|
| CH3CHO | CH3COOH | AcOEt | Others | |||
| 0.7 | 250 | 100 | 24 | 61 | 13 | 2 |
| 1.0 | 100 | 6 | 79 | 11 | 4 | |
| 1.5 | 100 | 4 | 78 | 9 | 9 | |
| 0.5 | 95 | 53 | 28 | 18 | 1 | |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falletta, E.; Rossi, M.; Teles, J.H.; Della Pina, C. Clean Transformation of Ethanol to Useful Chemicals. The Behavior of a Gold-Modified Silicalite Catalyst. Molecules 2016, 21, 379. https://doi.org/10.3390/molecules21030379
Falletta E, Rossi M, Teles JH, Della Pina C. Clean Transformation of Ethanol to Useful Chemicals. The Behavior of a Gold-Modified Silicalite Catalyst. Molecules. 2016; 21(3):379. https://doi.org/10.3390/molecules21030379
Chicago/Turabian StyleFalletta, Ermelinda, Michele Rossi, Joaquim Henrique Teles, and Cristina Della Pina. 2016. "Clean Transformation of Ethanol to Useful Chemicals. The Behavior of a Gold-Modified Silicalite Catalyst" Molecules 21, no. 3: 379. https://doi.org/10.3390/molecules21030379