1. Introduction
Marine fungi have played an important role for drug discovery as a source of structurally unique and biologically active secondary metabolites. Among marine fungi, sponge-derived fungi take up a large proportion of marine fungal diversity, and produce numerous new bioactive compounds which display promising biological and pharmacological properties, such as antiviral, antibacterial, antitumor, antifouling, anti-inflammatory, and immunomodulatory activities [
1,
2,
3,
4]. Recently, we reported 12 new chromone derivatives isolated from the sponge-derived fungus
Corynespora cassiicola XS-200900I7, collected from the Xisha Islands coral reef in the South China Sea [
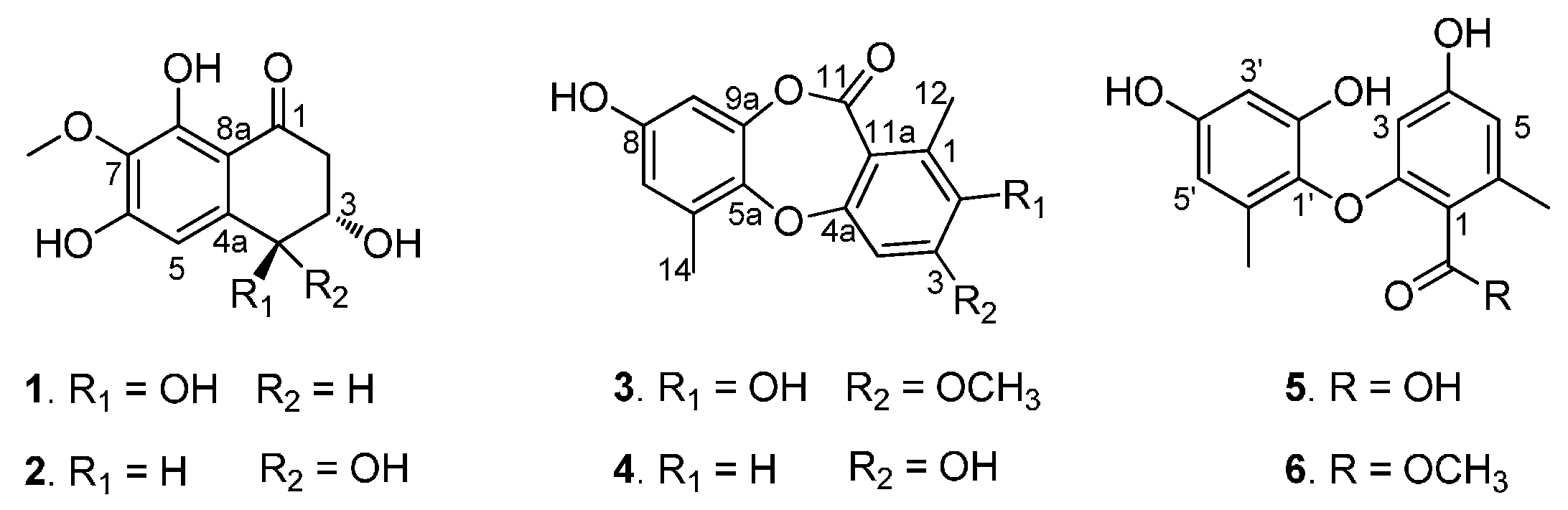
5], which encouraged us to continue to study this fungal strain. The further chemical investigation of this strain led to the isolation of two new naphthalenones, corynenones A and B (
1 and
2), and one new depsidone, corynesidone E (
3), together with three known compounds, corynesidone A (
4) [
6], corynethers A (
5) [
6] and B (
6) [
7] (
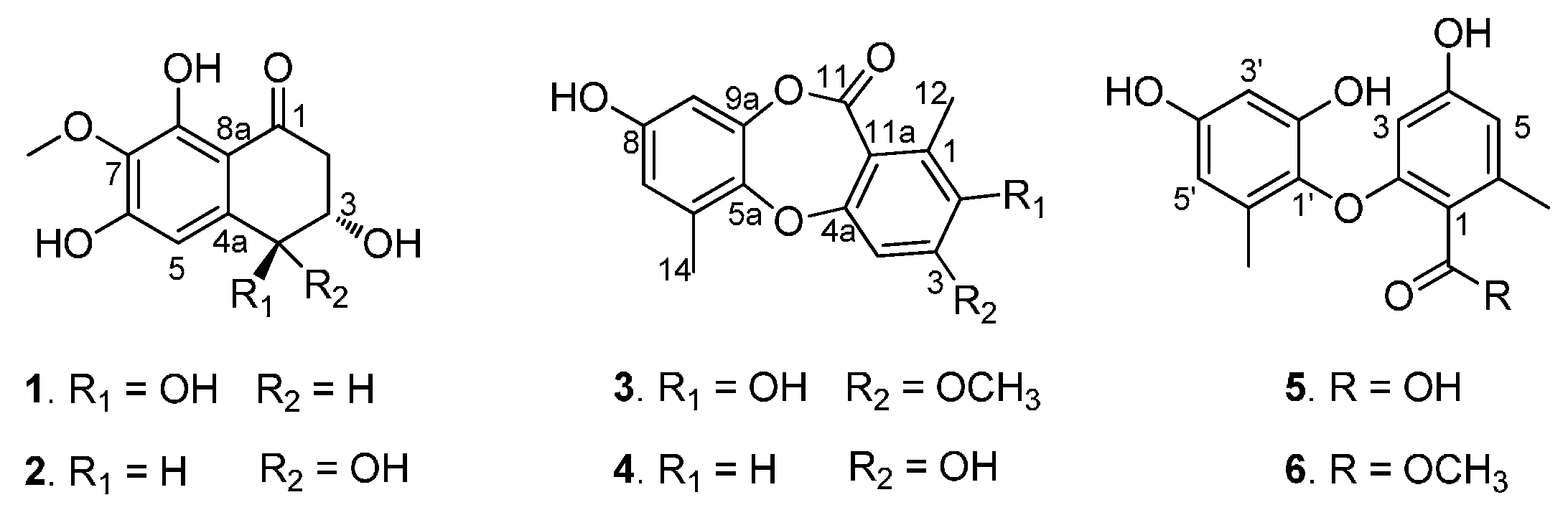
Figure 1). Herein we report the isolation, structure elucidation, and biological activities of these compounds.
Figure 1.
Compounds 1–6 isolated from the sponge-derived fungus Corynespora cassiicola.
Figure 1.
Compounds 1–6 isolated from the sponge-derived fungus Corynespora cassiicola.
2. Results
Corynenone A (
1) was isolated as a brown amorphous powder with the molecular formula of C
11H
12O
6, for which six degrees of unsaturation were required. The
1H-NMR spectrum exhibited signals for one hydrogen-bonded phenolic hydroxy group at δ
H 12.93 (s), one aromatic proton at δ
H 6.73 (s), two oxygenated methine protons at δ
H 4.54 (d,
J = 7.5 Hz) and δ
H 4.01 (m), and one methoxy group at δ
H 3.81 (s), together with one set of nonequivalent methylene protons at δ
H 2.94 (dd,
J = 17.0, 4.0 Hz) and δ
H 2.65 (dd,
J = 17.0, 9.0 Hz). In the
13C-NMR and DEPT spectra, five of the 11 carbons required by the molecular formula were identified as protonated centers, including three methines (one aromatic carbon, two oxygenated carbons) at δ
C 107.4, 73.1 and 71.4, one methoxy group at δ
C 60.5, and one methylene at δ
C 44.4. The six nonprotonated centers were separated into two categories: an α,β-unsaturated ketone at δ
C 202.1, five aromatic carbons at δ
C 157.9, 157.3, 142.7, 134.2, and 110.8. These spectroscopic features revealed that the parent skeleton of
1 was naphthalenone, and was very similar to 4-hydroxyscytalone previously isolated from
Pyricularia oryzae Cavara [
8]. The significant difference between these two compounds was the presence of a methoxy group signal in the
1H- and
13C-NMR spectra of
1. The downfield shift of C-7 in the
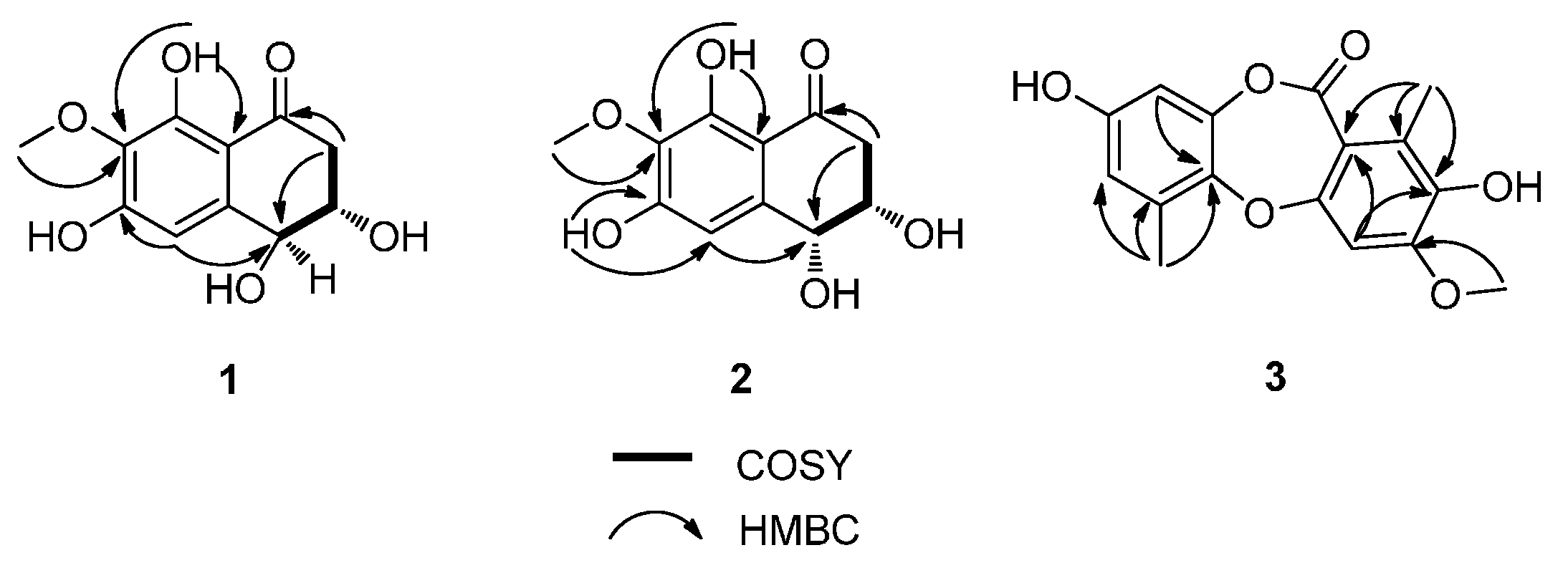
13C-NMR indicated that the methoxy group was attached to C-7, which was confirmed by the HMBC correlations from 7-OCH
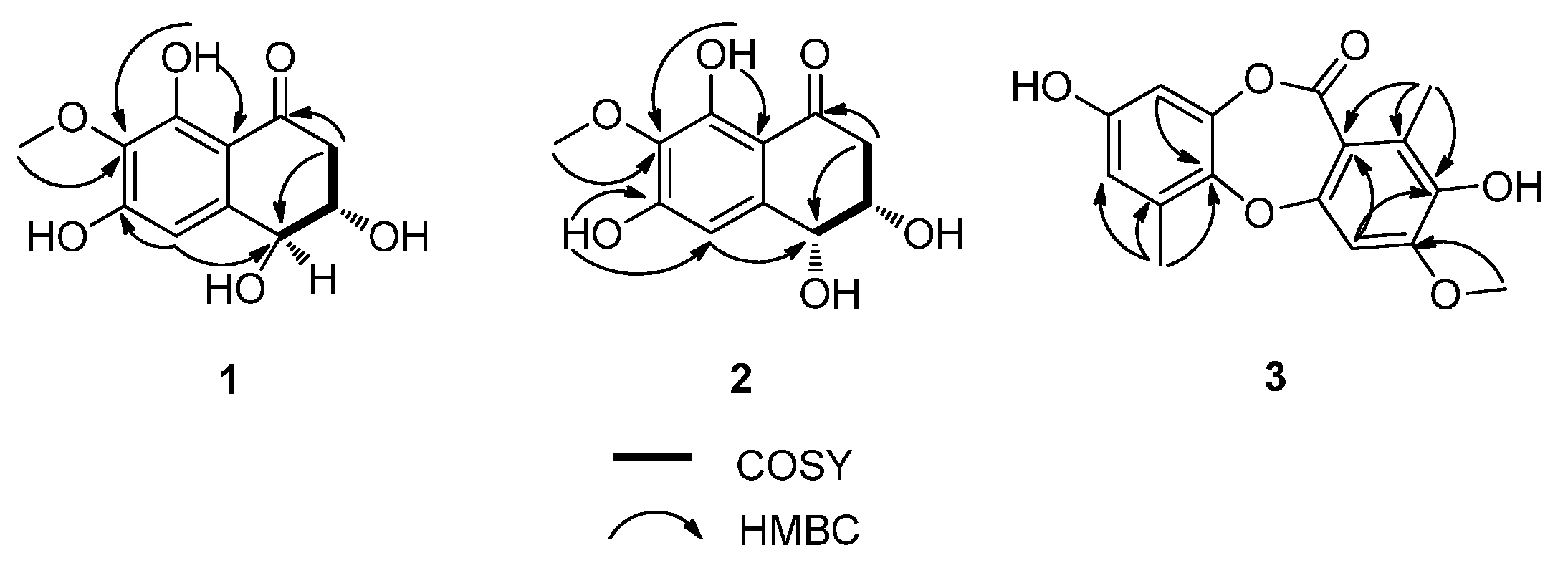
3 to C-7 and from H-5 to C-4 (
Figure 2). Detailed assignments for proton and carbon signals (
Table 1) were accomplished by analysis of 1D- and 2D-NMR data. The relative configurations of C-3 and C-4 were straightforwardly elucidated as
trans-arrangement based on the ax/ax coupling constant of the methine proton H-4 (δ
H 4.54) coupled to H-3 (δ
H 4.01) with a value of 7.50 Hz [
8,
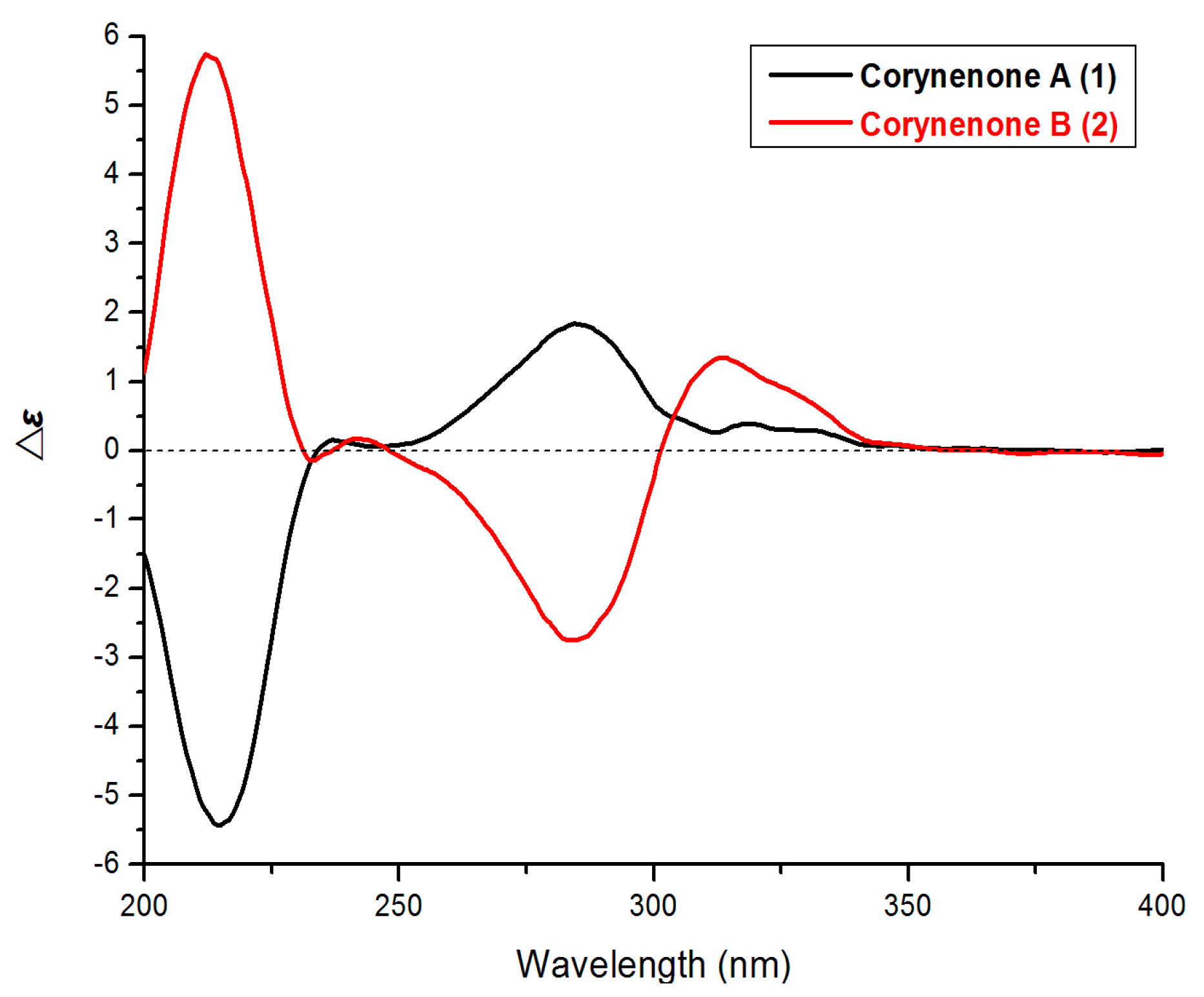
9], which suggested a half-chair conformation of the cyclohexenone ring. The absolute configuration of
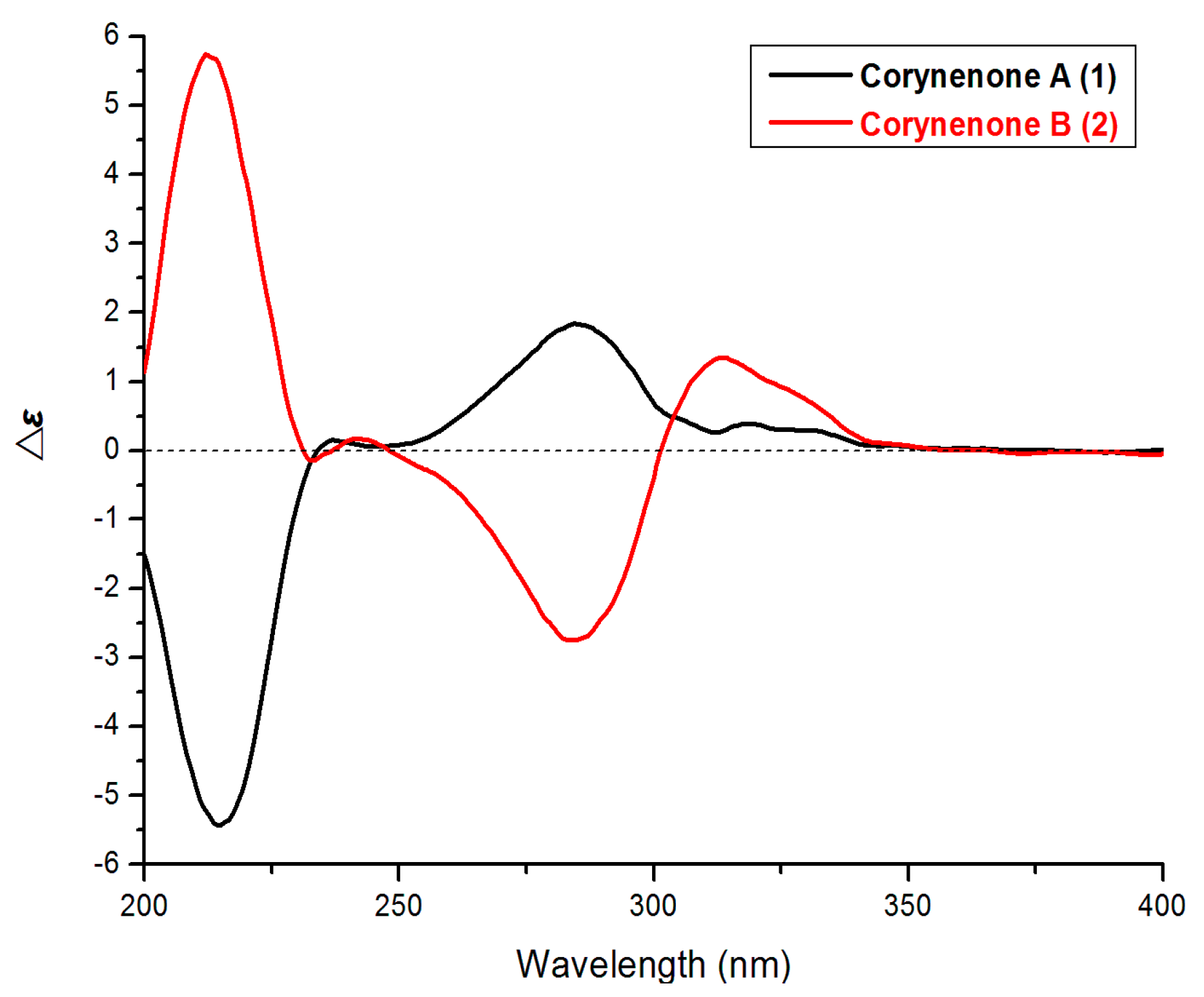
1 was determined by comparison its ECD spectrum with those of 3,4-diol naphthalenones described in the literature. For such naphthalenones, the first positive Cotton effect around 285 nm and second negative effect around 215 nm correspond to a 4
S configuration, whereas the first negative and second positive ones reflect a 4
R configuration [
9,
10]. Therefore, 4
S configuration was designed for
1 by its first positive and second negative Cotton effects at 280 and 214 nm (
Figure 3). Furthermore, based on the relative configuration, the absolute configuration of
1 was determined as 3
S,4
S.
Table 1.
1H- and 13C-NMR Data (500 and 125 MHz, resp. δ in ppm, J in Hz) of compounds 1 and 2.
Table 1.
1H- and 13C-NMR Data (500 and 125 MHz, resp. δ in ppm, J in Hz) of compounds 1 and 2.
| Position | 1 (Acetone-d6) | 2 (DMSO-d6) |
|---|
| δ(H) | δ(C) | δ(H) | δ(C) |
|---|
| 1 | | 202.1 | | 201.8 |
| 2 | 2.94 (dd, J = 17.0, 4.0)
2.65 (dd, J = 17.0, 9.0) | 44.4 | 2.81 (dd, J = 17.0, 3.0)
2.66 (dd, J = 17.0, 5.5) | 43.4 |
| 3 | 4.01, m | 71.4 | 4.10, brs | 69.1 |
| 4 | 4.54( d, J = 7.5) | 73.1 | 4.61( d, J = 3.0) | 69.2 |
| 4a | | 142.7 | | 141.9 |
| 5 | 6.73, s | 107.4 | 6.59, s | 107.2 |
| 6 | | 157.9 | | 157.4 |
| 7 | | 134.2 | | 133.0 |
| 8 | | 157.3 | | 156.4 |
| 8a | | 110.8 | | 109.2 |
| 3-OH | | | 4.97, s | |
| 4-OH | | | 5.39, brs | |
| 6-OH | | | 10.45, s | |
| 7-OCH3 | 3.81, s | 60.5 | 3.69, s | 59.7 |
| 8-OH | 12.93, s | | 12.81, s | |
Figure 2.
COSY Correlations and Key HMBCs of 1, 2, and 3.
Figure 2.
COSY Correlations and Key HMBCs of 1, 2, and 3.
Figure 3.
ECD spectra of 1 and 2.
Figure 3.
ECD spectra of 1 and 2.
Corynenone B (
2) was also obtained as a brown amorphous powder and has a molecular formula C
11H
12O
6 as deduced from its HRESIMS spectrum. Detailed analysis of the
1H- and
13C-NMR as well as 2D-NMR spectra of
1 and
2 (
Table 1,
Figure 2) revealed that these two compounds showed high similarity in NMR spectroscopic data. The significant difference between
1 and
2 was the chemical shifts of the oxymethines at C-3 and C-4 (δ
C 69.1 and 69.2 in
1 vs. δ
C 71.4 and 73.1 in
2) in the
13C-NMR spectra. This NMR feature implied that
1 and
2 might be diastereoisomers differing from each other by the configurations of the 3,4-diol centers. The small ax/ex coupling constant of H-4/H-3 (
J = 3.0 Hz) in
2 suggested a
cis relationship of C-3 and C-4 [
9], which also suggested a half-chair conformation of the cyclohexenone ring. The 3
S,4
R-configuration was determined by comparison of its ECD spectrum (
Figure 3) with that of (3
S,4
R)-4,8-dihydroxy-3-methoxy-3,4-dihydro-1(2
H)-naphthalenone [
9].
It should be noted that compounds
1 and
2 are diastereoisomers with the different ECD maxima at around 300–350 nm. The atomic spatial orientation and some little change of the half-chair conformation, especially the possible hydrogen bond between the
trans or
cis OH groups at C-3 and C-4 may effect the ECD spectra. In the literature [
9,
10], the absolute configurations at C-3 and C-4 of such naphthalenones were also determined by the Cotton effects at around 200–300 nm rather than above 300 nm.
Corynesidone E (
3) was obtained as a white, amorphous powder. Its molecular formula was deduced as C
16H
14O
6 by HRESIMS, thus revealing an increase in the molecular weight by 14 amu compared to the known compound corynesidone C, which was isolated from the endophytic fungus
C. cassiicola L36, derived from the leaf tissues of the Chinese mangrove medicinal plant
Laguncularia racemosa, collected from Hainan Island, China [
11]. The
1H- and
13C-NMR spectra of both
3 and corynesidone C were very similar, except for the presence of signals for one methoxy group in the spectra of
3. This was further confirmed by inspection of the HMBC spectrum revealing the correlations from 3-OCH
3 to C-3 (
Figure 2). Thus a methoxy group at C-3 was present in the structure of
3 instead of a hydroxy group at C-3 in corynesidone C. The structure of
3 was therefore determined as depict.
The structures of the known compounds, corynesidone A (
4) [
6], corynethers A (
5) [
6] and B (
6) [
7] were identified on the basis of their spectroscopic data and by comparison with those in the literature.
Compounds 1 and 2 were evaluated for their antifouling activities against the larval settlement of the barnacle Balanus amphitrite, but neither of the isolated compounds proved to be active. All of the isolated compounds were subjected to screening for their cytotoxic activities against the human promyelocytic leukemia HL-60, human leukemia K-562, human lung cancer A-549, and human cervical carcinoma HeLa cell lines. Only compound 4 exhibited moderate cytotoxic activity against HeLa cell line with an IC50 value of 15.2 μM. Compound 5 showed weak cytotoxic activity against HL-60 cells with IC50 values of 31.9 μM.
3. Experimental Section
3.1. General
Column chromatography (CC): silica gel (200–300 mesh; Qingdao Marine Chemical Group Co., Qingdao, China), octadecylsilyl silica gel (45–60 μm; Unicorn) and Sephadex LH-20 (Amersham Biosciences, Piscataway, NJ, USA). TLC: Precoated silica gel plates (G60, F-254; Yantai Zifu Chemical Group Co., Yantai, China). Prep. HPLC: Semi-preparative HPLC was performed on a Waters 1525 system coupled with a Waters 2996 photodiode array detector (Waters, Milford, MA, USA) using a C18 column (Kromasil, 5 μm, 10 × 250 mm). Optical rotations: JASCO P-1020 digital polarimeter (Jasco, Tokyo, Japan). UV spectra: Beckman DU 640 spectrophotometer (Beckman, Brea, CA, USA). ECD spectra: Jasco J-815-150S circular dichroism spectrometer (Jasco). IR spectra: Nicolet-Nexus-470 spectrometer (Nicolet, Glendale, WI, USA); KBr pellets; νmax in cm−1. NMR spectra: Agilent DD2 500 MHz NMR spectrometer (500 MHz for 1H and 125 MHz for 13C), using TMS as an internal standard (Agilent, Santa Clara, CA, USA). HR-ESI-MS of compounds 1 and 2 were measured on a Bruker Q-TOF maXIS spectrometer (Bruker, Billerica, MA, USA). ESI-MS and HR-ESI-MS of compound 3 were obtained from a Micromass Q-TOF spectrometer (Waters) and Thermo Scientific LTQ Orbitrap XL spectrometer (Thermo, Waltham, MA, USA).
3.2. Fungal Material
The fungal strain Corynespora cassiicola XS-200900I7 was isolated from a piece of fresh tissue from the inner part of an unidentified sponge (XS-2009001), which was collected from the Xisha Islands coral reef in the South China Sea in December 2009. The Fungus was identified as Corynespora cassiicola according to its morphological characteristics and a molecular biological protocol by 16s rRNA amplification and sequencing of the ITS region. The strain was deposited in the Key Laboratory of Marine Drugs, the Ministry of Education of China, School of Medicine and Pharmacy, Ocean University of China, Qingdao, PR China, with the GenBank (NCBI) accession number KM597051.
3.3. Extraction, Isolation and Characterization
The fungal strain was cultivated in sixty Erlenmeyer flasks solid medium (each containing 80 g of rice, 120 mL of H
2O, and 3.6 g natural sea salt from Yangkou saltern, China) at 28 °C for four weeks. The fermented solid medium was extracted three times with 400 mL of EtOAc for each Erlenmeyer flask. The combined EtOAc layers were evaporated to dryness under reduced pressure to give an EtOAc extract (18.6 g), which was subjected to vacuum liquid chromatography (VLC) on silica gel using step gradient elution with EtOAc–petroleum ether (0%–100%) and then with MeOH–EtOAc (0%–100%) to afford five fractions (Fr.1–Fr.5). Fr.2 was subjected to Sephadex LH-20 column chromatography (CH
2Cl
2/MeOH,
v/
v, 1:1) and further purified by using HPLC eluted with 60% MeOH–H
2O to obtain
3 (5.0 mg),
4 (7.0 mg), and
5 (82.0 mg). Fr.3 was first subjected to repeated silica gel CC (CH
2Cl
2–MeOH,
v/
v, 100:1), and then separated by Sephadex LH-20 CC (CH
2Cl
2–MeOH,
v/
v, 1:1) to yield
6 (3.0 mg). Fr.4 was applied to repeated Sephadex LH-20 CC (CH
2Cl
2/MeOH,
v/
v, 1:1) and further purified on HPLC with 5% MeCN–H
2O (with 0.1% trifluoroacetic acid) to give
1 (8.9 mg), and
2 (4.1 mg). The
1H-NMR,
13C-NMR, COSY, HMQC, HMBC, MS spectra of
1–
3 are available in
Supplementary Materials.
Corynenone A (
1)
: brown powder;
= 19.4 (
c 0.08, MeOH); UV (MeOH) λ
max (log ε) 221 (4.14), 290 (4.10); ECD (
c 2.08 mM, MeOH) 215 (−5.51), 284 (+1.88); IR (KBr) ν
max 3648, 2319, 1646, 1542, 1340, 1021 cm
−1;
1H- and
13C-NMR data,
Table 1; HRESIMS
m/
z 239.0561 [M − H]
− (calcd for C
11H
11O
6, 239.0561).
Corynenone B (
2)
: brown powder;
= 36.6 (
c 0.40, MeOH); UV (MeOH) λ
max (log ε) 220 (4.31), 290 (4.24); ECD (
c 2.08 mM, MeOH) 213 (+5.67), 284 (−2.75), 314 (+1.34); IR (KBr) ν
max 3607, 2364, 1697, 1536, 1447 cm
−1;
1H- and
13C-NMR data,
Table 1; HRESIMS
m/z 239.0564 [M − H]
− (calcd for C
11H
11O
6, 239.0561).
Corynesidone E (3): white powder; UV (MeOH) λmax (log ε) 230 (4.56), 274 (4.01); IR (KBr) νmax 3431, 2950, 1634, 1414, 1266, 1117 cm−1; 1H-NMR (CDCl3, 500 MHz) δ 6.86 (1H, s, H-4), 6.52 (1H, s, H-7), 6.52 (1H, s, H-9), 3.95 (3H, s, H-13), 2.39 (3H, s, H-14), 2.30 (3H, s, H-12). 13C-NMR (CDCl3, 125 MHz) δ 163.5 (C-11), 156.4 (C-4a), 155.3 (C-8), 151.5 (C-3), 145.8 (C-9a), 143.8 (C-5a), 143.0 (C-2), 132.0 (C-6), 127.5 (C-1), 115.0 (C-11a), 114.3 (C-7), 105.7 (C-9), 101.7 (C-4), 56.7 (C-13), 16.1 (C-14), 13.3 (C-12). ESIMS: 303.2 [M + H]+, 325.2 [M + Na]+, 627.3 [2M + Na]+. HRESIMS m/z 303.0868 [M + H]+ (calcd for C16H15O6, 303.0863).
3.4. Biological Assays
The antifouling activity against the larval settlement of the barnacle was determined using cyprids of
Balanus amphitrite Darwin based on the literature procedures [
12]. SeaNine 211™ (Rohm & Haas) was used as a positive control. The cytotoxic activity was tested using four human tumour cell lines, including the human promyelocytic leukemia HL-60, human leukemia K-562, human lung cancer A-549, and cervical carcinoma HeLa. The cytotoxicity against HL-60 andK-562 was evaluated by MTT method [
13], and the cytotoxicity against HeLa and A-549 was measured by SRB assay [
14]. Adriamycin was used as a positive control.





{kind=link}
{kind=link}
{kind=link}
{kind=link}