
Half-Sandwich Ru(II) Halogenido, Valproato and 4-Phenylbutyrato Complexes Containing 2,2′-Dipyridylamine: Synthesis, Characterization, Solution Chemistry and In Vitro Cytotoxicity





Abstract
:1. Introduction
2. Results and Discussion
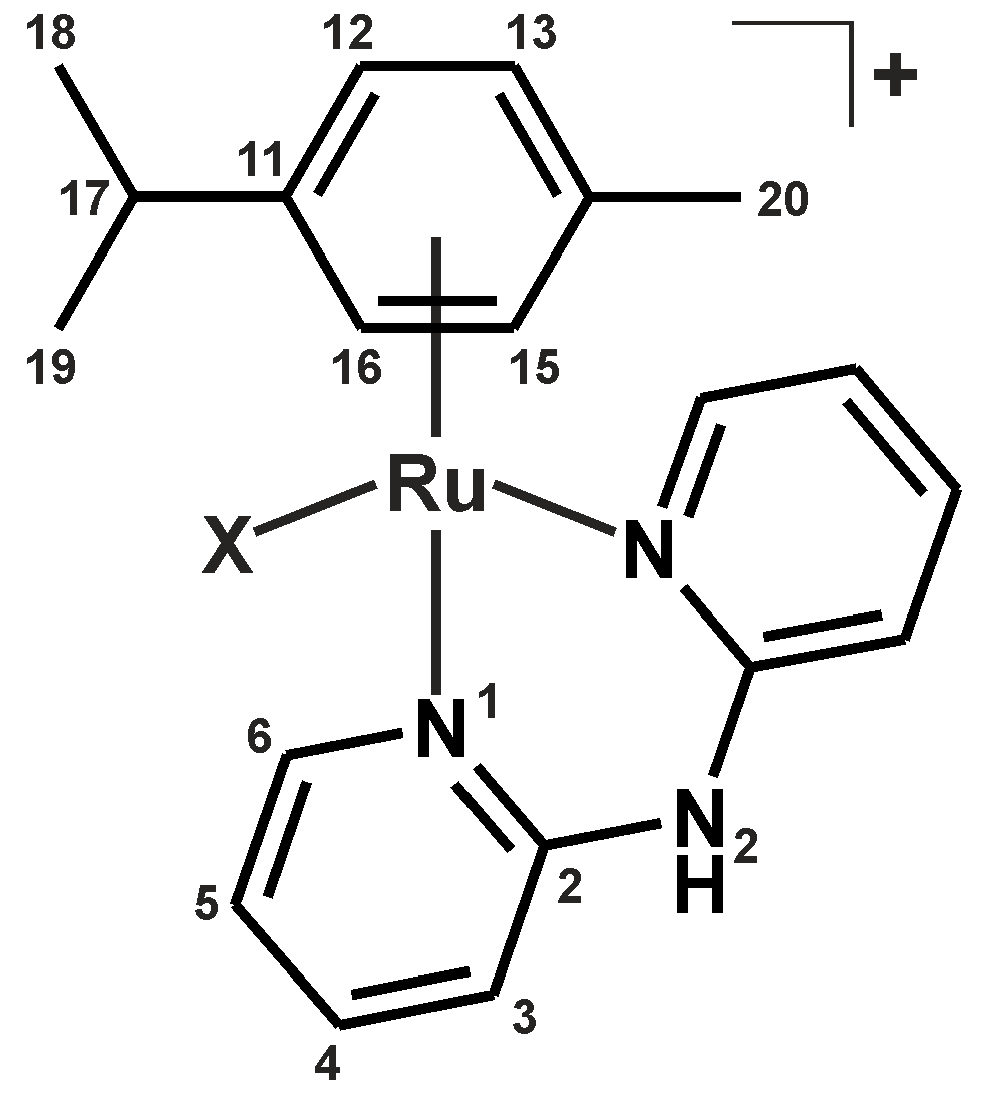
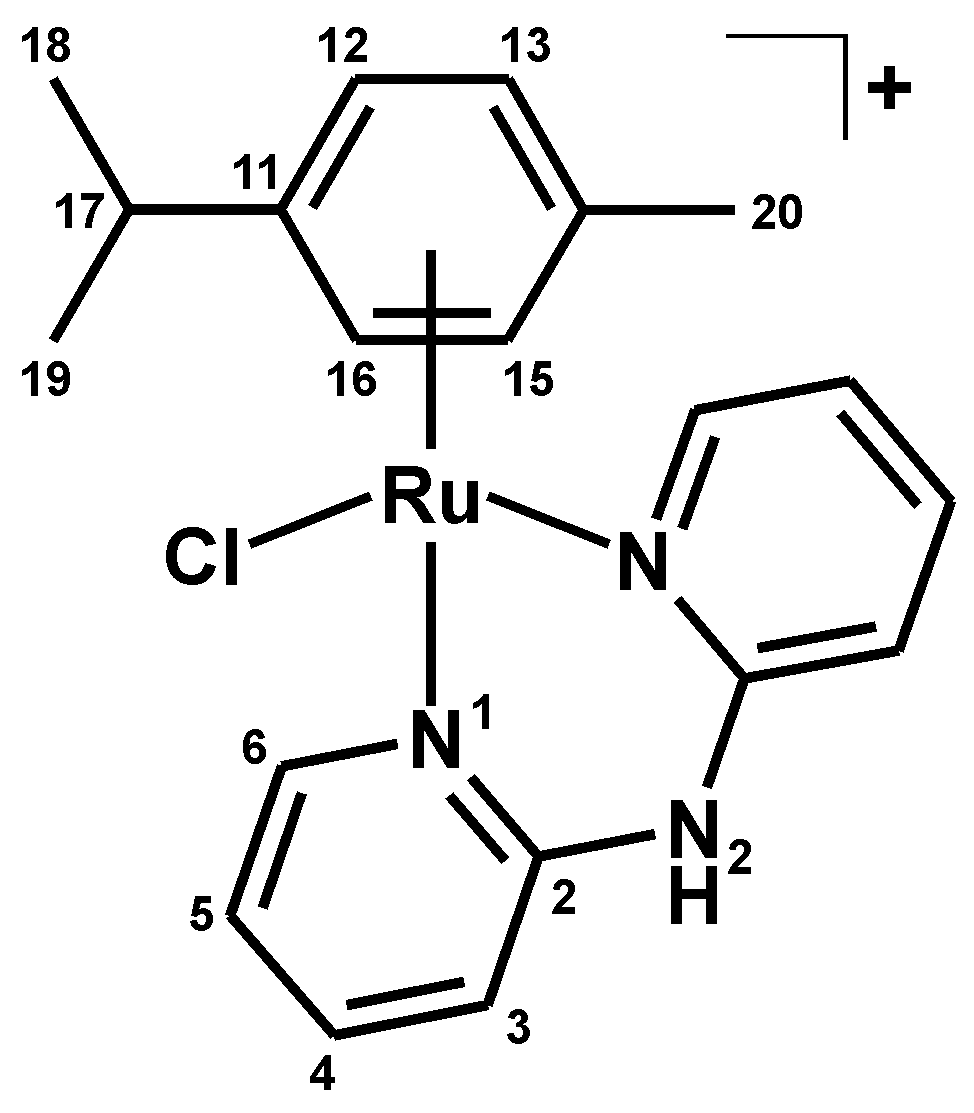
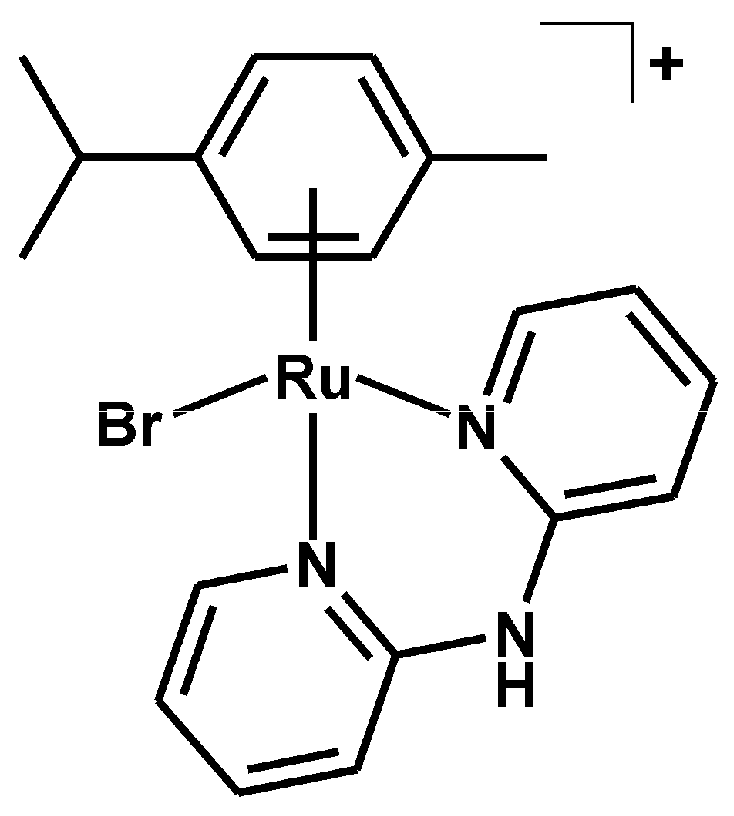
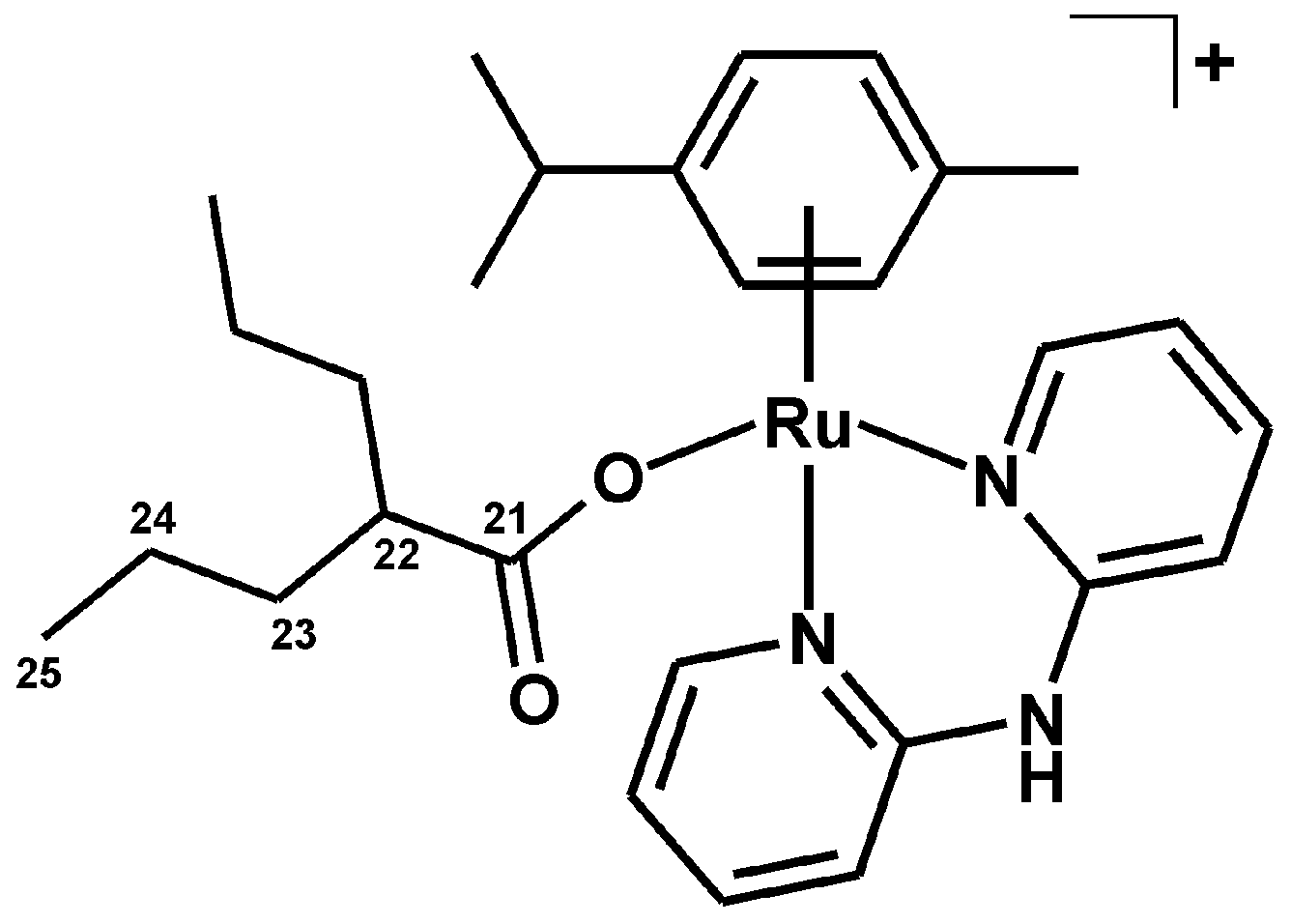
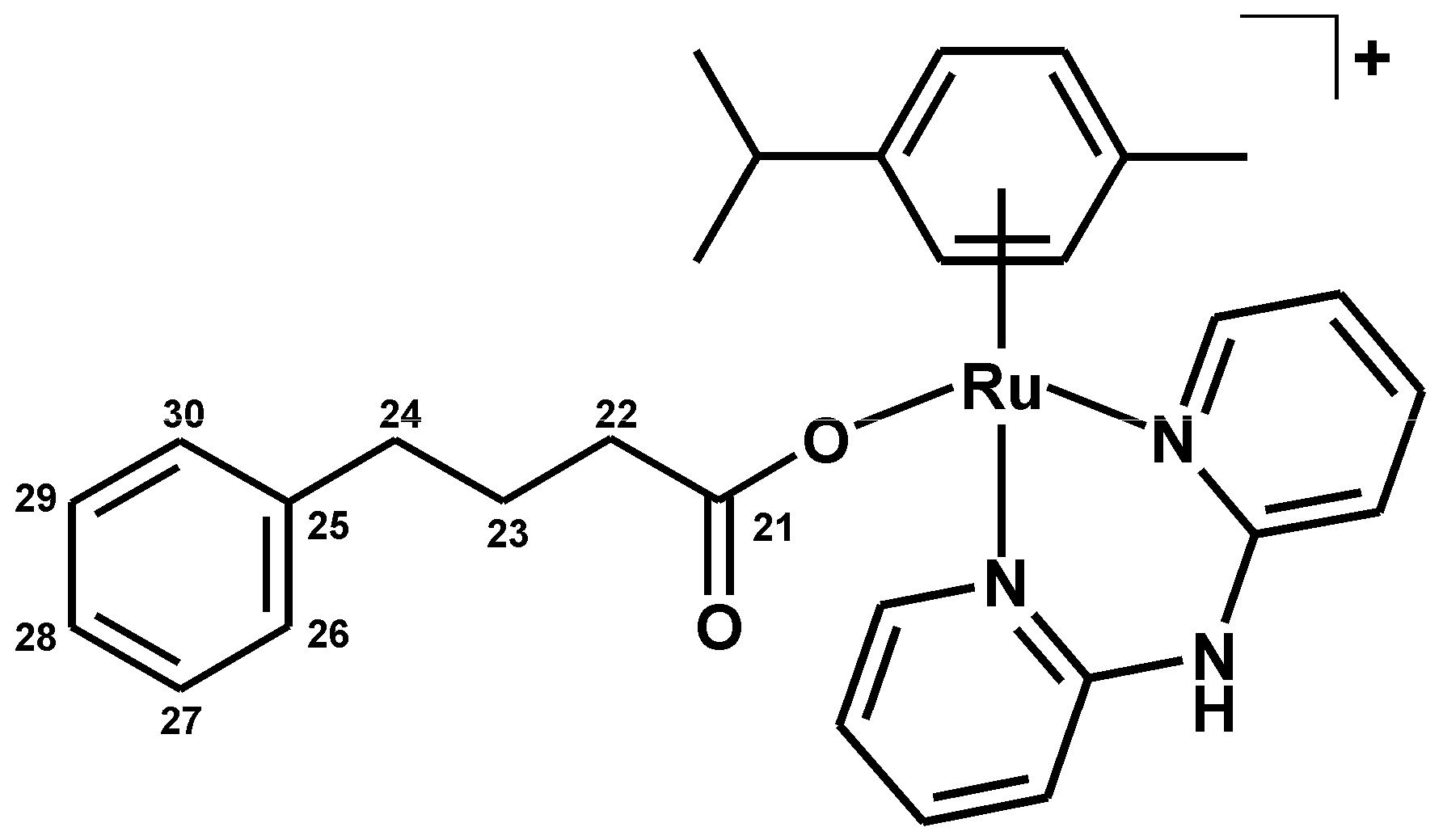
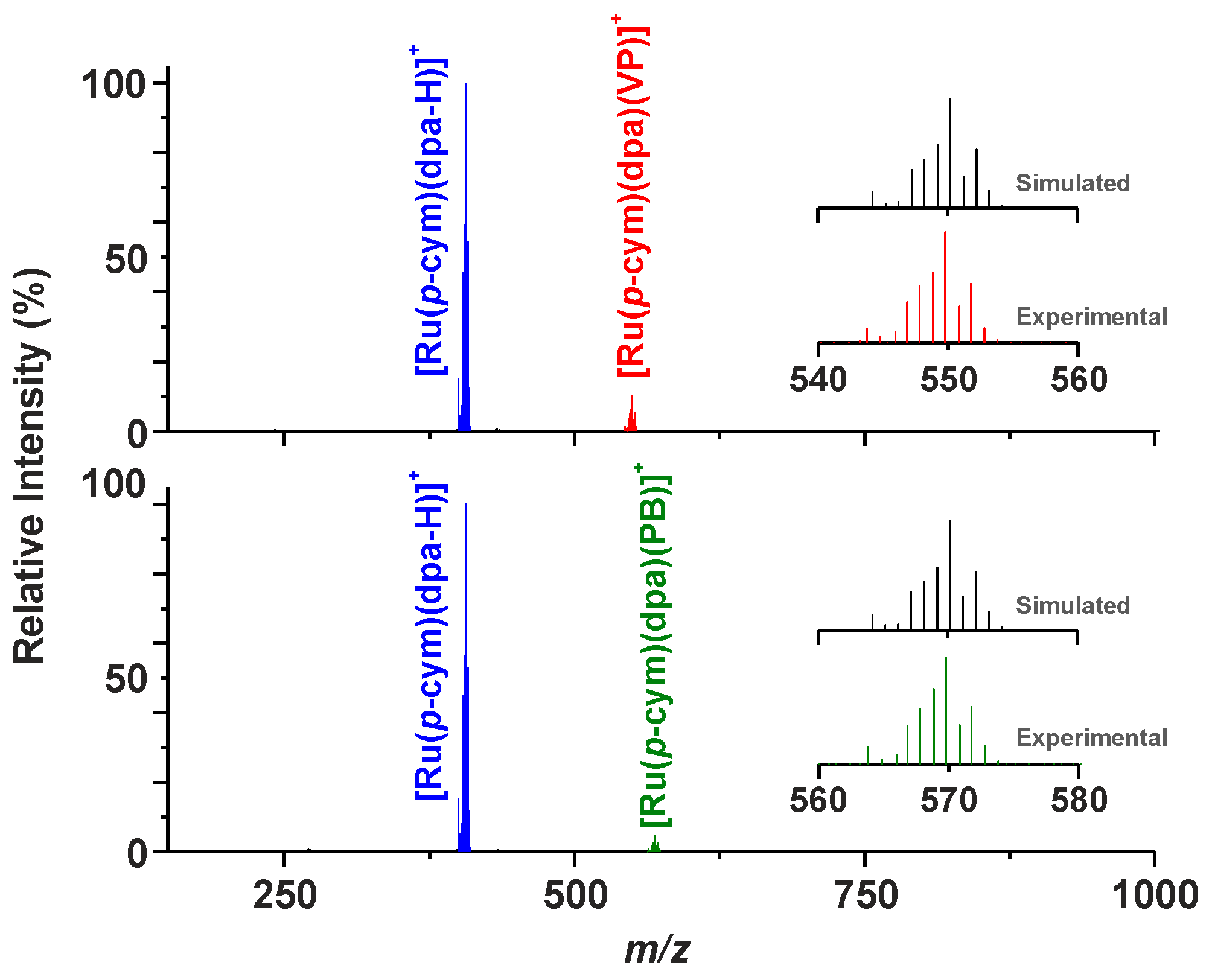
2.1. Synthesis and General Properties
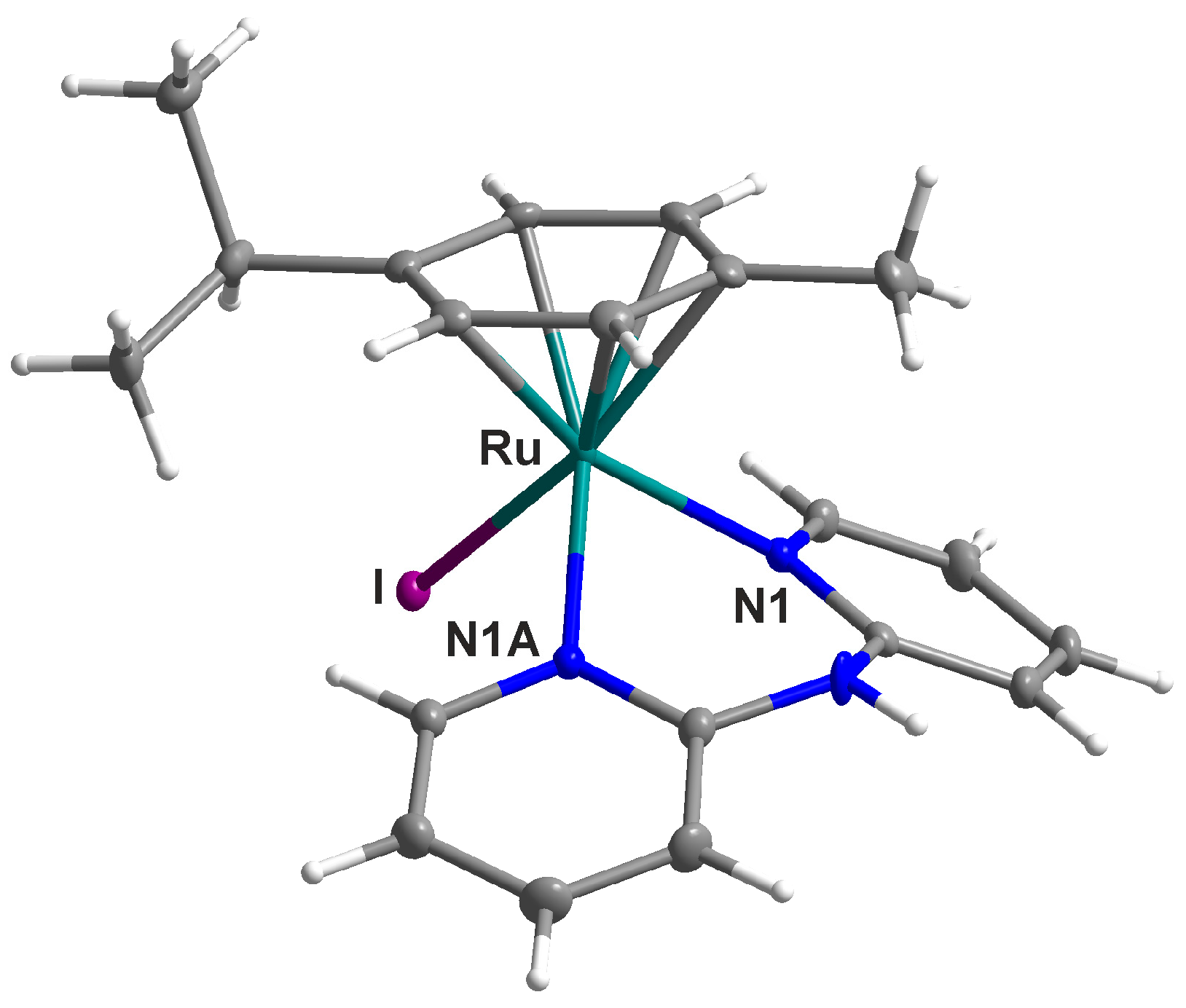
2.2. Single Crystal X-ray Analysis
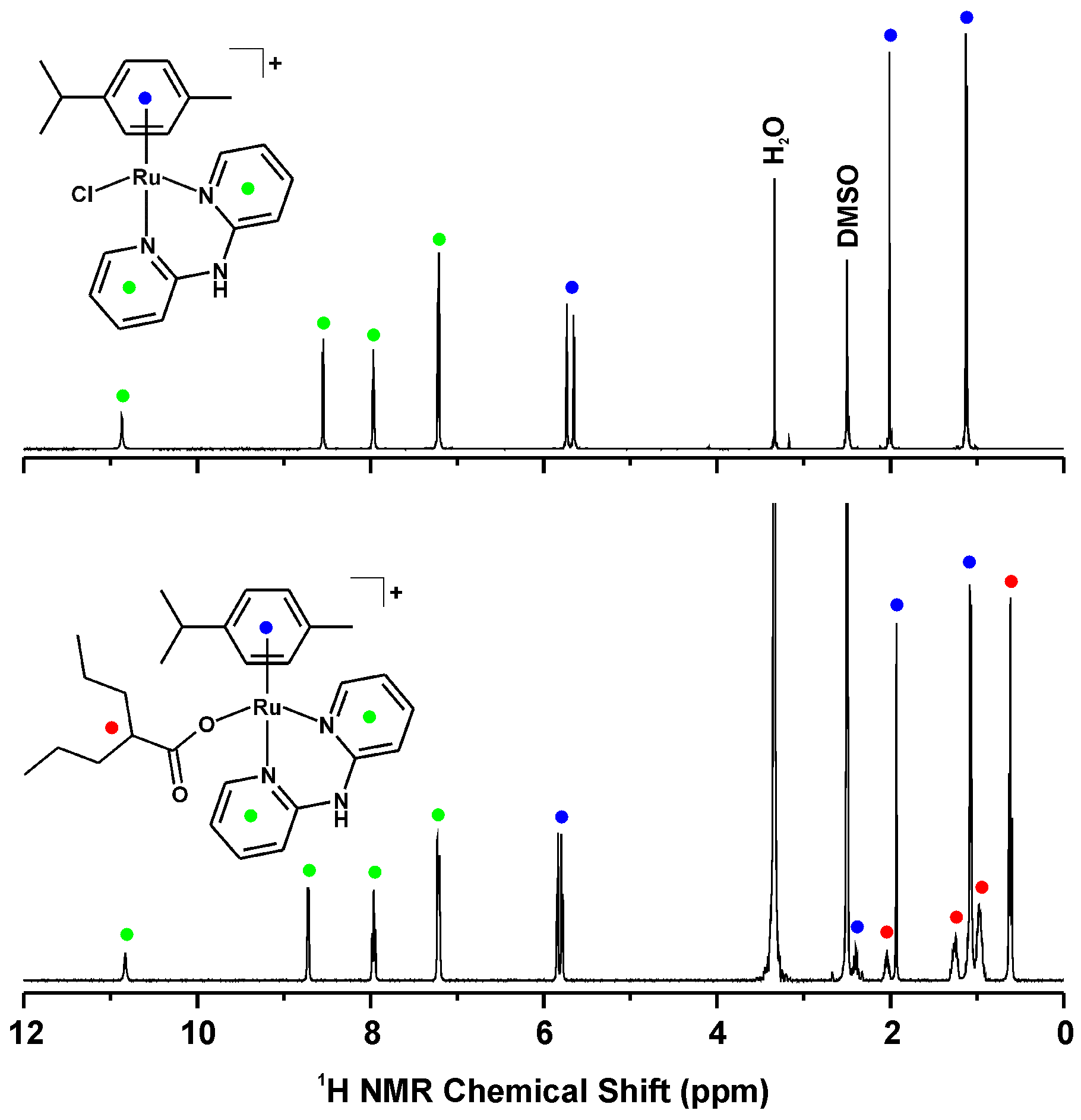
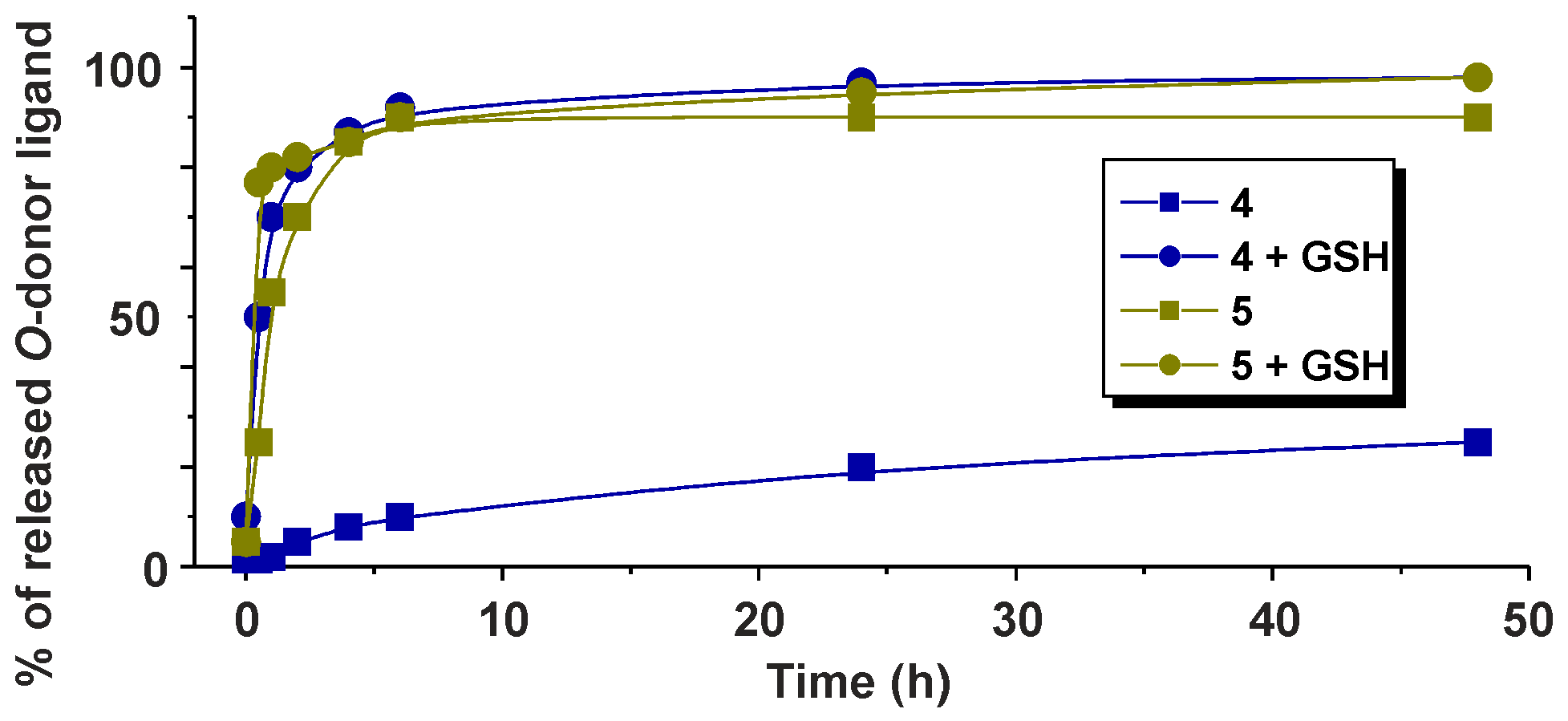
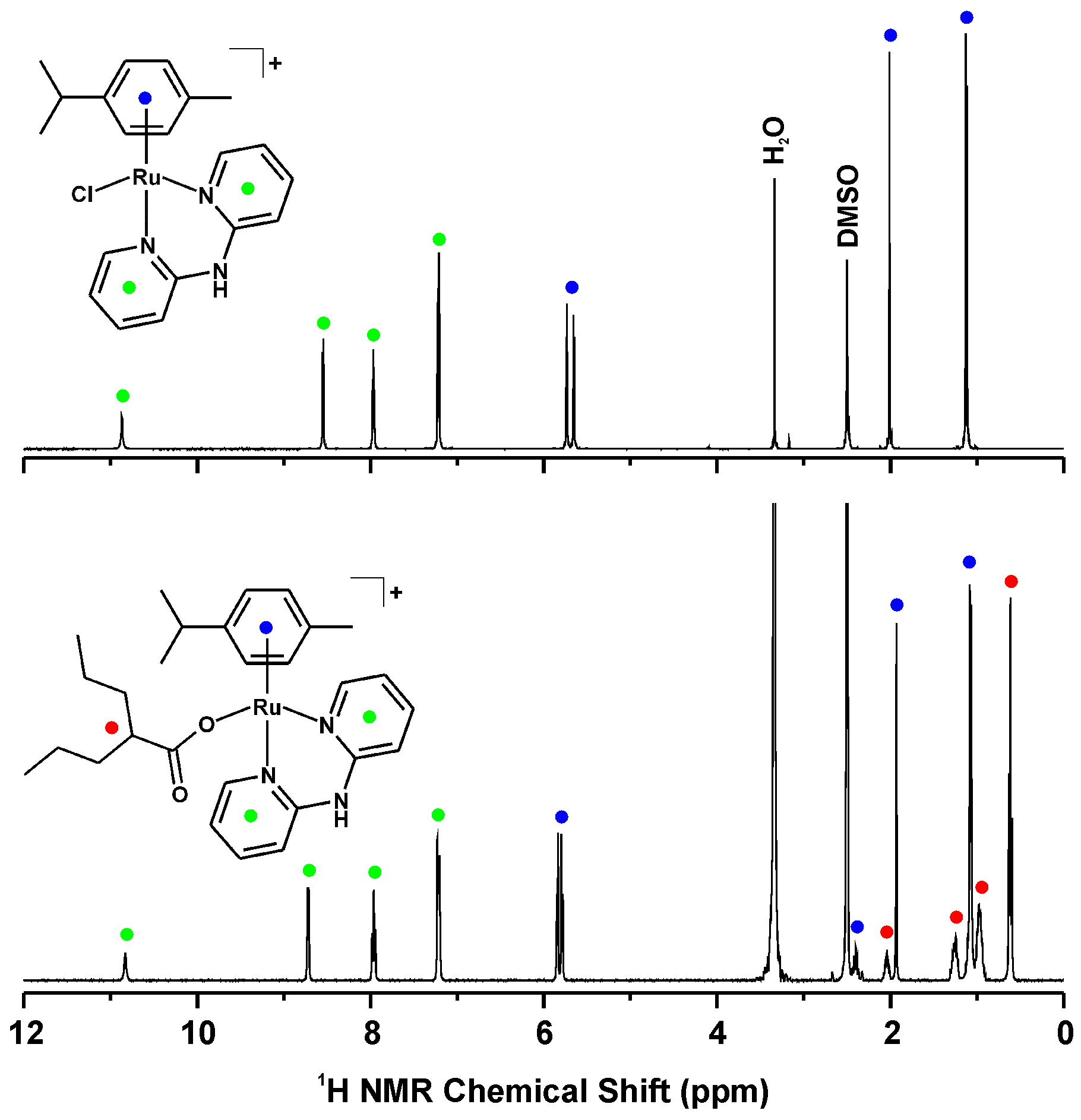
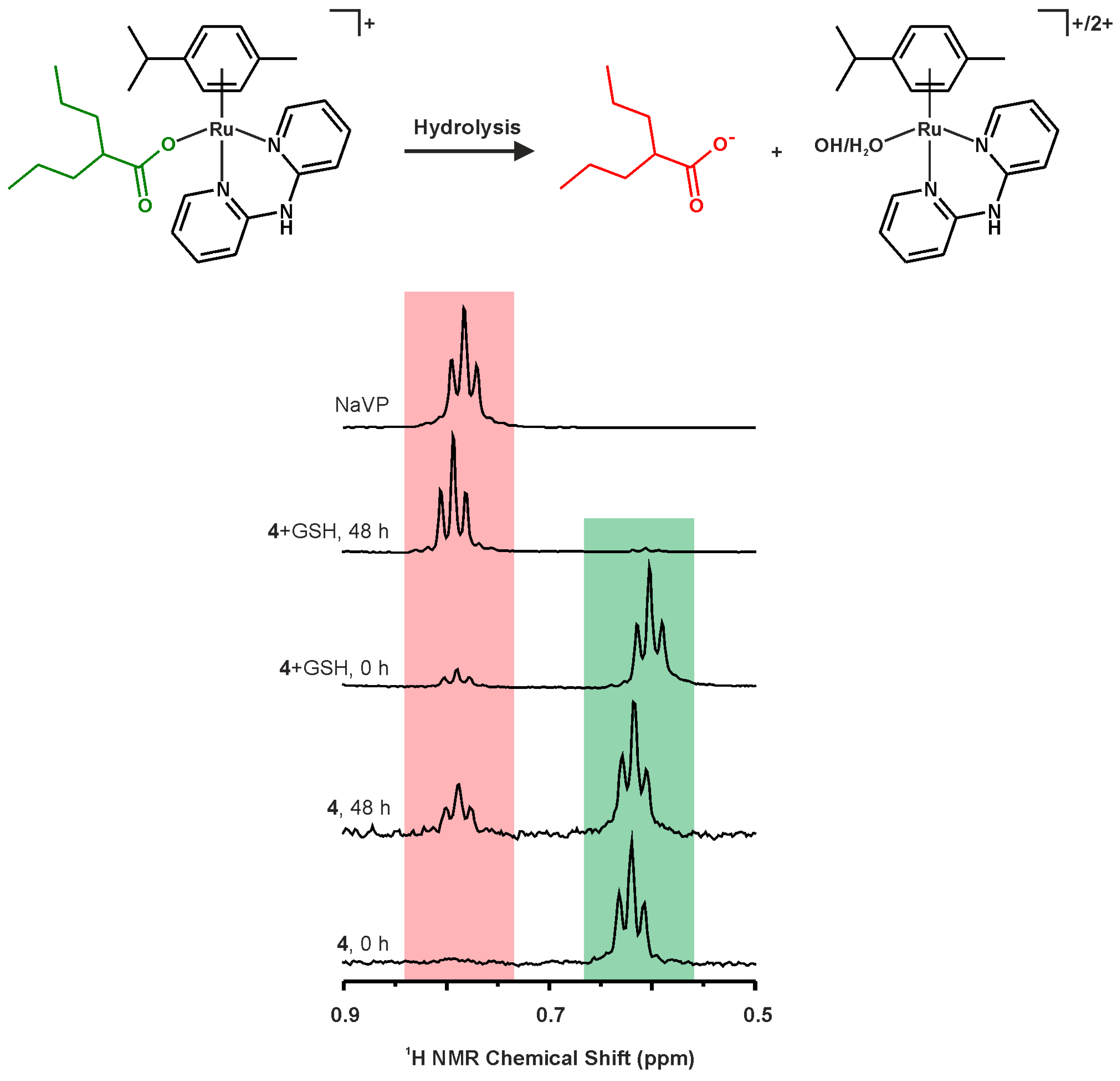
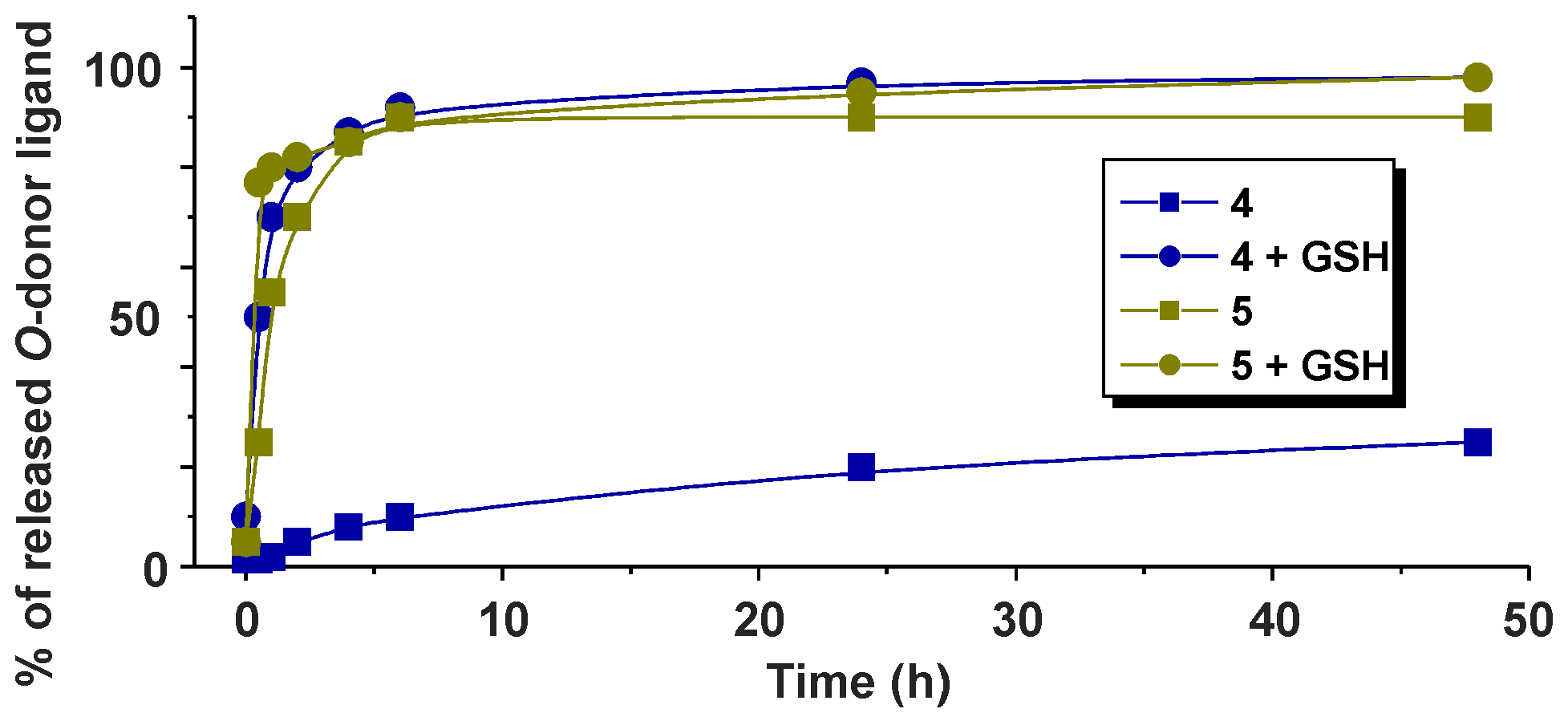
2.3. 1H-NMR Studies of Solution Chemistry and Interactions with Reduced Glutathione
2.4. In Vitro Cytotoxicity
3. Materials and Methods
3.1. Materials
3.2. Syntheses
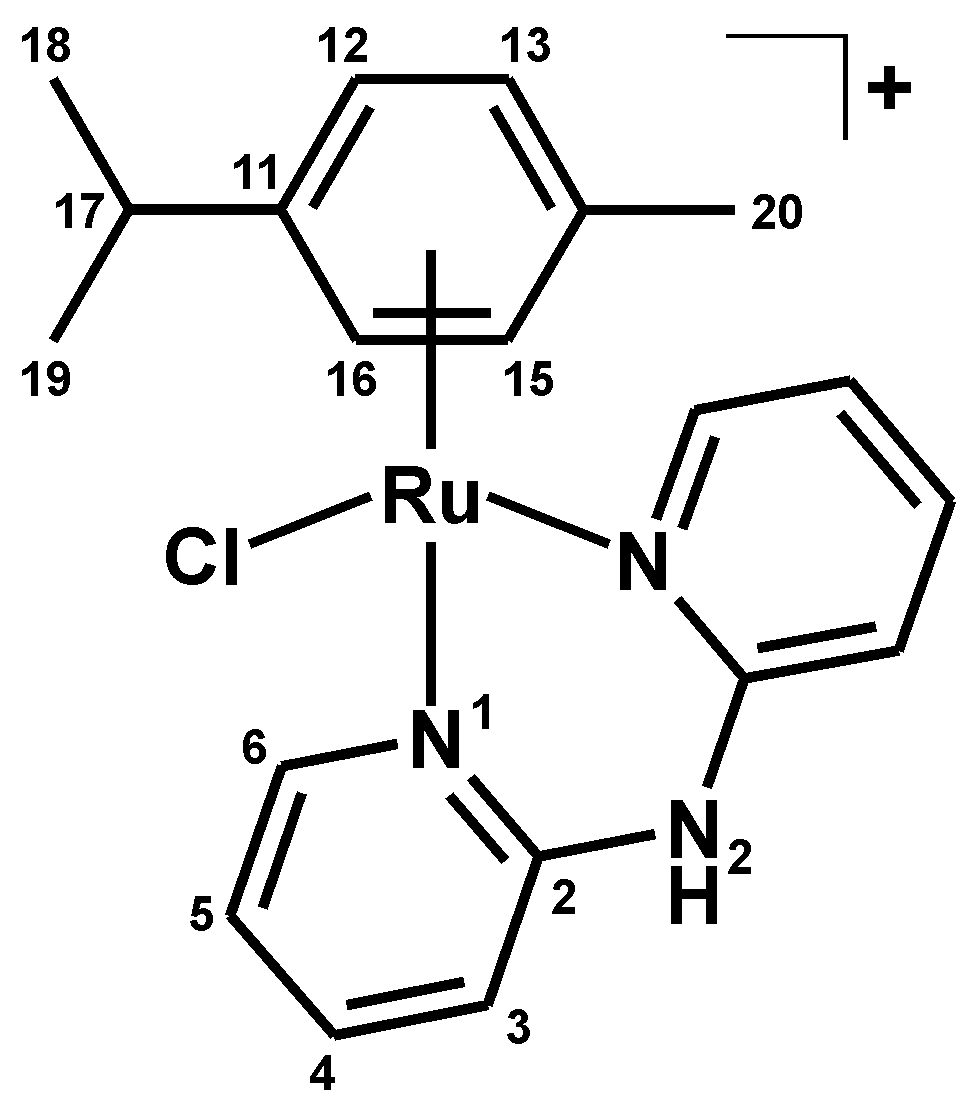
3.2.1. Synthesis of [Ru(η6-p-cym)(dpa)Cl]PF6 (1)
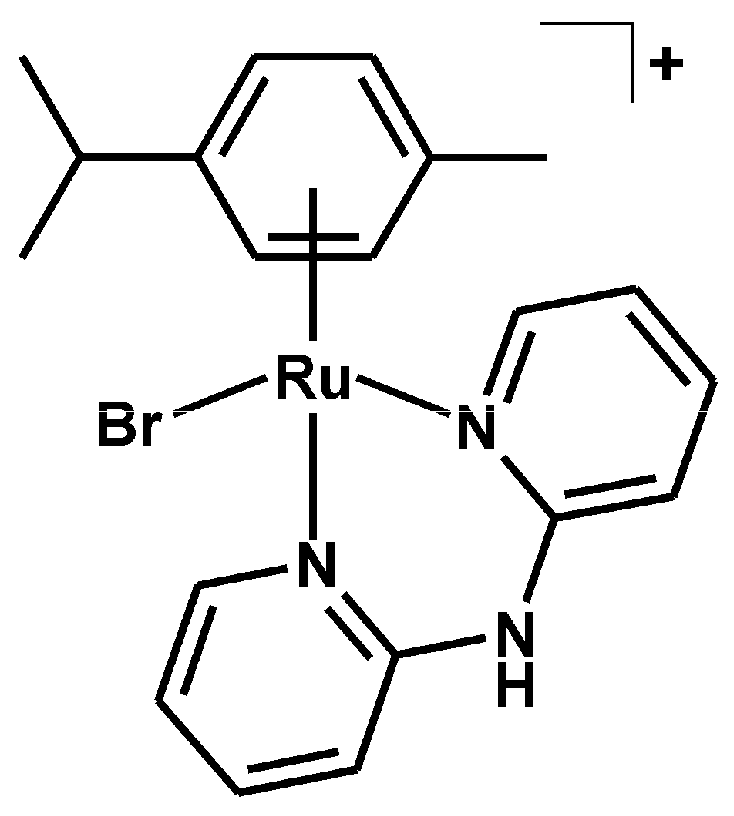
3.2.2. Synthesis of [Ru(η6-p-cym)(dpa)Br]PF6 (2)
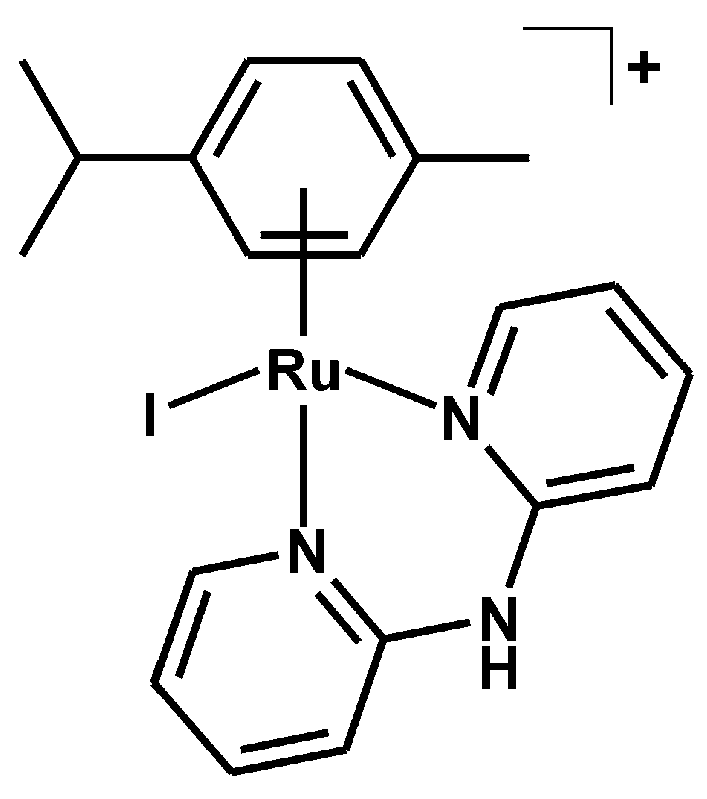
3.2.3. Synthesis of [Ru(η6-p-cym)(dpa)I]PF6 (3)
3.2.4. Synthesis of [Ru(η6-p-cym)(dpa)(VP)]PF6 (4)
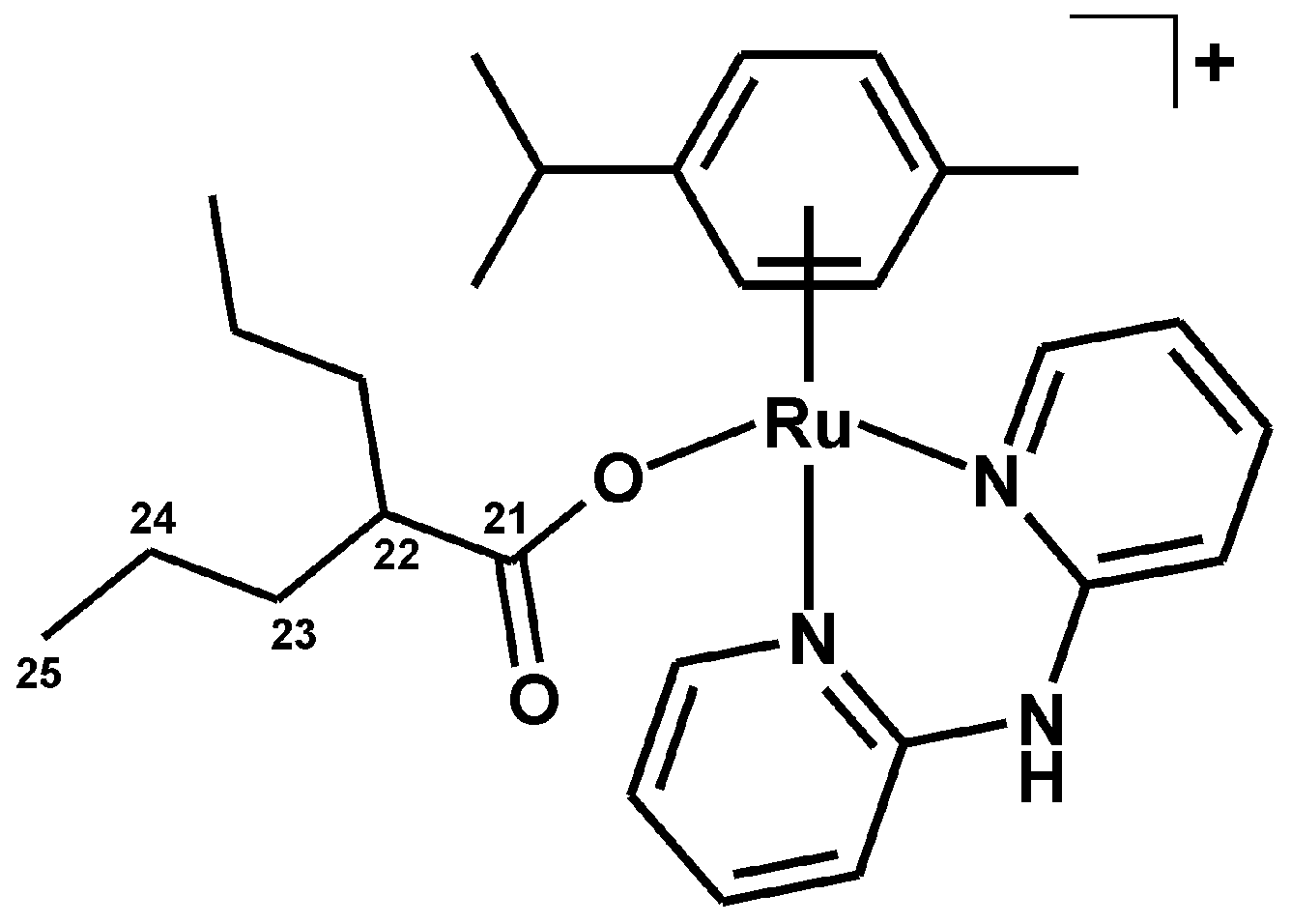
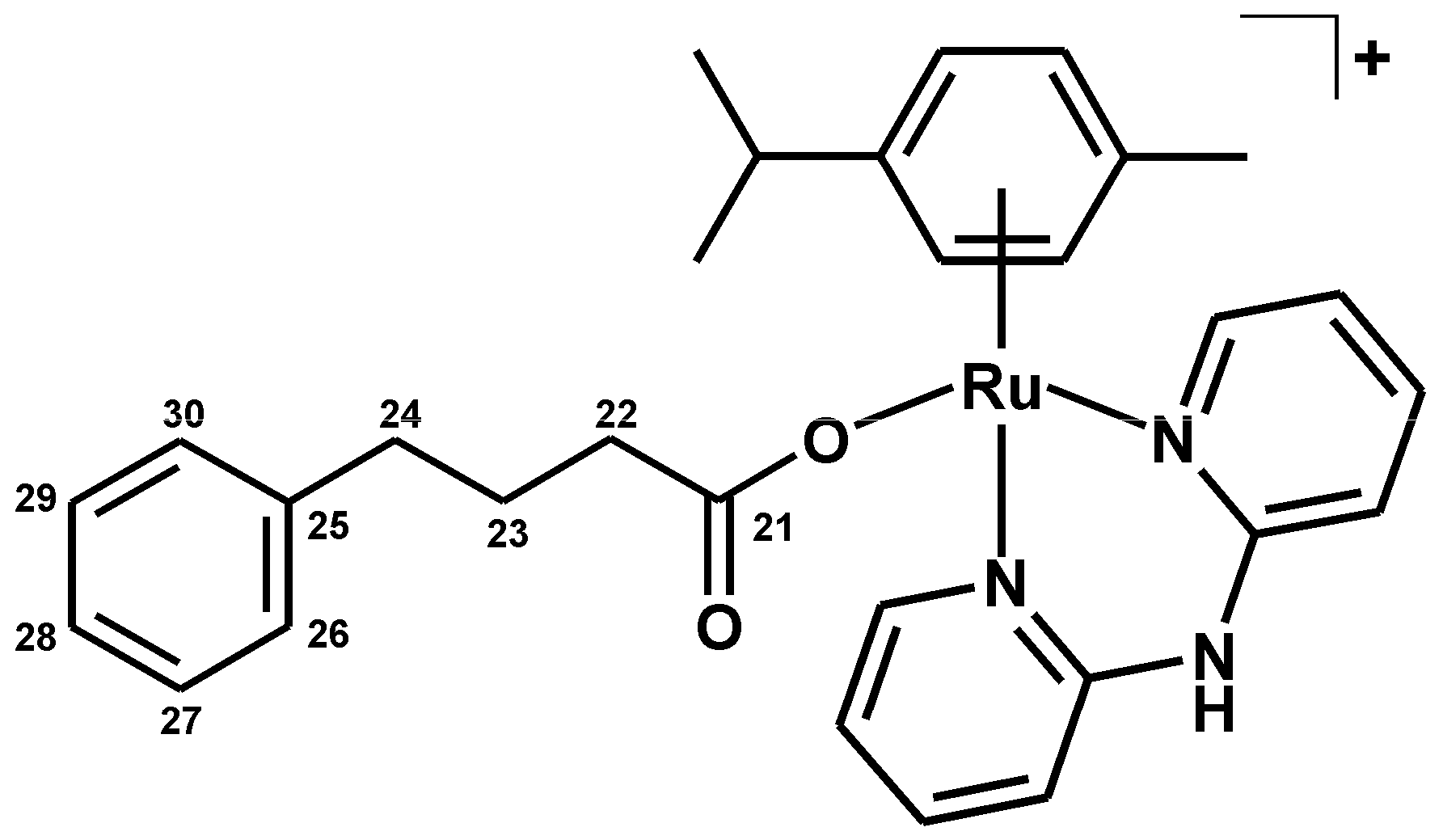
3.2.5. Synthesis of [Ru(η6-p-cym)(dpa)(PB)]PF6 (5)
3.3. Methods
3.4. 1H-NMR Studies of Aqueous Chemistry and Interactions with GSH
3.5. Cell Culture and In Vitro Cytotoxicity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Süss-Fink, G. Arene ruthenium complexes as anticancer agents. Dalton Trans. 2010, 39, 1673–1688. [Google Scholar] [CrossRef] [PubMed]
- Romero-Canelón, I.; Salassa, L.; Sadler, P.J. The contrasting activity of iodido versus chlorido ruthenium and osmium arene azo- and imino-pyridine anticancer complexes: Control of cell selectivity, cross-resistance, p53 dependence, and apoptosis pathway. J. Med. Chem. 2013, 56, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.T.; Wang, Z.; Hunsberger, J.G.; Chuang, D.M. Therapeutic potential of mood stabilizers lithium and valproic acid: Beyond bipolar disorder. Pharmacol. Rev. 2013, 65, 105–142. [Google Scholar] [CrossRef] [PubMed]
- Iannitti, T.; Palmieri, B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R&D 2011, 11, 227–249. [Google Scholar]
- Alessio, M.; Zanellato, I.; Bonarrigo, I.; Gabano, E.; Ravera, M.; Osella, D. Antiproliferative activity of Pt(IV)-bis(carboxylato) conjugates on malignant pleural mesothelioma cells. J. Inorg. Biochem. 2013, 129, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Kašpárková, J.; Kostrhunová, H.; Nováková, O.; Křikavová, R.; Vančo, J.; Trávníček, Z.; Brabec, V. A photoactivatable platinum(IV) complex targeting genomic DNA and histone deacetylases. Angew. Chem. Int. Ed. 2015, 54, 14478–14482. [Google Scholar] [CrossRef] [PubMed]
- Novohradsky, V.; Zerzankova, L.; Stepankova, J.; Vrana, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. Antitumor platinum(IV) derivatives of oxaliplatin with axial valproato ligands. J. Inorg. Biochem. 2014, 140, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Raveendran, R.; Braude, J.P.; Wexselblatt, E.; Novohradsky, V.; Stuchlikova, O.; Brabec, V.; Gandin, V.; Gibson, D. Pt(IV) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action. Chem. Sci. 2016, 7, 2381–2391. [Google Scholar] [CrossRef]
- Romain, C.; Gaillard, S.; Elmkaddem, M.K.; Toupet, L.; Fischmeister, C.; Thomas, C.M.; Renaud, J.L. New dipyridylamine ruthenium complexes for transfer hydrogenation of aryl ketones in water. Organometallics 2010, 29, 1992–1995. [Google Scholar] [CrossRef]
- Kaluderović, G.N.; Krajnović, T.; Momcilovic, M.; Stosic-Grujicic, S.; Mijatović, S.; Maksimović-Ivanić, D.; Hey-Hawkins, E. Ruthenium(II) p-cymene complex bearing 2,2′-dipyridylamine targets caspase 3 deficient MCF-7 breast cancer cells without disruption of antitumor immune response. J. Inorg. Biochem. 2015, 153, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Auzias, M.; Mattsson, J.; Therrien, B.; Süss-Fink, G. New dinuclear Ru2(CO)4 sawhorse-type complexes containing bridging carboxylato ligands. Z. Anorg. Allg. Chem. 2009, 635, 115–119. [Google Scholar] [CrossRef]
- Stodt, R.; Gencaslan, S.; Müller, I.M.; Sheldrick, W.S. Preparation, reactivity and peptide labelling Properties of (η6-arene)ruthenium(II) complexes with pendant carboxylate groups. Eur. J. Inorg. Chem. 2003, 1873–1882. [Google Scholar] [CrossRef]
- Stringer, T.; Therrien, B.; Hendricks, D.T.; Guzgay, H.; Smith, G.S. Mono- and dinuclear (η6-arene) ruthenium(II) benzaldehyde thiosemicarbazone complexes: Synthesis, characterization and cytotoxicity. Inorg. Chem. Commun. 2011, 14, 956–960. [Google Scholar] [CrossRef]
- Gupta, G.; Gloria, S.; Das, B.; Rao, K.M. Study of new mononuclear platinum group metal complexes containing η5 and η6-carbocyclic ligands and nitrogen based derivatives and formation of helices due to N–H···Cl interactions. J. Mol. Struct. 2010, 979, 205–213. [Google Scholar] [CrossRef]
- Kumar, P.; Singh, A.K.; Pandey, R.; Li, P.Z.; Singh, S.K.; Xu, Q.; Pandey, D.S. Synthesis, characterization and reactivity of arene ruthenium compounds based on 2,2′-dipyridylamine and di-2-pyridylbenzylamine and their applications in catalytic hydrogen transfer of ketones. J. Organomet. Chem. 2010, 695, 2205–2212. [Google Scholar] [CrossRef]
- Kubanik, M.; Holtkamp, H.; Sohnel, T.; Jamieson, S.M.F.; Hartinger, C.G. Impact of the halogen substitution pattern on the biological activity of organoruthenium 8-hydroxyquinoline anticancer agents. Organometallics 2015, 34, 5658–5668. [Google Scholar] [CrossRef]
- Pouchert, C.J. The Aldrich Library of Infrared Spectra (Ed. III); Aldrich Chemical Co.: Milwaukee, WI, USA, 1981. [Google Scholar]
- Govindaswamy, P.; Mozharivskyj, Y.A.; Kollipara, M.R. Syntheses, spectral and structural studies of Schiff base complexes of η5-pentamethylcyclopentadienyl rhodium and iridium. Polyhedron 2005, 24, 1710–1716. [Google Scholar] [CrossRef]
- Petruševski, G.; Naumov, P.; Jovanovski, G.; Bogoeva-Gaceva, G.; Ng, S.W. Solid-state forms of sodium valproate, active component of the anticonvulsant drug epilim. ChemMedChem 2008, 3, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Brittain, H.G. Vibrational spectroscopic studies of cocrystals and salts. 3. Cocrystal products formed by benzenecarboxylic acids and their sodium salts. Cryst. Growth Des. 2010, 10, 1990–2003. [Google Scholar] [CrossRef]
- Griffith, D.M.; Duff, B.; Suponitsky, K.Y.; Kavanagh, K.; Morgan, M.P.; Egan, D.; Marmion, C.J. Novel trans-platinum complexes of the histone deacetylase inhibitor valproic acid; synthesis, in vitro cytotoxicity and mutagenicity. J. Inorg. Biochem. 2011, 105, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Therrien, B.; Kim, J. [Bis-(2-pyrid-yl-κN)amine]chlorido(η6-hexa-methyl-benzene)-ruthenium(II) hexa-fluorido-phosphate dichloro-methane solvate. Acta Cryst. 2011, E67, m548. [Google Scholar]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Cryst. 2002, B58, 380–388. [Google Scholar] [CrossRef]
- Betanzos-Lara, S.; Novakova, O.; Deeth, R.J.; Pizarro, A.M.; Clarkson, G.J.; Liskova, B.; Brabec, V.; Sadler, P.J.; Habtemariam, A. Bipyrimidine ruthenium(II) arene complexes: Structure, reactivity and cytotoxicity. J. Biol. Inorg. Chem. 2012, 17, 1033–1051. [Google Scholar] [CrossRef] [PubMed]
- Sadler, P.; Morris, R.; Parsons, S.; Messenger, D. CCDC 276854: Experimental Crystal Structure Determination. CSD Commun. 2005. [Google Scholar] [CrossRef]
- Jolley, K.E.; Clarkson, G.J.; Wills, M. Tethered Ru(II) catalysts containing a Ru–I bond. J. Organomet. Chem. 2015, 776, 157–162. [Google Scholar] [CrossRef]
- Jamieson, E.R.; Lippard, S.J. Structure, recognition, and processing of Cisplatin−DNA adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.E.; Aird, R.E.; del Socorro Murdoch, P.; Chen, H.; Cummings, J.; Hughes, N.D.; Parsons, S.; Parkin, A.; Boyd, G.; Jodrell, D.I.; et al. Inhibition of cancer cell growth by ruthenium(II) arene complexes. J. Med. Chem. 2001, 44, 3616–3621. [Google Scholar] [CrossRef] [PubMed]
- Štarha, P.; Hanousková, L.; Trávníček, Z. Organometallic half-sandwich dichloridoruthenium(II) complexes with 7-azaindoles: Synthesis, characterization and elucidation of their anticancer inactivity against A2780 cell line. PLoS ONE 2015, 10, e0143871. [Google Scholar]
- Reedijk, J. Why does cisplatin reach guanine-N7 with competing S-donor ligands available in the cell? Chem. Rev. 1999, 99, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, J.; Habtemariam, A.; Bella, J.; Sadler, P.J. Competition between glutathione and guanine for a ruthenium(II) arene anticancer complex: Detection of a sulfenato intermediate. J. Am. Chem. Soc. 2005, 127, 17734–17743. [Google Scholar] [CrossRef] [PubMed]
- Govender, P.; Renfrew, A.K.; Clavel, C.M.; Dyson, P.J.; Therrien, B.; Smith, G.S. Antiproliferative activity of chelating N,O- and N,N-ruthenium(II) arene functionalised poly(propyleneimine) dendrimer scaffolds. Dalton Trans. 2011, 40, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Makhubela, B.C.E.; Meyer, M.; Smith, G.S. Evaluation of trimetallic Ru(II)- and Os(II)-arene complexes as potential anticancer agents. J. Organomet. Chem. 2014, 772–773, 229–241. [Google Scholar] [CrossRef]
- Tönnemann, J.; Risse, J.; Grote, Z.; Scopelliti, R.; Severin, K. Efficient and rapid synthesis of chlorido-bridged half-sandwich complexes of ruthenium, rhodium, and iridium by microwave heating. Eur. J. Inorg. Chem. 2013, 4558–4562. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef] [PubMed]
- Bruker. Apex3; Bruker AXS Inc.: Madison, WI, USA, 2015. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Brandenburg, K. Diamond Version 4.0.3; Crystal Impact GbR: Bonn, Germany, 2015. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–5 are available from the authors.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | N2–H | C3–H | C4–H | C5–H | C6–H |
|---|---|---|---|---|---|
| 1 | 1.21 | −0.52 | 0.34 | 0.38 | 0.34 |
| 2 | 1.22 | −0.54 | 0.33 | 0.36 | 0.41 |
| 3 | 1.24 | −0.56 | 0.31 | 0.34 | 0.50 |
| 4 | 1.17 | −0.51 | 0.34 | 0.39 | 0.52 |
| 5 | – | −0.54 | 0.32 | 0.36 | 0.56 |
| Empirical Formula | C20H23F6IN3PRu |
|---|---|
| Formula weight | 678.35 |
| Temperature (K) | 120(2) |
| Wavelength (Å) | 0.71073 |
| Crystal system | Triclinic, P-1 |
| Unit cell dimensions | |
| a (Å) | 8.708(3) |
| b (Å) | 10.275(4) |
| c (Å) | 13.349(5) |
| α (°) | 93.15(2) |
| β (°) | 105.564(15) |
| γ (°) | 94.245(17) |
| V (Å3) | 1143.9(8) |
| Z, Dcalc (g·cm−3) | 2, 1.969 |
| Absorption coefficient (mm−1) | 2.167 |
| Crystal size (mm) | 0.160 × 0.100 × 0.100 |
| F (000) | 660 |
| θ range for data collection (°) | 2.438 to 24.999 |
| Index ranges (h, k, l) | −10 ≤ h ≤ 10 |
| −12 ≤ k ≤ 12 | |
| −15 ≤ l ≤ 15 | |
| Reflections collected | 26913 |
| Independent reflections | 4035 [R(int) = 0.0393] |
| Data/restraints/parameters | 4035/1/295 |
| Goodness–of–fit on F2 | 1.052 |
| Final R indices [I > 2σ(I)] | R1 = 0.0202, wR2 = 0.0499 |
| R indices (all data) | R1 = 0.0241, wR2 = 0.0516 |
| Largest peak and hole (e·Å−3) | 0.585 and −0.871 |
| Parameter 2 | 1 | 3 |
|---|---|---|
| Ru1–X1 | 2.4148(7) | 2.7277(10) |
| Ru1–N1 | 2.107(2) | 2.101(2) |
| Ru1–N1A | 2.096(2) | 2.112(2) |
| Ru1–Cg | 1.6817(2) | 1.6895(6) |
| Ru1–C11 | 2.206(2) | 2.239(3) |
| Ru1–C12 | 2.202(2) | 2.197(3) |
| Ru1–C13 | 2.215(3) | 2.197(3) |
| Ru1–C14 | 2.237(2) | 2.210(3) |
| Ru1–C15 | 2.203(2) | 2.184(3) |
| Ru1–C16 | 2.169(3) | 2.212(3) |
| X1–Ru1–N1 | 87.04(7) | 87.52(6) |
| X1–Ru1–N1A | 87.26(6) | 88.46(6) |
| X1–Ru1–Cg | 127.32(6) | 127.54(2) |
| N1–Ru1–N1A | 82.30(8) | 84.47(8) |
| N1–Ru1–Cg | 128.72(6) | 127.27(6) |
| N1A–Ru1–Cg | 127.32(6) | 127.59(6) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štarha, P.; Trávníček, Z.; Křikavová, R.; Dvořák, Z. Half-Sandwich Ru(II) Halogenido, Valproato and 4-Phenylbutyrato Complexes Containing 2,2′-Dipyridylamine: Synthesis, Characterization, Solution Chemistry and In Vitro Cytotoxicity. Molecules 2016, 21, 1725. https://doi.org/10.3390/molecules21121725
Štarha P, Trávníček Z, Křikavová R, Dvořák Z. Half-Sandwich Ru(II) Halogenido, Valproato and 4-Phenylbutyrato Complexes Containing 2,2′-Dipyridylamine: Synthesis, Characterization, Solution Chemistry and In Vitro Cytotoxicity. Molecules. 2016; 21(12):1725. https://doi.org/10.3390/molecules21121725
Chicago/Turabian StyleŠtarha, Pavel, Zdeněk Trávníček, Radka Křikavová, and Zdeněk Dvořák. 2016. "Half-Sandwich Ru(II) Halogenido, Valproato and 4-Phenylbutyrato Complexes Containing 2,2′-Dipyridylamine: Synthesis, Characterization, Solution Chemistry and In Vitro Cytotoxicity" Molecules 21, no. 12: 1725. https://doi.org/10.3390/molecules21121725
APA StyleŠtarha, P., Trávníček, Z., Křikavová, R., & Dvořák, Z. (2016). Half-Sandwich Ru(II) Halogenido, Valproato and 4-Phenylbutyrato Complexes Containing 2,2′-Dipyridylamine: Synthesis, Characterization, Solution Chemistry and In Vitro Cytotoxicity. Molecules, 21(12), 1725. https://doi.org/10.3390/molecules21121725