
Enantioselective Analytical- and Preparative-Scale Separation of Hexabromocyclododecane Stereoisomers Using Packed Column Supercritical Fluid Chromatography

 ,
, 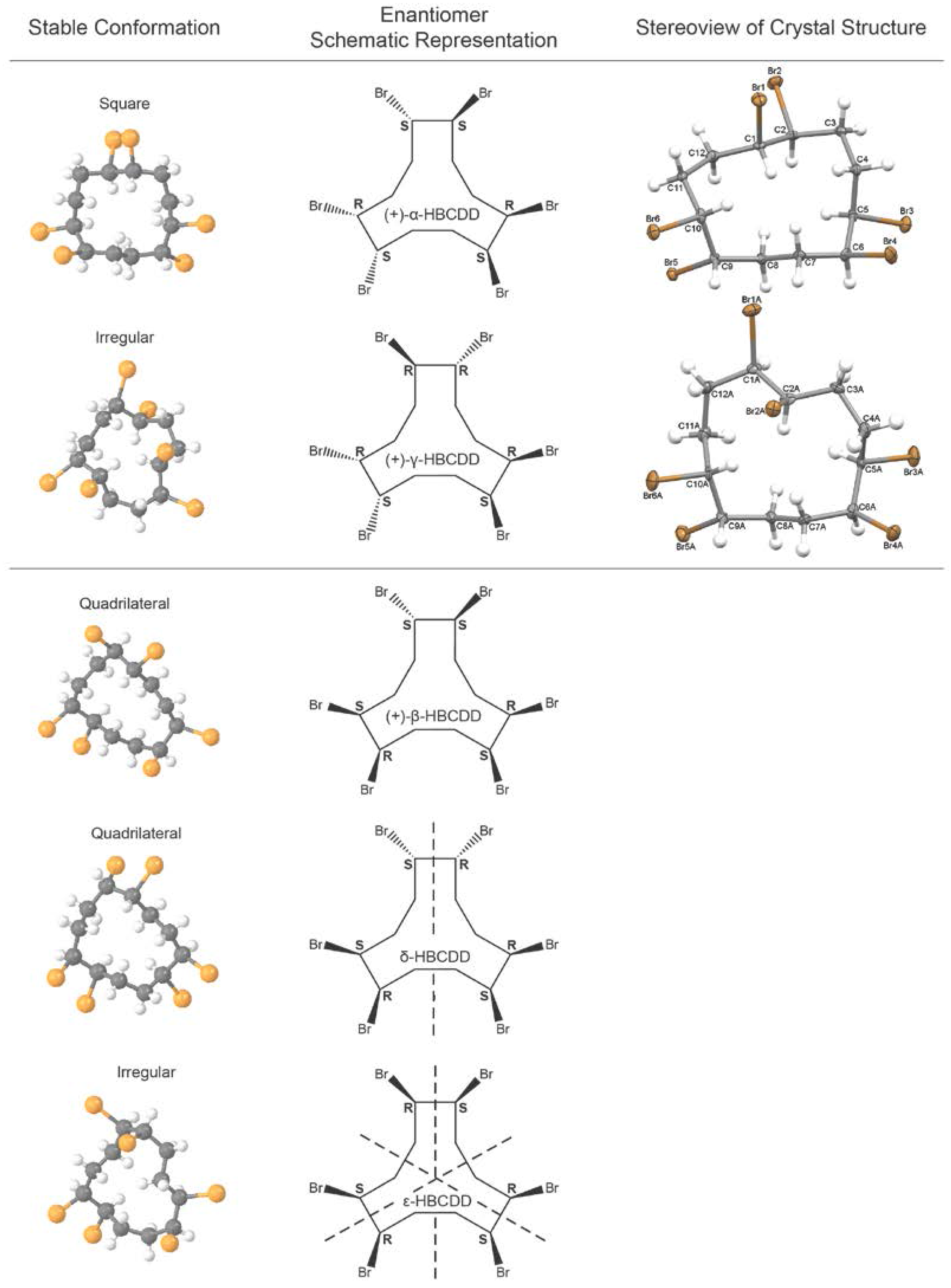{kind=link}
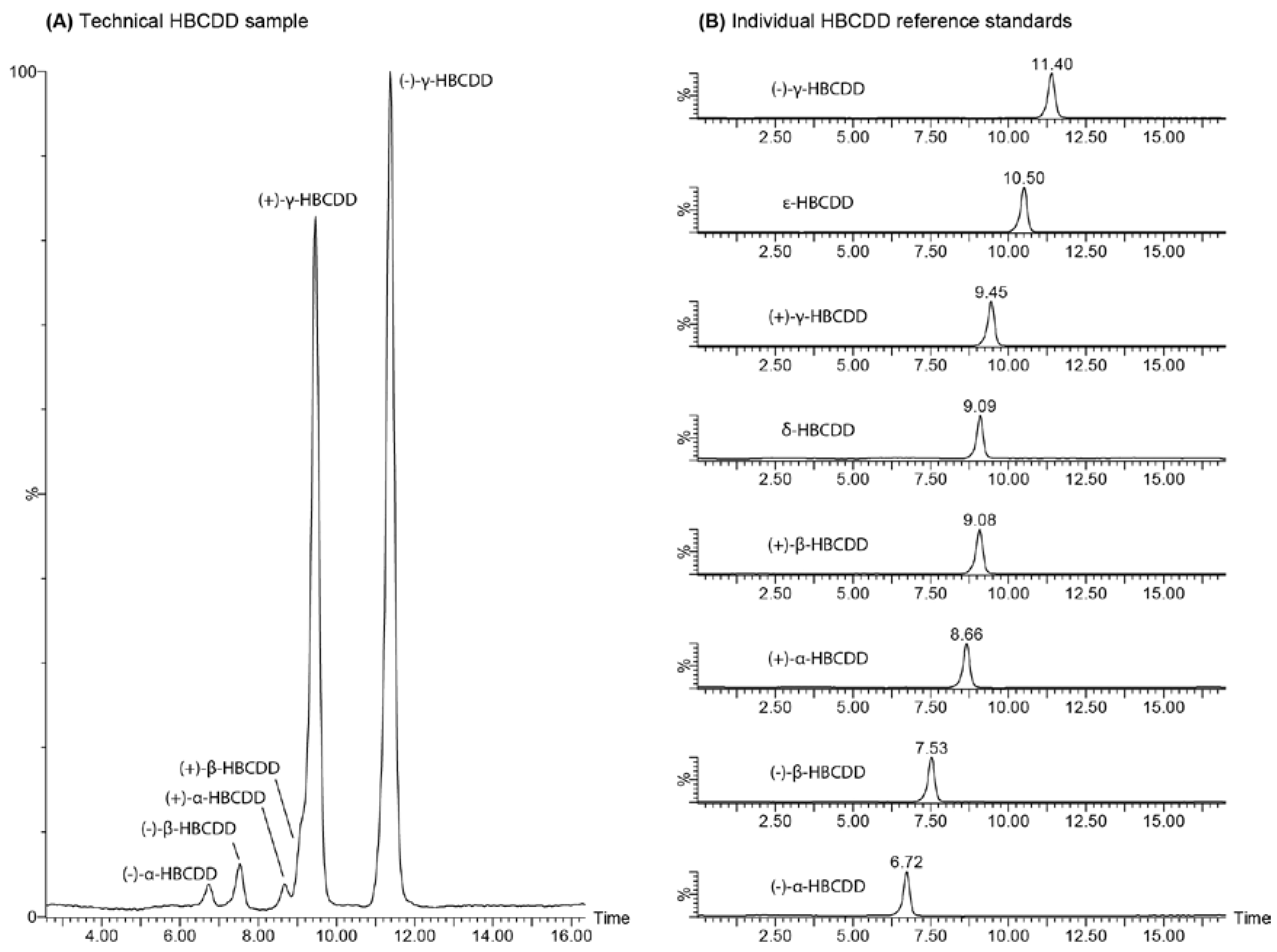{kind=link}
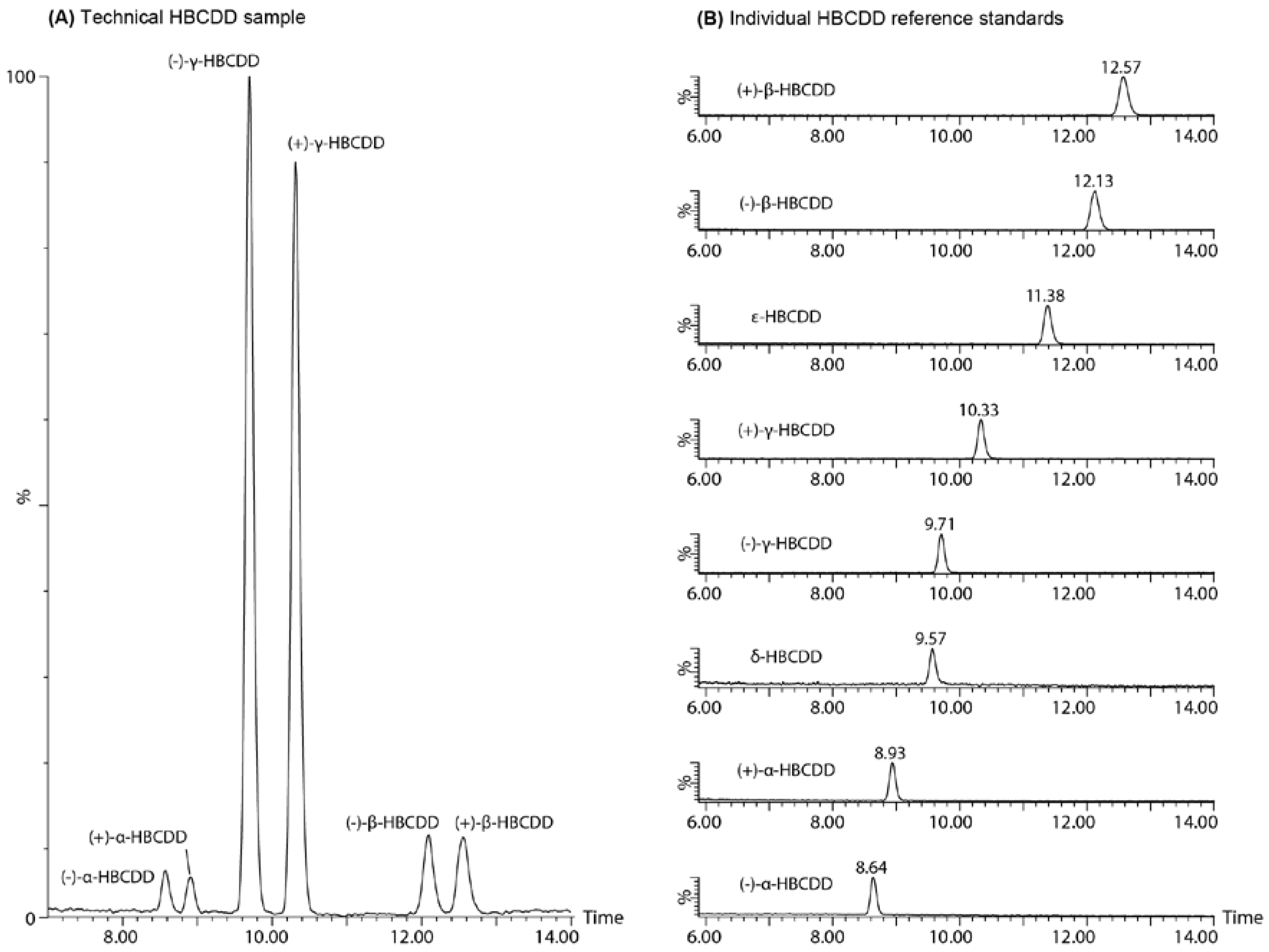{kind=link}
Abstract
:1. Introduction
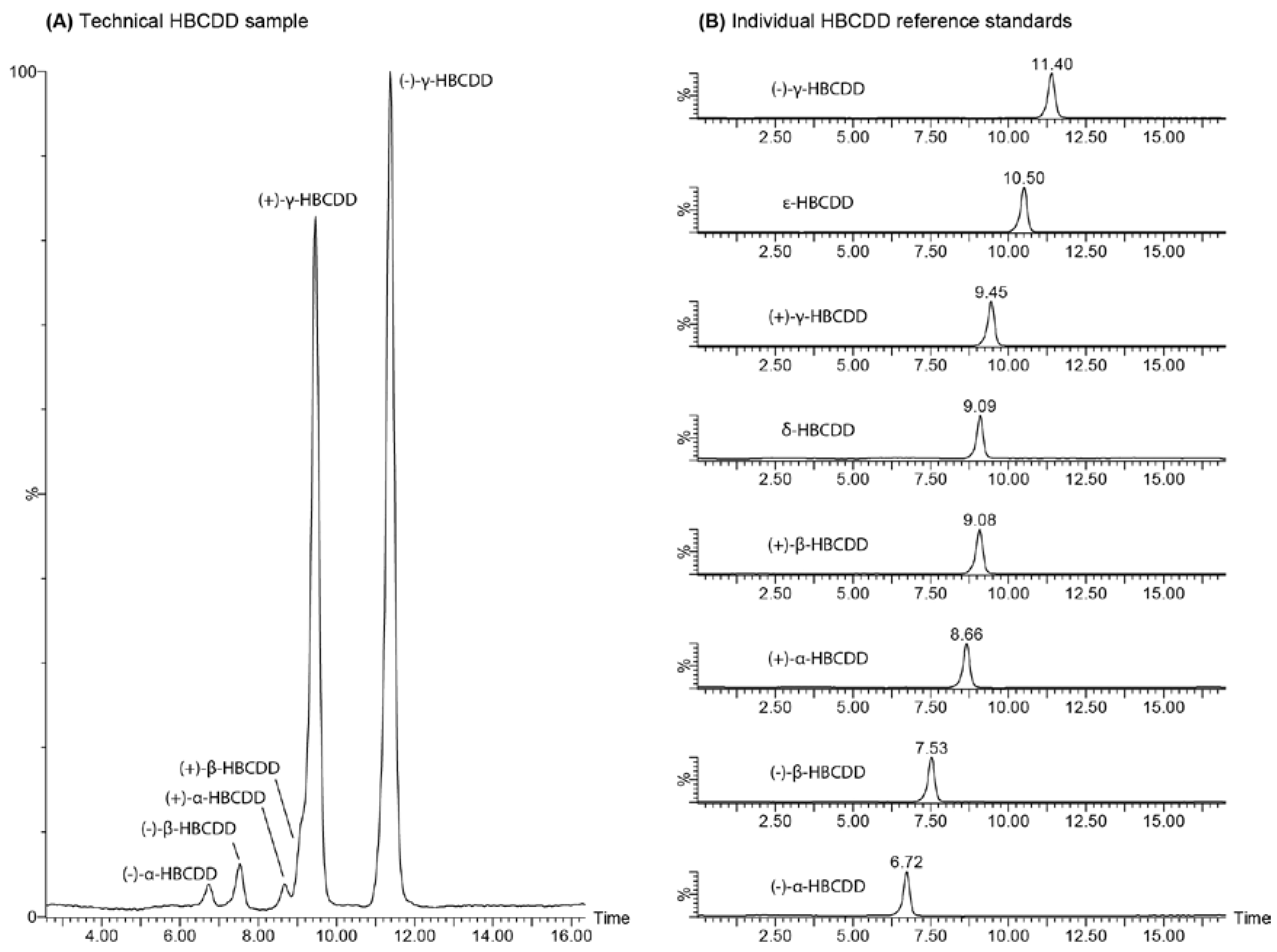
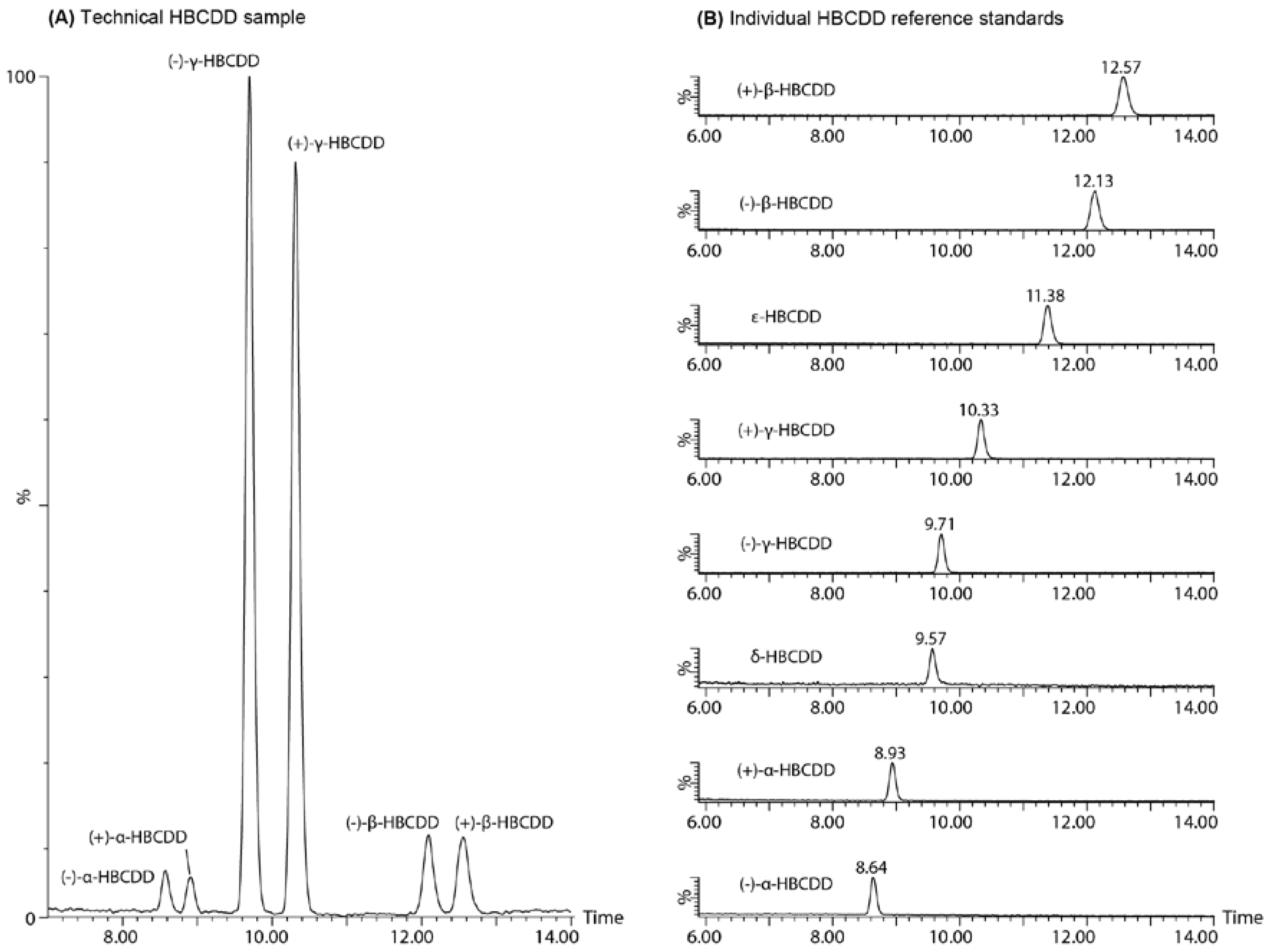
2. Results and Discussion
2.1. Preparative Separation of HBCDD Enantiomers
2.2. Characterization of Isolated HBCDD Enantiomers
2.3. Enantiomer Resolution
2.4. Chromatographic Considerations
3. Materials and Methods
3.1. Chemicals
3.2. Chromatographic Systems and Conditions
3.2.1. Isolation of Enantiomerically Pure (+)- and (−)-α-, β-, and γ-HBCDD by Preparative-Scale pSFC
3.2.2. Analytical pSFC-MS Analysis of the HBCDD Enantiomers
3.2.3. LC-MS Analysis of the HBCDD Enantiomers
3.2.4. X-ray Diffraction Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alaee, M.; Arias, P.; Sjodin, A.; Bergman, A. An overview of commercially used brominated flame retardants, their applications, their use patterns in different countries/regions and possible modes of release. Environ. Int. 2003, 29, 683–689. [Google Scholar] [CrossRef]
- Covaci, A.; Gerecke, A.C.; Law, R.J.; Voorspoels, S.; Kohler, M.; Heeb, N.V.; Leslie, H.; Allchin, C.R.; de Boer, J. Hexabromocyclododecanes (HBCDs) in the environment and humans: A review. Environ. Sci. Technol. 2006, 40, 3679–3688. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, R.W.; Gonsior, S.; MacGregor, J.A.; Desjardins, D.; Ariano, J.; Friederich, U. Fate and effect of hexabromocyclododecane in the environment. Organohalog. Compd. 2004, 66, 2300–2305. [Google Scholar]
- Koch, C.; Schmidt-Koetters, T.; Rupp, R.; Sures, B. Review of hexabromocyclododecane (HBCD) with a focus on legislation and recent publications concerning toxicokinetics and -dynamics. Environ. Pollut. 2015, 199, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Law, R.J.; Covaci, A.; Harrad, S.; Herzke, D.; Abdallah, M.A.E.; Femie, K.; Toms, L.-M.L.; Takigami, H. Levels and trends of PBDEs and HBCDs in the global environment: Status at the end of 2012. Environ. Int. 2014, 65, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Heeb, N.V.; Graf, H.; Schweizer, W.B.; Lienemann, P. Thermally-induced transformation of hexabromocyclo dodecanes and isobutoxypenta bromocyclododecanes in flame-proofed polystyrene materials. Chemosphere 2010, 80, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Harrad, S.; Abdallah, M.A.E.; Covaci, A. Causes of variability in concentrations and diastereomer patterns of hexabromocyclododecanes in indoor dust. Environ. Int. 2009, 35, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Vorkamp, K.; Bester, K.; Riget, F.F. Species-Specific Time Trends and Enantiomer Fractions of Hexabromocyclododecane (HBCD) in Biota from East Greenland. Environ. Sci. Technol. 2012, 46, 10549–10555. [Google Scholar] [CrossRef] [PubMed]
- Borga, K.; Bidleman, T.F. Enantiomer fractions of organic chlorinated pesticides in arctic marine lee fauna, zooplankton, and benthos. Environ. Sci. Technol. 2005, 39, 3464–3473. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Li, D.; Shen, R.; Wang, X.; Shi, D. Mechanisms of hexabromocyclododecanes induced developmental toxicity in marine medaka (Oryzias melastigma) embryos. Aquat. Toxicol. 2014, 152, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, F.; Xu, C.; Liu, W.; Wen, S.; Xu, Y. Cytotoxicity evaluation of three pairs of hexabromocyclododecane (HBCD) enantiomers on Hep G2 cell. Toxicol. In Vitro 2008, 22, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Esslinger, S.; Becker, R.; Jung, C.; Schroeter-Kermani, C.; Bremser, W.; Nehls, I. Temporal trend (1988–2008) of hexabromocyclododecane enantiomers in herring gull eggs from the german coastal region. Chemosphere 2011, 83, 161–167. [Google Scholar] [CrossRef] [PubMed]
- He, M.-J.; Luo, X.-J.; Yu, L.-H.; Liu, J.; Zhang, X.-L.; Chen, S.-J.; Chen, D.; Mai, B.-X. Tetrabromobisphenol-A and Hexabromocyclododecane in Birds from an E-Waste Region in South China: Influence of Diet on Diastereoisomer- and Enantiomer-Specific Distribution and Trophodynamics. Environ. Sci. Technol. 2010, 44, 5748–5754. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, R.; Becker, R.; Esslinger, S.; Nehls, I. Enantiomer-specific analysis of hexabromocyclododecane in fish from Etnefjorden (Norway). Chemosphere 2010, 80, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-X.; Luo, X.-J.; Mo, L.; He, M.-J.; Zhang, Q.; Chen, S.-J.; Zou, F.-S.; Mai, B.-X. Hexabromocyclododecane in terrestrial passerine birds from e-waste, urban and rural locations in the Pearl River Delta, South China: Levels, biomagnification, diastereoisomer- and enantiomer-specific accumulation. Environ. Pollut. 2012, 171, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Janak, K.; Covaci, A.; Voorspoels, S.; Becher, G. Hexabromocyclododecane in marine species from the Western Scheldt Estuary: Diastereoisomer- and enantiomer-specific accumulation. Environ. Sci. Technol. 2005, 39, 1987–1994. [Google Scholar] [CrossRef] [PubMed]
- Tomy, G.T.; Pleskach, K.; Oswald, T.; Halldorson, T.; Helm, P.A.; Macinnis, G.; Marvin, C.H. Enantioselective bioaccumulation of hexabromocyclododecane and congener-specific accumulation of brominated diphenyl ethers in an eastern Canadian Arctic marine food web. Environ. Sci. Technol. 2008, 42, 3634–3639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, F.; Luo, C.; Wen, S.; Zhang, X.; Xu, Y. Bioaccumulative characteristics of hexabromocyclododecanes in freshwater species from an electronic waste recycling area in China. Chemosphere 2009, 76, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Lin, L.; Yan, C.; Zhang, X. Diastereoisomer- and Enantiomer-Specific Accumulation, Depuration, and Bioisomerization of Hexabromocyclododecanes in Zebrafish (Danio rerio). Environ. Sci. Technol. 2012, 46, 11040–11046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, H.; Ruan, Y. Enantiomer-specific accumulation, depuration, metabolization and isomerization of hexabromocyclododecane (HBCD) diastereomers in mirror carp from water. J. Hazard. Mater. 2014, 264, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.A.-E.; Uchea, C.; Chipman, J.K.; Harrad, S. Enantioselective Biotransformation of Hexabromocyclododecane by in Vitro Rat and Trout Hepatic Sub-Cellular Fractions. Environ. Sci. Technol. 2014, 48, 2732–2740. [Google Scholar] [CrossRef] [PubMed]
- Heeb, N.V.; Schweizer, W.B.; Mattrel, P.; Haag, R.; Gerecke, A.C.; Schmid, P.; Zennegg, M.; Vonmont, H. Regio- and stereoselective isomerization of hexabromocyclododecanes (HBCDs): Kinetics and mechanism of gamma- to alpha-HBCD isomerization. Chemosphere 2008, 73, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Esslinger, S.; Becker, R.; Mueller-Belecke, A.; Bremser, W.; Iung, C.; Nehls, I. HBCD Stereoisomer Pattern in Mirror Carps Following Dietary Exposure to Pure gamma-HBCD Enantiomers. J. Agric. Food Chem. 2010, 58, 9705–9710. [Google Scholar] [CrossRef] [PubMed]
- Esslinger, S.; Becker, R.; Maul, R.; Nehls, I. Hexabromocyclododecane Enantiomers: Microsomal Degradation and Patterns of Hydroxylated Metabolites. Environ. Sci. Technol. 2011, 45, 3938–3944. [Google Scholar] [CrossRef] [PubMed]
- Guerra, P.; Eliarrat, E.; Barcelo, D. Enantiomeric specific determination of hexabromocyclododecane by liquid chromatography-quadrupole linear ion trap mass spectrometry in sediment samples. J. Chromatogr. A 2008, 1203, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Marvin, C.H.; MacInnis, G.; Alaee, M.; Arsenault, G.; Tomy, G.T. Factors influencing enantiomeric fractions of hexabromocyclododecane measured using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Dodder, N.G.; Peck, A.M.; Kucklick, J.R.; Sander, L.C. Analysis of hexabromocyclododecane diastereomers and enantiomers by liquid chromatography/tandem mass spectrometry: Chromatographic selectivity and ionization matrix effects. J. Chromatogr. A 2006, 1135, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Bester, K.; Vorkamp, K. A two-dimensional HPLC separation for the enantioselective determination of hexabromocyclododecane (HBCD) isomers in biota samples. Anal. Bioanal. Chem. 2013, 405, 6519–6527. [Google Scholar] [CrossRef] [PubMed]
- Mullin, L.; Burgess, J.A.; Jogsten, I.E.; Geng, D.; Aubin, A.; van Bavel, B. Rapid separation of hexabromocyclododecane diastereomers using a novel method combining convergence chromatography and tandem mass spectrometry. Anal. Methods 2015, 7, 2950–2958. [Google Scholar] [CrossRef]
- Vanwasen, U.; Swaid, I.; Schneider, G.M. Physicochemical Principles and Applications of Supercritical Fluid Chromatography (SFC). Angew. Chem. Int. Ed. 1980, 19, 575–587. [Google Scholar] [CrossRef]
- Morita, A.; Kajimoto, O. Solute Solvent Interaction in Nonpolar Supercritical Fluid—A Clustering Model and Size Distribution. J. Phys. Chem. 1990, 94, 6420–6425. [Google Scholar] [CrossRef]
- Koeppen, R.; Becker, R.; Emmerling, F.; Jung, C.; Nehls, I. Enantioselective preparative HPLC separation of the HBCD—Stereoisomers from the technical product and their absolute structure elucidation using X-ray crystallography. Chirality 2007, 19, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Heeb, N.V.; Schweizer, W.B.; Mattrel, P.; Haag, R.; Gerecke, A.C.; Kohler, M.; Schmid, P.; Zennegg, M.; Wolfensberger, M. Solid-state conformations and absolute configurations of (+) and (-)alpha-, beta-, and gamma-hexabromocyclododecanes (HBCDs). Chemosphere 2007, 68, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, G.; Konstantinov, A.; Marvin, C.H.; MacInnis, G.; McAlees, A.; McCrindle, R.; Riddell, N.; Tomy, G.T.; Yeo, B. Synthesis of the two minor isomers, delta- and epsilon-1,2,5,6,9,10-hexabromocyclododecane, present in commercial hexabromocyclododecane. Chemosphere 2007, 68, 887–892. [Google Scholar] [CrossRef] [PubMed]
- De Klerck, K.; Mangelings, D.; Heyden, Y.V. Supercritical fluid chromatography for the enantioseparation of pharmaceuticals. J. Pharm. Biomed. Anal. 2012, 69, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ng, S.C.; Tan, T.T.Y.; Wang, Y. Recent development of cyclodextrin chiral stationary phases and their applications in chromatography. J. Chromatogr. A 2012, 1269, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.G.A.; Collins, C.H. Super/Subcritical Fluid Chromatography with Packed Columns: State of the Art and Applications. Quim. Nova 2014, 37, 1047–U210. [Google Scholar] [CrossRef]
- Chankvetadze, B. Recent developments on polysaccharide-based chiral stationary phases for liquid-phase separation of enantiomers. J. Chromatogr. A 2012, 1269, 26–51. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds may be available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riddell, N.; Mullin, L.G.; Van Bavel, B.; Ericson Jogsten, I.; McAlees, A.; Brazeau, A.; Synnott, S.; Lough, A.; McCrindle, R.; Chittim, B. Enantioselective Analytical- and Preparative-Scale Separation of Hexabromocyclododecane Stereoisomers Using Packed Column Supercritical Fluid Chromatography. Molecules 2016, 21, 1509. https://doi.org/10.3390/molecules21111509
Riddell N, Mullin LG, Van Bavel B, Ericson Jogsten I, McAlees A, Brazeau A, Synnott S, Lough A, McCrindle R, Chittim B. Enantioselective Analytical- and Preparative-Scale Separation of Hexabromocyclododecane Stereoisomers Using Packed Column Supercritical Fluid Chromatography. Molecules. 2016; 21(11):1509. https://doi.org/10.3390/molecules21111509
Chicago/Turabian StyleRiddell, Nicole, Lauren Gayle Mullin, Bert Van Bavel, Ingrid Ericson Jogsten, Alan McAlees, Allison Brazeau, Scott Synnott, Alan Lough, Robert McCrindle, and Brock Chittim. 2016. "Enantioselective Analytical- and Preparative-Scale Separation of Hexabromocyclododecane Stereoisomers Using Packed Column Supercritical Fluid Chromatography" Molecules 21, no. 11: 1509. https://doi.org/10.3390/molecules21111509