
Anti-Complementary Components of Helicteres angustifolia


Abstract
:1. Introduction
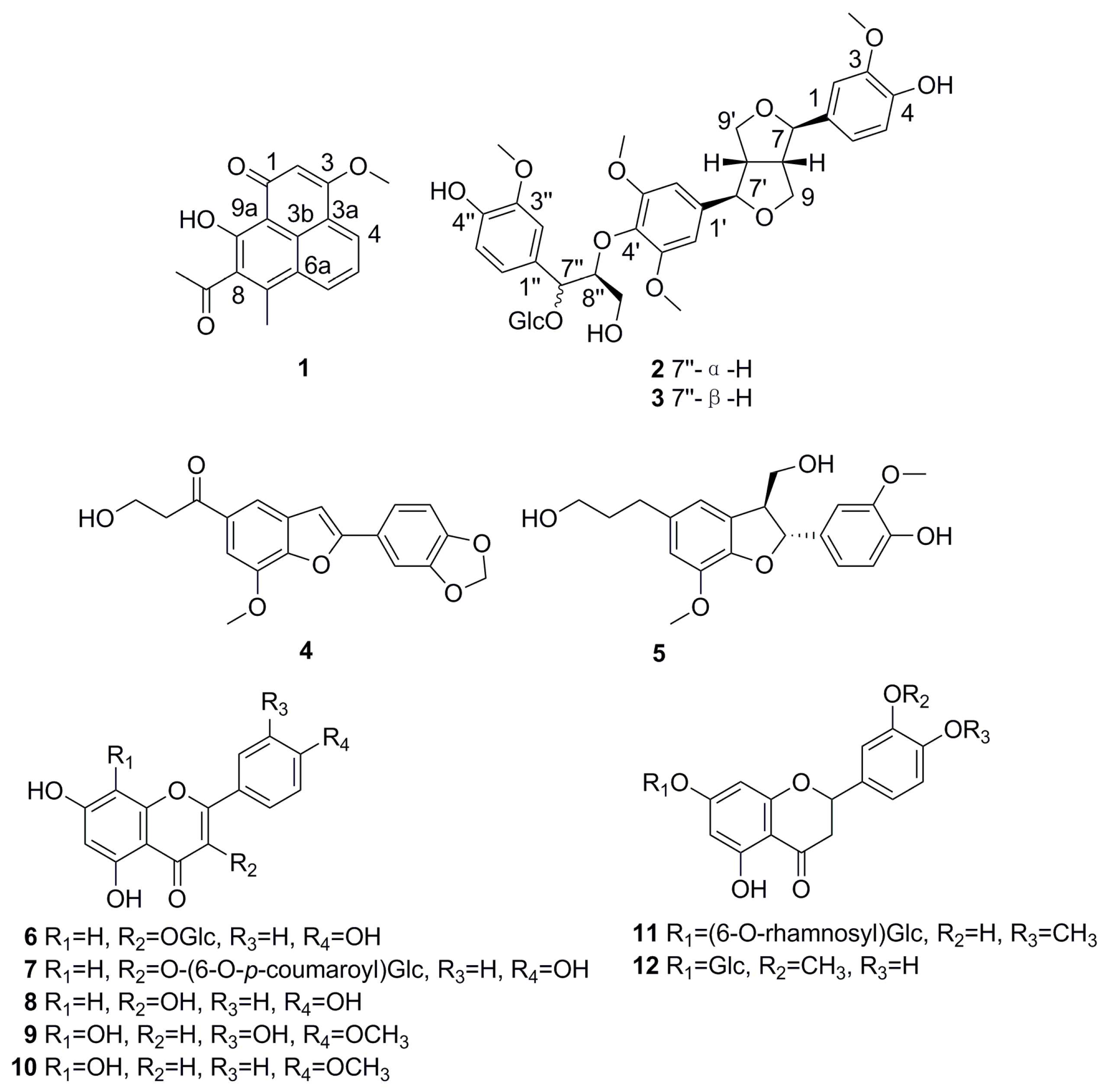
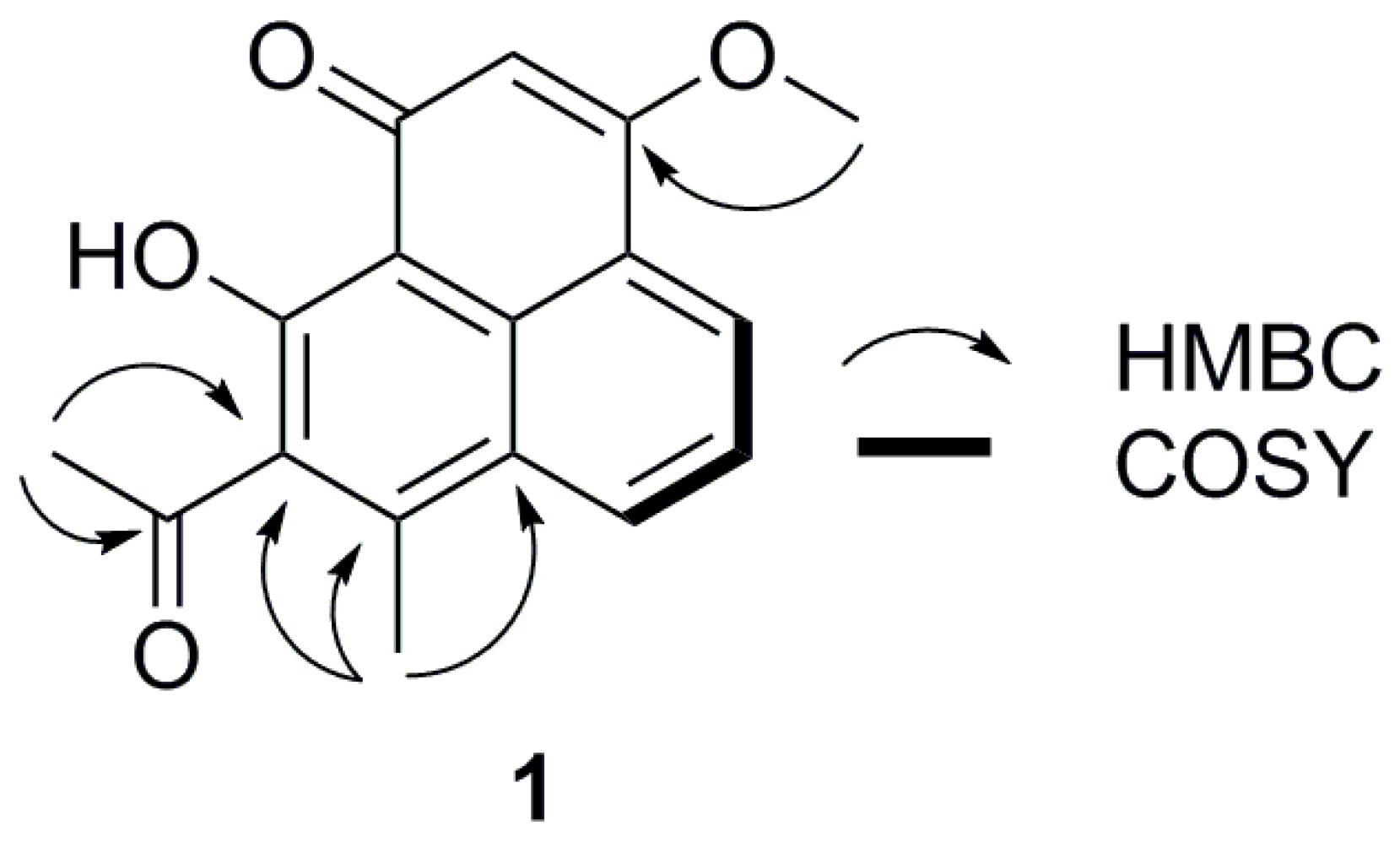
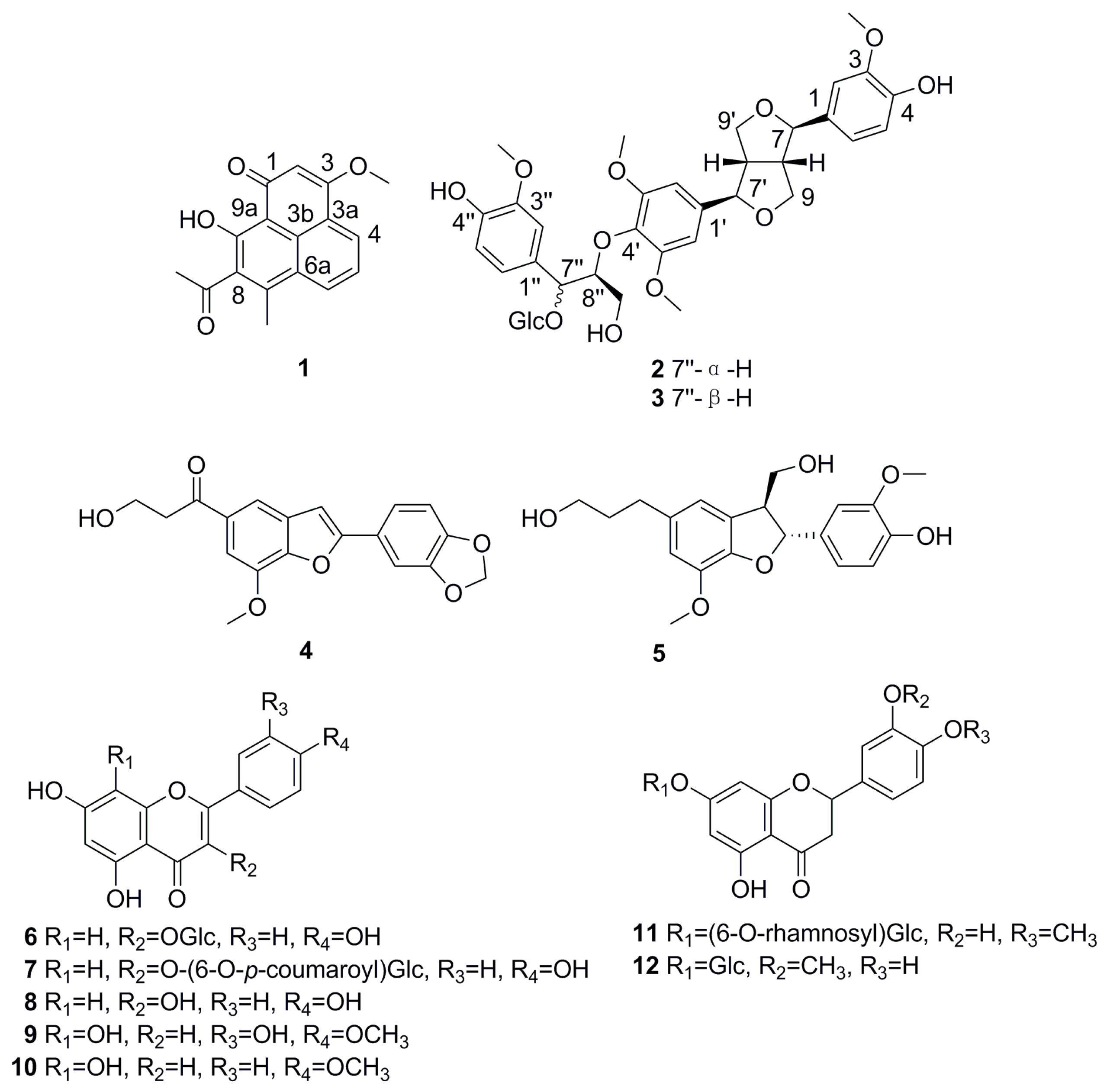
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectroscopic Data
3.5. Acid Hydrolysis of Compounds 2 and 3
3.6. Anti-Complementary Activity Assay Against CP and AP
3.7. Identification of the Targets in the Complement Activation Cascade
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carroll, M.C.; Fisher, M.B. Complement and the immune response. Curr. Opin. Immunol. 1997, 9, 64–69. [Google Scholar] [CrossRef]
- Morgan, B.P.; Harris, C.L. Complement therapeutics; history and current progress. Mol. Immunol. 2003, 40, 159–170. [Google Scholar] [CrossRef]
- Zhu, H.W.; Di, H.Y.; Zhang, Y.Y.; Zhang, J.W.; Chen, D.F. A protein-bond polysaccharide from the stem bark of Eucommia ulmoides and its anti-complementary effect. Carbohydr. Res. 2009, 344, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Mollnes, T.E.; Song, W.C.; Lambris, J.D. Complement in inflammatory tissue damage and disease. Trends Immunol. 2002, 23, 61–64. [Google Scholar] [CrossRef]
- Vovel, C.W.; Fritzinger, D.C. Cobra venom factor: Structure, function, and humanization for therapeutic complement depletion. Toxicon 2010, 56, 1198–1222. [Google Scholar]
- Thorgersen, E.B.; Ghebremariam, Y.T.; Thurman, J.M.; Fung, M.; Nielsen, E.W.; Holers, V.M.; Kotwal, G.J.; Mollnes, T.E. Candidate inhibitors of porcine complement. Mol. Immunol. 2007, 44, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.C.; Ricklin, D.; Lambris, J.D. Recent developments in low molecular weight complement inhibitors. Mol. Immunol. 2009, 47, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M. Current and future pharmacologic complement inhibitors. Hematol/Oncol. Clin. N. Am. 2015, 29, 561–582. [Google Scholar] [CrossRef] [PubMed]
- Jiangsu New Medical College. Dictionary of Chinese Herb Medicines; Shanghai Scientific and Technologic Press: Shanghai, China, 1996; pp. 178–179. [Google Scholar]
- Chen, W.L.; Tang, W.D.; Lou, L.G.; Zhao, W.M. Pregnane, coumarin and lupane derivatives and cyto toxic constituents from Helicters angustifolia. Phytochemistry 2006, 67, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.H.; Chen, C.M.; Lee, S.W.; Chen, Z.T. Cytotoxic triterpenoids from the root bark of Helicters angustifolia. Chem. Biodivers. 2008, 5, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.D.; Huang, Z.S.; Bao, Y.D.; An, L.K.; Ma, L.; Gu, L.Q. Two new sesquiterpenoids from Helicters angustifolia. Chin. Chem. Lett. 2005, 16, 49–52. [Google Scholar]
- Chen, M.; Muchersie, E.; Luo, C.; Forrester, J.V.; Xu, H.P. Inhibition of the alternative pathway of complement activation reduces inflammation in experimental autoimmune uveoretinitis. Eur. J. Immunol. 2010, 40, 2870–2881. [Google Scholar] [CrossRef] [PubMed]
- Ignatius, A.; Schoengraf, P.; Kreja, L.; Liedert, A.; Rechnagel, S.; Kandert, S.; Brenner, R.E.; Schneider, M.; Lambris, J.D. Complement C3a and C5a modulate osteoclast formation and inflammatory response of osteoblasts in synergism with IL-1β. J. Cell. Biochem. 2011, 112, 2594–2605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chen, D.F. Anticomplementary principles of a Chinese multiherb remedy for the treatment and prevention of SARS. J. Ethnopharmacol. 2008, 117, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhang, Y.Y.; Zhang, J.W.; Chen, D.F. Isolation and characterization of an anti-complementary polysaccharide D3-S1 from the roots of Bupleurum smithii. Int. Immunopharmacol. 2007, 7, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Matsumoto, T. Ungewöhnliche wichterle-reaktionen. Eine neue synthese von phenalenonen. Bull. Chem. Soc. Jpn. 1975, 48, 2935–2938. [Google Scholar] [CrossRef]
- Ernst-Russell, M.A.; Chai, C.L.L.; Elix, J.A.; McCarthy, P.M. Myeloconone A2, a new phenalenone from the lichen Myeloconis erumpens. Aust. J. Chem. 2000, 53, 1011–1013. [Google Scholar] [CrossRef]
- Tohru, K.; Satoko, M.; Shigetoshi, K.; Takaaki, T. Studies on the constituents of medicinal and related plants in Sri Lanka. III. Novel sesquilignans from Hedyotis lawsoniae. Chem. Pharm. Bull. 1985, 33, 1444–1451. [Google Scholar]
- Yukinori, M.; Tsuyoshi, T. Studies on constituents of Scutellaria species X IX: lignan glycosides of roots of Scutellaria baicalensis Georgi. Nat. Med. 1998, 52, 82–86. [Google Scholar]
- Bardet, M.; Robert, D.; Lundquist, K.; Von-Unge, S. Distribution of erythro and threo forms of different types of β-O-4 structures in aspen lignin by carbon-13 NMR using the 2D INADEQUATE experiment. Magn. Reson. Chem. 1998, 36, 597–600. [Google Scholar] [CrossRef]
- Talaptra, B.; Ray, T.; Talapatra, S.K. Structures of three new norlignans from Machilus glaucescens and their partial syntheses. Chem. Nat. Prod. 1978, 2, 200–201. [Google Scholar]
- Kuang, H.; Xia, Y.; Yang, B.; Wang, Q.; Lu, S. Lignan constituents from Chloranthus japonicus Sieb. Arch. Pharm. Res. 2009, 32, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Wu, X.; Wang, Y.; Ye, W.C.; Zhang, Q.W. Studies on the flavonoids from the flowers of Hylocereus undatus. J. Chin. Med. Mater. 2011, 34, 712–716. [Google Scholar]
- Zhong, H.J.; Chen, J.J.; Wang, H.Y.; Luo, S.D. Chemical investigation on Potentilla griffithii var. velutina. Chin. Tradit. Herb. Drugs. 2000, 31, 488–490. [Google Scholar]
- Xia, P.F.; Song, S.; Feng, Z.M.; Zhang, P.C. Chemical constituents from leaves of Sterculia foetida. Zhong Guo Zhong Yao Za Zhi 2009, 34, 2604–2606. [Google Scholar]
- Yang, N.Y.; Duan, J.A.; Li, P.; Qian, S.H. Studies on flavonoids of Lysimachia christinae Hance. Chin. Pharm. J. 2006, 4, 1621–1624. [Google Scholar]
- Kong, D.Y.; Luo, S.G.; Li, H.T.; Lei, X.H. Studies on chemical components of Viscum coloratum L. Pharm. Ind. 1987, 18, 123–127. [Google Scholar]
- Song, W.H.; Chen, Z.H.; Chen, D.F. Anticomplement monoterpenoid glucosides from the root bark of Paeonia suffruticosa. J. Nat. Prod. 2014, 77, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Di, H.Y.; Zhang, Y.Y.; Chen, D.F. An anti-complementary polysaccharide from the roots of Bupleurum chinense. Int. J. Biol. Macromol. 2013, 58, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.


{kind=link}
{kind=link}
{kind=link}
| No. | δH | δC | No. | δH | δC |
|---|---|---|---|---|---|
| 1 | 181.1 | 7 | 144.1 | ||
| 2 | 6.66 s | 100.2 | 8 | 136.3 | |
| 3 | 166.6 | 9 | 174.2 | ||
| 3a | 120.8 | 9a | 106.5 | ||
| 3b | 127.1 | 3-OCH3 | 3.92 s | 56.4 | |
| 4 | 8.43 d (7.8) | 127.9 | 7-CH3 | 2.58 s | 16.0 |
| 5 | 7.58 t (7.8) | 123.7 | 8-COCH3 | 204.1 (CO) | |
| 6 | 8.24 d (7.8) | 130.8 | 2.72 s | 32.1 (CH3) | |
| 6a | 125.4 | 9-OH | 17.36 s |
| No. | 2 | 3 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | 133.7 | 133.7 | ||
| 2 | 6.97 d (1.8) | 111.0 | 6.97 d (1.4) | 111.0 |
| 3 | 149.1 | 149.1 | ||
| 4 | 147.4 a | 147.4 a | ||
| 5 | 6.79 d (8.1) | 116.1 | 6.79 d (8.1) | 116.1 |
| 6 | 6.83 dd (8.1, 1.7) | 120.1 | 6.83 dd (8.0, 1.3) | 120.1 |
| 7 | 4.72 d (4.8) | 87.5 | 4.73 d (3.6) | 87.5 |
| 8 | 3.14 ddd (11.3, 8.3, 4.9) | 55.3 b | 3.13–3.15 overlapped | 55.3 b |
| 9 | 4.24–4.30 overlapped, 3.88–3.91 overlapped | 72.9 c | 4.27–4.31 overlapped, 3.88–3.91 overlapped | 72.9 c |
| 1′ | 138.9 | 138.9 | ||
| 2′,6′ | 6.64 s | 104.2 | 6.66 s | 104.1 |
| 3′,5′ | 154.3 | 154.5 | ||
| 4′ | 136.2 | 136.4 | ||
| 7′ | 4.75 d (4.6) | 87.2 | 4.76 d (3.0) | 87.3 |
| 8′ | 3.14 ddd (11.3, 8.3, 4.9) | 55.7 b | 3.13–3.15 overlapped | 55.7 b |
| 9′ | 4.24–4.30 overlapped, 3.88–3.91 overlapped | 72.8 c | 4.27–4.31 overlapped, 3.88–3.91 overlapped | 72.7 c |
| 1′′ | 132.4 | 130.8 | ||
| 2′′ | 7.03 d (1.7) | 112.7 | 7.24 brs | 113.1 |
| 3′′ | 148.4 | 148.7 | ||
| 4′′ | 147.1 a | 147.1 a | ||
| 5′′ | 6.72 d (8.1) | 115.3 | 6.80 d (8.0) | 115.5 |
| 6′′ | 6.87 dd (8.1, 1.7) | 121.9 | 6.92 dd (8.0, 1.3) | 122.0 |
| 7′′ | 5.15 d (6.2) | 82.3 | 5.28 d (3.2) | 77.7 |
| 8′′ | 4.42–4.44 m | 86.6 | 4.26–4.27 m | 86.8 |
| 9′′ | 4.05 dd (12.4, 4.0) 3.76 dd (12.4, 3.0) | 61.5 | 3.88–3.91 overlapped 3.44 dd (11.4, 4.5) | 61.4 |
| β-d-glc | ||||
| 1′′′ | 4.67 d (7.8) | 104.4 | 4.21 d (7.5) | 101.0 |
| 2′′′ | 3.27-3.29 m | 75.7 | 3.33–3.34 m | 75.2 |
| 3′′′ | 3.38 d (9.0) | 78.1 | 3.28–3.31 overlapped | 77.7 |
| 4′′′ | 3.30–3.32 m | 71.5 | 3.28–3.31 overlapped | 71.9 |
| 5′′′ | 3.19 ddd (9.6, 5.4, 2.5) | 77.8 | 3.12–3.13 m | 77.8 |
| 6′′′ | 3.73 dd (11.9, 2.3) 3.60 dd (11.8, 5.4) | 62.7 | 3.83–3.85 m 3.69 dd (11.9, 5.9) | 62.7 |
| 3-OCH3 | 3.87 s | 56.41 d | 3.88 s | 56.37 d |
| 3′,5′-OCH3 | 3.80 s | 56.6 | 3.74 s | 56.5 |
| 3′′-OCH3 | 3.83 s | 56.43 d | 3.85 s | 56.42 d |
| Compound | CH50 (mM) a | AP50 (mM) a |
|---|---|---|
| 1 | 0.744 ± 0.099 | >1 |
| 2 | 0.419 ± 0.043 | >1 |
| 3 | 0.249 ± 0.021 | >1 |
| 4 | 0.040 ± 0.009 | 0.105 ± 0.015 |
| 5 | 0.009 ± 0.002 **,b | 0.021 ± 0.003 **,b |
| 6 | 0.877 ± 0.081 | >1 |
| 7 | 0.143 ± 0.019 | 0.335 ± 0.040 |
| 8 | 0.147 ± 0.022 | 0.311 ± 0.033 |
| 9 | 0.232 ± 0.25 | 0.501 ± 0.065 |
| 10 | 0.511 ± 0.043 | 0.984 ± 0.107 |
| 11 | >1 | >1 |
| 12 | 0.351 ± 0.033 | 0.556 ± 0.061 |
| Heparin c | 0.026 ± 0.005 | 0.055 ± 0.008 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, X.; Lu, Y.; Cheng, Z.-H.; Chen, D.-F. Anti-Complementary Components of Helicteres angustifolia. Molecules 2016, 21, 1506. https://doi.org/10.3390/molecules21111506
Yin X, Lu Y, Cheng Z-H, Chen D-F. Anti-Complementary Components of Helicteres angustifolia. Molecules. 2016; 21(11):1506. https://doi.org/10.3390/molecules21111506
Chicago/Turabian StyleYin, Xiang, Yan Lu, Zhi-Hong Cheng, and Dao-Feng Chen. 2016. "Anti-Complementary Components of Helicteres angustifolia" Molecules 21, no. 11: 1506. https://doi.org/10.3390/molecules21111506