3.3. Synthetic Procedures
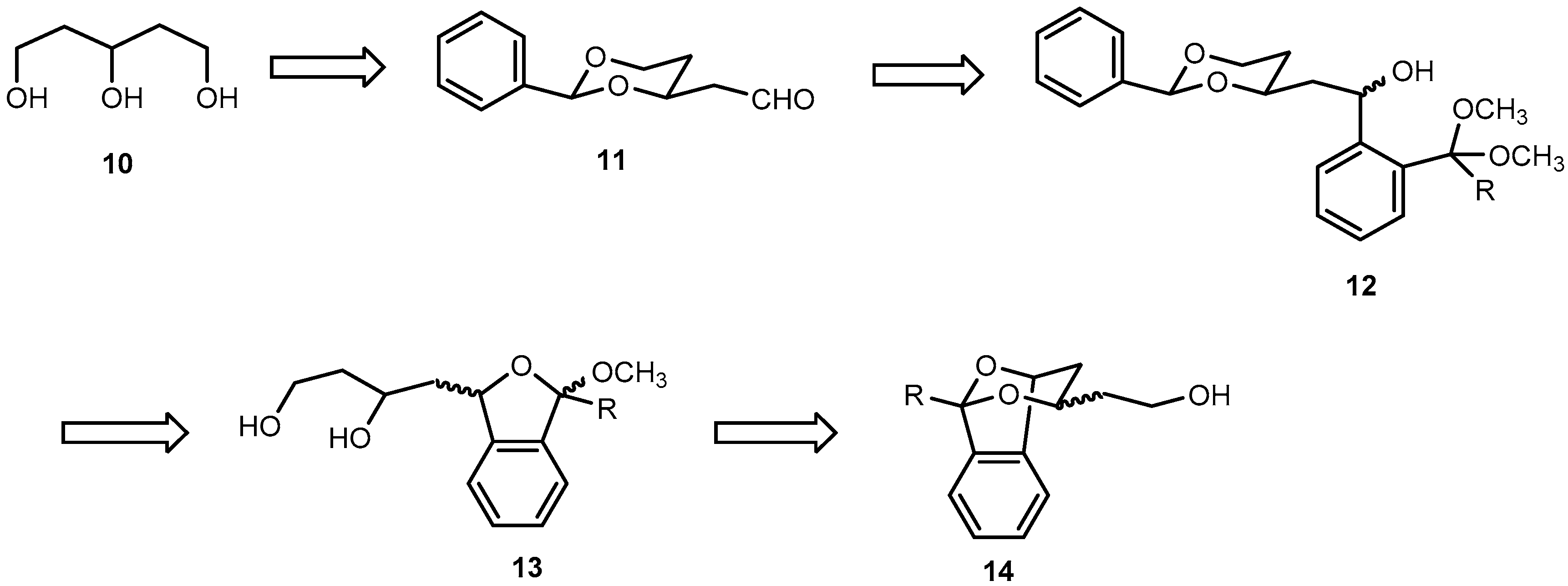
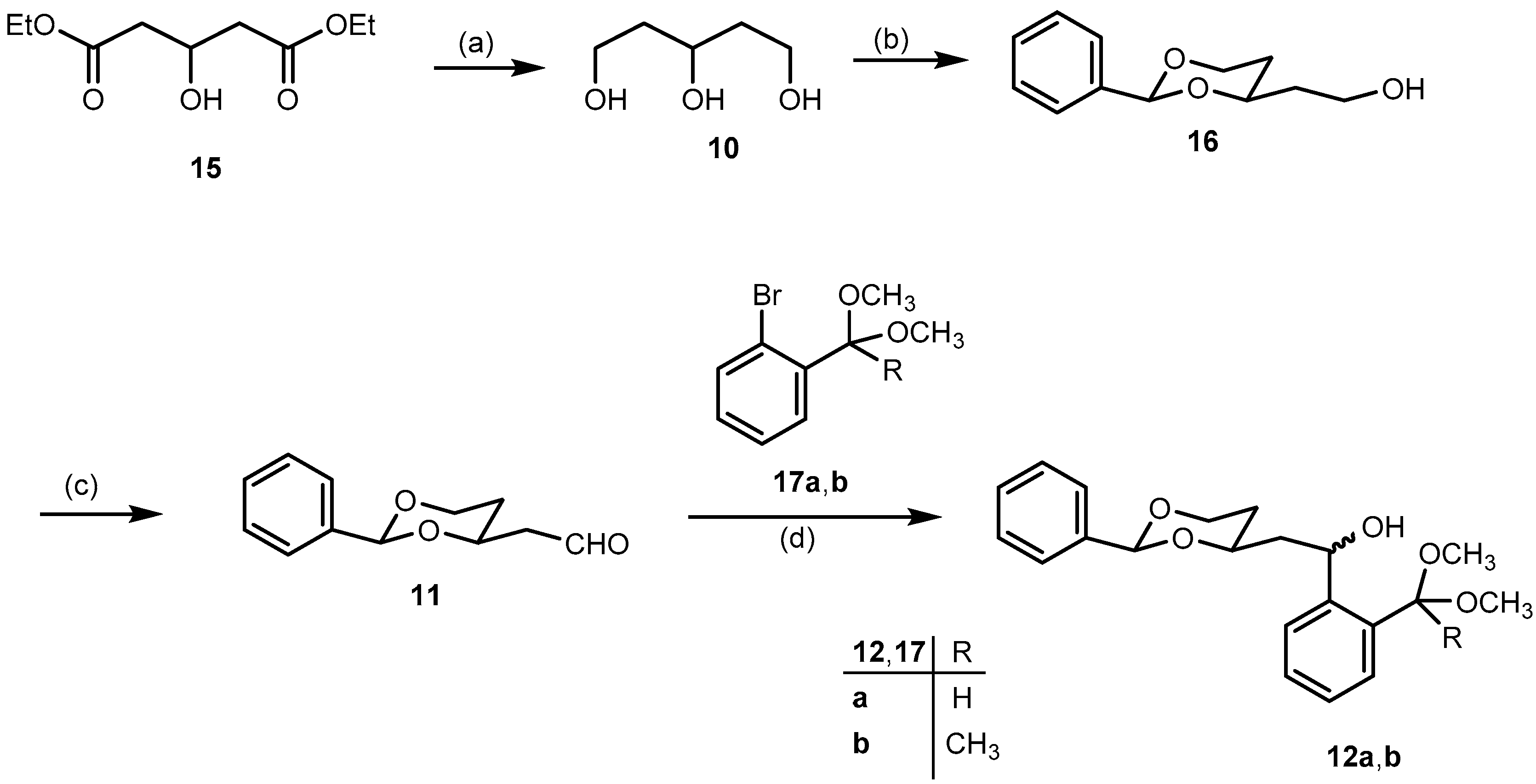
Pentane-1,3,5-triol (
10) [
12]. Under N
2 atmosphere and ice cooling (0 °C), diethyl 3-hydroxyglutarate (
15, 5.31 g, 26 mmol) in dry THF (10 mL) was added dropwise to a suspension of LiAlH
4 (2.43 g, 64 mmol) in dry THF (60 mL). The reaction mixture was heated at 66 °C for 72 h. The excess of LiAlH
4 was quenched with cold water (50 mL) under ice cooling until a completely white precipitate was observed. A solution of H
2SO
4 (1/4 conc.) was added dropwise to the mixture under ice cooling until the precipitate dissolved completely (pH 1). The reaction mixture was then heated to reflux for 12 h and the THF was removed under reduced pressure. The resulting aqueous mixture was alkalized with NH
3 solution (25%) until a gelatinous precipitate of Al(OH)
3 was observed (pH 10). The precipitate was removed by suction filtration, and the residue was washed with water (3 × 50 mL). The filtrate was concentrated in vacuo, and the residue was heated to reflux in CH
3OH (150 mL) for 4 h. The mixture was then filtered to remove the insoluble Li
2SO
4. The residue was washed with CH
3OH (3 × 50 mL). The combined filtrate was concentrated in vacuo, and the crude product was purified by fc (Ø = 6 cm, h = 15 cm, ethyl acetate:methanol = 9:1, V = 20 mL,
Rf = 0.21). Colorless oil, yield 2.9 g (97%). C
5H
12O
3 (120.1). Exact mass (APCI):
m/
z = 121.0871, calcd. 121.0859 for C
5H
12O
3 [MH]
+.
1H-NMR (400 MHz, CD
3OD): δ [ppm] = 1.58–1.75 (m, 4H, C
H2CHOHC
H2), 3.69 (t,
J = 6.5 Hz, 4H, C
H2OH), 3.87 (tt,
J = 8.4/4.5 Hz, 1H, C
HOH). Signals for the OH protons are not observed in the spectrum.
13C-NMR (100 MHz (CD
3)
2SO): δ [ppm] = 40.6 (2C,
CH
2CHOH), 58.3 (2C,
CH
2OH), 65.2 (1C,
CHOH). FTIR (neat): ν (cm
−1) = 3298 (O-H), 2940, 2886 (C-H), 1042 (C-O).
2-[(2RS,4SR)-2-Phenyl-1,3-dioxan-4-yl]ethanol (16). p-Toluenesulfonic acid (50 mg, 0.3 mmol) was added to a solution of pentane-1,3,5-triol (10, 0.64 g, 5.3 mmol) and benzaldehyde (1.1 mL, 1.1 g, 11 mmol) in dry CH2Cl2 (25 mL). Then anhydrous Na2SO4 (3.80 g, 27 mmol) was added and the reaction mixture was stirred at r.t. for 24 h. The reaction mixture was filtered to remove Na2SO4, and the residue was washed with CH2Cl2 (3 × 10 mL). The combined filtrate was washed with saturated aqueous solution of NaHCO3 (3 × 10 mL). The aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layer was washed with brine (10 mL) and dried (Na2SO4). The solvent was removed in vacuo, and the crude product was purified by fc (Ø = 3 cm, h = 18 cm, cyclohexane:ethyl acetate = 1:1, V = 20 mL, Rf = 0.25). Colorless oil, yield 0.90 g (85%). C12H16O3 (208.3). Exact mass (APCI): m/z = 209.1155, calcd. 209.1172 for C12H16O3 [MH]+. 1H-NMR (400 MHz, CD2Cl2): δ [ppm] = 1.52 (dtd, J = 13.3/2.5/1.4 Hz, 1H, 5-Heq), 1.79 (dddd, J = 14.5/6.6/5.2/4.0 Hz, 1H, CH2CH2OH), 1.83–1.90 (m, 2H, CH2CH2OH (1H), 5-Hax (1H), 2.07 (t, J = 4.6 Hz, 1H, CH2OH), 3.73–3.84 (m, 2H, CH2OH), 3.98 (ddd, J = 12.4/11.4/2.6 Hz, 1H, 6-Hax), 4.10 (tdd, J = 11.1/4.0/2.4 Hz, 1H, 4-Hax), 4.25 (ddd, J = 11.4/5.1/1.4 Hz, 1H, 6-Heq), 5.53 (s, 1H, 2-Hax), 7.32–7.40 (m, 3H, Harom), 7.43–7.50 (m, 2H, Harom). 13C-NMR (100 MHz, CD2Cl2): δ [ppm] = 31.8 (1C, C-5dioxane), 38.9 (1C, CH2CH2OH), 60.4 (1C, CH2OH), 67.5 (1C, C-6dioxane), 76.7 (1C, C-4dioxane), 101.7 (1C, C-2dioxane), 126.6 (2C, Carom), 128.7 (2C, Carom), 129.2 (1C, Carom), 139.5 (1C, Cqarom). FTIR (neat): ν (cm−1) = 3410, 3383 (O-H), 2947, 2859 (C-H), 1454, 1400, 1366 (C=Carom), 1215, 1138, 1099 (C-O), 748, 698 (C-Harom monosubst).
2-[(2RS,4RS)-2-Phenyl-1,3-dioxan-4-yl)acetaldehyde (11). Under N2 atmosphere, a solution of oxalyl chloride in CH2Cl2 (3.0 mL, 6.0 mmol) was added in one portion to dry CH2Cl2 (10 mL) and the resulting solution was cooled down to −78 °C. Then a solution of dry (CH3)2SO (0.6 mL, 8.0 mmol) in dry CH2Cl2 (10 mL) was added dropwise (10 mL/h via a syringe pump). The reaction mixture was stirred at −78 °C for 15 min. Then a solution of alcohol 16 (0.84 g, 4.0 mmol) in dry CH2Cl2 (10 mL) was added dropwise (10 mL/h via a syringe pump), and the mixture was stirred at −78 °C for another 20 min. Finally, Et3N (2.9 mL, 20 mmol) was added in one portion, and the mixture was stirred at −78 °C for additional 10 min. The mixture was allowed to warm to r.t. The reaction mixture was diluted with Et2O (20 mL) and the precipitate was filtered off. The residue was washed with Et2O (2 × 20 mL) and the combined filtrate was concentrated in vacuo. The crude product was purified by fc (Ø = 3 cm, h = 17 cm, cyclohexane:ethyl acetate = 8:2, V = 20 mL, Rf = 0.30). Colorless oil, yield 0.70 g (92%). C12H14O3 (206.2). Exact mass (APCI): m/z = 207.1008, calcd. 207.1016 for C12H15O3 [MH]+. 1H-NMR (400 MHz, CD2Cl2): δ [ppm] = 1.61 (dtd, J = 13.3/2.5/1.4 Hz, 1H, 5-Heq), 1.86 (dddd, J = 13.2/12.2/11.3/5.0 Hz, 1H, 5-Hax), 2.60 (ddd, J = 16.8/4.8/1.6 Hz, 1H, CH2CHO), 2.74 (ddd, J = 16.8/7.6/2.4 Hz, 1H, CH2CHO), 3.95–4.04 (m, 1H, 6-Hax), 4.25 (ddd, J = 11.5/5.0/1.4 Hz, 1H, 6-Heq), 4.42 (dddd, J = 11.2/7.5/4.8/2.5 Hz, 1H, 4-Hax), 5.55 (s, 1H, 2-Hax), 7.30–7.41 (m, 3H, Harom), 7.41–7.50 (m, 2H, Harom), 9.81 (dd, J = 2.4/1.6 Hz, 1H, CH2CHO). 13C-NMR (100 MHz, CD2Cl2): δ [ppm] = 31.6 (1C, C-5dioxane), 50.0 (1C, CH2CHO), 67.3 (1C, C-6dioxane), 72.8 (1C, C-4dioxane), 101.7 (1C, C-2dioxane), 126.6 (2C, Carom), 128.6 (2C, Carom), 129.3 (1C, Carom), 139.2 (1C, Cqarom), 200.8 (1C, CH2CHO). FTIR (neat): ν (cm−1) = 2855 (C-H), 1721 (C=O), 1238, 1142, 1099 (C-O), 752, 698 (C-Harom monosubst).
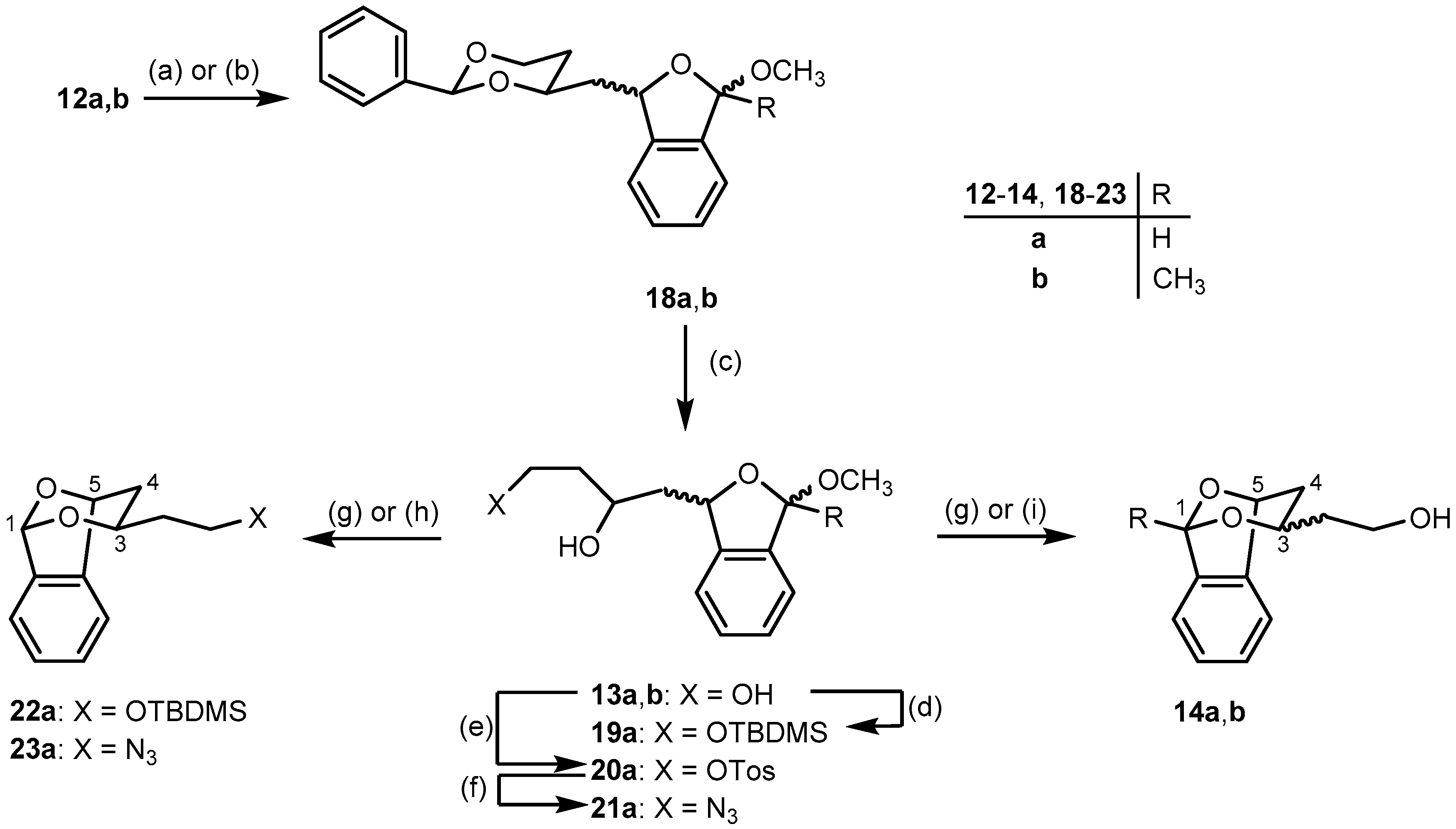
2-{(1RS)-2-[(2RS,4RS)- and (2SR,4SR)-4-Phenyl-1,3-dioxan-2-yl]-1-hydroxyethyl}benzaldehyde dimethyl acetal (12a). Under N2 atmosphere, a solution of bromoacetal 17a (0.72 g, 3.1 mmol) in dry THF (10 mL) was cooled down to −78 °C. Then 1.6 M n-butyllithium in n-hexanes (2.0 mL, 3.1 mmol) was added dropwise over 15 min, and the reaction mixture was stirred at −78 °C for 10 min. Then a solution of aldehyde 11 (0.43 g, 2.1 mmol) in dry THF (5 mL) was added dropwise over 15 min. The reaction mixture was stirred at −78 °C for 2 h, and was then allowed to warm to r.t. overnight. Under ice cooling, water (15 mL) was added dropwise to the reaction mixture. THF was removed in vacuo from the mixture and the resulting aqueous layer was extracted with CHCl3 (3 × 20 mL). The combined organic layer was washed with brine (15 mL), dried (Na2SO4,), filtered, and the solvent was removed in vacuo. The crude product was purified by fc (Ø = 3 cm, h = 18 cm, cyclohexane: ethyl acetate = 8:2, V = 20 mL, Rf = 0.26). Pale yellow oil, yield 0.70 g (93%), mixture of two diastereomers (2:1). C21H26O5 (358.4). Exact Mass (APCI): m/z = 325.1437, calcd. 325.1434 for C20H21O4 [M − HOCH3 − H]+; m/z = 295.1325, calcd. 295.1329 for C19H19O3 [M − HOCH3 − OCH3]+. MS (ESI): m/z = 381 [M + Na]+, 295 [M − HOCH3 − OCH3]+. 1H-NMR (400 MHz, CD3OD-d4): δ [ppm] = 1.54 (dtd, J = 13.3/2.6/1.4 Hz, 0.33H, 5-Heqo), 1.59 (dtd, J = 13.3/2.5/1.4 Hz, 0.67H, 5-Heqm), 1.68–1.97 (m, 2.34H, CH2CHOHm (2 × 0.67H), 5-Haxo+m (1H)), 2.14 (dt, J = 13.9/7.5 Hz, 2 × 0.33H, CH2CHOH°), 3.18 (s, 3 × 0.67H, OCH3m), 3.22 (s, 3 × 0.33H, OCH3o), 3.27 (s, 3 × 0.67H, OCH3m), 3.27 (s, 3 × 0.33H, OCH3o), 3.84–4.04 (m, 2H, 6-Heqo (0.33H), 4-Haxm (0.67H), 6-Haxo+m (1H)), 4.12–4.28 (m, 1H, 4-Haxo (0.33H), 6-Heqm (0.67H)), 5.36 (dd, J = 7.9/5.8 Hz, 0.67H, CHOHm), 5.42–5.47 (m, 1H, CHOHo (0.33H), (CH(OCH3)2m (0.67H)), 5.51 (s, 0.33H, 2-Haxo), 5.57 (s, 0.67H, 2-Haxm), 5.59 (s, 0.33H, CH(OCH3)2o), 7.20–7.29 (m, 2 × 0.67H, Harom), 7.30–7.40 (m, 4H, Harom), 7.44–7.48 (m, 2H, Harom), 7.49–7.55 (m, 0.67H, Harom), 7.59 (ddd, J = 7.8/4.1/1.3 Hz, 1H, Harom). Ratio of diastereomers 67:33. o = minor isomer; m = major isomer. 13C-NMR (151 MHz, DMSO-d6): δ [ppm] = 30.6m (0.67C, C-5dioxane), 31.4o (0.33C, C-5dioxane), 45.3m (0.67C, CH2CHOH), 46.2o (0.33C, CH2CHOH), 53.3m (2 × 0.67C, OCH3), 53.5o (2 × 0.33C, OCH3), 63.5o (0.33C, CHOH), 64.1m (0.67C, CHOH), 66.3o+m (1C, C-6dioxane), 73.4o (0.33C, C-4dioxane), 74.4m (0.67C, C-4dioxane), 100.2o (0.33C, CH(OCH3)2), 100.3m (0.67C, CH(OCH3)2), 100.97m (0.67C, C-2dioxane), 101.0o (0.33C, C-2dioxane), 125.6m (0.67C, Carom), 125.7o (0.33C, Carom), 126.06m (0.67C, Carom), 126.08m (2 × 0.67C, Carom), 126.10o (2 × 0.33C, Carom), 126.13o (0.33C, Carom), 126.3o (0.33C, Carom), 126.5m (0.67C, Carom), 127.9m (0.67C, Carom), 128.0m (2 × 0.67C, Carom), 128.4o (0.33C, Carom), 128.5o+m (1C, Carom), 128.53m (0.67C, Carom), 133.9o (0.33C, Cqarom), 134.1m (0.67C, Cqarom), 139.0m (0.67C, Cqarom), 139.2o (0.33C, Cqarom), 144.2m (0.67C, Cqarom), 144.8o (0.33C, Cqarom). Ratio of diastereomers 67:33. o = minor isomer; m = major isomer. FTIR (neat): ν (cm−1) = 3460 (O-H), 2947 (C-H), 1454 (C=Caromatic), 1099 (C-O), 752, 698 (C-Harom mono- and 1,2-disubst).
1-{2-[(1RS)-2-[(2RS,4RS)- and (2SR,4SR)-2-Phenyl-1,3-dioxan-4-yl]-1-hydroxyethyl]phenyl}ethanone dimethyl ketal (12b). Under N2 atmosphere, a solution of 2′-bromoacetophenone dimethyl ketal (17b, 0.95 g, 3.7 mmol) in dry THF (12 mL) was cooled down to −78 °C. Then 2.5 M n-butyllithium in n-hexanes (1.5 mL, 3.7 mmol) was added dropwise over 15 min. The reaction mixture was stirred at −78 °C for 10 min. A solution of aldehyde 11 (0.51 g, 2.5 mmol) in dry THF (6 mL) was added dropwise over 18 min. The reaction mixture was stirred at −78 °C for 2 h, then it was allowed to warm to r.t. overnight. Under ice cooling, water (15 mL) was added dropwise to the reaction mixture. THF was removed in vacuo from the mixture and the resulting aqueous layer was extracted with chloroform (3 × 20 mL). The combined organic layer was washed with brine (15 mL), dried (Na2SO4), filtered, and the solvent removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 15 cm, cyclohexane:ethyl acetate = 8:2, V = 20 mL, Rf = 0.40). Pale yellow oil, yield 0.89 g (97%), mixture of two diastereomers (2:1). C22H28O5 (372.5). Exact Mass (APCI): m/z = 310.1536, calcd. 310.1569 for C20H22O3 [M − HOCH3 − OCH3 + H]+. MS (ESI): m/z = 395 [M + Na]+. 1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.46 (s, 3 × 0.33H, CH3C(OCH3)2o), 1.50 (s, 3 × 0.67H, CH3C(OCH3)2m), 1.53–1.79 (m, 3.34H, 5-Heqo+m (1H), 5-Haxo+m (1H), CH2CHOHm (2 × 0.67H)), 1.91–2.02 (m, 2 × 0.33H, CH2CHOHo), 3.01 (s, 3 × 0.67H, OCH3m), 3.07 (2s, 6 × 0.33H, OCH3o), 3.10 (s, 3 × 0.67H, OCH3m), 3.93 (qd, J = 11.4/2.7 Hz, 1H, 6-Haxo+m), 4.05–4.25 (m, 2H, 4-Haxo+m (1H), 6-Heqo+m (1H)), 4.97 (2d, J = 4.8 Hz, 1H, CHOHo+m), 5.34 (ddd, J = 10.0/5.0/3.0 Hz, 0.67H, CHOHm), 5.51 (s, 1H, 2-Haxm (0.67H), CHOHo (0.33H)), 5.55 (s, 0.33H, 2-Haxo), 7.18 (dtd, J = 8.3/7.1/1.5 Hz, 2 × 0.67H, Haromm), 7.25–7.36 (m, 4H, Haromo+m), 7.36–7.41 (m, 2 × 0.67H, Haromm), 7.44 (ddd, J = 8.0/6.5/1.8 Hz, 2 × 0.67H, Haromm), 7.60 (td, J = 8.0/1.4 Hz, 1H, Haromo+m). Ratio of diastereomers 67:33. o = minor isomer; m = major isomer. 13C-NMR (101 MHz, DMSO-d6): δ [ppm] = 25.7o (0.33C, CH3), 25.9m (0.67C, CH3), 30.9m (0.67C, C-5dioxane), 32.0o (0.33C, C-5dioxane), 45.9m (0.67C, CH2CHOH), 46.8o (0.33C, CH2CHOH), 48.85m (2 × 0.67C, OCH3), 48.88o (2 × 0.33C, OCH3), 63.8o (0.33C, CHOH), 64.4m (0.67C, CHOH), 66.77m (0.67C, C-6dioxane), 66.80o (0.33C, C-6dioxane), 74.1o (0.33C, C-4dioxane), 75.2m (0.67C, C-4dioxane), 100.6o (0.33C, C-2dioxane), 100.9m (0.67C, C-2dioxane), 102.1o (0.33C, C(OCH3)2), 102.3m (0.67C, C(OCH3)2), 126.5o (2 × 0.33C, Carom), 126.54m (2 × 0.67C, Carom), 126.6o (0.33C, Carom), 126.61o (0.33C, Carom), 126.7m (0.67C, Carom), 126.8m (0.67C, Carom), 128.15o (0.33C, Carom), 128.16m (0.67C, Carom), 128.2m (0.67C, Carom), 128.24o (0.33C, Carom), 128.3o (2 × 0.33C, Carom), 128.34m (2 × 0.67C, Carom), 128.8o (0.33C, Carom), 128.9m (0.67C, Carom), 138.5o (0.33C, Cqarom), 138.7m (0.67C, Cqarom), 139.5m (0.67C, Cqarom), 139.7o (0.33C, Cqarom), 144.9m (0.67C, Cqarom), 145.6o (0.33C, Cqarom). Ratio of diastereomers 67:33. o = minor isomer; m = major isomer. FTIR (neat): ν (cm−1) = 3487 (OH), 2924, 2851 (C-H), 1450 (C=Caromatic), 1099 (C-O), 760, 698 (C-Harom mono- and 1,2-disubst).
1-Methoxy-3-[(2-phenyl-1,3-dioxan-4-yl)methyl]-1,3-dihydro-2-benzofuran (18a) (four diastereomers). A solution of p-toluenesulfonic acid in toluene (0.5 mg/mL, 18 mL) was added to alcohols 12a (0.33 g, 0.9 mmol). The reaction mixture was stirred vigorously at r.t. for 2 h. Saturated aqueous solution of NaHCO3 (30 mL) was added and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layer was washed with brine (20 mL), dried (Na2SO4), filtered, and the solvent was removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 15 cm, cyclohexane: ethyl acetate = 9:1, V = 10 mL, Rf = 0.39). Colorless oil, yield 0.27 g (89%), mixture of four diastereomers (2:2:1:1). C20H22O4 (326.4). Exact Mass (APCI): m/z = 295.1330, calcd. 295.1329 for C19H19O3 [M − OCH3]+; m/z = 221.0973, calcd. 221.1172 for C13H17O3 [M − PhCO]+. MS (ESI): m/z = 349 [M + Na]+, 295 [M – OCH3]+, 277 [M − OCH3 − H2O]+, 221 [M − PhCO]+. 1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.59–2.02 (m, 3.34H, 5-Heqo+m (1H), 5-Haxo+m (1H), CHCH2CHo+m+o (2 × 0.67H)), 2.04–2.27 (m, 2 × 0.33H, CHCH2CHm), 3.33 (s, 3 × 0.17H, OCH3o), 3.34 (s, 3 × 0.33H, OCH3m), 3.40 (s, 3 × 0.17H, OCH3o), 3.41 (s, 3 × 0.33H, OCH3m), 3.95–4.08 (m, 1.34H, 6-Haxo+m (1H), 4-Haxo (2 × 0.17H)), 4.16–4.34 (m, 1.66H, 4-Haxm (2 × 0.33H), 6-Heqo+m (1H)), 5.34 (dt, J = 10.4/4.6 Hz, 0.50H, 3-Hbfo+m), 5.43–5.50 (m, 0.50H, 3-Hbfo+m), 5.58 (s, 0.33H, 2-Haxm), 5.61 (s, 0.33H, 2-Haxm), 5.66 (2s, 2 × 0.17H, 2-Haxo), 6.12 (s, 0.33H, 1-Hbfm), 6.14 (s, 0.17H, 1-Hbfo), 6.20 (d, J = 2.1 Hz, 0.17H, 1-Hbfo), 6.22 (d, J = 2.1 Hz, 0.33H, 1-Hbfm), 7.34–7.45 (m, 16 × 0.33H + 18 × 0.17H, Harom), 7.48–7.54 (m, 2 × 0.33H, Harom). Ratio of diastereomers 33:33:17:17. o = minor isomer; m = major isomer; bf = benzofuran. 13C-NMR (101 MHz, DMSO-d6): δ [ppm] = 31.0m (0.33C, C-5dioxane), 31.2m (0.33C, C-5dioxane), 32.1o (2 × 0.17C, C-5dioxane), 41.9m (0.33C, CHCH2CH), 43.0o (0.17C, CHCH2CH), 43.5m (0.33C, CHCH2CH), 44.7o (0.17C, CHCH2CH), 54.0o (0.17C, OCH3), 54.1m (0.33C, OCH3), 54.3o (0.17C, OCH3), 54.5m (0.33C, OCH3), 66.6o+m (0.50C, C-6dioxane), 66.7m (0.33C, C-6dioxane), 67.4o (0.17C, C-6dioxane), 73.8m (0.33C, C-4dioxane), 74.0o (0.17C, C-4dioxane), 74.1m (0.33C, C-4dioxane), 74.2o (0.17C, C-4dioxane), 78.8o (0.17C, C-3), 79.0o (0.17C, C-3), 79.03m (0.33C, C-3), 79.3m (0.33C, C-3), 100.5m (0.33C, C-2dioxane), 100.6m (0.33C, C-2dioxane), 100.7o (0.17C, C-2dioxane), 100.74o (0.17C, C-2dioxane), 106.2o (0.17C, C-1), 106.3m (0.33C, C-1), 106.6o (0.17C, C-1), 106.7m (0.33C, C-1), 121.6o (0.17C, Carom), 121.8o (0.17C, Carom), 121.84m (0.33C, Carom), 122.0m (0.33C, Carom), 123.4m (2 × 0.33C, Carom), 123.42o (2 × 0.17C, Carom), 126.4m (2 × 0.33C, Carom), 126.42m (2 × 0.33C, Carom), 126.5o (2 × 0.17C, Carom), 126.53o (2 × 0.17C, Carom), 128.06m (0.33C, Carom), 128.10o+m (0.50C, Carom), 128.2o (0.17C, Carom), 128.26m ( 2 × 0.33C, Carom), 128.3m (2 × 0.33C, Carom), 128.38o (2 × 0.17C, Carom), 128.39o (2 × 0.17C, Carom), 128.8m (0.33C, Carom), 128.81o (2 × 0.17C, Carom), 128.9m (0.33C, Carom), 129.6o+m (0.50C, Carom), 129.61o+m (0.50C, Carom), 137.7o (0.17C, Cqarom), 137.74o (0.17C, Cqarom), 137.8m (2 × 0.33C, Cqarom), 139.3o (2 × 0.17C, Cqarom), 139.34m (0.33C, Cqarom), 139.4m (0.33C, Cqarom), 143.3m (0.33C, Cqarom), 143.4m (0.33C, Cqarom), 143.5o (0.17C, Cqarom), 143.51o (0.17C, Cqarom). Ratio of diastereomers 33:33:17:17. o = minor isomer; m = major isomer. FTIR (neat): ν (cm−1) = 2947, 2858 (C-H), 1458 (C=Caromatic), 1096 (C-O), 752, 698 (C-Harom mono- and 1,2-disubst).
1-Methoxy-1-methyl-3-[(2-phenyl-1,3-dioxan-4-yl)methyl]-1,3-dihydro-2-benzofuran (18b) (four diastereomers). A solution of p-toluenesulfonic acid in toluene (25 mL, 1 mg/mL) was added to a solution of alcohols 12b (0.89 g, 2.4 mmol) in cyclohexane (25 mL). The reaction mixture was stirred vigorously at r.t. for 2 h. Saturated aqueous solution of NaHCO3 (30 mL) was added and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layer was washed with brine (20 mL), dried (Na2SO4), filtered, and the solvent was removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 15 cm, cyclohexane:ethyl acetate = 9:1, V = 10 mL, Rf = 0.40). Pale yellow oil, yield 0.58 g (71%), mixture of four diastereomers (2:2:1:1). C21H24O4 (340.4). MS (ESI): m/z = 657 [2(M − OCH3) + K]+, 363 [M + Na]+, 309 [M − OCH3]+. 1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.55–1.65 (m, 3.66H, CH3C(OCH3)o+m (3H), 5-Heqm (2 × 0.33H)), 1.70–1.83 (m, 1.34H, 5-Heqo (2 × 0.17H), 5-Haxo+m (1H)), 1.91–2.19 (m, 2H, CHCH2CHo+m), 2.82 (s, 3 × 0.17H, OCH3o), 2.84 (s, 1.5H, OCH3o+m), 2.94 (s, 3 × 0.33H, OCH3m), 3.94 (dd, J = 11.7/2.9 Hz, 2 × 0.33H, 6-Hax m), 3.97–4.04 (m, 0.67H, 6-Haxo (2 × 0.17H), 6-Heqm (0.33H)), 4.12–4.16 (m, 2 × 0.33H, 4-Haxm), 4.19 (ddd, J = 8.9/5.6/3.2 Hz, 0.33H + 2 × 0.17H, 6-Heq o+m+o), 4.23 (dt, J = 7.1, 2.1 Hz, 2 × 0.17H, 4-Haxo), 5.24 (ddd, J = 16.6/9.6/3.5 Hz, 2 × 0.17H, 3-Hbfo), 5.39–5.47 (m, 2 × 0.33H, 3-Hbfm), 5.52 (s, 0.33H, 2-Haxm), 5.60 (s, 0.33H, 2-Haxm), 5.63 (2s, 2 × 0.17H, 2-Haxo), 7.23 (qd, J = 3.8/1.6 Hz, 1H, Haromo+m), 7.26–7.43 (m, 7H, Haromo+m), 7.46 (ddd, J = 7.4/3.8/1.9 Hz, 1H, Haromo+m). Ratio of diastereomers 33:33:17:17. o = minor isomer; m = major isomer; bf = benzofuran. 13C-NMR (101 MHz, DMSO-d6): δ [ppm] = 26.1 m (0.33C, CH3C(OCH3)), 26.2o (0.17C, CH3C(OCH3)), 26.8m (0.33C, CH3C(OCH3)), 27.1 o (0.17C, CH3C(OCH3)), 30.3m (0.33C, C-5dioxane), 30.5o (2 × 0.17C, C-5dioxane), 31.5m (0.33C, C-5dioxane), 41.6m (0.33C, CHCH2CH), 41.8m (0.33C, CHCH2CH), 42.4o (0.17C, CHCH2CH), 43.2o (0.17C, CHCH2CH), 48.9m (0.33C, OCH3), 48.99o (0.17C, OCH3), 49.2m (0.33C, OCH3), 49.26o (0.17C, OCH3), 66.0o+m (1C, C-6dioxane), 73.1m (0.33C, C-4dioxane), 73.37o (0.17C, C-4dioxane), 73.4o (0.17C, C-4dioxane), 73.44m (0.33C, C-4dioxane), 76.6m (0.33C, C-3), 76.66o (0.17C, C-3), 78.4o (0.17C, C-3), 78.7m (0.33C, C-3), 99.8m (0.33C, C-2dioxane), 100.0o+m (0.50C, C-2dioxane), 100.1o (0.17C, C-2dioxane), 109.8o+m (0.50C, C-1), 110.1m (0.33C, C-1), 110.2o (0.17C, C-1), 121.0o (0.17C, Carom), 121.2o+m (0.50C, Carom), 121.4m (0.33C, Carom), 121.8o (0.17C, Carom), 121.83o (0.17C, Carom), 121.9m (2 × 0.33C, Carom), 125.7m (2 × 0.33C, Carom), 125.8m (2 × 0.33C, Carom), 125.9o (2 × 0.17C, Carom), 126.0o (2 × 0.17C, Carom), 127.6m (2 × 0.33C, Carom), 127.7m (2 × 0.33C, Carom), 127.77m (4 × 0.33C, Carom), 127.8o (0.17C, Carom), 127.9o (0.17C, Carom), 128.1m (0.33C, Carom), 128.2m (0.33C, Carom), 128.3o (0.17C, Carom), 128.31o (0.17C, Carom), 128.8o+m+m (2 × 0.33C + 0.17C, Carom), 128.9o (0.17C, Carom), 138.6m (2 × 0.33C, Cqarom), 138.7o (2 × 0.17C, Cqarom), 138.72o (0.17C, Cqarom), 138.8m (0.33C, Cqarom), 139.0o (0.17C, Cqarom), 139.08m (0.33C, Cqarom), 142.3m (0.33C, Cqarom), 142.4o (0.17C, Cqarom), 142.7m (0.33C, Cqarom), 142.8o (0.17C, Cqarom). Ratio of diastereomers 33:33:17:17. o = minor isomer; m = major isomer. FTIR (neat): ν (cm−1) = 2932, 2855 (C-H), 1454 (C=Carom), 1099 (C-O), 748, 698 (C-Harom mono- and 1,2-disubst).
4-(3-Methoxy-1,3-dihydro-2-benzofuran-1-yl)butane-1,3-diol (13a) (four diastereomers). A solution of 2-benzofuran 18a (0.32 g, 1.3 mmol) in a mixture of methanol and water (20 mL, 4:1) was treated with p-toluenesulfonic acid (0.17 g, 1.0 mmol). The reaction mixture was stirred at r.t. for 48 h. A saturated aqueous solution of NaHCO3 (10 mL) was added. Methanol was evaporated in vacuo and the resulting aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was washed with brine (10 mL), dried (Na2SO4), filtered, and the solvent was removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 13 cm, ethyl acetate (100%), V = 10 mL, Rf = 0.44). Pale yellow oil, yield 0.27 g (87%), mixture of four diastereomers (2:2:1:1). C13H18O4 (238.3). Exact Mass (APCI): m/z = 413.2047, calcd. 413.1959 for C24H30O6 [2(M − HOCH3) + H]+; m/z = 207.1090, calcd. 207.1016 for C12H15O3 [M − OCH3]+. MS (ESI): m/z = 467 [M + C12H14O3 + Na]+, 261 [M + Na]+, 207 [M − OCH3]+, 189 [M − H2O − OCH3]+. 1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.33–1.98 (m, 4H, CH2CH2OHo+m (2H), CH2CHOHo+m (2H)), 3.24 (s, 3 × 0.33H, OCH3m), 3.28 (s, 3 × 0.17H, OCH3o), 3.27 (s, 3 × 0.17H, OCH3o), 3.31 (s, 3 × 0.33H, OCH3m), 3.46–3.57 (m, 2H, CH2OHo+m), 3.84–3.97 (m, 1H, CHOHo+m), 4.29–4.38 (m, 1H, CH2OHo+m), 4.48 (d, J = 5.1 Hz, 0.33H, CHOHm), 4.52 (d, J = 5.1 Hz, 0.33H, CHOHm), 4.60 (d, J = 2.5 Hz, 0.17H, CHOHo), 4.62 (d, J = 2.6 Hz, 0.17H, CHOHo), 5.22 (dd, J = 7.4/6.2 Hz, 0.33H, 1-Hbfm), 5.27 (dd, J = 10.4/2.5 Hz, 0.17H, 1-Hbfo), 5.34 (ddd, J = 7.4/5.1/2.0 Hz, 0.33H, 1-Hbfm), 5.40 (dt, J = 10.2/2.3 Hz, 0.17H, 1-Hbfo), 6.01 (s, 0.33H, 3-Hbfm), 6.03 (s, 0.17H, 3-Hbfo), 6.09 (2s, Hz, 0.50H, 3-Hbfo+m), 7.24–7.41 (m, 4H, Harom). Ratio of diastereomers 33:33:17:17. o = minor isomer; m = major isomer; bf = benzofuran. 13C-NMR (101 MHz, DMSO-d6): δ [ppm] = 39.8m (2 × 0.33C, CH2CH2OH), 41.1o (0.17C, CH2CH2OH), 41.14o (0.17C, CH2CH2OH), 44.0m (0.33C, CH2CHOH), 44.7o (0.17C, CH2CHOH), 45.5m (0.33C, CH2CHOH), 46.3o (0.17C, CH2CHOH), 53.5m (0.33C, OCH3), 53.7o (2 × 0.17C, OCH3), 53.9m (0.33C, OCH3), 58.0o (0.17C, CH2OH), 58.05o+m (0.50C, CH2OH), 58.06m (0.33C, CH2OH), 64.3o (0.17C, CHOH), 64.7o (0.17C, CHOH), 64.72m (0.33C, CHOH), 64.8m (0.33C, CHOH), 79.2o (0.17C, C-1), 79.6o (0.17C, C-1), 79.7m (0.33C, C-1), 80.0m (0.33C, C-1), 105.6m (0.33C, C-3), 105.7o (0.17C, C-3), 106.0o (0.17C, C-3), 106.1m (0.33C, C-3), 121.0o (0.17C, Carom), 121.2o (0.17C, Carom), 121.3m (0.33C, Carom), 121.6m (0.33C, Carom), 122.9m (2 × 0.33C, Carom), 123.0o (2 × 0.17C, Carom), 127.5o (0.17C, Carom), 127.55m (0.33C, Carom), 127.59o (0.17C, Carom), 127.6m (0.33C, Carom), 129.1m (0.33C, Carom), 129.13o (0.17C, Carom), 129.14m (0.33C, Carom), 129.2o (0.17C, Carom), 137.3o (0.17C, Cqarom), 137.4m (0.33C, Cqarom), 137.42m (0.33C, Cqarom), 137.5o (0.17C, Cqarom), 143.46m (0.33C, Cqarom), 143.50m (0.33C, Cqarom), 143.7o (0.17C, Cqarom), 143.8o (0.17C, Cqarom). Ratio of diastereomers 33:33:17:17. o = minor isomer; m = major isomer. FTIR (neat): v (cm−1) = 3375 (O-H), 2936, 2889 (C-H), 1431 (C=Caromatic), 1080 (C-O), 752 (C-Harom 1,2-disubst).
4-(3-Methoxy-3-methyl-1,3-dihydro-2-benzofuran-1-yl)butane-1,3-diol (13b) (four diastereomers). A solution of 2-benzofuran 18b (0.22 g, 0.6 mmol) in a mixture of methanol and water (12 mL, 4:1) was treated with p-toluenesulfonic acid (40.0 mg, 0.2 mmol). The reaction mixture was stirred at r.t. for 72 h. Saturated aqueous solution of NaHCO3 (10 mL) was added. Methanol was removed in vacuo and the resulting aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was washed with brine (10 mL), dried (Na2SO4), filtered, and the solvent was removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 10 cm, ethyl acetate (100%), V = 10 mL, Rf = 0.40). Yellow oil, yield 88.4 mg (55%), mixture of four diastereomers (2:2:1:1). C14H20O4 (252.31). MS (ESI): m/z = 463 [2(M − HOCH3) +Na]+, 275 [M + Na]+, 203 [M − OCH3 − H2O]+, 221 [M − OCH3]+, 1H-NMR (400 MHz, CD3OD): δ [ppm] = 1.55–2.07 (m, 7H, CH3o+m (3H), CH2CHOHCH2o+m (4H)), 2.94 (s, 3 × 0.33H*, OCH3m), 2.95 (s, 3 × 0.17H*, OCH3o), 3.01 (s, 3 × 0.17H*, OCH3o), 3.04 (s, 3 × 0.33H*, OCH3m), 3.41–3.62 (m, 0.33H, CH2CH2OHm), 3.65–3.87 (m, 3 × 0.33H + 4 × 0.17H, CH2CH2OHo+m), 3.95–4.25 (m, 1H*, CH2CHOHo+m), 5.17–5.36 (m, 0.50H, 1-Hbfo+m), 5.38–5.48 (m, 0.33H, 1-Hbfm), 5.53 (dd, J = 10.2/2.3 Hz, 0.17H, 1-Hbfo), 7.20–7.49 (m, 4H, Haromo+m). Ratio of diastereomers 33:33:17:17. Signals of the OH protons are not observed in the spectrum. * The integrals of these signals are slightly lower than expected. o = minor isomer; m = major isomer; bf = benzofuran. 13C-NMR (101 MHz, CD3OD): δ [ppm] = 26.7o (0.17C, CCH3(OCH3)), 26.73m (0.33C, CCH3(OCH3)), 27.7m (0.33C, CCH3(OCH3)), 27.8o (0.17C, CCH3(OCH3)), 40.2m (0.33C, CH2CH2OH), 40.4m (0.33C, CH2CH2OH), 41.6o (0.17C, CH2CH2OH), 41.63o (0.17C, CH2CH2OH), 45.0m (0.33C, CH2CHOH), 45.6o (0.17C, CH2CHOH), 45.8m (0.33C, CH2CHOH), 46.6o (0.17C, CH2CHOH), 50.1m (2 × 0.33C, OCH3), 50.5o (0.17C, OCH3), 50.6o (0.17C, OCH3), 60.2o+m (1C, CH2CH2OH), 67.0o (2 × 0.17C, CH2CHOH), 68.0m (0.33C, CH2CHOH), 68.1m (0.33C, CH2CHOH), 80.6m (0.33C, C-1), 81.4o (2 × 0.17C, C-1), 82.4m (0.33C, C-1), 111.9m (0.33C, C-3), 112.1m (0.33C, C-3), 112.5o (2 × 0.17C, C-3), 122.0m (0.33C, Carom), 122.3o (0.17C, Carom), 122.34m (0.33C, Carom), 122.5o (0.17C, Carom), 123.26o (0.17C, Carom), 123.3m (0.33C, Carom), 123.4o (0.17C, Carom), 123.42m (0.33C, Carom), 129.1o (0.17C, Carom), 129.2m (0.33C, Carom), 129.3o (0.17C, Carom), 129.33m (0.33C, Carom), 130.3o (0.17C, Carom), 130.36o (0.17C, Carom), 130.4m (2 × 0.33C, Carom), 140.1o (0.17C, Cqarom), 140.2o (0.17C, Cqarom), 140.4m (0.33C, Cqarom), 140.6m (0.33C, Cqarom), 144.2o (0.17C, Cqarom), 144.6m (2 × 0.33C, Cqarom), 144.9o (0.17C, Cqarom). Ratio of diastereomers 33:33:17:17. o = minor isomer; m = major isomer. FTIR (neat): ν (cm−1) = 3395 (O-H), 2935, 2881 (C-H), 1431, (C=Caromatic), 1041 (C-O), 764 (C-Harom 1,2-disubst).
[(tert-Butyldimethylsilyl)oxy]-1-(3-methoxy-1,3-dihydro-2-benzofuran-1-yl)butan-2-ol (19a) (four diastereomers). In DMF (0.50 mL), diols 13a (34.1 mg, 0.14 mmol) were treated with imidazole (29.2 mg, 0.43 mmol) and tert-butyldimethylsilyl chloride (22.2 mg, 0.14 mmol). The reaction mixture was stirred at r.t. for 24 h, diluted with EtOAc (10 mL) and washed with water (4 × 5 mL). The organic phase was separated, dried (Na2SO4), filtered and was concentrated under reduced pressure. The crude product was purified by fc (Ø = 2 cm, h = 10 cm, cyclohexane: ethyl acetate (8:2), V = 10 mL, Rf = 0.44). Colorless oil, yield 26.6 mg (58%). C19H32O4Si (352.5). Ratio of diastereomers 33:33:17:17. 1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 0.02–0.04 (m, 6H, (CH3)2tBuSio+m), 0.86–0.87 (m, 9H, Me2 (C(CH3)3Sio+m), 1.43–1.95 (m, 4H, CH2CHOHCH2o+m), 3.28 (s, 3 × 0.33H, OCH3m), 3.29 (s, 3 × 0.17H*, OCH3o), 3.32 (s, 3 × 0.17H *, OCH3o), 3.34 (s, 3 × 0.33H, OCH3m), 3.68–3.78 (m, 2H, CH2OHo+m), 3.90–3.96 (m, 1H, CHOHo+m), 4.51 (d, J = 5.3 Hz, 0.33H, CHOHm), 4.53 (d, J = 5.3 Hz, 0.33H, CHOHm), 4.63 (d, J = 3.3 Hz, 0.17H*, CHOHo), 4.64 (d, J = 3.2 Hz, 0.17H *, CHOHo), 5.26 (dd, J = 7.4/6.4 Hz, 0.33H, 1-Hbfm), 5.31 (dd, J = 10.4/2.4 Hz, 0.17H, 1-Hbfo), 5.34–5.39 (dd, J = 10.4/3.7 Hz, 0.33H, 1-Hbfm), 5.43 (dd, J = 11.4/1.4 Hz, 0.17H, 1-Hbfo), 6.04 (s, 0.33H, 3-Hbfm), 6.06 (s, 0.17H, 3-Hbfo), 6.12 (2s, 0.5H, 3-Hbfo+m), 7.28–7.44 (m, 4H, Harom). Ratio of diastereomers 33:33:17:17. * Signals were slightly lower than expected. o = minor isomer, m = major isomer, bf = benzofuran.
[3-Hydroxy-4-(3-methoxy-1,3-dihydro-2-benzofuran-1-yl)butyl-] 4-methylbenzenesulfonate (20a) (four diastereomers). A solution of diols 13a (0.20 g, 0.84 mmol) in dry CH2Cl2 (4 mL) was treated with Bu2SnO (10.7 mg, 0.04 mmol), Et3N (176 μL, 1.3 mmol) and p-toluenesulfonyl chloride (0.18 g, 0.9 mmol). The reaction mixture was heated to reflux for 16 h. The solvent was removed under reduced pressure at 30 °C. The crude product was purified by fc (Ø = 2 cm, h = 12 cm, cyclohexane: ethyl acetate (7:3), V = 10 mL, Rf = 0.36 (1st spot); 0.19 (2nd spot)). Colorless oil, yield 0.16 g (51%), mixture of four diastereomers (2:2:1:1). C20H24O6S (392.5). MS (ESI): m/z = 361 [M − OCH3]+, 343 [M − H2O − OCH3]+, 171 [M − C13H17O3]+. 1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.41–2.21 (m, 4H, CH2CH2OSO2PhCH3o+m (2H), CH2CHOHo+m (2H)), 2.41 (2s, 3Ho+m, CH3Ph), 3.26 (s, 3 × 0.33H, OCH3m), 3.28 (s, 3 × 0.17H*, OCH3o), 3.30 (s, 3 × 0.33H, OCH3o), 3.68–3.77 (m, 2 × 0.17H, CH2CHOHo), 3.81 (dt, J = 10.2/5.5 Hz, 2 × 0.33H, CH2CHOHm), 3.88–4.19 (m, 2H CH2CH2OSO2PhCH3o+m), 4.70–4.82 (m, 2 × 0.33H, CH2CHOHm), 4.87–5.01 (m, 2 × 0.17H, CH2CHOHo), 5.18 (dd, J = 8.1/5.3 Hz, 0.33H, 1-Hbfm), 5.22–5.27 (m, 0.17H, 1-Hbfo), 5.29 (ddd, J = 7.3/4.6/1.9 Hz, 0.33H, 1-Hbfm), 5.33–5.40 (m, 0.17H, 1-Hbfo), 6.03 (s, 0.33H, 3-Hbfm), 6.05 (s, 0.17H, 3-Hbfo), 6.08 (d, J = 2.0 Hz, 0.33H, 3-Hbfm), 6.10 (d, J = 2.0 Hz, 0.17H, 3-Hbfo), 7.24–7.31 (m, 1H, Haromo+m), 7.31–7.42 (m, 10 × 0.33H, Haromo+m), 7.47 (dd, J = 8.3/3.1 Hz, 2H, Haromo+m), 7.78 (dt, J = 8.3/2.1 Hz, 5 × 0.33H, Haromo+m). Ratio of diastereomers 33:33:17:17. * A signal (3 × 0.17H) for the OCH3 moiety of the fourth isomer cannot be observed in the spectrum, because the water peak of DMSO-d6 is overlapping with this signal. o = minor isomer; m = major isomer; bf = benzofuran. 13C-NMR (151 MHz, DMSO-d6): δ [ppm] = 28.7o (2 × 0.17C, CH3Ph), 29.6m (0.33C, CH3Ph), 30.9m (0.33C, CH3Ph), 35.2m (2 × 0.33C, CH2CH2OSO2PhCH3), 35.3o (2 × 0.17C, CH2CH2OSO2PhCH3), 43.1m (0.33C, CH2CHOH), 43.9o (0.17C, CH2CHOH), 44.7m (0.33C, CH2CHOH), 45.6o (0.17C, CH2CHOH), 53.2m (0.33C, OCH3), 53.7m (0.33C, OCH3), 54.6o (2 × 0.17C, OCH3), 63.2o (2 × 0.17C, CH2CHOH), 63.3m (2 × 0.33C, CH2CHOH), 68.1o (2 × 0.17C, CH2CH2OSO2PhCH3), 68.2m (2 × 0.33C, CH2CH2OSO2PhCH3), 79.0o (2 × 0.17C, C-1), 79.2m (2 × 0.33C, C-1), 105.4m (0.33C, C-3), 105.5o (0.17C, C-3), 105.8o (0.17C, C-3), 105.9m (0.33C, C-3), 120.9o+m (0.50C, Carom), 121.2o+m (0.50C, Carom), 122.7o+m+o (0.33C + 2 × 0.17C, Carom), 122.71m (2 × 0.33C, Carom), 127.3o+m (2C, Carom), 127.33o+m (0.50C, Carom), 127.4o+m (0.50C, Carom), 128.8o+m (0.50C, Carom), 128.9o+m (0.50C, Carom), 129.9o+m (2C, Carom), 132.2m (0.33C, Cqarom), 137.0o+m (0.50C, Cqarom), 137.06o (0.17C, Cqarom), 142.9m (0.33C, Cqarom), 142.92m (0.33C, Cqarom), 143.0o (0.17C, Cqarom), 143.2o (0.17C, Cqarom), 144.5o+m (1C, Cqarom). Ratio of diastereomers 33:33:17:17. o = minor isomer; m = major isomer. FTIR (neat): ν (cm−1) = 3506 (O-H), 2920 (C-H), 1173 (RO-SO2R1), 1096(C-O), 752, 737, 664 (C-Harom1,2- and 1,4-disubst).
4-Azido-1-(3-methoxy-1,3-dihydro-2-benzofuran-1-yl)butan-2-ol (21a) (four diastereomers). A solution of tosylates 20a (46.6 mg, 0.12 mmol) in dry DMF (1 mL) was treated with NaN3 (23.2 mg, 0.36 mmol). The reaction mixture was heated to 90 oC for 3 h. The solvent was removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 12 cm, cyclohexane: ethyl acetate (7:3), V = 10 mL, Rf = 0.48). Pale yellow oil, yield 28.7 mg (92%), mixture of four diastereomers (2:2:1:1). C13H17N3O3 (263.3). Exact Mass (APCI): m/z = 234.1121, calcd. 234.1125 for C13H16NO3 [M – N2 – H]+; m/z = 204.1027, calcd. 204.1019 for C12H14NO2 [M – N2 – OCH3]+. 1H-NMR (400 MHz, CDCl3): δ [ppm] = 1.64–2.16 (m, 4H, CH2CHOHCH2o+m), 3.26–3.74 (m, 5H, OCH3o+m (3H), CH2CH2N3o+m (2H)), 3.87–4.06 (m, 2 × 0.17H, CH2CHOHo), 4.20 (dddd, J = 9.5/6.9/5.1/2.2 Hz, 2 × 0.33H, CH2CHOHm), 5.35–5.42 (m, 0.33H, 1-Hbfm), 5.46 (dd, J = 7.8/3.3 Hz, 0.17H, 1-Hbfo), 5.54 (dt, J = 10.4/2.5 Hz, 0.33H, 1-Hbfm), 5.63 (d, J = 7.6 Hz, 0.17H, 1-Hbfo), 6.11 (s, 0.17H, 3-Hbfo), 6.16 (s, 0.33H, 3-Hbfm), 6.18 (d, J = 2.0 Hz, 0.17H, 3-Hbfo), 6.20 (d, J = 2.1 Hz, 0.33H, 3-Hbfm), 7.17–7.22 (m, 1H, Harom), 7.35–7.42 (m, 3H, Harom). Ratio of diastereomers 33:33:17:17. Signals due to the OH protons are not observed in the spectrum. o = minor isomer; m = major isomer; bf = benzofuran. 13C-NMR (101 MHz, CDCl3): δ [ppm] = 36.7o (0.17C, CH2CH2N3), 36.8o (0.17C, CH2CH2N3), 36.84m (0.33C, CH2CH2N3), 36.9m (0.33C, CH2CH2N3), 42.4o (0.17C, CH2CHOH), 43.4m (0.33C, CH2CHOH), 44.1o (0.17C, CH2CHOH), 44.6m (0.33C, CH2CHOH), 48.5m (0.33C, CH2CH2N3), 48.6m (0.33C, CH2CH2N3), 48.8o (2 × 0.17C, CH2CH2N3), 55.1o+m (0.50C, OCH3), 55.7m (0.33C, OCH3), 56.1o (0.17C, OCH3), 66.5o (0.17C, CH2CHOH), 66.7o (0.17C, CH2CHOH), 68.8m (0.33C, CH2CHOH), 69.0m (0.33C, CH2CHOH), 81.1o (0.17C, C-1), 81.6o (0.17C, C-1), 83.8m (0.33C, C-1), 84.0m (0.33C, C-1), 107.1o (0.17C, C-3), 107.3m (0.33C, C-3), 108.2o+m (0.50C, C-3), 121.27m (0.33C, Carom), 121.30o (0.17C, Carom), 121.5m (0.33C, Carom), 121.53o (0.17C, Carom), 123.6o (0.17C, Carom), 123.62m (0.33C, Carom), 123.64m (0.33C, Carom), 123.7o (0.17C, Carom), 128.6o (0.17C, Carom), 128.7o+m (0.50C, Carom), 128.8m (0.33C, Carom), 130.0o+m (0.50C, Carom), 130.04o+m (0.50C, Carom), 137.2m (0.33C, Cqarom), 137.3m (0.33C, Cqarom), 137.9o (0.17C, Cqarom), 138.0o (0.17C, Cqarom), 142.5o (0.17C, Cqarom), 142.7o (0.17C, Cqarom), 142.8m (0.33C, Cqarom), 142.9m (0.33C, Cqarom). Ratio of diastereomers 33:33:17:17. o = minor isomer; m = major isomer. FTIR (neat): ν (cm−1) = 3464 (O-H), 2932 (C-H), 2091 (N=N=N), 1084 (C-O), 752 (C-Harom 1,2-disubst).
2-[(1RS,3RS,5RS)- and (1RS,3SR, 5RS)-1,5-Epoxy-1,3,4,5-tetrahydro-2-benzoxepin-3-yl]ethan-1-ol (14a). A solution of p-toluenesulfonic acid in toluene (4 mL, 0.5mg/mL) was added to diols 13a (49.2 mg, 0.21 mmol). The reaction mixture was heated to 85 °C for 72 h. Saturated aqueous solution of NaHCO3 (10 mL) was added and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layer was dried (Na2SO4), filtered, and the solvent was removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 12 cm, CH2Cl2:ethyl acetate (6:4), V = 10 mL, Rf =0.46 (1st isomer), 0.35 (2nd isomer)). Colorless solid, yield 7.23 mg (17%), mixture of two diastereomers (1:1). C12H14O3 (206.2). Exact mass (APCI): m/z = 207.1014, calcd. 207.1016 for C12H15O3 [MH]+. 1H-NMR (400 MHz, DMSO-d6): δ [ppm] = 1.52–1.93 (m, 2.5H, 4-Heq (1H), CH2CH2OH (1.5H)), 2.00 (m, 1.5H, CH2CH2OH (0.5H), 4-Hax (1H)), 3.66–3.81 (m, 2H, CH2CH2OH), 3.82–3.93 (m, 1H, 3-Hax (0.5H), 3-Heq (0.5H)), 4.61 (d, J = 4.4 Hz, 0.5H, CH2CH2OH), 4.73 (d, J = 5.7 Hz, 0.5H, CH2CH2OH), 5.33 (d, J = 10.8 Hz, 0.5H, 5-Heq), 5.41 (dd, J = 7.9/3.6 Hz, 0.5H, 5-Heq), 6.05 (s, 0.5H, 1-Heq), 6.09 (s, 0.5H, 1-Heq), 7.26–7.44 (m, 4H, Harom). Ratio of diastereomers 50:50. FTIR (neat): ν (cm−1) = 3372 (O-H), 2874 (C-H), 1462 (C=Carom), 1080 (C-O), 748 (C-Harom 1,2-disubst).
2-[{(1RS,3RS,5RS)- and (1RS,3SR,5RS)-1-Methyl}-1,5-epoxy-1,3,4,5-tetrahydro-2-benzoxepin-3-yl]ethan-1-ol (14b). A solution of diols 13b (50.0 mg, 0.2 mmol) in a mixture of toluene/AcOH (4:1, 5mL) was stirred at r.t. for 72 h. Saturated aqueous Solution of NaHCO3 (5 mL) was added. The mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layer was washed with brine (5 mL), dried (Na2SO4), filtered, and the solvent was removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 10 cm, ethyl acetate/cyclohexane (1:1)), V = 10 mL, Rf = 0.45 (1st spot) and 0.25 (2nd spot)). Yellow oil, yield 2.6 mg (6%) mixture of two diastereomers (1:1). C14H20O4 (220.27). 1H-NMR (400 MHz, CDCl3): δ [ppm] = 1.26 (2s, 3H, 6 × 0.5H, CH3), 1.62–1.89 (m, 1H, 4-Heq (0.5H) *, CH2CH2OH (0.5H)), 1.89–2.15 (m, 1.5H, CH2CH2OH), 2.17–2.33 (m, 1H, 2 × 0.5H, 4-Hax), 3.88 (dd, J = 12.0/4.7 Hz, 1H**, 2 × 0.5H, CH2CH2OH), 3.94–3.98 (m, 1H **, 2 × 0.5H, CH2CH2OH), 4.01–4.09 (m, 0.5H, 3-Hax), 4.08–4.14 (m, 0.5H, 3-Hax), 5.31–5.36 (m, 0.5H, 5-Heq), 5.67 (dd, J = 6.1/2.7 Hz, 0.5H, 5-Heq), 7.19 (m, 1H, 2 × 0.5H, Harom), 7.29–7.47 (m, 3H, 6 × 0.5H, Harom). Ratio of diastreoisomers 50:50. * Signals for one isomer is overlapping with other signals including the signal for the residual water in CDCl3. ** Signals are slightly higher than expected due to impurities. Due to the low amount of 18b the 13C-NMR, IR and MS spectra could not be recorded.
(1RS,3SR,5RS)-3-[2-(tert-Butyldimethylsilyloxy)ethyl]-1,5-epoxy-1,3,4,5-tetrahydro-2-benzoxepine (22a). In toluene (5 mL), silyl ethers 19a (26.6 mg, 0.08 mmol) were treated with 0.1 M HCl in AcOH (15 μL) and was heated to 65 °C for 60 h. The reaction mixture was diluted with EtOAc (10 mL) and was washed with saturated aqueous solution of NaHCO3 (3 × 5 mL). The pooled organic phase was dried (Na2SO4), filtered, and the solvent was removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 10 cm, cyclohexane: ethyl acetate (9:1), V = 10 mL, Rf = 0.48). Colorless oil, yield 4.5 mg (19%). C18H28O3Si (320.5). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] = −0.12 (s, 3H, (CH3)2tBuSi), −0.14 (s, 3H, (CH3)2tBuSi), 0.68 (s, 9H, Me2 (C(CH3)3Si), 1.44 (ddd, J = 13.8/7.7/1.9 Hz, 1H, 4-Heq), 1.50–1.62 (m, 2H, CH2CH2OTBDMS), 2.01 (ddd, J = 13.2/11.2/3.7 Hz, 1H, 4-Hax), 3.20–3.28 (m, 1H, 3-Hax), 3.44–3.53 (m, 2H, CH2OTBDMS), 5.23 (dd, J = 3.7/1.8 Hz, 1H, 5-Heq), 6.12 (s, 1H, 1-Heq), 7.25–7.37 (m, 4H, Harom).
(1RS,3SR,5RS)-3-(2-Azidoethyl)-1,5-epoxy-1,3,4,5-tetrahydro-2-benzoxepine (23a). A solution of p-toluenesulfonic acid in toluene (5 mL, 0.5 mg/mL) was added to azide 21a (69.8 mg, 0.27 mmol). The reaction mixture was heated to 85 °C for 72 h. Saturated aqueous solution of NaHCO3 (10 mL) was added and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layer was dried (Na2SO4), filtered, and the solvent was removed in vacuo. The crude product was purified by fc (Ø = 2 cm, h = 12 cm, cyclohexane: ethyl acetate (8:2), V = 10 mL, Rf = 0.45). Yellow oil, yield 7.7 mg (13%). C12H13N3O2 (231.3). After fc purification, only one diastereomer was isolated. MS (ESI): m/z =424 [2(M – OCH3) + Na + H]+, 232 [MH]+, 204 [M –N2 + H]+. 1H-NMR (400 MHz, CD2Cl2): δ [ppm] = 1.54–1.66 (m, 2H, 4-Heq (1H), CH2CH2N3 (1H)), 1.76 (ddt, J = 14.5/8.7/5.8 Hz, 1H, CH2CH2N3), 2.17 (ddd, J = 13.0/11.1/3.7 Hz, 1H, 4-Hax), 3.22–3.35 (m, 3H, CH2CH2N3 (2H), 3-Hax (1H)), 5.22 (dd, J = 3.8/1.8 Hz, 1H, 5-Heq), 6.11 (s, 1H, 1-Heq), 7.28–7.40 (m, 4H, Harom). 13C-NMR (151 MHz, CDCl3): δ [ppm] = 35.1 (1C, CH2CH2N3), 37.6 (1C, C-4), 47.2 (1C, CH2CH2N3), 65.3 (1C, C-3), 77.9 (1C, C-5), 100.8 (1C, C-1), 119.6 (1C, Carom), 121.3 (1C, Carom), 128.0 (1C, Carom), 128.6 (1C, Carom), 138.7 (1C, Cqarom), 143.0 (1C, Cqarom). FTIR (neat): ν (cm−1) = 2874 (C-H), 2095 (N=N=N), 1015 (C-O), 756 (C-Harom 1,2-disubst).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}