
Structural Characterization of Flavonoid Glycoconjugates and Their Derivatives with Mass Spectrometric Techniques



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
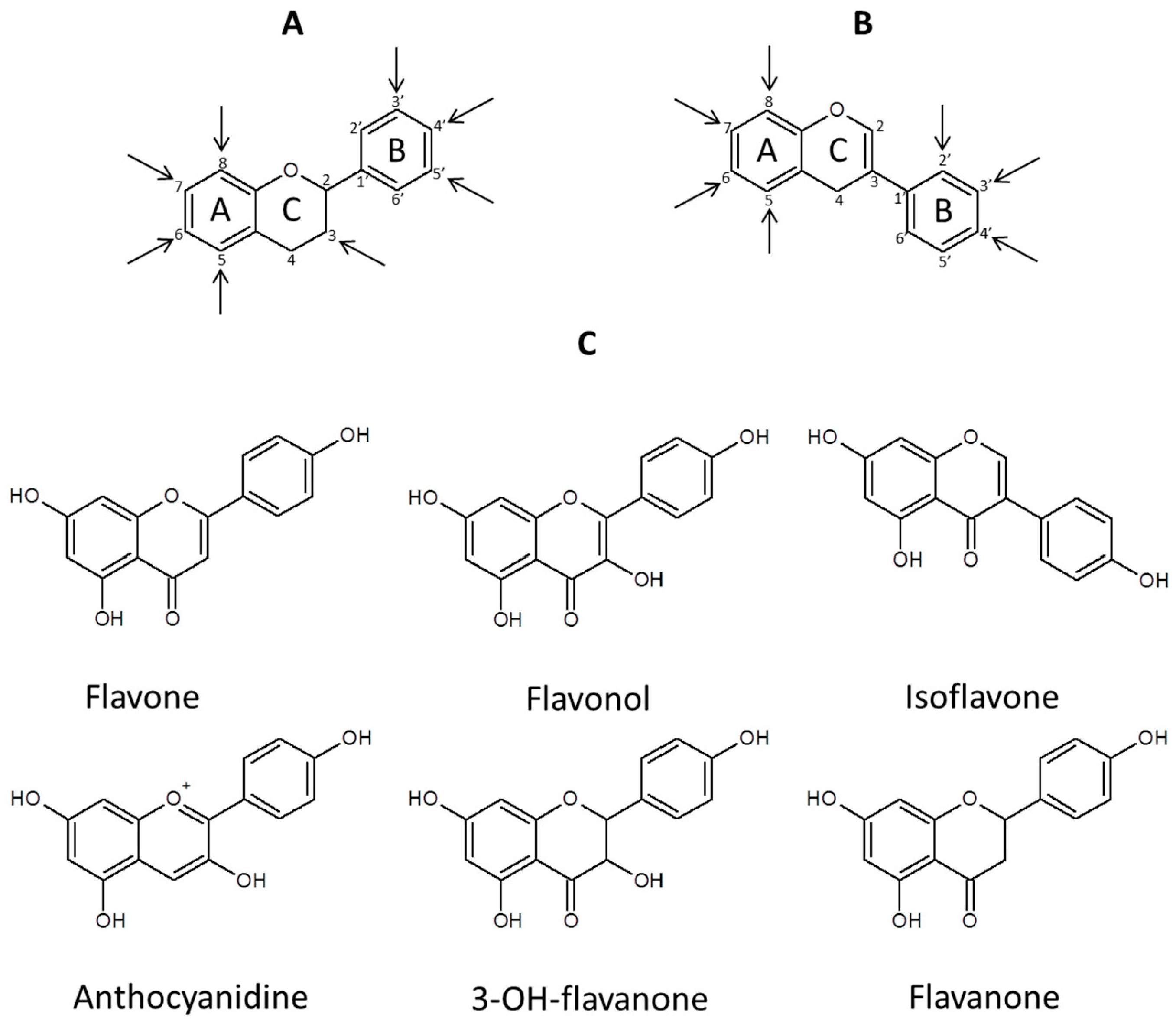
2. Concept/Workflow for Flavonoid Aglycones and Their Derivatives Analysis, Instrumental Requirements
- (1)
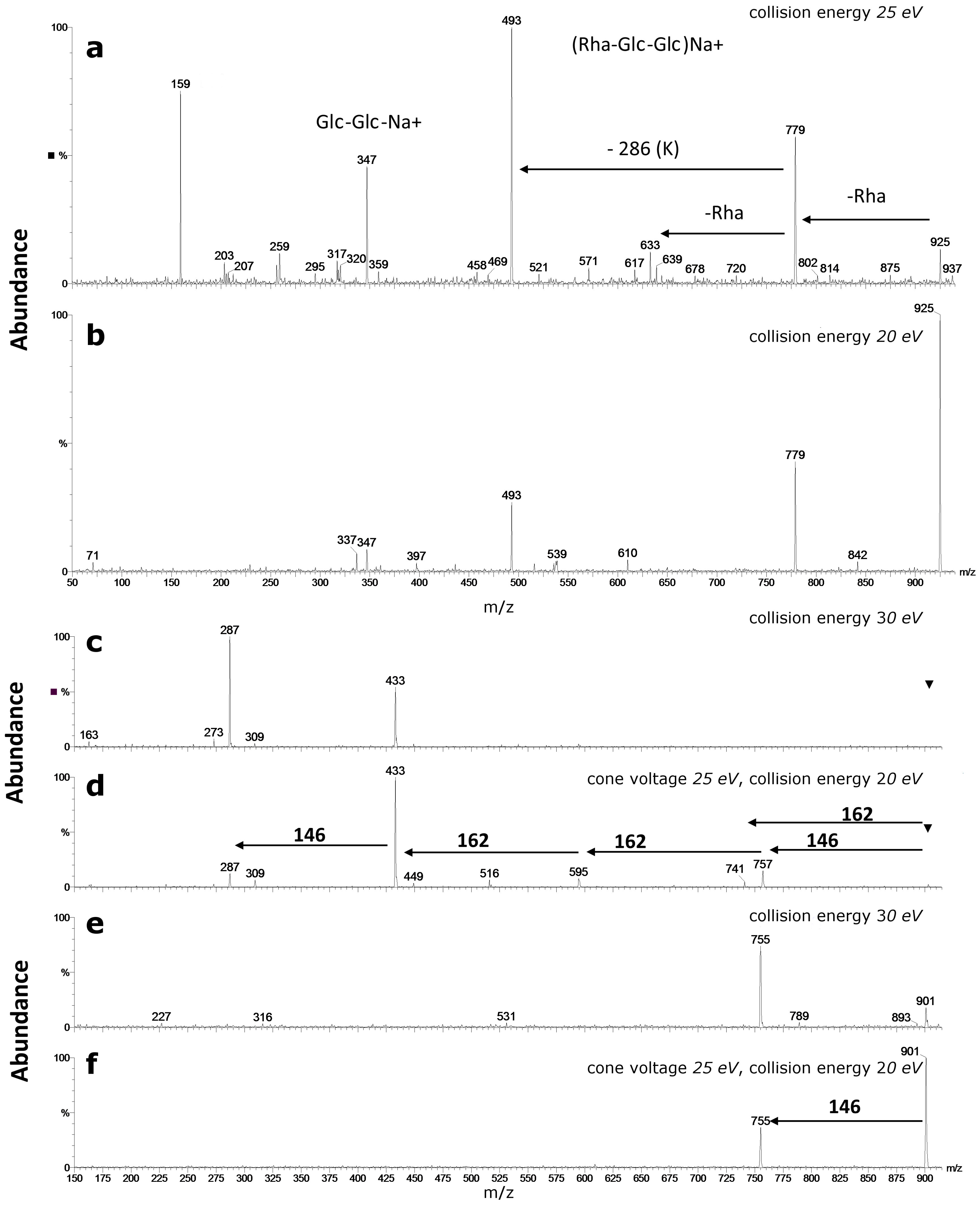
- Differentiation of sugar substitution pattern of flavonoid oligoglycosides is a difficult structural problem. Elucidation of sugar pattern on an aglycone is rather impossible if conducted exclusively with mass spectrometric approach. However, application of postcolumn addition of metal salts during LC-MS experiments can help to solve this problem, at least to some extent.
- (2)
- Acylation of flavonoid glycosides with aliphatic (e.g., acetic or malonic) or aromatic (e.g., benzoic or phenylpropenoic acids derivatives) carboxylic acids is a frequently observed structural feature. Detection of this type of substitution and identification of the acyl group may be achieved with the use of high-resolution mass spectrometers, most frequently with q-ToF analyzers (resolution FWHM = 40,000) or various types of Orbitrap analyzers, (FWHM of 70,000 or more). Analysis of acyl placement is not possible with MS methods. However, differentiation of substitution can be achieved by observation of various retention times of eluted positional isomers.
- (3)
- Structural characterization, fingerprinting, semi-quantitative analysis and visualization within tissues of glycoconjugates of flavonoids and especially anthocyanins may be also conducted using matrix-assisted laser desorption ionization (MALDI) of the samples. This technique is an alternative to chromatography based analytical methods.
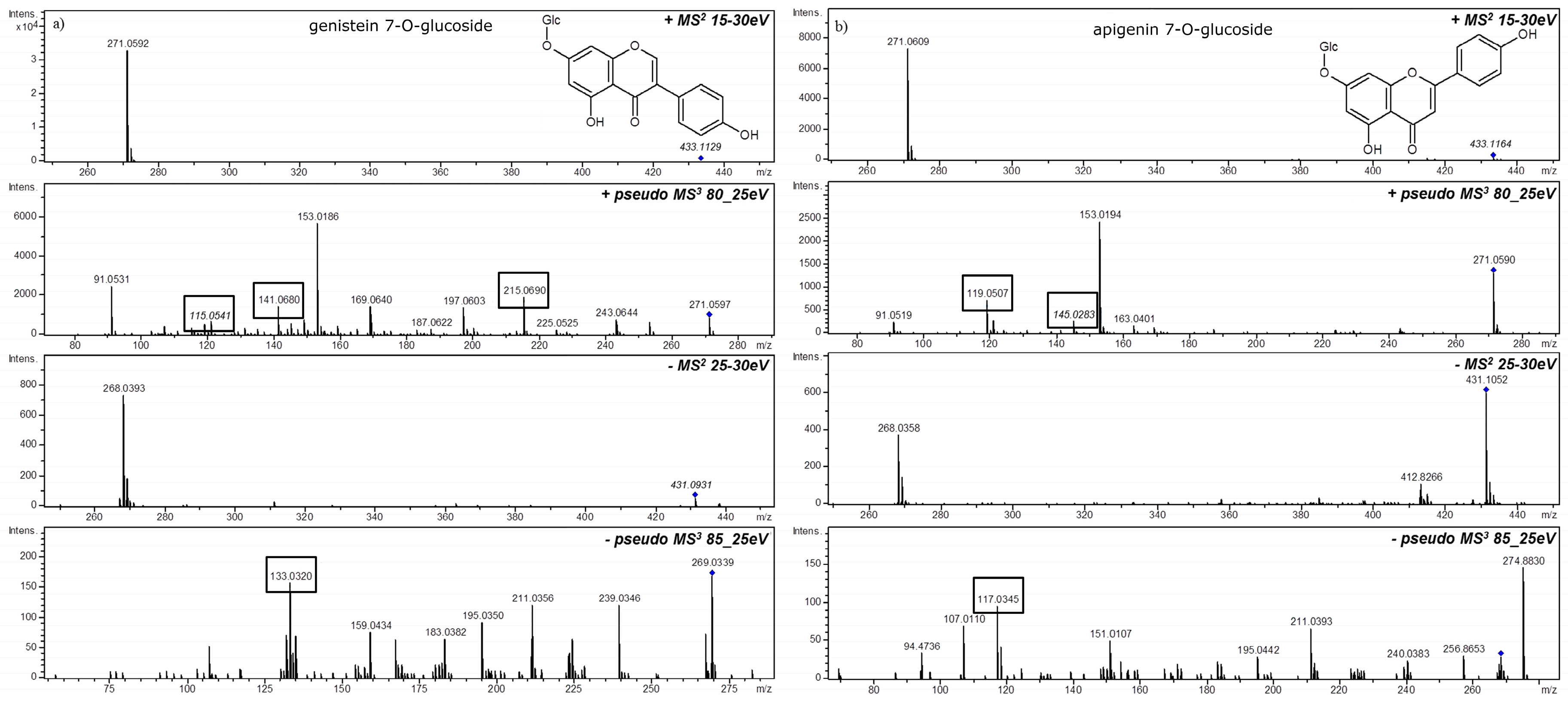
3. MSn in Flavonoid Analysis
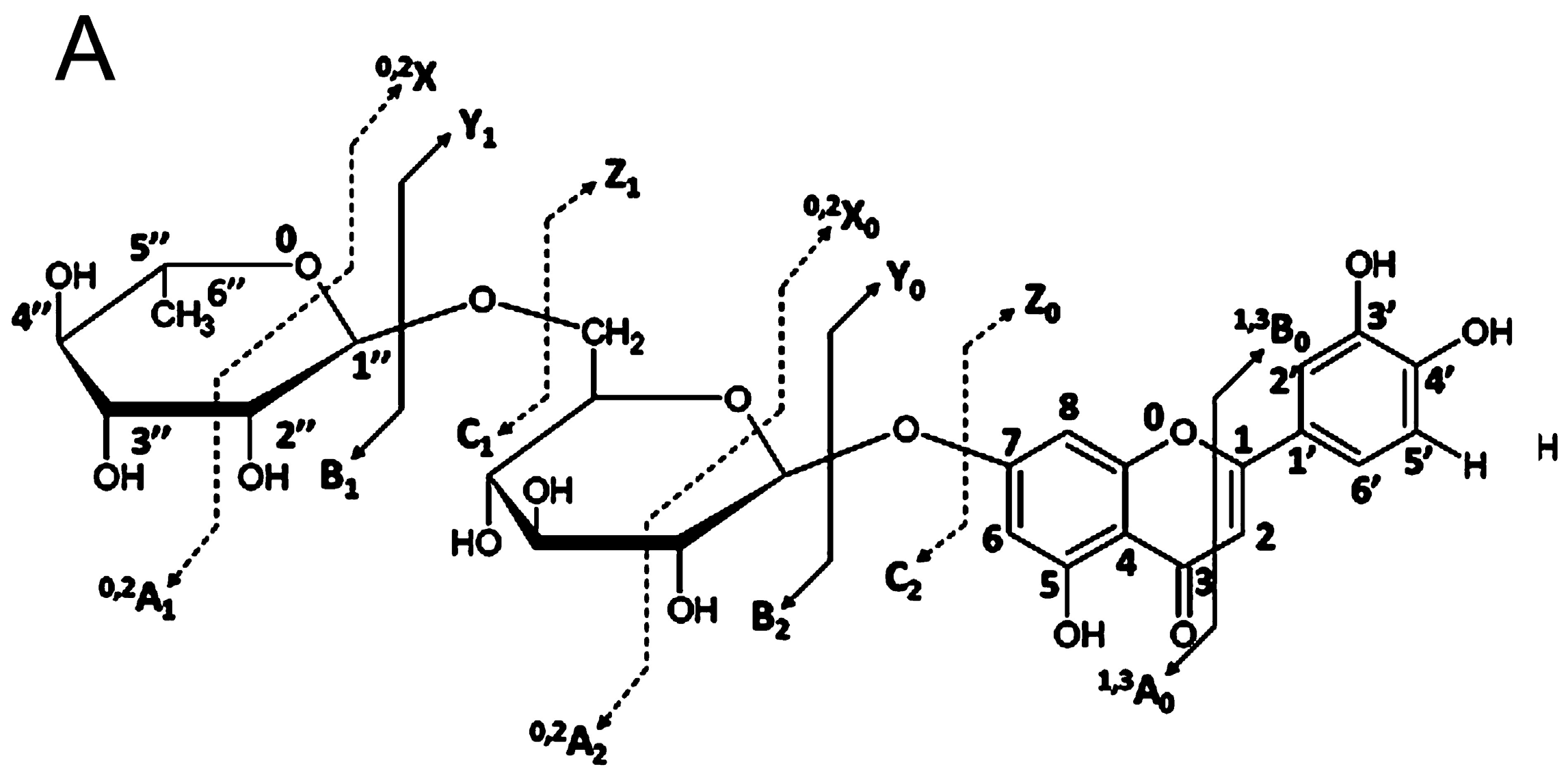
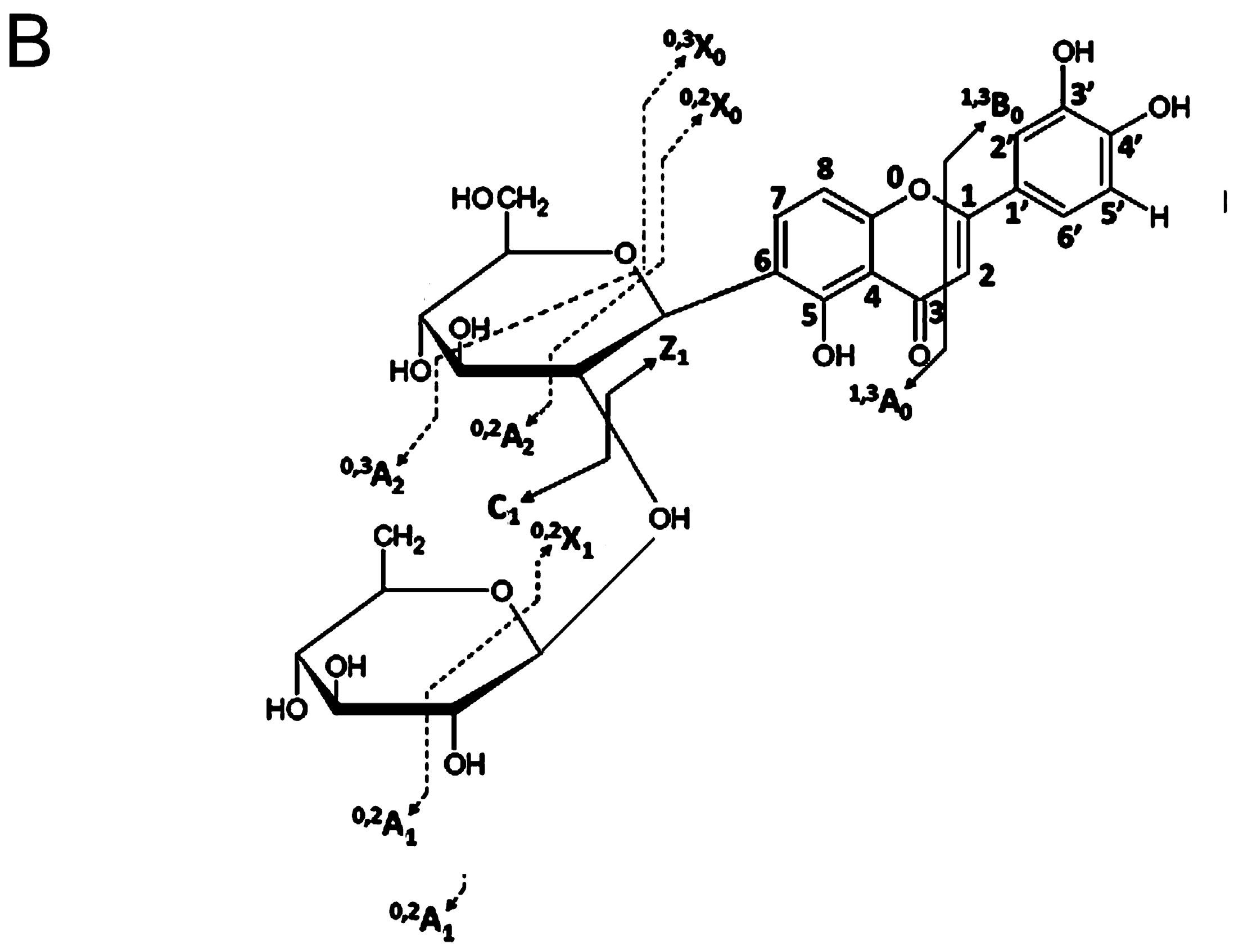
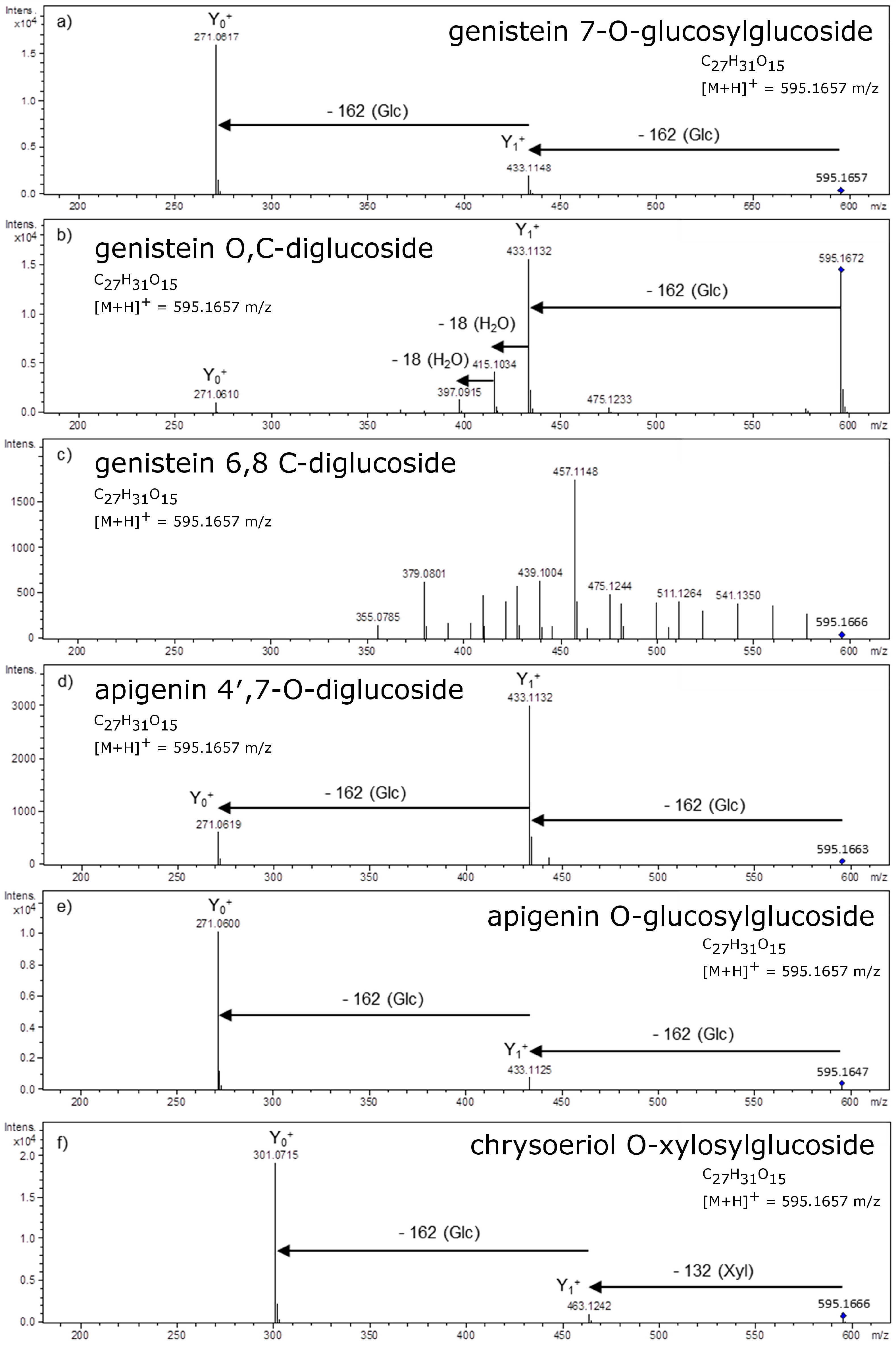
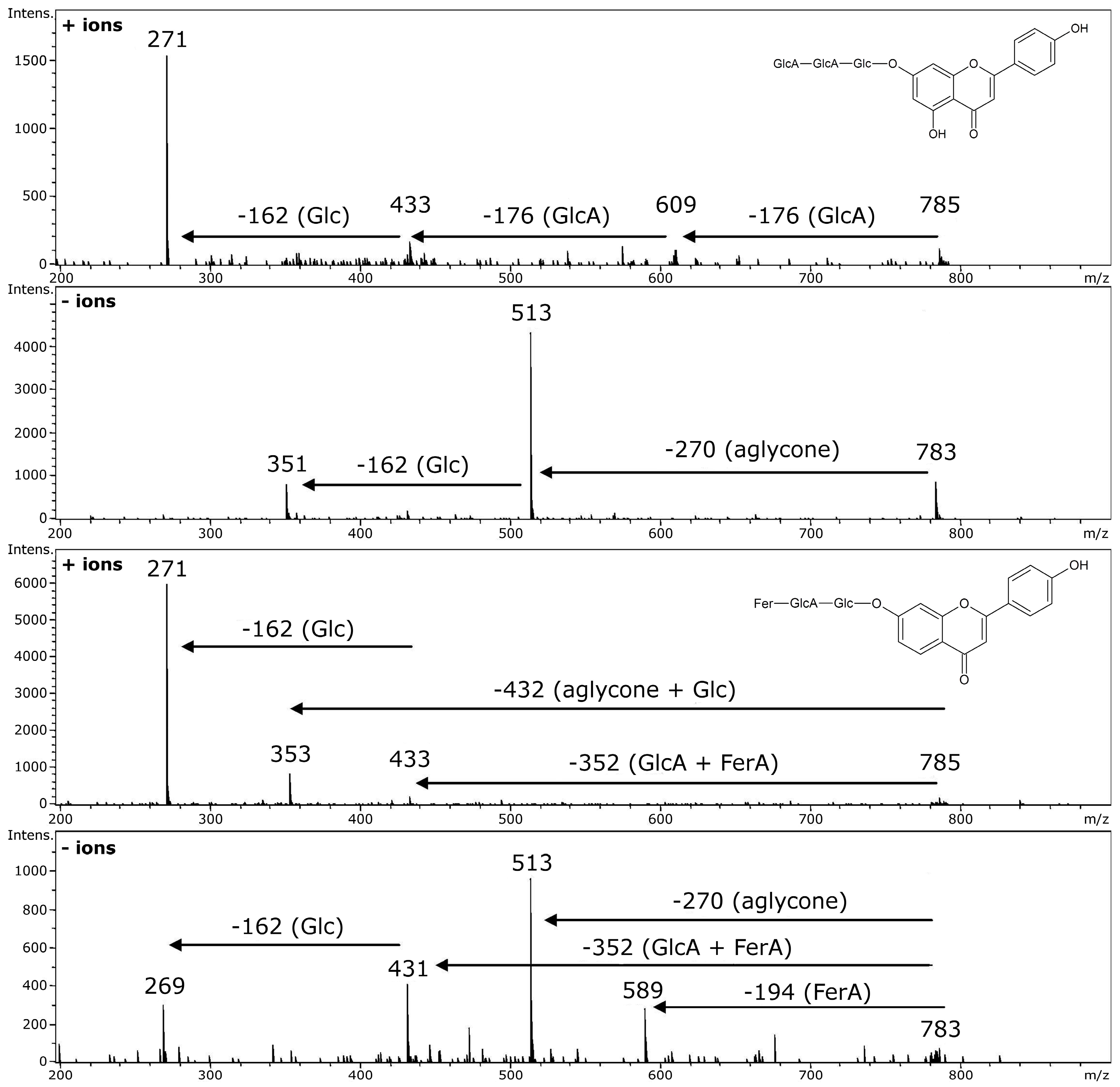
4. Glycosidic Part of Flavonoid Glycoconjugates
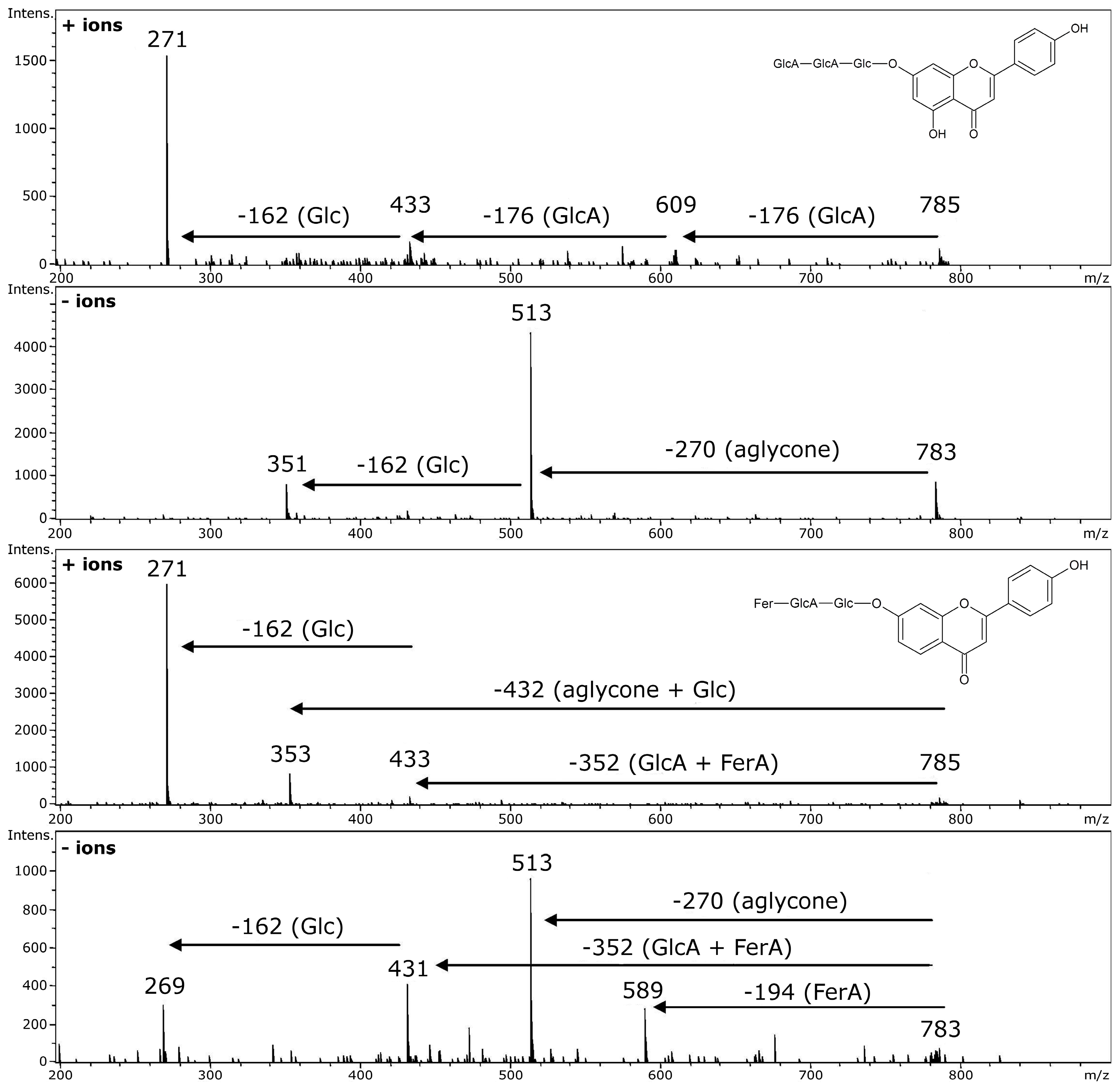
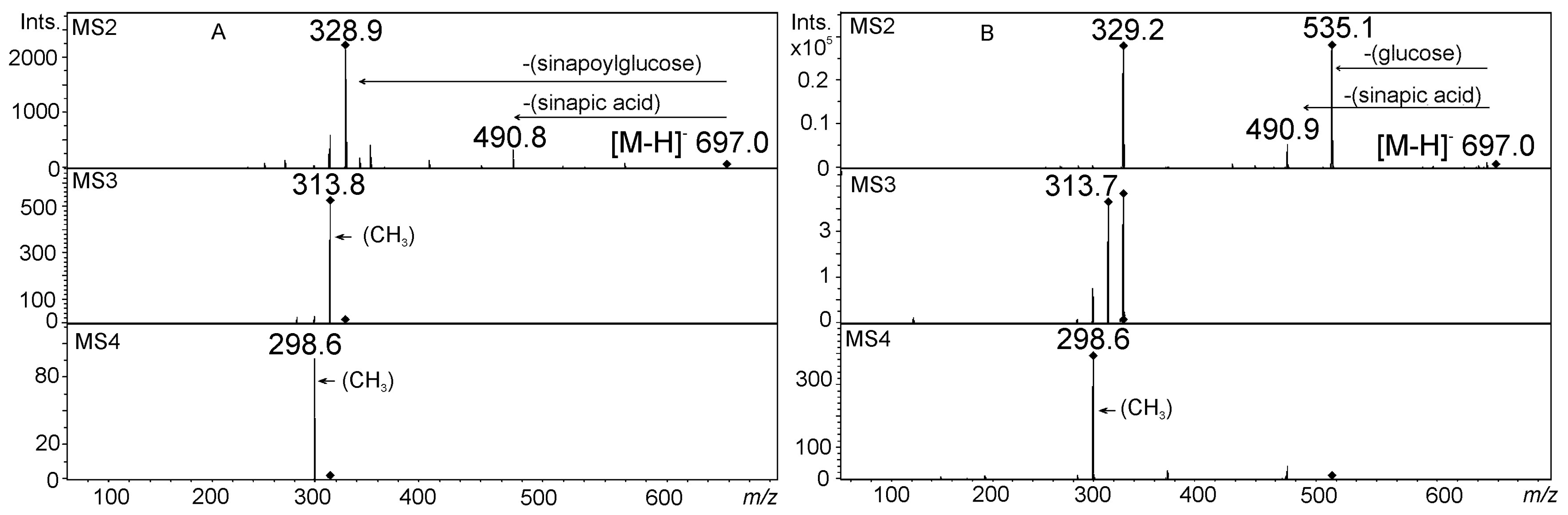
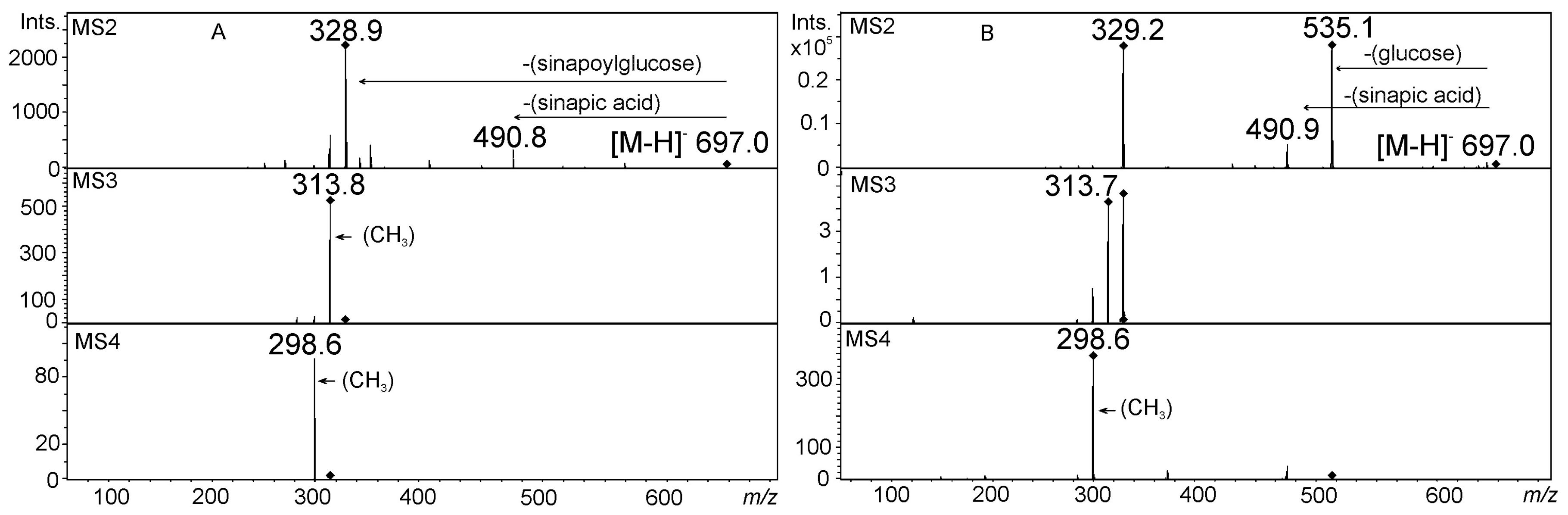
5. Acylated Flavonoid Glycoconjugates
6. Analysis of Flavonoids by MALDI Mass Spectrometry
7. Databases of Plant Metabolite MS Data
8. Summary
Acknowledgments
Conflicts of Interest
References
- Gross, J.H. Principles of Ionization and Ion Dissociation. In Mass Spectrometry; Springer: Berlin/Heidelberg, Germany, 2011; pp. 21–66. [Google Scholar]
- Stobiecki, M.; Kachlicki, P.; Jeleñ, H. Mass Spectrometry in Agriculture, Food, and Flavors: Selected Applications. In Mass Spectrometry Handbook; Lee, M.S., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 529–558. [Google Scholar]
- Walhout, M.; Vidal, M.; Dekker, J. Handbook of Systems Biology: Concepts and Insights; Elsever/Academic Press: London, UK, 2013; pp. 45–196. [Google Scholar]
- Bassalo, M.C.; Liu, R.; Gill, R.T. Directed evolution and synthetic biology applications to microbial systems. Curr. Opin. Biotechnol. 2016, 39, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Han, N.; Cleto, S.; Cao, J.; Purcell, O.; Shah, K.A.; Lee, K.; Ram, R.; Lu, T.K. Synthetic biology and microbioreactor platforms for programmable production of biologics at the point-of-care. Nat. Commun. 2016, 7, 12211. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.A.; Strack, D. Phytochemistry meets genome analysis, and beyond. Phytochemistry 2003, 62, 815–816. [Google Scholar] [CrossRef]
- Andersen, O.M.; Markham, K.R. Flavonoids. Chemistry, Biochemistry and Applications; CRC, Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Piasecka, A.; Jedrzejczak-Rey, N.; Bednarek, P. Secondary metabolites in plant innate immunity: Conserved function of divergent chemicals. New Phytol. 2015, 206, 948–964. [Google Scholar] [CrossRef] [PubMed]
- Bednarek, P. Chemical warfare or modulators of defence responses—The function of secondary metabolites in plant immunity. Curr. Opin. Plant Biol. 2012, 15, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Nijveldt, R.J.; van Nood, E.; van Hoorn, D.E.; Boelens, P.G.; van Norren, K.; van Leeuwen, P.A. Flavonoids: A review of probable mechanisms of action and potential applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [PubMed]
- Kroon, P.A.; Clifford, M.N.; Crozier, A.; Day, A.J.; Donovan, J.L.; Manach, C.; Williamson, G. How should we assess the effects of exposure to dietary polyphenols in vitro? Am. J. Clin. Nutr. 2004, 80, 15–21. [Google Scholar] [PubMed]
- Kay, C.D. The future of flavonoid research. Br. J. Nutr. 2010, 104, S91–S95. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Mateos, A.; Vauzour, D.; Krueger, C.G.; Shanmuganayagam, D.; Reed, J.; Calani, L.; Mena, P.; Del Rio, D.; Crozier, A. Bioavailability, bioactivity and impact on health of dietary flavonoids and related compounds: An update. Arch. Toxicol. 2014, 88, 1803–1853. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, A.; Venter, P.; Pasch, H. Recent advances and trends in the liquid-chromatography–mass spectrometry analysis of flavonoids. J. Chromatogr. A 2016, 1430, 16–78. [Google Scholar] [CrossRef] [PubMed]
- Forcisi, S.; Moritz, F.; Kanawati, B.; Tziotis, D.; Lehmann, R.; Schmitt-Kopplin, P. Liquid chromatography-mass spectrometry in metabolomics research: Mass analyzers in ultra high pressure liquid chromatography coupling. J. Chromatogr. A 2013, 1292, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, X.; Wang, S.; Yu, J.; Qin, Y.; Zhang, X.; Xie, F. Systematic screening and characterization of glycosides in tobacco leaves by liquid chromatography with atmospheric pressure chemical ionization tandem mass spectrometry using neutral loss scan and product ion scan. J. Sep. Sci. 2015, 38, 4029–4035. [Google Scholar] [CrossRef] [PubMed]
- Garmón-Lobato, S.; Abad-García, B.; Sánchez-Ilárduya, M.B.; Romera-Fernández, M.; Berrueta, L.A.; Gallo, B.; Vicente, F. Improvement using chemometrics in ion mobility coupled to mass spectrometry as a tool for mass spectrometry fragmentation studies: Flavonoid aglycone cases. Anal. Chim. Acta 2013, 771, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, G.B.; Raes, K.; Coelus, S.; Struijs, K.; Smagghe, G.; Van Camp, J. Ultra(high)-pressure liquid chromatography-electrospray ionization-time-of-flight-ion mobility-high definition mass spectrometry for the rapid identification and structural characterization of flavonoid glycosides from cauliflower waste. J. Chromatogr. A 2014, 1323, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Groessl, M.; Azzollini, A.; Eugster, P.J.; Plet, B.; Wolfender, J.-L.; Knochenmuss, R. Comparison of UHPLC-ESI-MS and Hadamard transform atmospheric pressure ion mobility-ESI-MS for rapid profiling of isomeric flavonoids. Chimia (Aarau) 2014, 68, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Kachlicki, P.; Einhorn, J.; Muth, D.; Kerhoas, L.; Stobiecki, M. Evaluation of glycosylation and malonylation patterns in flavonoid glycosides during LC/MS/MS metabolite profiling. J. Mass Spectrom. 2008, 43, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Marczak, L.; Stobiecki, M.; Jasinski, M.; Oleszek, W.; Kachlicki, P. Fragmentation pathways of acylated flavonoid diglucuronides from leaves of Medicago truncatula. Phytochem. Anal. 2010, 21, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Stobiecki, M.; Staszków, A.; Piasecka, A.; Garcia-Lopez, P.M.; Zamora-Natera, F.; Kachlicki, P. LC-MSMS profiling of flavonoid conjugates in wild Mexican lupine, Lupinus reflexus. J. Nat. Prod. 2010, 73, 1254–1260. [Google Scholar] [CrossRef] [PubMed]
- Wojakowska, A.; Piasecka, A.; García-López, P.M.; Zamora-Natera, F.; Krajewski, P.; Marczak, Ł.; Kachlicki, P.; Stobiecki, M. Structural analysis and profiling of phenolic secondary metabolites of Mexican lupine species using LC–MS techniques. Phytochemistry 2013, 92, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, A.; Sawikowska, A.; Krajewski, P.; Kachlicki, P. Combined mass spectrometric and chromatographic methods for in-depth analysis of phenolic secondary metabolites in barley leaves. J. Mass Spectrom. 2015, 50, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Milman, B.L. General principles of identification by mass spectrometry. TrAC Trends Anal. Chem. 2015, 69, 24–33. [Google Scholar] [CrossRef]
- Jaroszewski, J.W. Hyphenated NMR methods in natural products research, part 1: Direct hyphenation. Planta Med. 2005, 71, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Jaroszewski, J.W. Hyphenated NMR methods in natural products research, Part 2: HPLC-SPE-NMR and other new trends in NMR hyphenation. Planta Med. 2005, 71, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Seger, C.; Sturm, S.; Stuppner, H. Mass spectrometry and NMR spectroscopy: Modern high-end detectors for high resolution separation techniques--state of the art in natural product HPLC-MS, HPLC-NMR, and CE-MS hyphenations. Nat. Prod. Rep. 2013, 30, 970–987. [Google Scholar] [CrossRef] [PubMed]
- Braunberger, C.; Zehl, M.; Conrad, J.; Wawrosch, C.; Strohbach, J.; Beifuss, U.; Krenn, L. Flavonoids as chemotaxonomic markers in the genus Drosera. Phytochemistry 2015, 118, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wang, Y.; Bai, L.; Xia, B.; Li, X.; Tang, Y.; Xu, P.; Xue, M. Structural Elucidation of In Vivo Metabolites of Isobavachalcone in Rat by LC–ESI-MSn and LC–NMR. J. Pharm. Biomed. Anal. 2015, 104, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Lommen, A.; Godejohann, M.; Venema, D.P.; Hollman, P.C.H.; Spraul, M. Application of Directly Coupled HPLC−NMR−MS to the Identification and Confirmation of Quercetin Glycosides and Phloretin Glycosides in Apple Peel. Anal. Chem. 2000, 72, 1793–1797. [Google Scholar] [CrossRef] [PubMed]
- Yim, S.-H.; Lee, Y.J.; Park, K.D.; Lee, I.-S.; Shin, B.A.; Jung, D.W.; Williams, D.R.; Kim, H.J. Phenolic Constituents from the Flowers of Hamamelis japonica Sieb. et Zucc. Nat. Prod. Sci. 2015, 21, 162–169. [Google Scholar]
- Stafford, G. Ion trap mass spectrometry: A personal perspective. J. Am. Soc. Mass Spectrom. 2002, 13, 589–596. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, D.H.; Liu, K.-H.; Oh, T.K.; Lee, C.H. Identification of flavonoids using liquid chromatography with electrospray ionization and ion trap tandem mass spectrometry with an MS/MS library. Rapid Commun. Mass Spectrom. 2005, 19, 3539–3548. [Google Scholar] [CrossRef] [PubMed]
- Kachlicki, P.; Marczak, Ł.; Kerhoas, L.; Einhorn, J.; Stobiecki, M. Profiling isoflavone conjugates in root extracts of lupine species with LC/ESI/MSn systems. J. Mass Spectrom. 2005, 40, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Ferreres, F.; Gil-Izquierdo, A.; Andrade, P.B.; Valentão, P.; Tomás-Barberán, F.A. Characterization of C-glycosyl flavones O-glycosylated by liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2007, 1161, 214–223. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-K.; Yao, Y.-Y.; Chang, Y.-N. Characterization of Anthocyanins in Perilla frutescens var. acuta Extract by Advanced UPLC-ESI-IT-TOF-MSn Method and Their Anticancer Bioactivity. Molecules 2015, 20, 9155–9169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, J.; Duan, J.; Liang, Z.; Zhang, L.; Huo, Y.; Zhang, Y. Quantitative and qualitative analysis of flavonoids in leaves of Adinandra nitida by high performance liquid chromatography with UV and electrospray ionization tandem mass spectrometry detection. Anal. Chim. Acta 2005, 532, 97–104. [Google Scholar] [CrossRef]
- Kang, J.; Hick, L.A.; Price, W.E. A fragmentation study of isoflavones in negative electrospray ionization by MSn ion trap mass spectrometry and triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Fridén, M.E.; Sjöberg, P.J.R. Strategies for differentiation of isobaric flavonoids using liquid chromatography coupled to electrospray ionization mass spectrometry. J. Mass Spectrom. 2014, 49, 646–663. [Google Scholar] [CrossRef] [PubMed]
- Ferreres, F.; Llorach, R.; Gil-Izquierdo, A. Characterization of the interglycosidic linkage in di-, tri-, tetra- and pentaglycosylated flavonoids and differentiation of positional isomers by liquid chromatography/electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 2004, 39, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Muth, D.; Marsden-Edwards, E.; Kachlicki, P.; Stobiecki, M. Differentiation of isomeric malonylated flavonoid glyconjugates in plant extracts with UPLC-ESI/MS/MS. Phytochem. Anal. 2008, 19, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Wojakowska, A.; Muth, D.; Narożna, D.; Mądrzak, C.; Stobiecki, M.; Kachlicki, P. Changes of phenolic secondary metabolite profiles in the reaction of narrow leaf lupin (Lupinus angustifolius) plants to infections with Colletotrichum lupini fungus or treatment with its toxin. Metabolomics 2013, 9, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Wojakowska, A.; Perkowski, J.; Góral, T.; Stobiecki, M. Structural characterization of flavonoid glycosides from leaves of wheat (Triticum aestivum L.) using LC/MS/MS profiling of the target compounds. J. Mass Spectrom. 2013, 48, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Abrankó, L.; Szilvássy, B. Mass spectrometric profiling of flavonoid glycoconjugates possessing isomeric aglycones. J. Mass Spectrom. 2014, 50, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wang, C.; Yang, L.; Luo, H.; Fan, W.; Zi, C.; Dong, F.; Hu, J.; Zhou, J. C-dideoxyhexosyl flavones from the stems and leaves of Passiflora edulis Sims. Food Chem. 2013, 136, 94–99. [Google Scholar] [CrossRef] [PubMed]
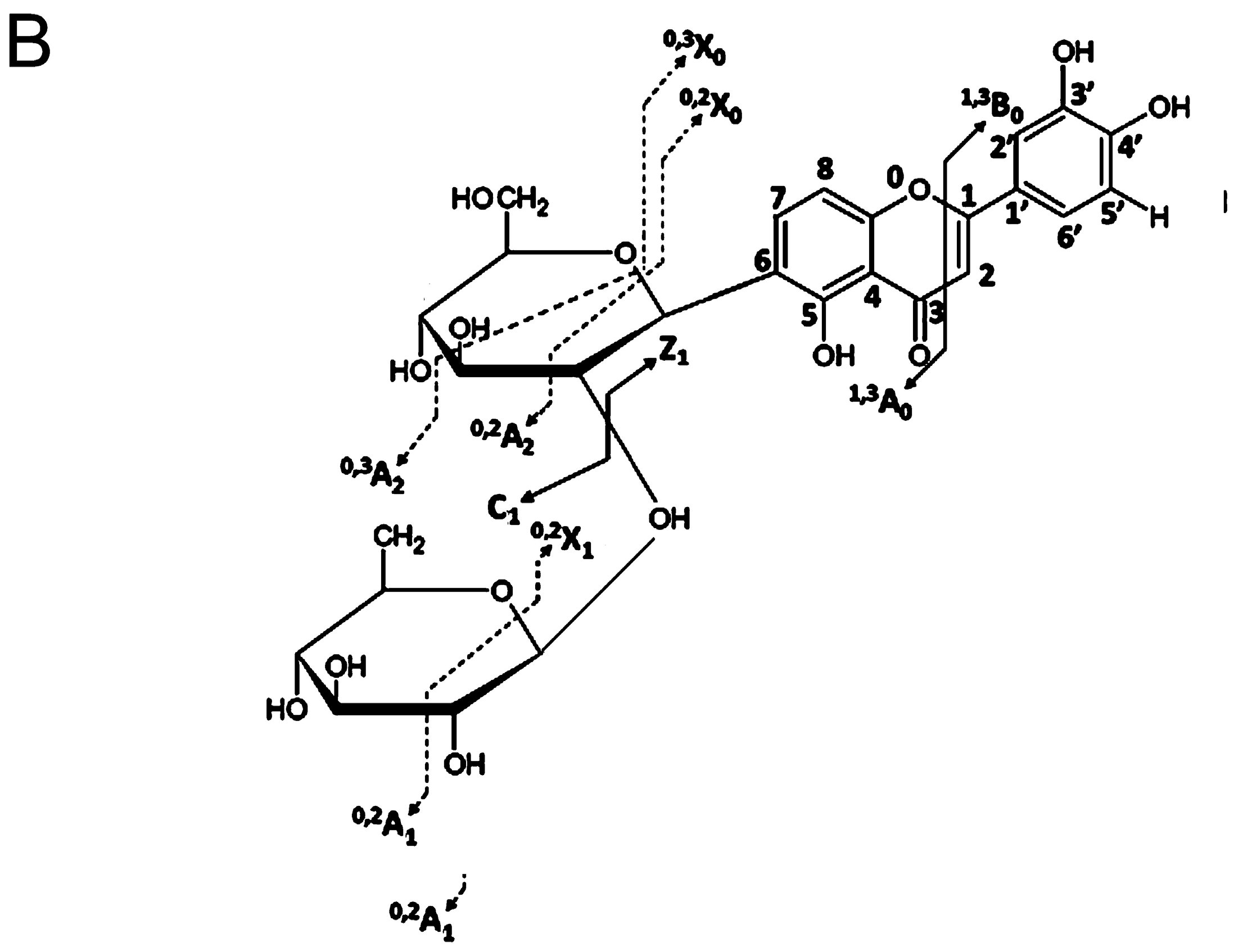
- Domon, B.; Costello, C.E. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
- Vukics, V.; Guttman, A. Structural characterization of flavonoid glycosides by multi-stage mass spectrometry. Mass Spectrom. Rev. 2008, 29, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Simirgiotis, M.J.; Schmeda-Hirschmann, G.; Bórquez, J.; Kennelly, E.J. The Passiflora tripartita (Banana Passion) fruit: A source of bioactive flavonoid C-glycosides isolated by HSCCC and characterized by HPLC–DAD–ESI/MS/MS. Molecules 2013, 18, 1672–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farag, M.A.; Otify, A.; Porzel, A.; Michel, C.G.; Elsayed, A.; Wessjohann, L.A. Comparative metabolite profiling and fingerprinting of genus Passiflora leaves using a multiplex approach of UPLC-MS and NMR analyzed by chemometric tools. Anal. Bioanal. Chem. 2016, 408, 3125–3143. [Google Scholar] [CrossRef] [PubMed]
- Kamel, M.S. Flavone C-glycosides from Lupinus hartwegii. Phytochemistry 2003, 63, 449–452. [Google Scholar] [CrossRef]
- Benayad, Z.; Gómez-Cordovés, C.; Es-Safi, N.E. Identification and quantification of flavonoid glycosides from fenugreek (Trigonella foenum-graecum) germinated seeds by LC–DAD–ESI/MS analysis. J. Food Compos. Anal. 2014, 35, 21–29. [Google Scholar] [CrossRef]
- Zehrmann, N.; Zidorn, C.; Ganzera, M. Analysis of rare flavonoid C-glycosides in Celtis australis L. by micellar electrokinetic chromatography. J. Pharm. Biomed. Anal. 2010, 51, 1165–1168. [Google Scholar] [CrossRef] [PubMed]
- Obmann, A.; Werner, I.; Presser, A.; Zehl, M.; Swoboda, Z.; Purevsuren, S.; Narantuya, S.; Kletter, C.; Glasl, S. Flavonoid C- and O-glycosides from the Mongolian medicinal plant Dianthus versicolor Fisch. Carbohydr. Res. 2011, 346, 1868–1875. [Google Scholar] [CrossRef] [PubMed]
- Muharini, R.; Wray, V.; Lai, D.; Proksch, P. New flavone C-glycosides from leaves of Sarcotheca griffithii (Hook F) Hallier F. Phytochem. Lett. 2014, 9, 26–32. [Google Scholar] [CrossRef]
- Sharaf, M.; Skiba, A.; Weglarz, Z.; El-Ansari, M.A. Two flavonol 5-O-glycosides from the roots of Leuzea carthamoides. Fitoterapia 2001, 72, 940–942. [Google Scholar] [CrossRef]
- Ferreres, F.; Andrade, P.B.; Valentão, P.; Gil-Izquierdo, A. Further knowledge on barley (Hordeum vulgare L.) leaves O-glycosyl-C-glycosyl flavones by liquid chromatography-UV diode-array detection-electrospray ionisation mass spectrometry. J. Chromatogr. A 2008, 1182, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Chen, W.; Wang, W.; Zhang, H.; Liu, X.; Luo, J. Comprehensive profiling and natural variation of flavonoids in rice. J. Integr. Plant Biol. 2014, 56, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Muzashvili, T.S.; Georgiev, M.I. Advances in the biotechnological glycosylation of valuable flavonoids. Biotechnol. Adv. 2014, 32, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Ateba, S.B.; Njamen, D.; Gatterer, C.; Scherzer, T.; Zehl, M.; Kählig, H.; Krenn, L. Rare phenolic structures found in the aerial parts of Eriosema laurentii De Wild. Phytochemistry 2016, 128, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Prescott, T.A.K.; Kite, G.C.; Porter, E.A.; Veitch, N.C. Highly glycosylated flavonols with an O-linked branched pentasaccharide from Iberis saxatilis (Brassicaceae). Phytochemistry 2013, 88, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Cuyckens, F.; Ma, Y.L.; Pocsfalvi, G.; Claeysi, M. Tandem mass spectral strategies for the structural characterization of flavonoid glycosides. Analusis 2000, 28, 888–895. [Google Scholar] [CrossRef]
- Lu, L.; Song, F.-R.; Tsao, R.; Jin, Y.-R.; Liu, Z.-Q.; Liu, S.-Y. Studies on the homolytic and heterolytic cleavage of kaempferol and kaempferide glycosides using electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-Z.; Qiao, X.; Bo, T.; Wang, Q.; Guo, D.-A.; Ye, M. Low energy induced homolytic fragmentation of flavonol 3-O-glycosides by negative electrospray ionization tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2014, 28, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Cuyckens, F.; Claeys, M. Determination of the glycosylation site in flavonoid mono-O-glycosides by collision-induced dissociation of electrospray-generated deprotonated and sodiated molecules. J. Mass Spectrom. 2005, 40, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Cuyckens, F.; Rozenberg, R.; de Hoffmann, E.; Claeys, M. Structure characterization of flavonoid O-diglycosides by positive and negative nano-electrospray ionization ion trap mass spectrometry. J. Mass Spectrom. 2001, 36, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-L.; Cuyckens, F.; Van den, H.H.; Claeys, M. Mass spectrometric methods for the characterisation and differentiation of isomericO-diglycosyl flavonoids. Phytochem. Anal. 2001, 12, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Es-Safi, N.-E.; Kerhoas, L.; Ducrot, P.-H. Application of positive and negative electrospray ionization, collision-induced dissociation and tandem mass spectrometry to a study of the fragmentation of 6-hydroxyluteolin 7-O-glucoside and 7-O-glucosyl-(1→3)-glucoside. Rapid Commun. Mass Spectrom. 2005, 19, 2734–2742. [Google Scholar] [CrossRef] [PubMed]
- Stobiecki, M.; Skirycz, A.; Kerhoas, L.; Kachlicki, P.; Muth, D.; Einhorn, J.; Mueller-Roeber, B. Profiling of phenolic glycosidic conjugates in leaves of Arabidopsis thaliana using LC/MS. Metabolomics 2007, 2, 197–219. [Google Scholar] [CrossRef]
- Ferreres, F.; Silva, B.M.; Andrade, P.B.; Seabra, R.M.; Ferreira, M.A. Approach to the study of C-glycosyl flavones by ion trap HPLC-PAD-ESI/MS/MS: Application to seeds of quince (Cydonia oblonga). Phytochem. Anal. 2003, 14, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Waridel, P.; Wolfender, J.L.; Ndjoko, K.; Hobby, K.R.; Major, H.J.; Hostettmann, K. Evaluation of quadrupole time-of-flight tandem mass spectrometry and ion-trap multiple-stage mass spectrometry for the differentiation of C-glycosidic flavonoid isomers. J. Chromatogr. A 2001, 926, 29–41. [Google Scholar] [CrossRef]
- Cuyckens, F.; Claeys, M. Mass spectrometry in the structural analysis of flavonoids. J. Mass Spectrom. 2004, 39, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fiol, M.; Adermann, S.; Neugart, S.; Rohn, S.; Mügge, C.; Schreiner, M.; Krumbein, A.; Kroh, L.W. Highly glycosylated and acylated flavonols isolated from kale (Brassica oleracea var. sabellica)—Structure–antioxidant activity relationship. Food Res. Int. 2012, 47, 80–89. [Google Scholar] [CrossRef]
- Pedras, M.S.C.; Zheng, Q.-A. Metabolic responses of Thellungiella halophila/salsuginea to biotic and abiotic stresses: Metabolite profiles and quantitative analyses. Phytochemistry 2010, 71, 581–589. [Google Scholar] [CrossRef] [PubMed]
- De Rijke, E.; de Kanter, F.; Ariese, F.; Brinkman, U.A.T.; Gooijer, C. Liquid chromatography coupled to nuclear magnetic resonance spectroscopy for the identification of isoflavone glucoside malonates in T. pratense L. leaves. J. Sep. Sci. 2004, 27, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Ozarowski, M.; Mikolajczak, P.L.; Piasecka, A.; Kachlicki, P.; Kujawski, R.; Bogacz, A.; Bartkowiak-Wieczorek, J.; Szulc, M.; Kaminska, E.; Kujawska, M.; et al. Influence of the Melissa officinalis Leaf Extract on Long-Term Memory in Scopolamine Animal Model with Assessment of Mechanism of Action. Evid. Based Complement. Altern. Med. 2016, 2016, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.H.; Gusev, A.I. Small molecule analysis by MALDI mass spectrometry. Anal. Bioanal. Chem. 2002, 373, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Marczak, Ł.; Kachlicki, P.; Koźniewski, P.; Skirycz, A.; Krajewski, P.; Stobiecki, M. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry monitoring of anthocyanins in extracts from Arabidopsis thaliana leaves. Rapid Commun. Mass Spectrom. 2008, 22, 3949–3956. [Google Scholar] [CrossRef] [PubMed]
- Guinan, T.; Kirkbride, P.; Pigou, P.E.; Ronci, M.; Kobus, H.; Voelcker, N.H. Surface-assisted laser desorption ionization mass spectrometry techniques for application in forensics. Mass Spectrom. Rev. 2015, 34, 627–640. [Google Scholar] [CrossRef] [PubMed]
- March, R.E.; Lewars, E.G.; Stadey, C.J.; Miao, X.-S.; Zhao, X.; Metcalfe, C.D. A comparison of flavonoid glycosides by electrospray tandem mass spectrometry. Int. J. Mass Spectrom. 2006, 248, 61–85. [Google Scholar] [CrossRef]
- Wang, J.; Sporns, P. MALDI-TOF MS Analysis of Food Flavonol Glycosides. J. Agric. Food Chem. 2000, 48, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, P.; Dong, Y.; Strupat, K.; Vrhovsek, U.; Mattivi, F. Combining intensity correlation analysis and MALDI imaging to study the distribution of flavonols and dihydrochalcones in Golden Delicious apples. J. Exp. Bot. 2012, 63, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Lopes, N.; Silva, D. Application of MALDI Mass Spectrometry in Natural Products Analysis. Planta Med. 2016, 82, 671–689. [Google Scholar] [CrossRef] [PubMed]
- Petković, M.; Petrović, B.; Savić, J.; Bugarčić, Ž.D.; Dimitrić-Marković, J.; Momić, T.; Vasić, V. Flavonoids as matrices for MALDI-TOF mass spectrometric analysis of transition metal complexes. Int. J. Mass Spectrom. 2010, 290, 39–46. [Google Scholar] [CrossRef]
- Hölscher, D.; Shroff, R.; Knop, K.; Gottschaldt, M.; Crecelius, A.; Schneider, B.; Heckel, D.G.; Schubert, U.S.; Svatoš, A. Matrix-free UV-laser desorption/ionization (LDI) mass spectrometric imaging at the single-cell level: Distribution of secondary metabolites of Arabidopsis thaliana and Hypericum species. Plant J. 2009, 60, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cha, S.; Yeung, E.S. Colloidal Graphite-Assisted Laser Desorption/Ionization MS and MSn of Small Molecules. Direct Profiling and MS Imaging of Small Metabolites from Fruits. Anal. Chem. 2007, 79, 6575–6584. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Gross, M.L. Ionic-liquid matrices for quantitative analysis by MALDI-TOF mass spectrometry. J. Am. Soc. Mass Spectrom. 2004, 15, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Tholey, A.; Heinzle, E. Ionic (liquid) matrices for matrix-assisted laser desorption/ionization mass spectrometry—applications and perspectives. Anal. Bioanal. Chem. 2006, 386, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Madeira, P.J.A.; Florêncio, M.H. Flavonoid-matrix cluster ions in MALDI mass spectrometry. J. Mass Spectrom. 2009, 44, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Keki, S.; Deak, G.; Zsuga, M. Fragmentation study of rutin, a naturally occurring flavone glycoside cationized with different alkali metal ions, using post-source decay matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 2001, 36, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Frison-Norrie, S.; Sporns, P. Identification and Quantification of Flavonol Glycosides in Almond Seedcoats Using MALDI-TOF MS. J. Agric. Food Chem. 2002, 50, 2782–2787. [Google Scholar] [CrossRef] [PubMed]
- Kim, S. Getting the most out of PubChem for virtual screening. Expert Opin. Drug Discov. 2016, 11, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Little, J.L.; Williams, A.J.; Pshenichnov, A.; Tkachenko, V. Identification of “Known Unknowns” Utilizing Accurate Mass Data and ChemSpider. J. Am. Soc. Mass Spectrom. 2012, 23, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Mochamad Afendi, F.; Kawsar Parvin, A.; Ono, N.; Tanaka, K.; Hirai Morita, A.; Sato, T.; Sugiura, T.; Altaf-Ul-Amin, M.; Kanaya, S. KNApSAcK Metabolite Activity Database for Retrieving the Relationships Between Metabolites and Biological Activities. Plant Cell Physiol. 2014, 55, e7. [Google Scholar] [CrossRef] [PubMed]
- Köhl, K.I.; Basler, G.; Lüdemann, A.; Selbig, J.; Walther, D. A plant resource and experiment management system based on the Golm Plant Database as a basic tool for omics research. Plant Methods 2008, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Dreher, K. Putting The Plant Metabolic Network pathway databases to work: Going offline to gain new capabilities. Methods Mol. Biol. 2014, 1083, 151–171. [Google Scholar] [PubMed]
- Kanehisa, M. KEGG Bioinformatics Resource for Plant Genomics and Metabolomics. Methods Mol. Biol. 2016, 1374, 55–70. [Google Scholar] [PubMed]
- Kale, N.S.; Haug, K.; Conesa, P.; Jayseelan, K.; Moreno, P.; Rocca-Serra, P.; Nainala, V.C.; Spicer, R.A.; Williams, M.; Li, X.; et al. MetaboLights: An Open-Access Database Repository for Metabolomics Data. Curr. Protoc. Bioinform. 2016, 53, 14.13.1–14.13.18. [Google Scholar]
- Kite, G.C.; Veitch, N.C. Assigning glucose or galactose as the primary glycosidic sugar in 3-O-mono-, di- and triglycosides of kaempferol using negative ion electrospray and serial mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 3125–3132. [Google Scholar] [CrossRef] [PubMed]
- Kite, G.C.; Veitch, N.C. Identification of common glycosyl groups of flavonoid O-glycosides by serial mass spectrometry of sodiated species. Rapid Commun. Mass Spectrom. 2011, 25, 2579–2590. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, J.; Wilson, S.; Her, G.R.; Stobiecki, M.; Goldman, P. Urinary organic acid profiles in fatty Zucker rats: Indications for impaired oxidation of butyrate and hexanoate. Metabolism 1990, 39, 1012–1020. [Google Scholar] [CrossRef]


© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kachlicki, P.; Piasecka, A.; Stobiecki, M.; Marczak, Ł. Structural Characterization of Flavonoid Glycoconjugates and Their Derivatives with Mass Spectrometric Techniques. Molecules 2016, 21, 1494. https://doi.org/10.3390/molecules21111494
Kachlicki P, Piasecka A, Stobiecki M, Marczak Ł. Structural Characterization of Flavonoid Glycoconjugates and Their Derivatives with Mass Spectrometric Techniques. Molecules. 2016; 21(11):1494. https://doi.org/10.3390/molecules21111494
Chicago/Turabian StyleKachlicki, Piotr, Anna Piasecka, Maciej Stobiecki, and Łukasz Marczak. 2016. "Structural Characterization of Flavonoid Glycoconjugates and Their Derivatives with Mass Spectrometric Techniques" Molecules 21, no. 11: 1494. https://doi.org/10.3390/molecules21111494