
Synthesis and Antifungal Activity against Fusarium oxysporum of Some Brassinin Analogs Derived from l-tryptophan: A DFT/B3LYP Study on the Reaction Mechanism

 ,
, 

Abstract
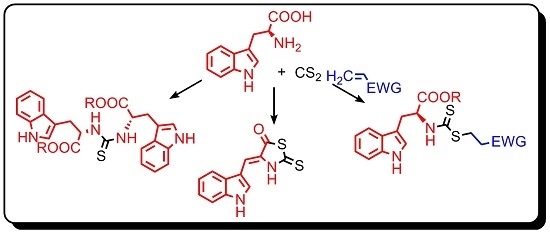
:
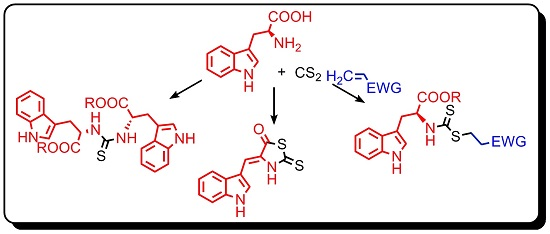
1. Introduction
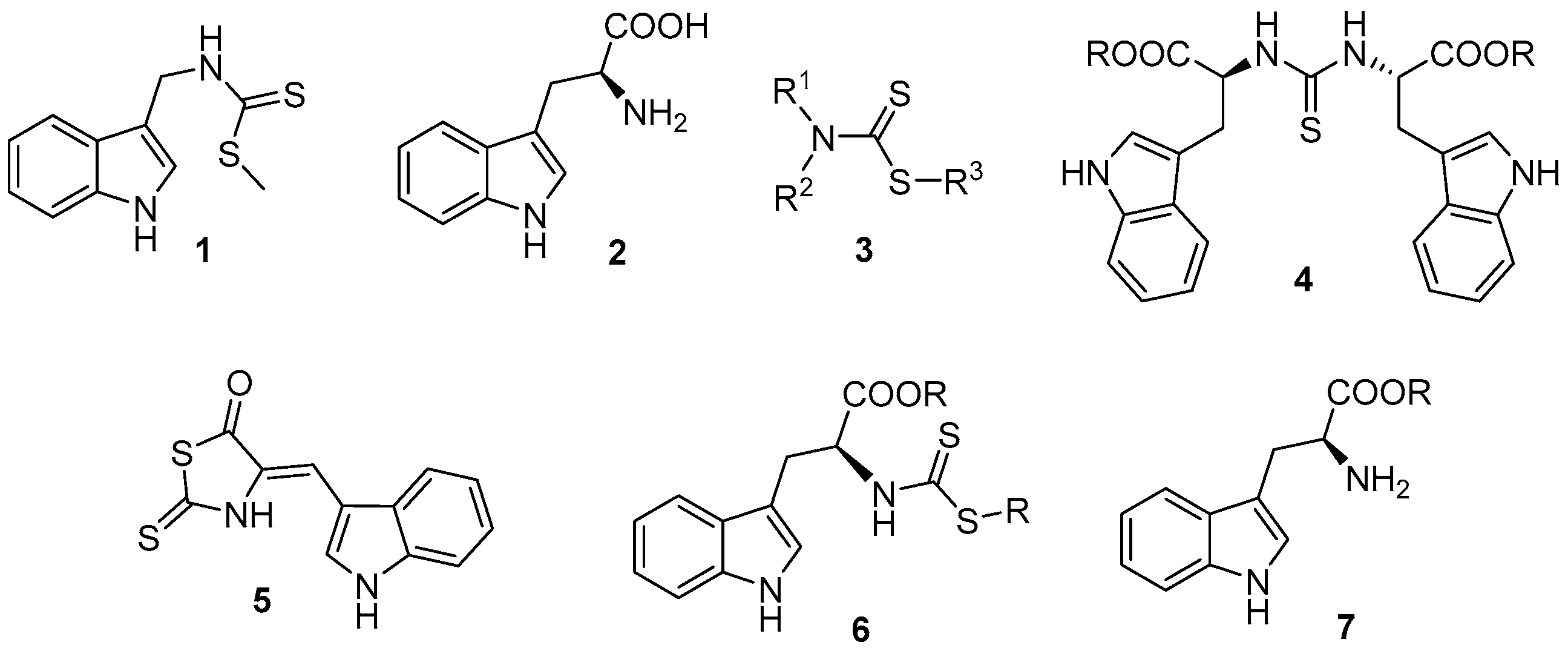
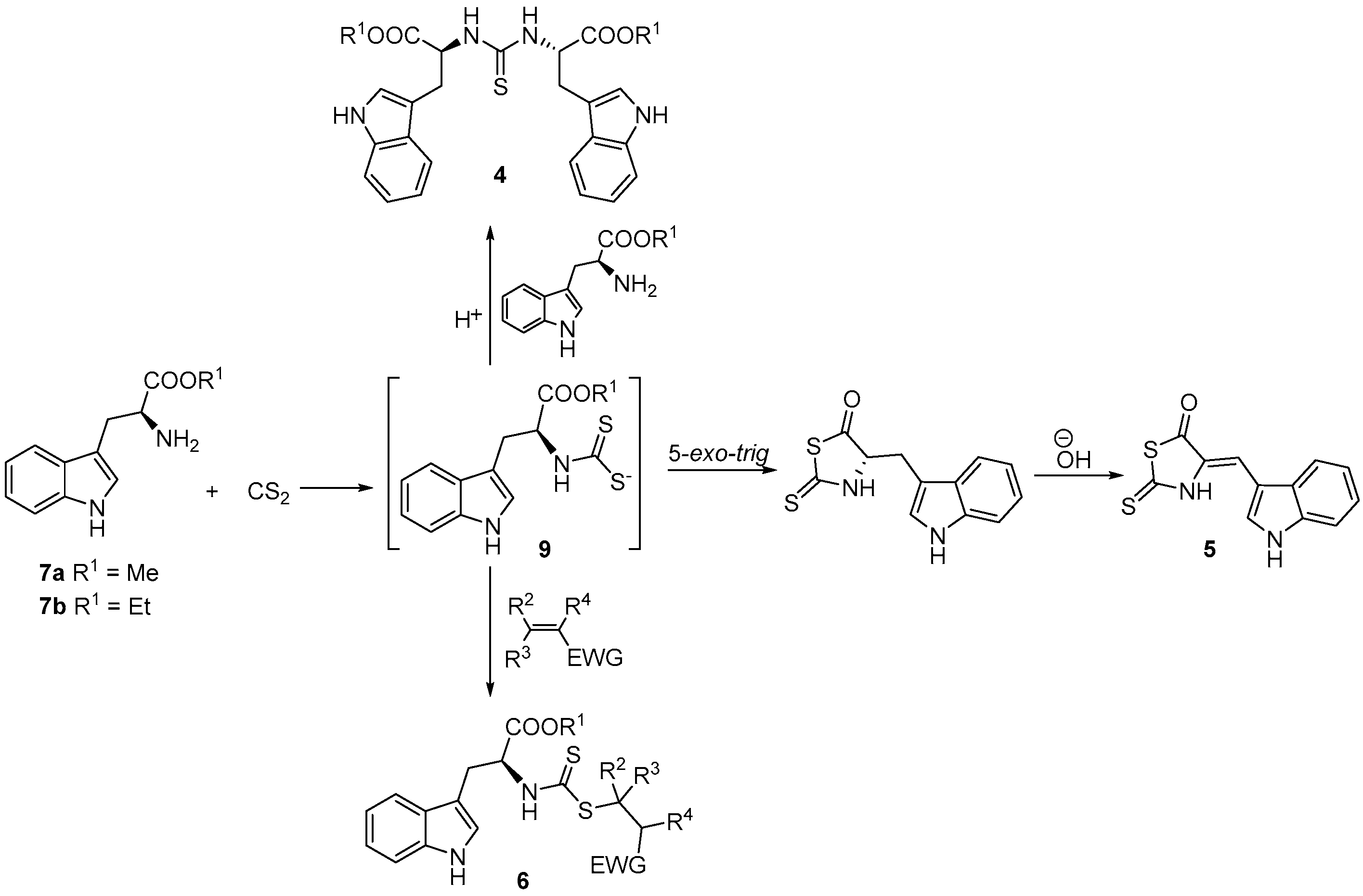
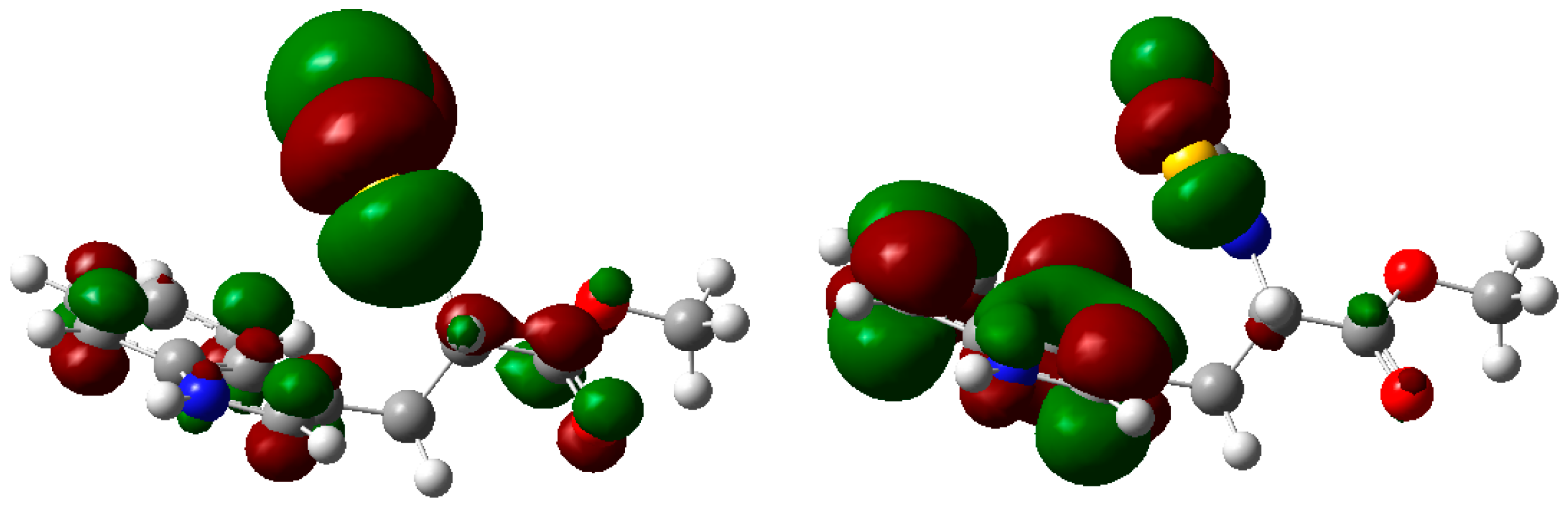
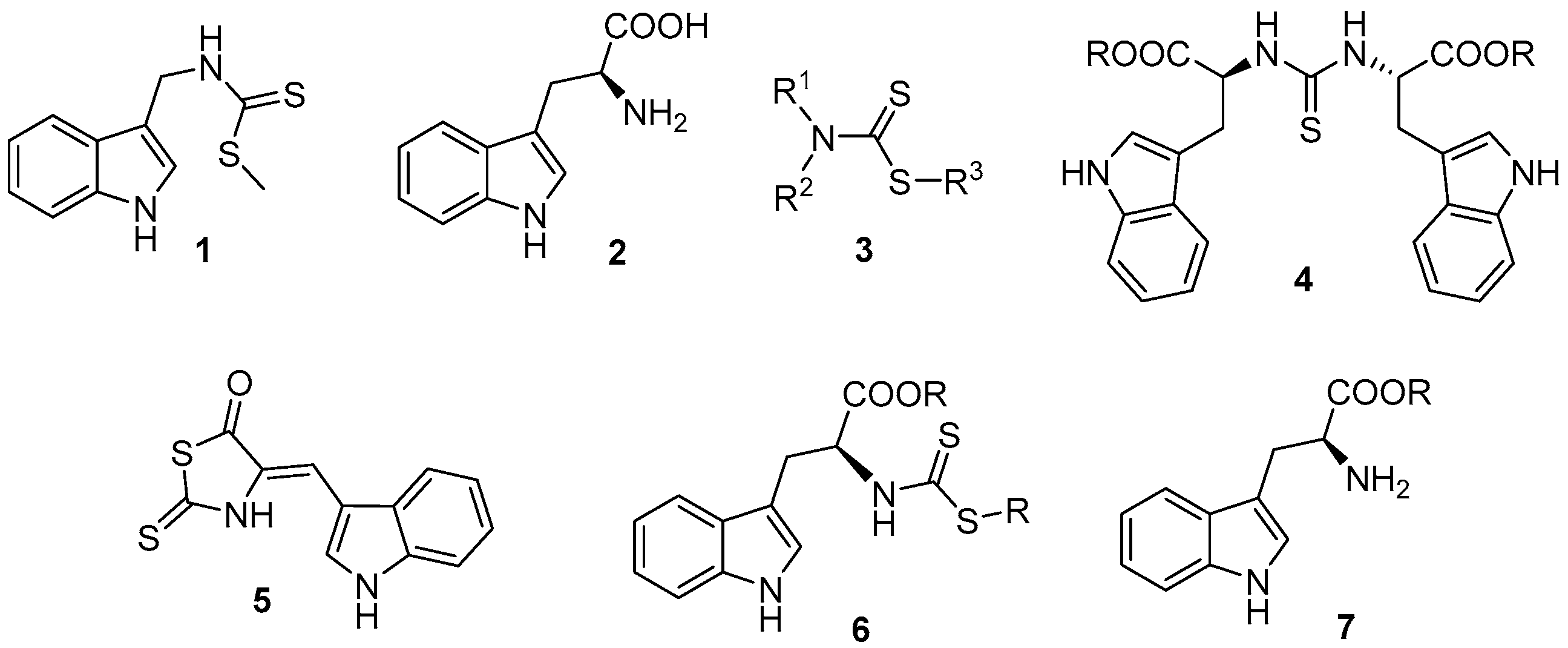
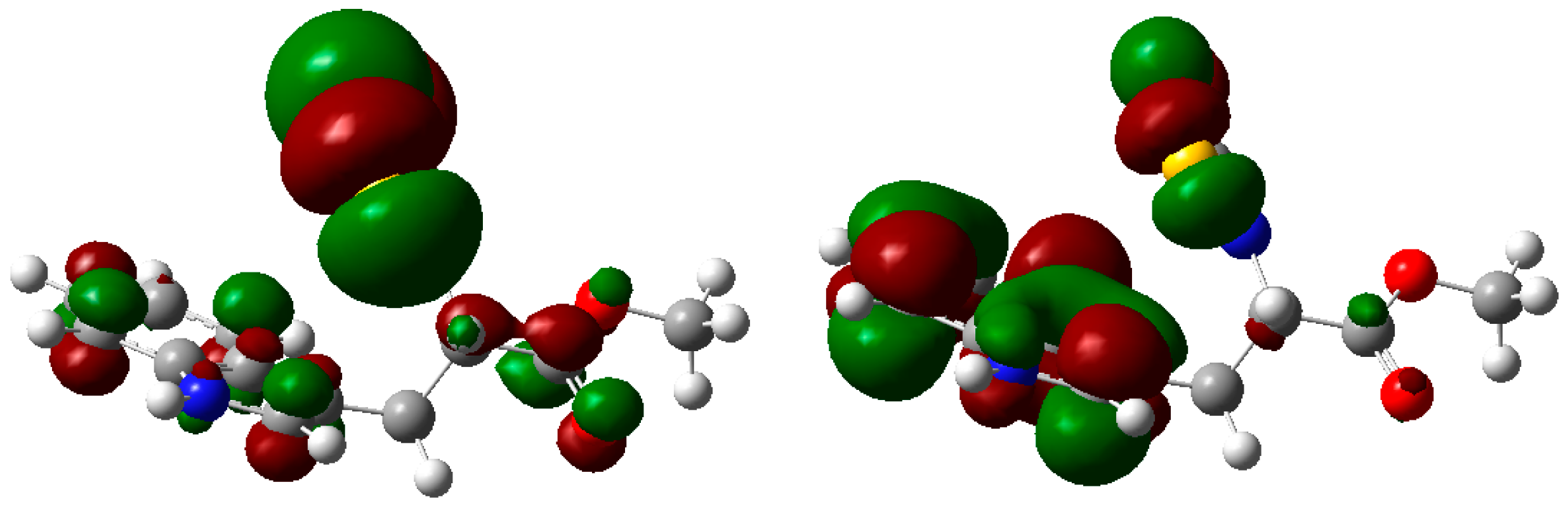
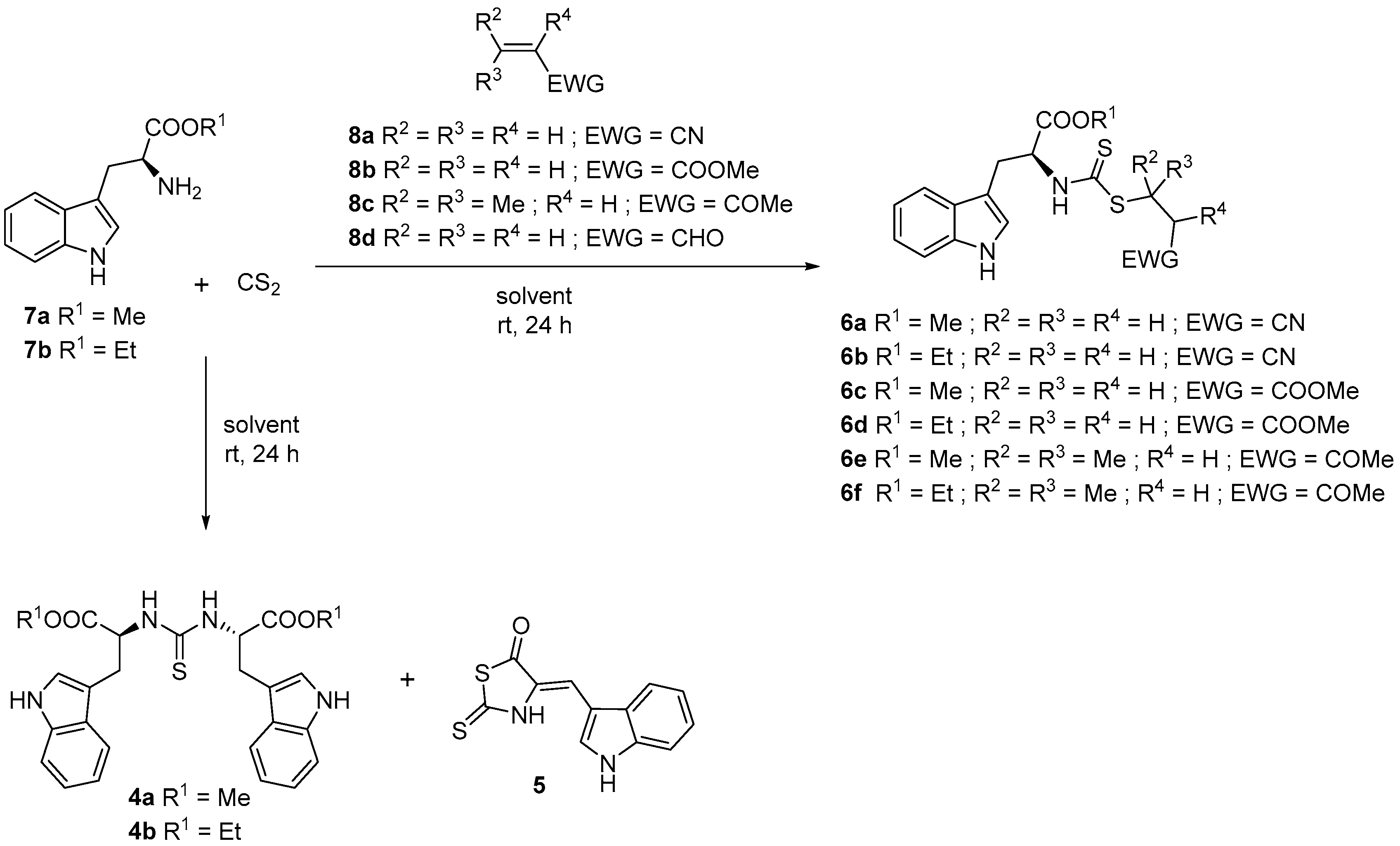
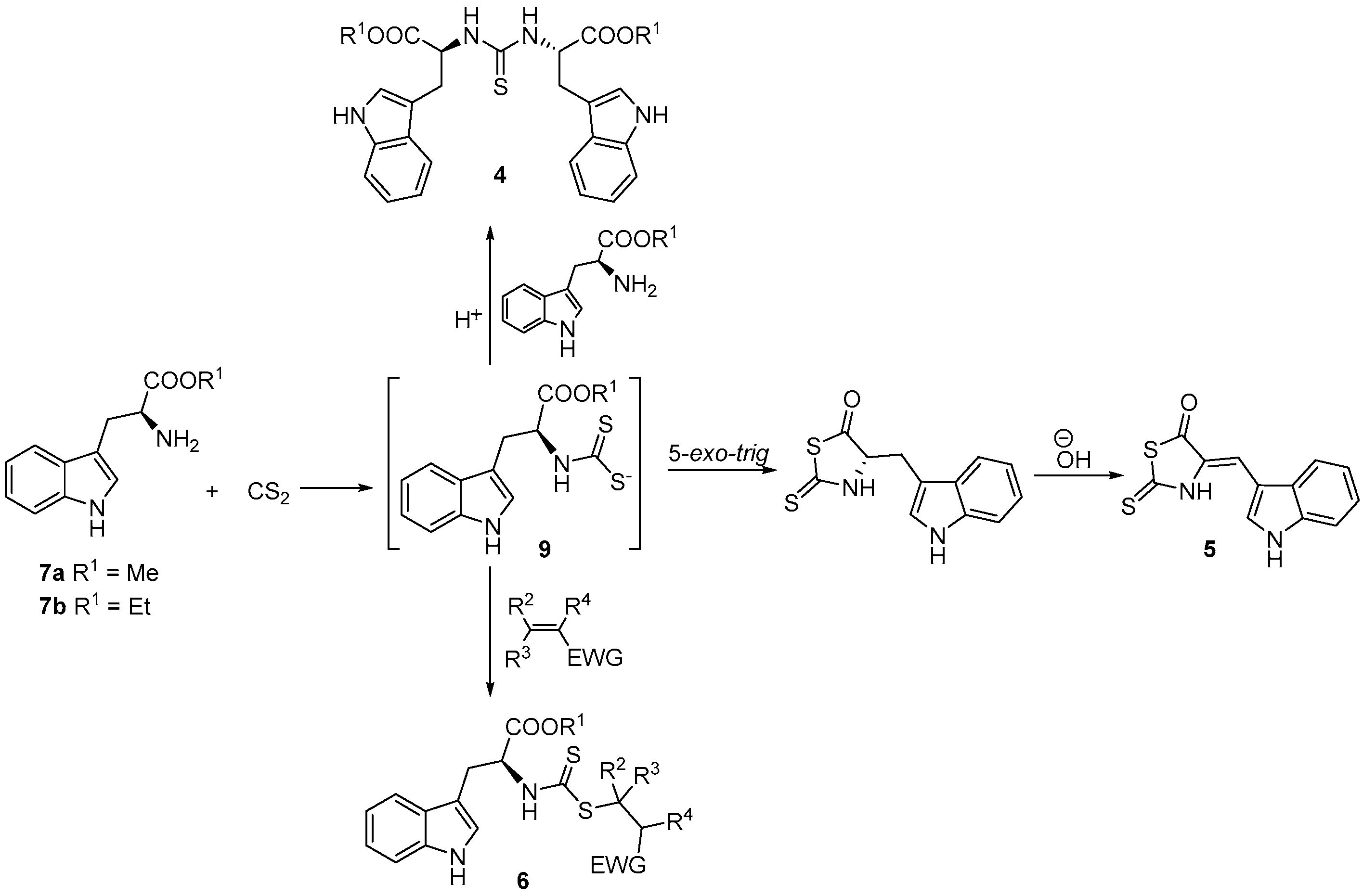
2. Results and Discussion
3. Materials and Methods
3.1. General Information
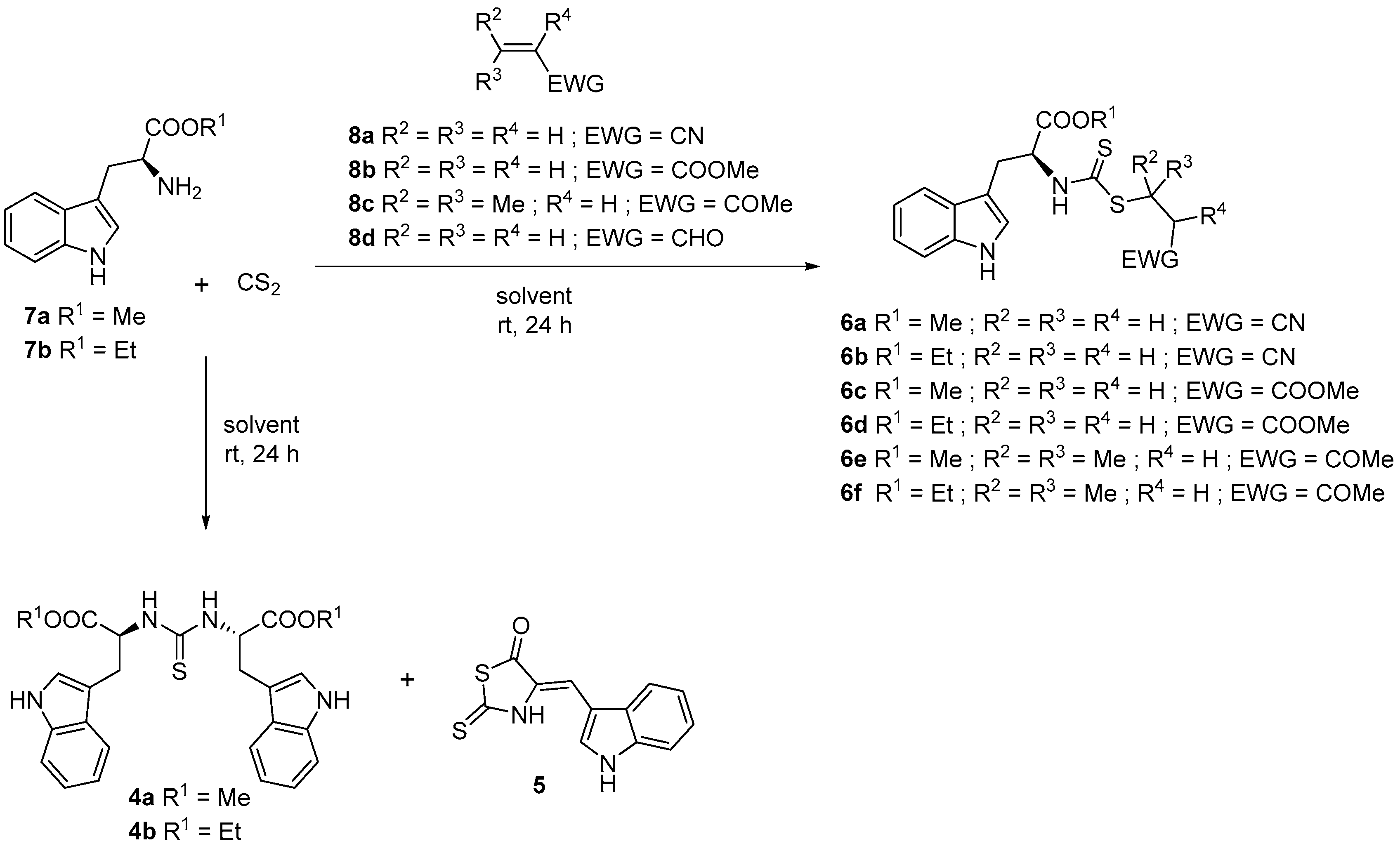
3.2. General Procedure for the Synthesis of Alkyl Esters (7a–d)
3.3. General Procedure for the Synthesis of N,N′-Dialkylthioureas (4a–b)
3.4. Synthesis of 4-[(1H-Indol-3-yl)methylene]-2-sulfanylidene-1,3-thiazolidin-5-one (5)
3.5. General Procedure for the Synthesis of Alkyl (2S)-3-(1H-indol-3-yl)-2-{[(alkylsulfanyl)carbonothioyl]amino}propanoate (6a–f)
3.6. Antifungal Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pedras, M.S.C.; Minic, Z. The phytoalexins brassilexin and camalexin inhibit cyclobrassinin hydrolase, a unique enzyme from the fungal pathogen Alternaria brassicicola. Bioorg. Med. Chem. 2014, 22, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Minic, Z.; Hossain, S. Discovery of inhibitors and substrates of brassinin hydrolase: Probing selectivity with dithiocarbamate bioisosteres. Bioorg. Med. Chem. 2012, 20, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Minic, Z.; Sarma-Mamillapalle, V.K.; Suchy, M. Discovery of inhibitors of brassinin oxidase based on the brassilexin and wasalexin scaffolds. Bioorg. Med. Chem. 2010, 18, 2456–2453. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Zoran, M.; Sarma-Mamillapalle, V.K. Substrate specificity and inhibition of brassinin hydrolases, detoxifying enzymes from the plant pathogens Leptosphaeria maculans and Alternaria brassicicola. FEBS J. 2009, 276, 7412–7428. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Jha, M.; Minic, Z.; Okeola, O.G. Isosteric probes provide structural requirements essential for detoxification of the phytoalexin brassinin by the fungal pathogen Leptosphaeria maculans. Bioorg. Med. Chem. 2007, 15, 6054–6061. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Gadagi, R.S.; Jha, M.; Sarma-Mamillapalle, V.K. Detoxification of the phytoalexin brassinin by isolates of Leptosphaeria maculans pathogenic on brown mustard involves an inducible hydrolase. Phytochemistry 2007, 68, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.C.; Hossain, M. Design, synthesis, and evaluation of potential inhibitors of brassinin glucosyl transferase, a phytoalexin detoxifying enzyme from Sclerotinia sclerotiorum. Bioorg. Med. Chem. 2007, 15, 5981–5996. [Google Scholar] [CrossRef] [PubMed]
- Chripkova, M.; Drutovic, D.; Pilatova, M.; Mikes, J.; Budovska, M.; Vaskova, J.; Broggini, M.; Mirossay, M.; Mojzis, J. Brassinin and its derivatives as potential anticancer agents. Toxicol. In Vitro 2014, 28, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Matos, A.; Abrantes, L.; Viana, A.; Jin, G. Antibody oriented immobilization on gold using the reaction between carbon disulfide and amine groups and its application in immunosensing. Langmuir 2012, 28, 17718–17725. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Shen, Y. Transition-metal-catalyzed reactions of propargylamine with carbon dioxide and carbon disulfide. J. Org. Chem. 2002, 67, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.; Dolman, S. Isothiocyanates from tosyl chloride mediated decomposition of in situ generated dithiocarbamic acid salts. J. Org. Chem. 2007, 72, 3969–3971. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-L.; Liu, B.-L. Reaction of carbon disulfide and o-phenylenediamine catalyzed by tertiary amines in a homogeneous solution. Ind. Eng. Chem. Res. 1995, 34, 3688–3695. [Google Scholar] [CrossRef]
- Bardajee, G.R.; Sadraei, S.; Taimoory, S.; Abtin, E. One-pot synthesis of dithiocarbamates under solvent-free conditions in the presence of KF/Al2O3. Asian J. Biochem. Pharm. Res. 2011, 1, 178–184. [Google Scholar]
- Azizi, N.; Aryanasab, F.; Saidi, M. Straightforward and highly efficient catalyst-free one-pot synthesis of dithiocarbamates under solvent-free conditions. Org. Lett. 2006, 8, 5275–5277. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sha, Y. A convenient synthesis of amino acid methyl esters. Molecules 2008, 13, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Software Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Sample Availability: Samples of the compounds are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 a (mM) ± SD |
|---|---|
| 4a | 0.49 ± 0.09 |
| 4b | 0.76 ± 0.23 |
| 4c | 1.5 ± 0.5 |
| 4d | 1.1 ± 0.2 |
| 5 | 1.8 ± 0.9 |
| 6a | 2.5 ± 0.7 |
| 6b | 0.16 ± 0.05 |
| 6c | 2.1 ± 0.8 |
| 6d | 1.1 ± 0.7 |
| 6e | 0.59 ± 0.21 |
| 6f | 1.7 ± 0.5 |
| 7a | 3.1 ± 1.7 |
| 7b | >50 |
| 7c | 7.9 ± 3.8 |
| 7d | 0.76 ± 0.14 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quiroga, D.; Becerra, L.D.; Sadat-Bernal, J.; Vargas, N.; Coy-Barrera, E. Synthesis and Antifungal Activity against Fusarium oxysporum of Some Brassinin Analogs Derived from l-tryptophan: A DFT/B3LYP Study on the Reaction Mechanism. Molecules 2016, 21, 1349. https://doi.org/10.3390/molecules21101349
Quiroga D, Becerra LD, Sadat-Bernal J, Vargas N, Coy-Barrera E. Synthesis and Antifungal Activity against Fusarium oxysporum of Some Brassinin Analogs Derived from l-tryptophan: A DFT/B3LYP Study on the Reaction Mechanism. Molecules. 2016; 21(10):1349. https://doi.org/10.3390/molecules21101349
Chicago/Turabian StyleQuiroga, Diego, Lili Dahiana Becerra, John Sadat-Bernal, Nathalia Vargas, and Ericsson Coy-Barrera. 2016. "Synthesis and Antifungal Activity against Fusarium oxysporum of Some Brassinin Analogs Derived from l-tryptophan: A DFT/B3LYP Study on the Reaction Mechanism" Molecules 21, no. 10: 1349. https://doi.org/10.3390/molecules21101349
APA StyleQuiroga, D., Becerra, L. D., Sadat-Bernal, J., Vargas, N., & Coy-Barrera, E. (2016). Synthesis and Antifungal Activity against Fusarium oxysporum of Some Brassinin Analogs Derived from l-tryptophan: A DFT/B3LYP Study on the Reaction Mechanism. Molecules, 21(10), 1349. https://doi.org/10.3390/molecules21101349