Inhibition of Uterine Contractility by Thalidomide Analogs via Phosphodiesterase-4 Inhibition and Calcium Entry Blockade

 ,
,
Abstract
:1. Introduction
2. Results
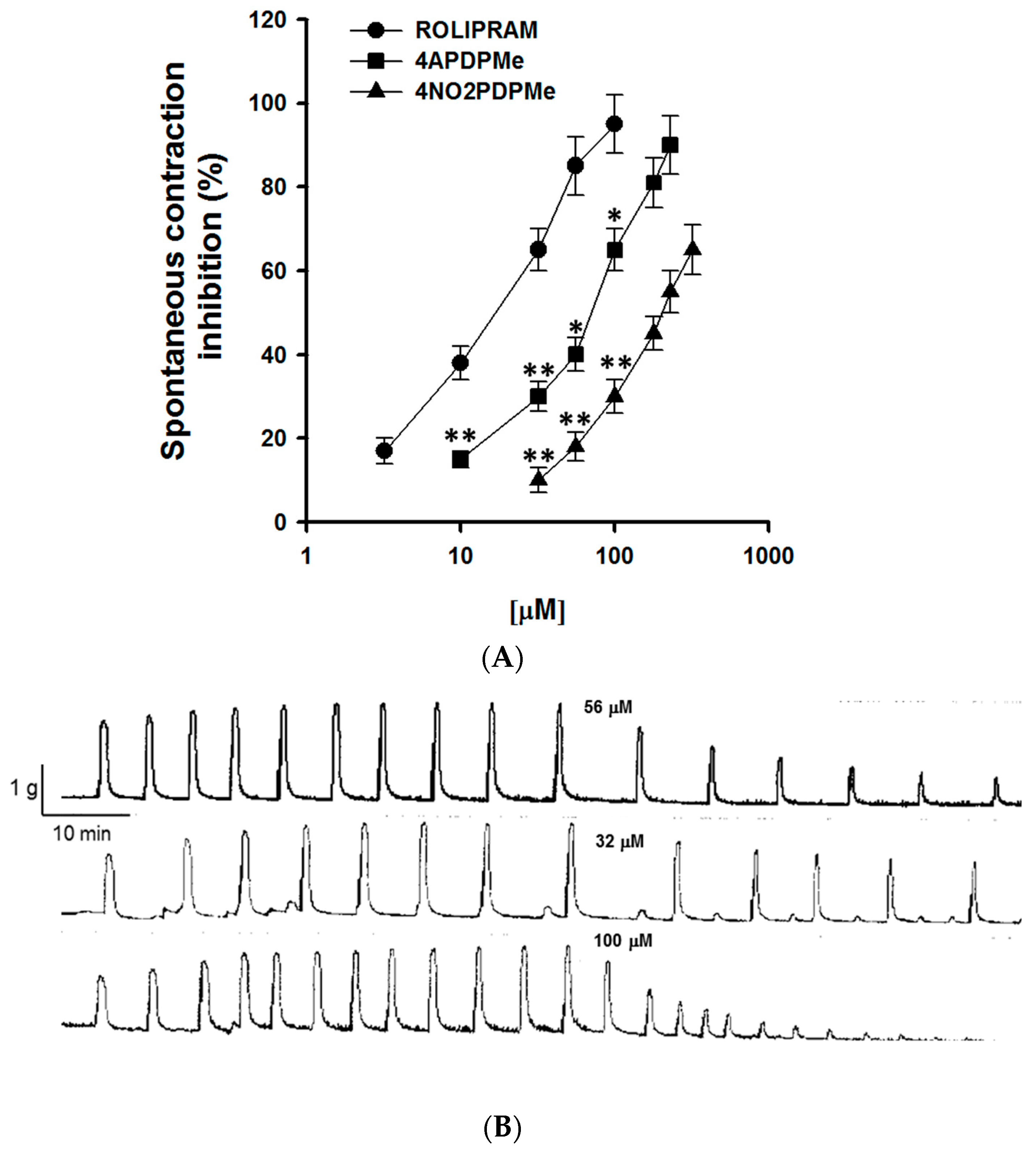
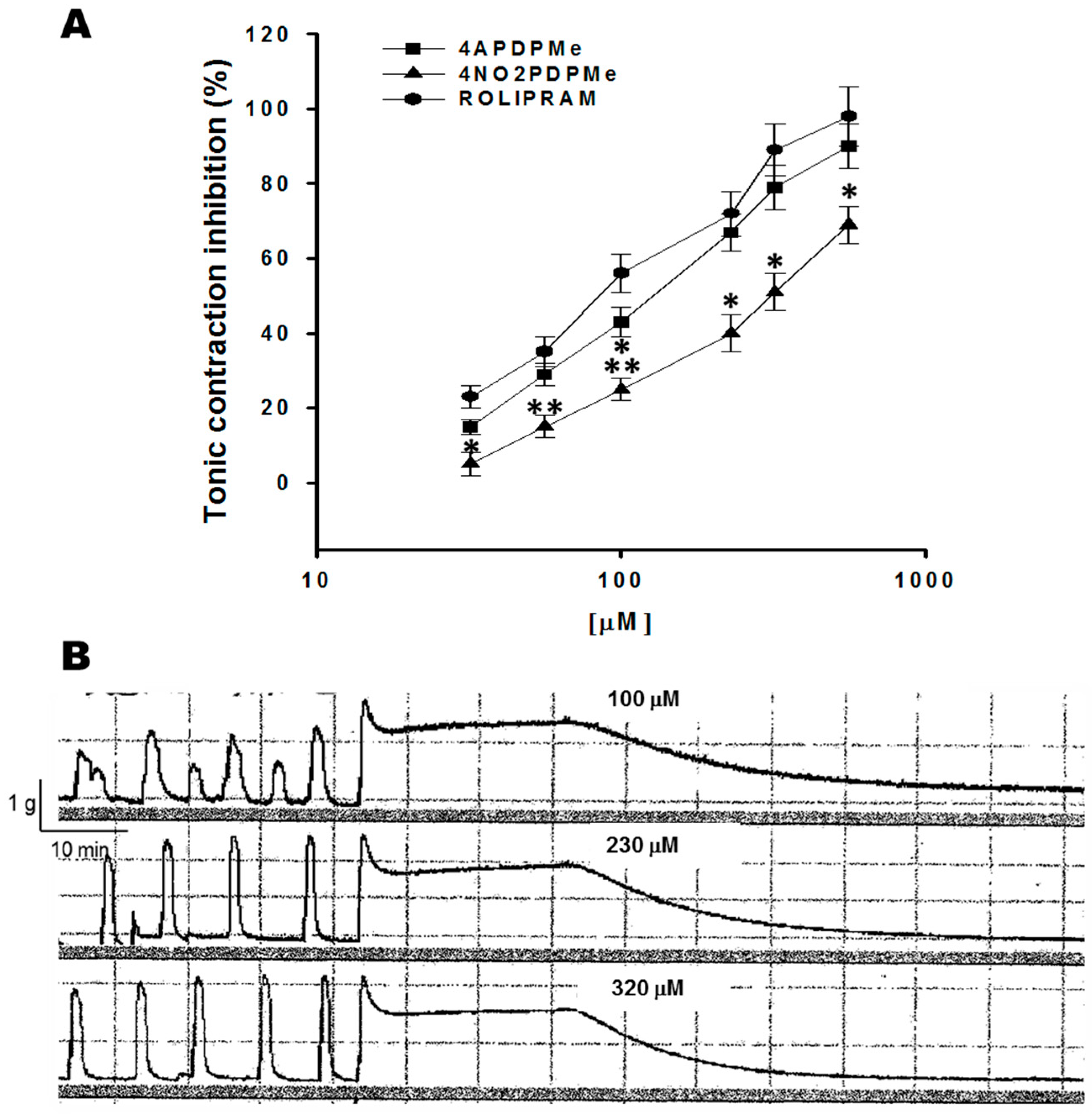
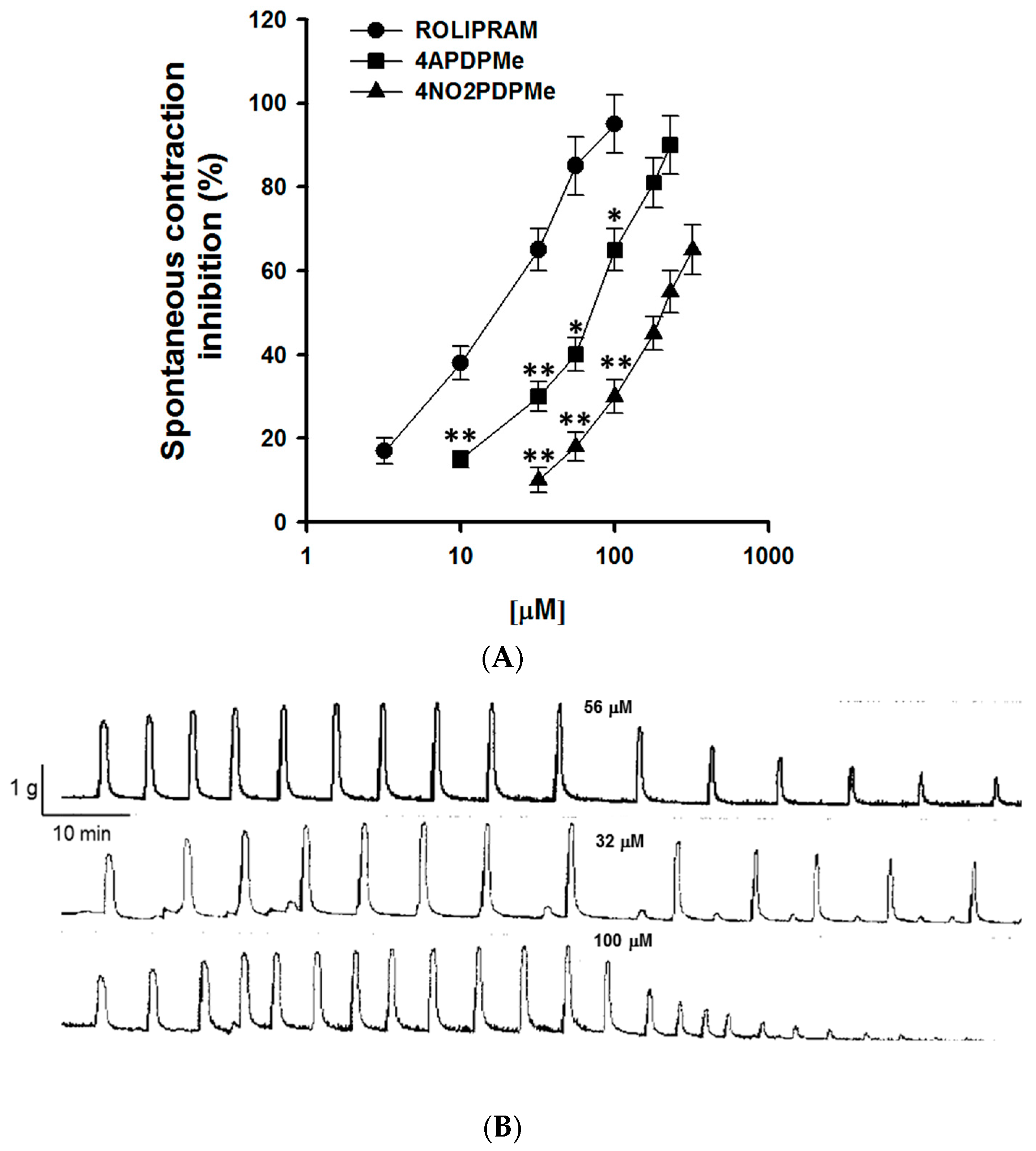
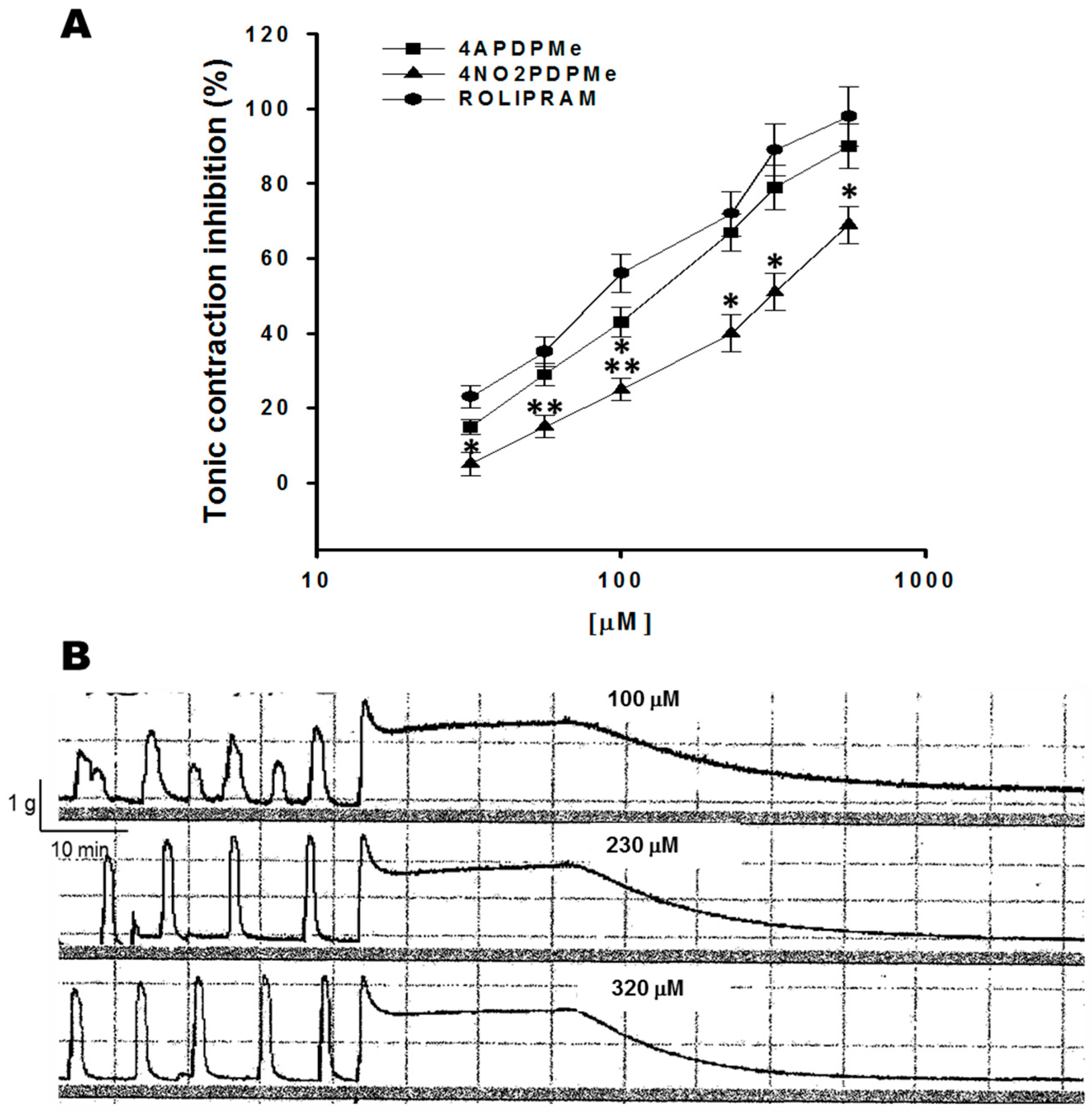
2.1. Thalidomide Analogs and Rolipram Inhibit Spontaneous and Tonic Contractions
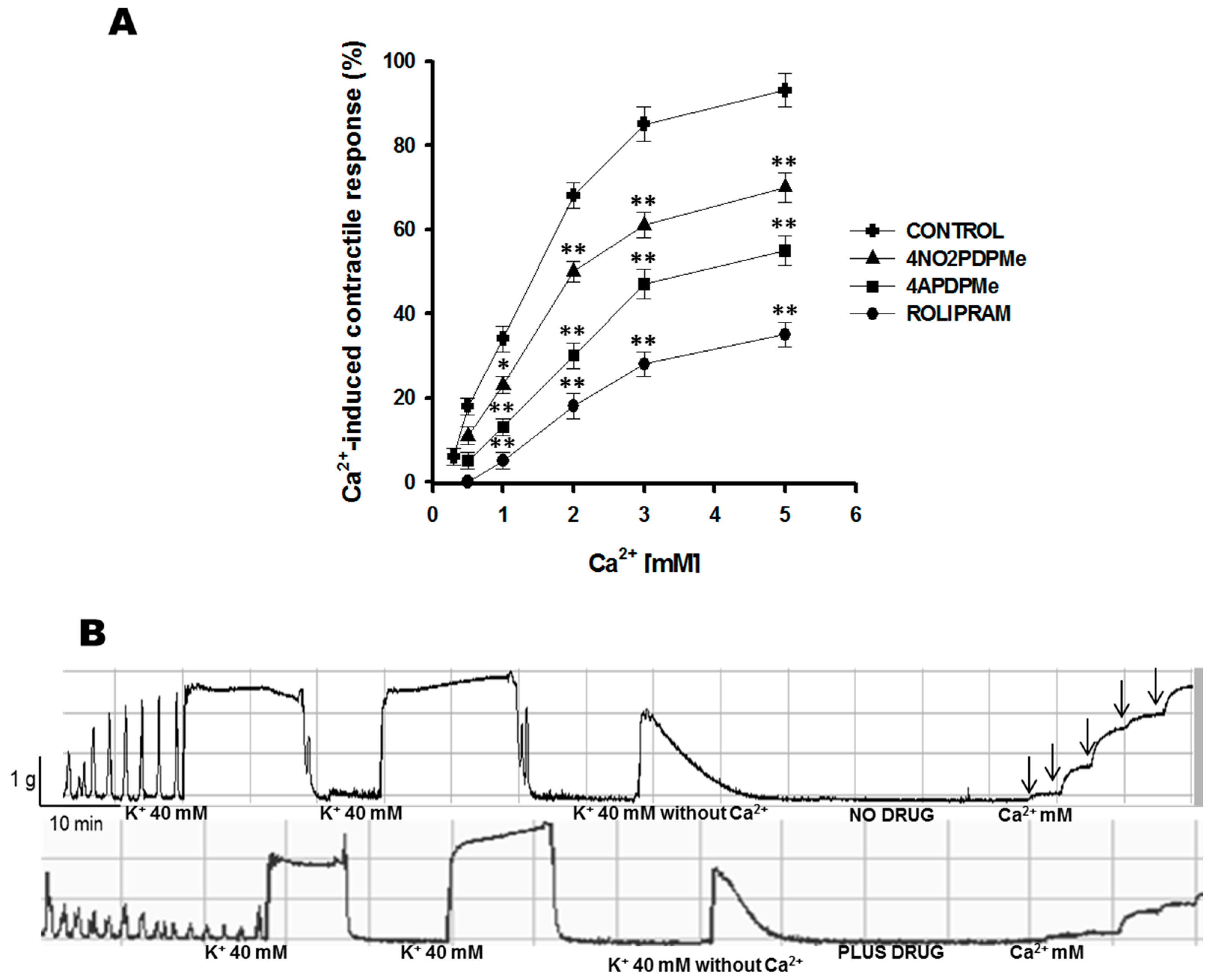
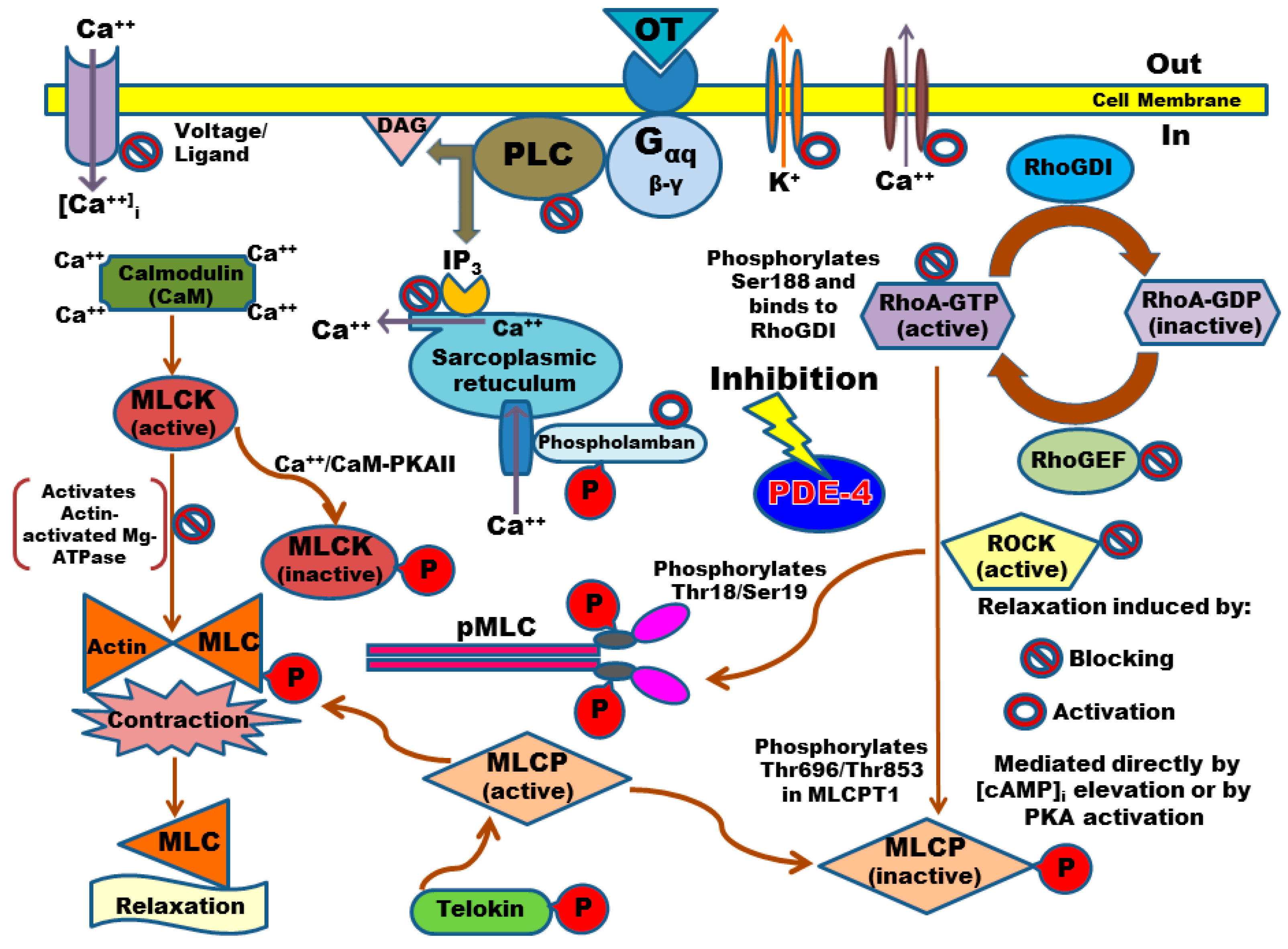
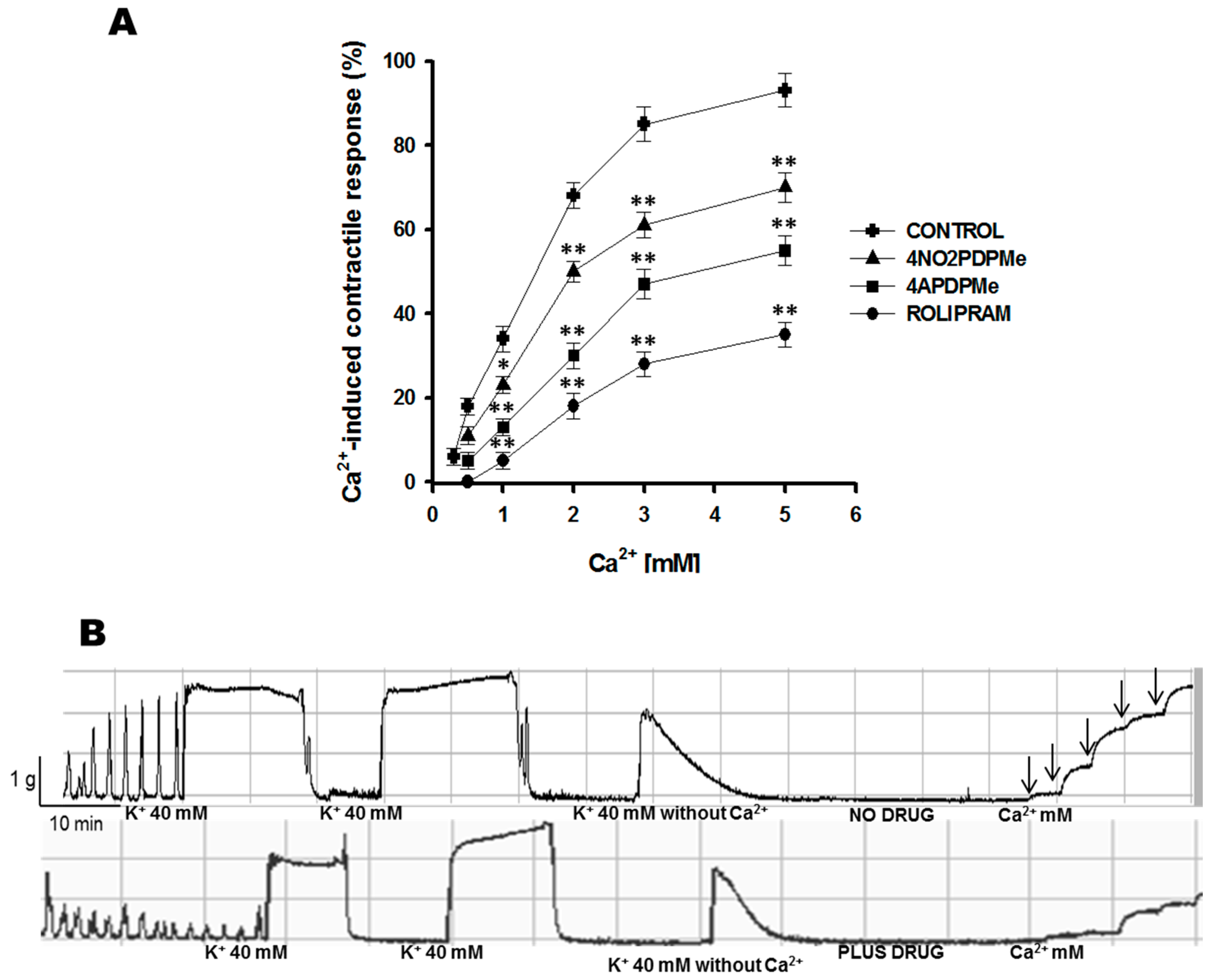
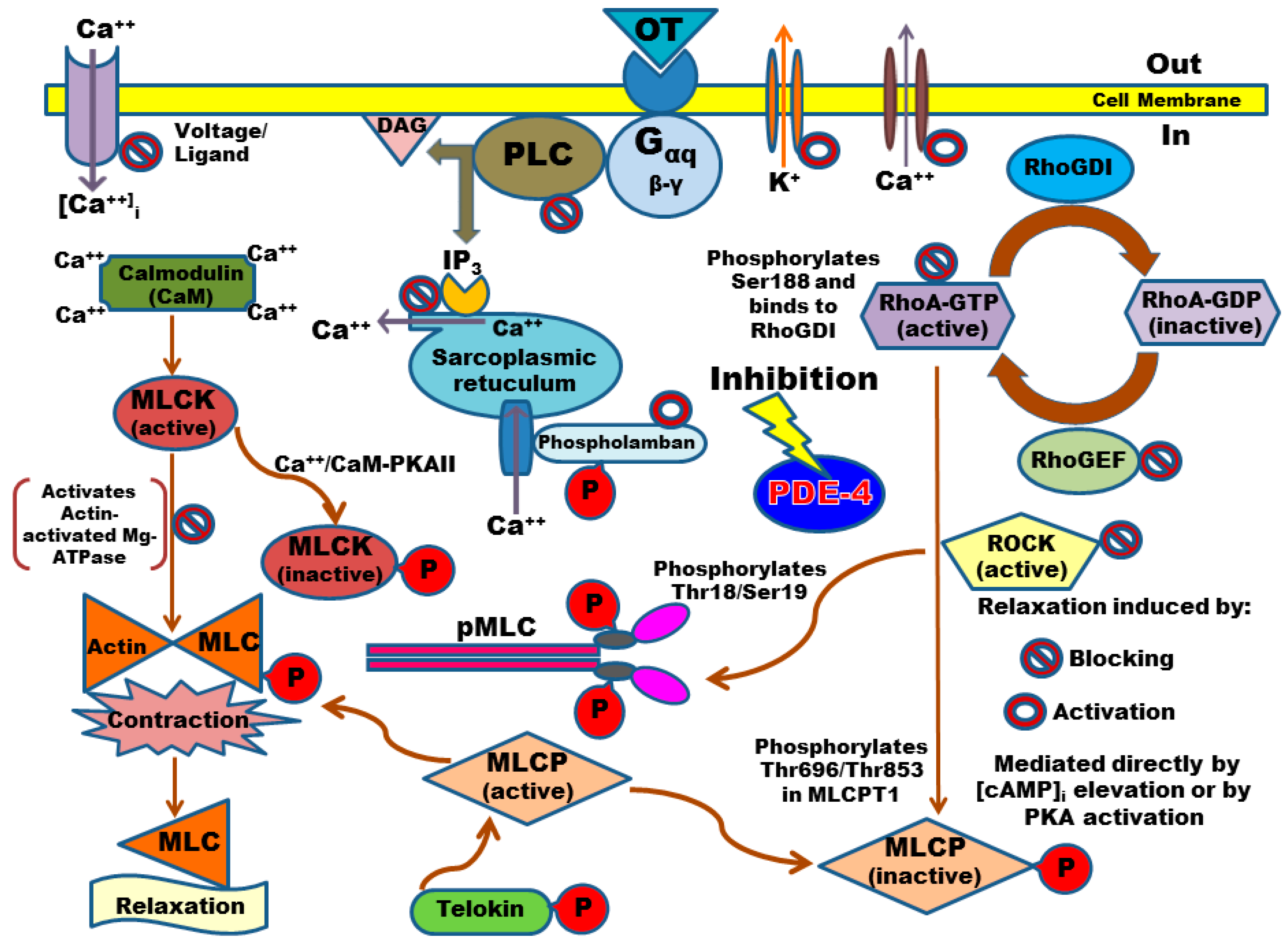
2.2. Calcium Entry Blockade as a Possible Uterus–Relaxant Mechanism of Thalidomide Analogs and Rolipram
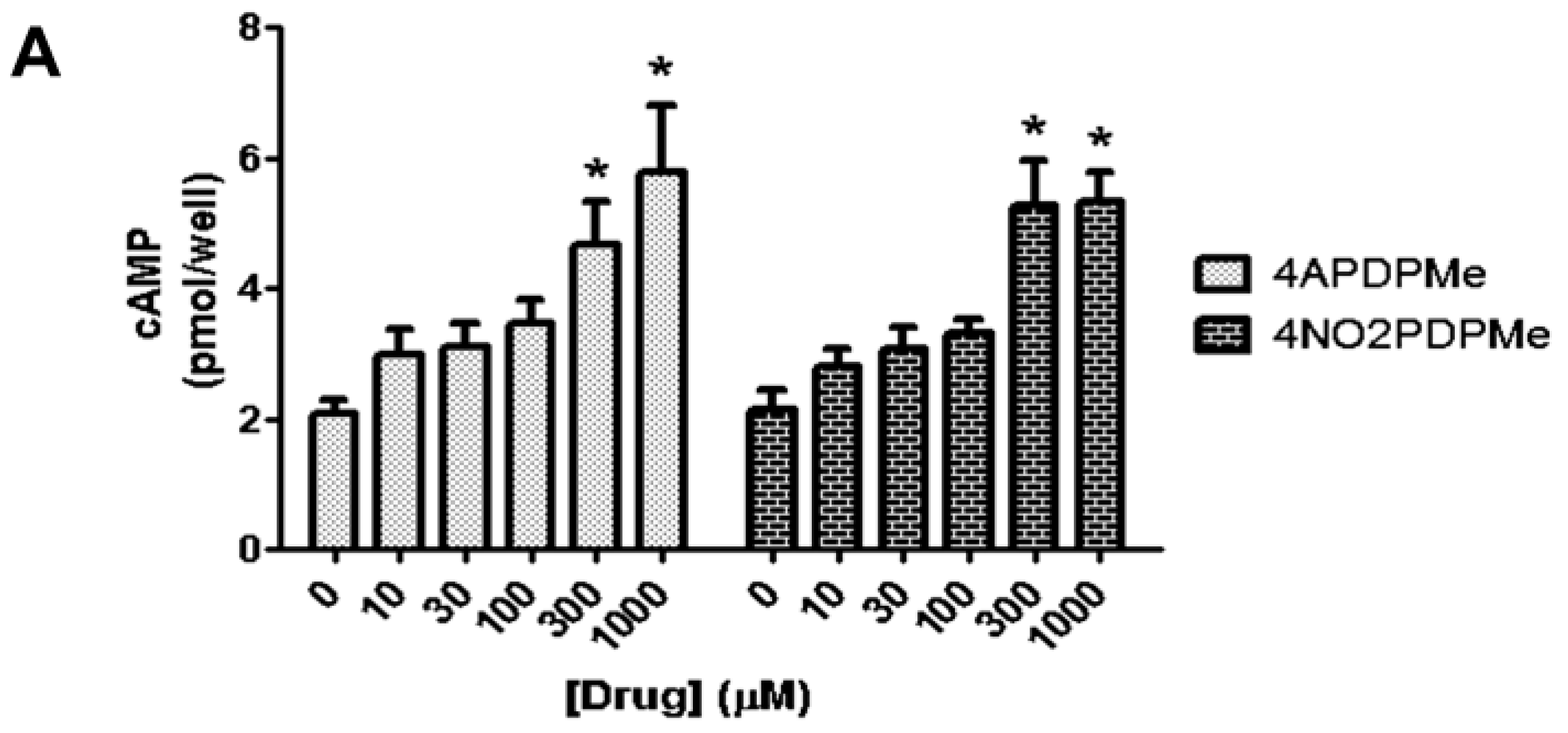
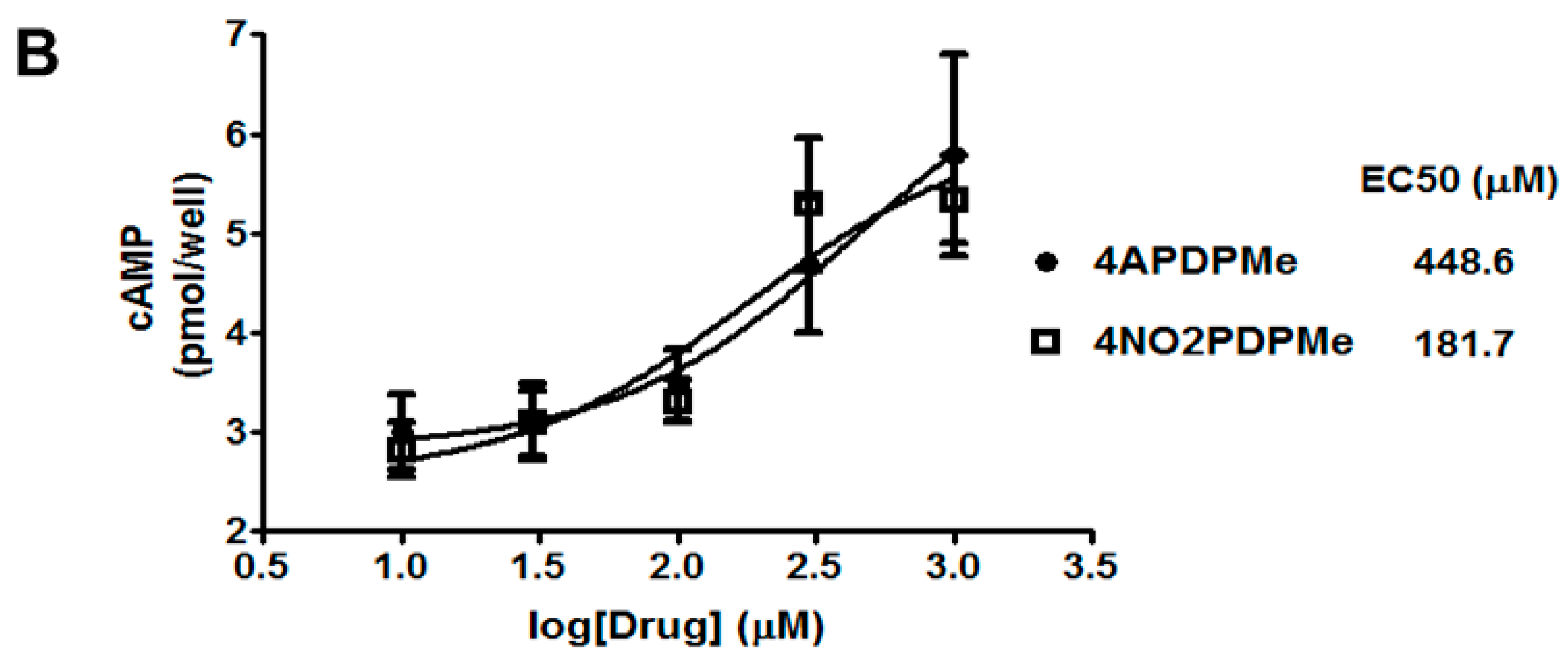
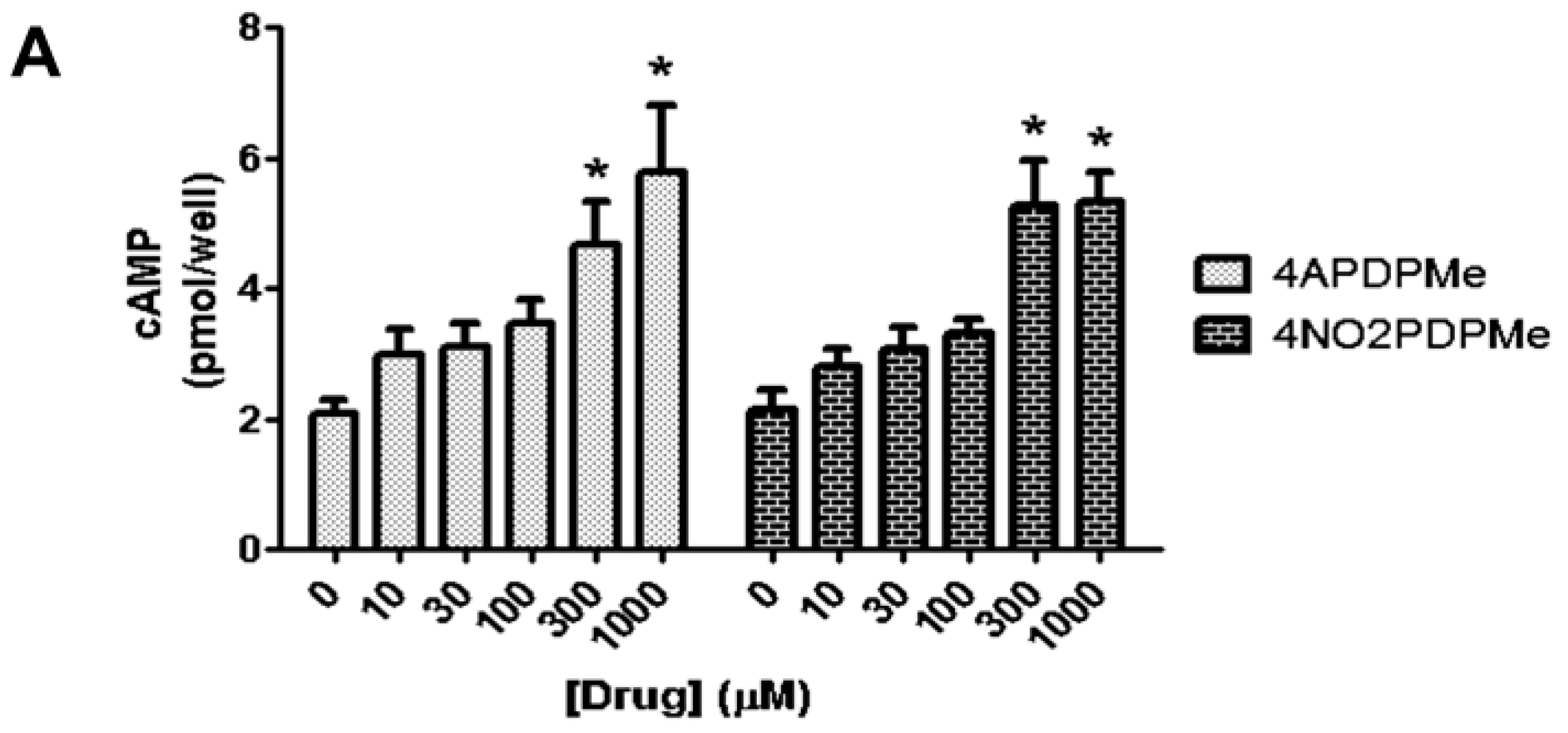
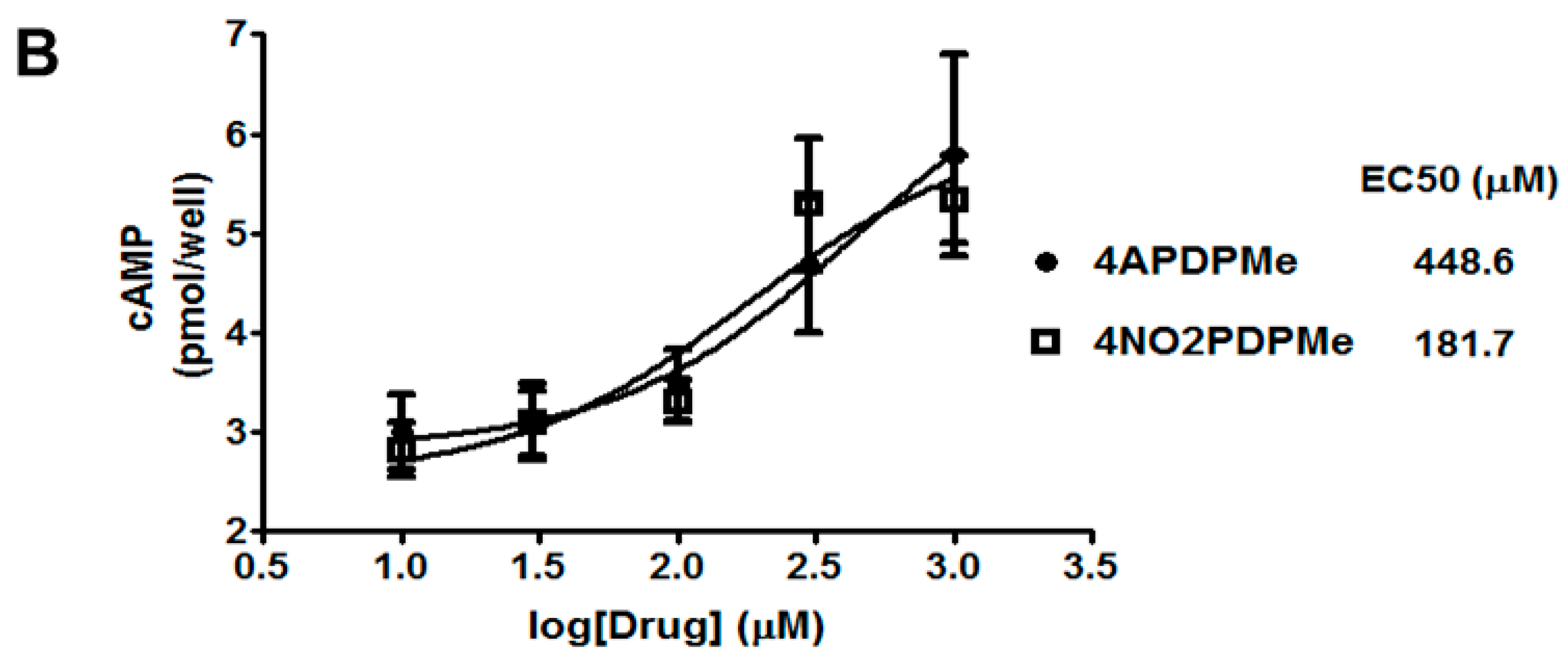
2.3. Thalidomide Analogs Increase cAMP Levels in HeLa Cells
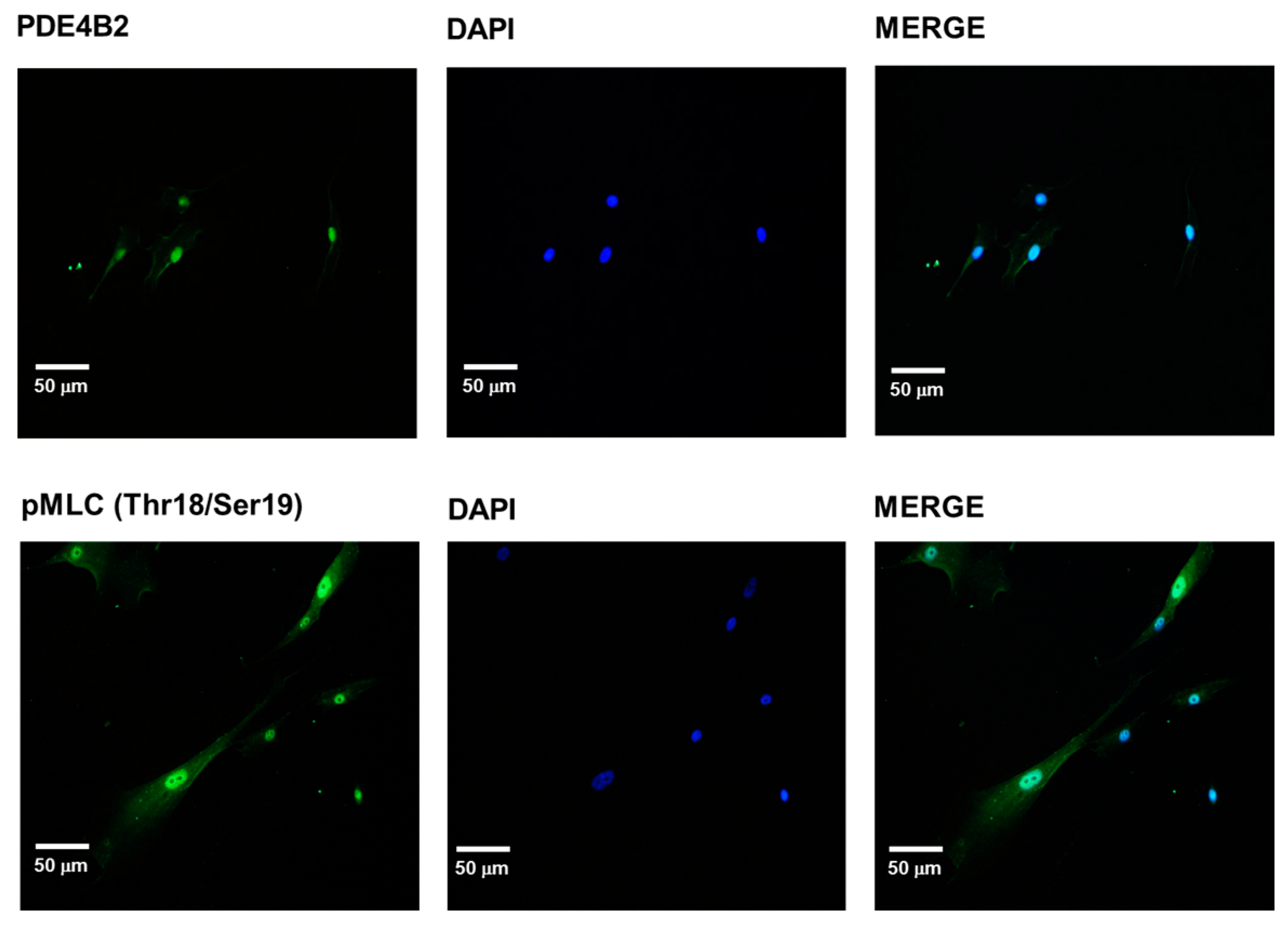
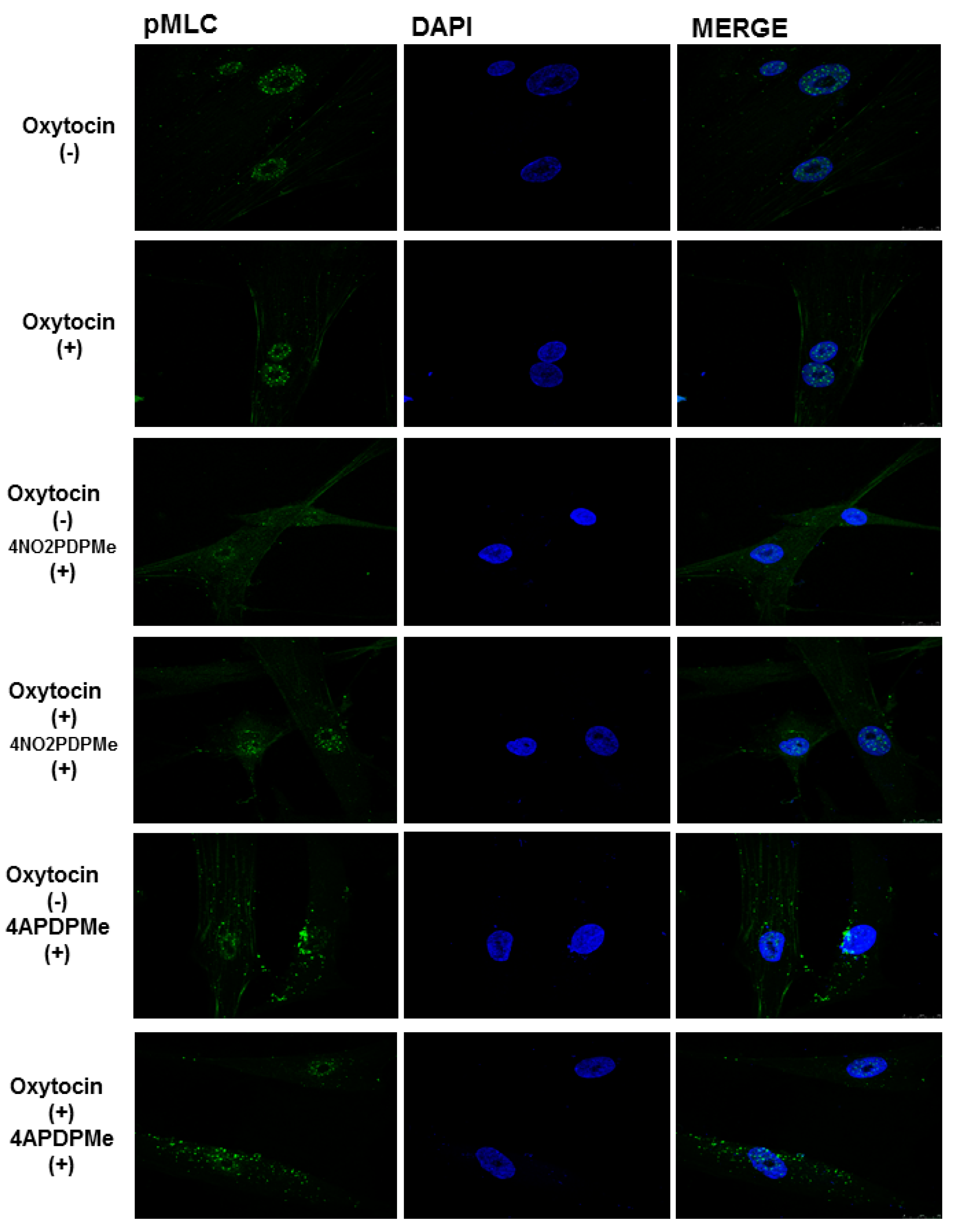
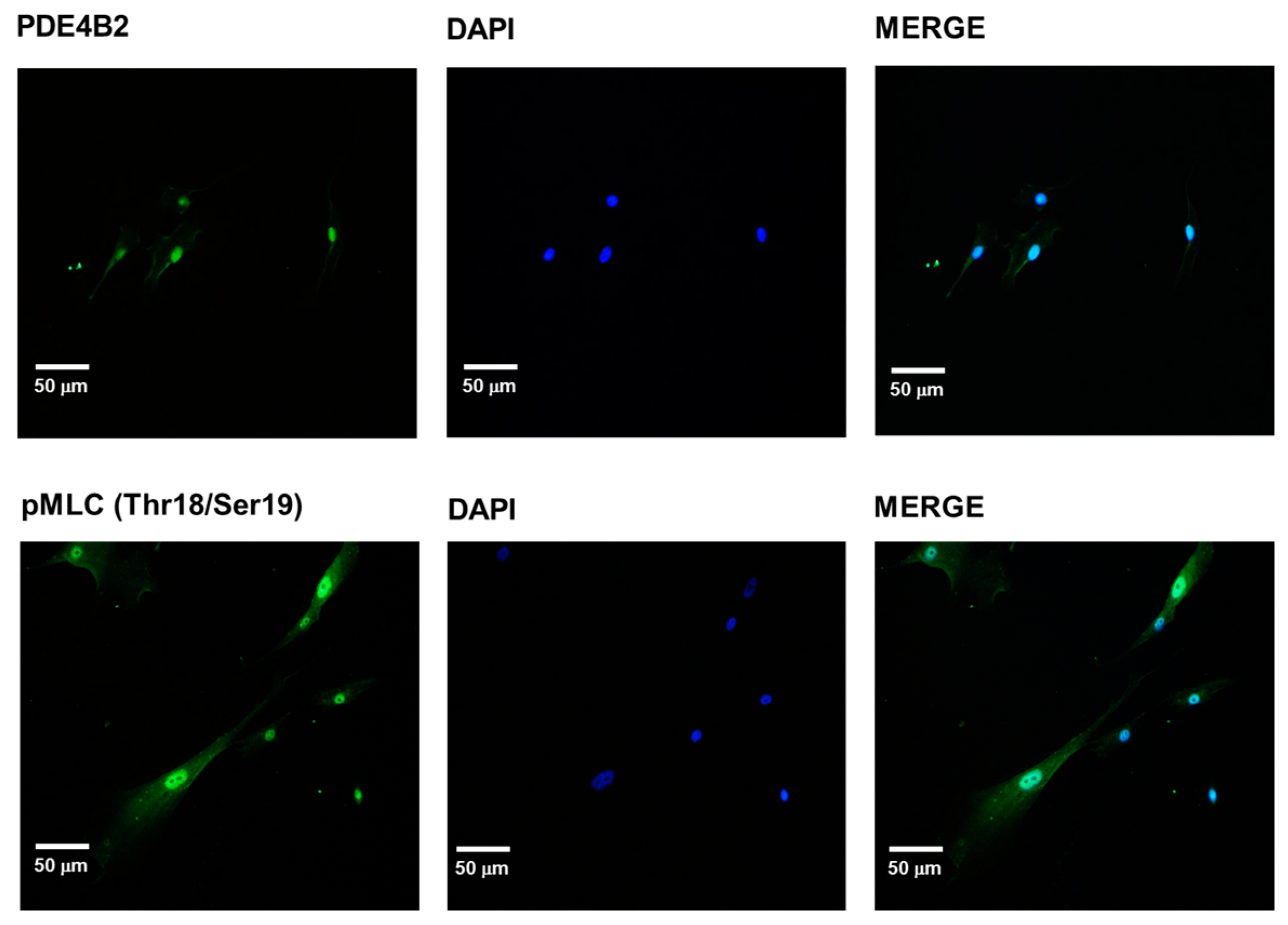
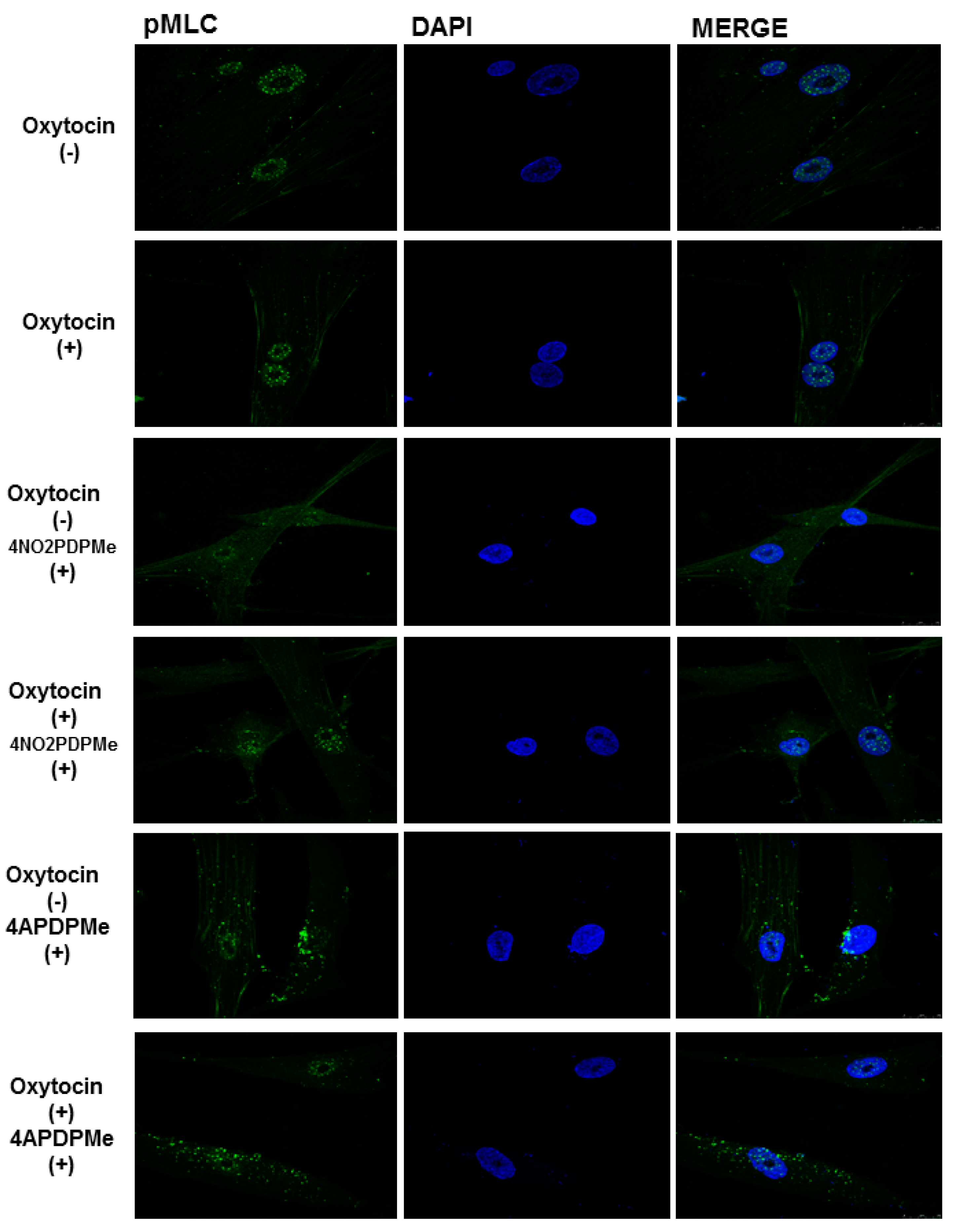
2.4. PDE-4B2 and pMLC Expression in UtSMC, and the Effects of Thalidomide Analogs on OT–Induced pMLC Production
3. Discussion
4. Materials and Methods
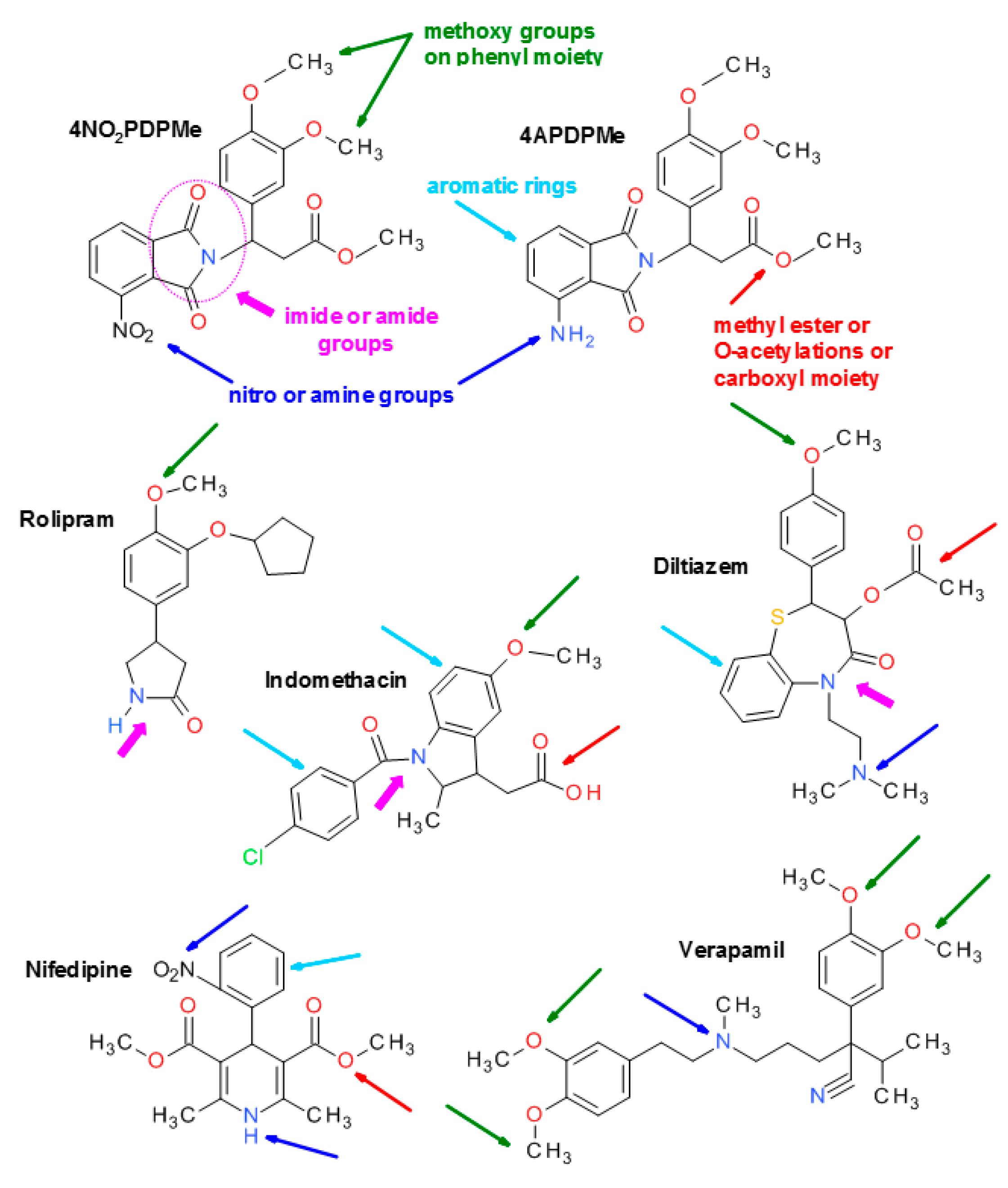
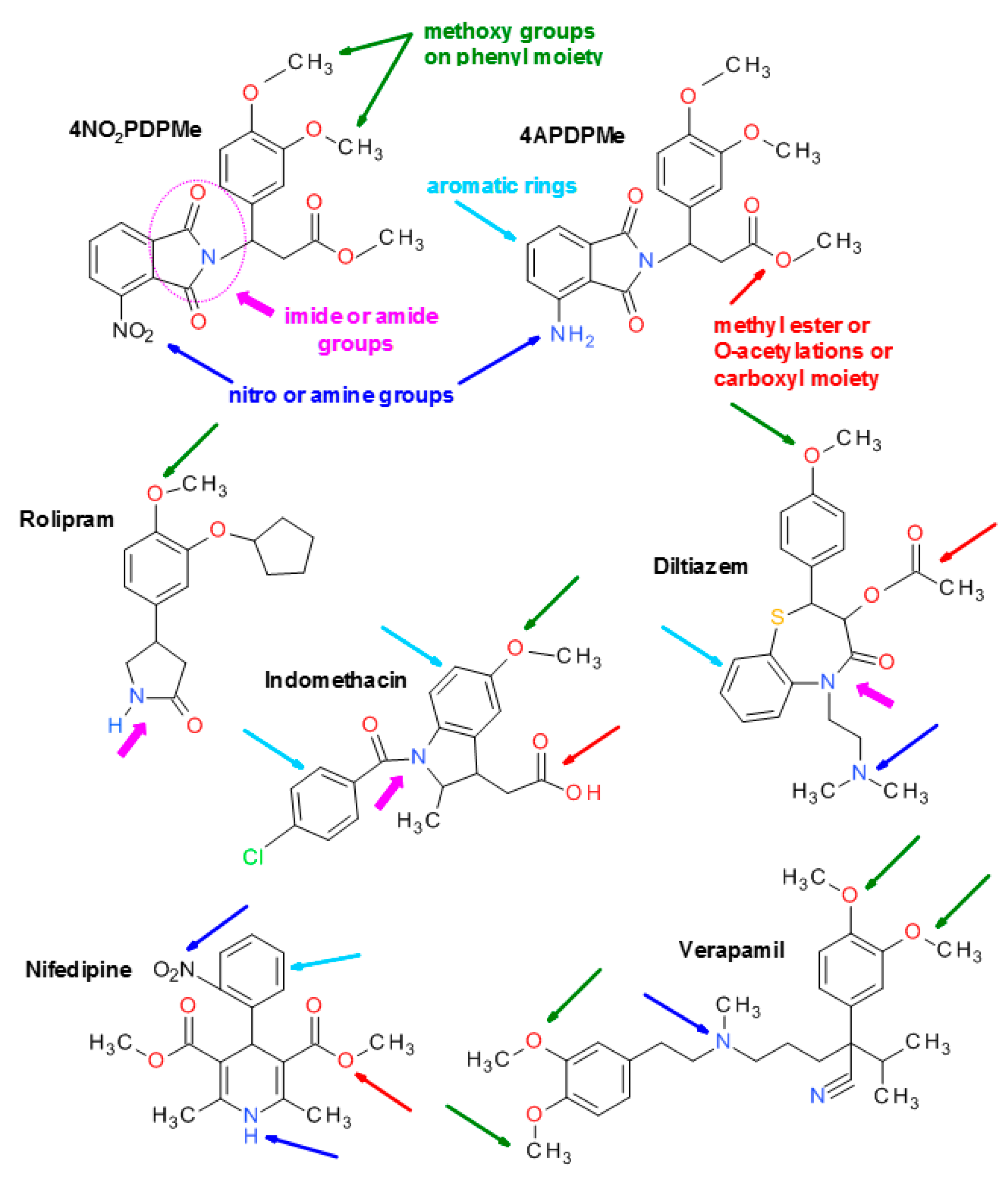
4.1. Thalidomide Analogs
4.2. Chemicals and Solutions
4.3. Tissue Collection
4.4. In Vitro Functional Studies with Human Myometrial Strips
4.4.1. Inhibitory Effect of 4APDPMe, 4NO2PDPMe, and Rolipram on Spontaneous Contraction
4.4.2. Inhibitory Effect of 4APDPMe, 4NO2PDPMe, and Rolipram on Tonic Contraction Induced by High Levels of K+
4.4.3. Calcium Entry Blockade by 4APDPMe, 4NO2PDPMe, and Rolipram
4.5. Measurement of cAMP Accumulation in Intact Cells
4.6. Immunocytochemistry
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lopez Bernal, A. Overview. Preterm labour: Mechanisms and management. BMC Pregnancy Childbirth 2007, 7 (Suppl. 1), S2. [Google Scholar] [CrossRef] [PubMed]
- Reich, E.S. Pre-term births on the rise. Nature 2012, 485, 20. [Google Scholar] [CrossRef] [PubMed]
- Norwitz, E.R.; Robinson, J.N. A systematic approach to the management of preterm labor. Semin. Perinatol. 2001, 25, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Vause, S.; Johnston, T. Management of preterm labour. Arch. Dis. Child. Fetal. Neonatal. Ed. 2000, 83, F79–F85. [Google Scholar] [CrossRef] [PubMed]
- Bastek, J.A.; Gomez, L.M.; Elovitz, M.A. The role of inflammation and infection in preterm birth. Clin. Perinatol. 2011, 38, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Peltier, M.R. Immunology of term and preterm labor. Reprod. Biol. Endocrinol. 2003, 1, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Bernal, A. The regulation of uterine relaxation. Semin. Cell. Dev. Biol. 2007, 18, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.A.; Bernal, A.L. The regulation of myosin phosphatase in pregnant human myometrium. Biochem. Soc. Trans. 2012, 40, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Challis, J.R.; Sloboda, D.M.; Alfaidy, N.; Lye, S.J.; Gibb, W.; Patel, F.A.; Whittle, W.L.; Newnham, J.P. Prostaglandins and mechanisms of preterm birth. Reproduction 2002, 124, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Price, S.A.; Bernal, A.L. Uterine quiescence: The role of cyclic amp. Exp. Physiol. 2001, 86, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Lopez Bernal, A. Cyclic amp signalling pathways in the regulation of uterine relaxation. BMC Pregnancy Childbirth 2007, 7 (Suppl. 1), S10. [Google Scholar] [CrossRef] [PubMed]
- Mehats, C.; Oger, S.; Leroy, M.J. Cyclic nucleotide phosphodiesterase-4 inhibitors: A promising therapeutic approach to premature birth? Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 117 (Suppl. 1), S15–S17. [Google Scholar] [CrossRef] [PubMed]
- Mehats, C.; Schmitz, T.; Oger, S.; Herve, R.; Cabrol, D.; Leroy, M.J. PDE4 as a target in preterm labour. BMC Pregnancy Childbirth 2007, 7 (Suppl. 1), S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, T.; Souil, E.; Herve, R.; Nicco, C.; Batteux, F.; Germain, G.; Cabrol, D.; Evain-Brion, D.; Leroy, M.J.; Mehats, C. PDE4 inhibition prevents preterm delivery induced by an intrauterine inflammation. J. Immunol. 2007, 178, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Vasta, V.; Beavo, J. Functions and pharmacological inhibitors of cyclic nucleotide phosphodiesterases. Celltransmissions 2004, 20, 1–8. [Google Scholar]
- Houslay, M.D.; Schafer, P.; Zhang, K.Y. Keynote review: Phosphodiesterase-4 as a therapeutic target. Drug. Discov. Today 2005, 10, 1503–1519. [Google Scholar] [CrossRef]
- Hashimoto, Y. Thalidomide as a multi-template for development of biologically active compounds. Arch. Pharm. 2008, 341, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Teo, S.K. Properties of thalidomide and its analogues: Implications for anticancer therapy. AAPS J. 2005, 7, E14–E19. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Martínez, E. Thalidomide and its analogs: A potential immunomodulatory alternative for treating liver diseases and cirrhosis. In Liver Cirrhosis: Causes, Diagnosis and Treatment; Michelli, M.L., Ed.; Nova Science Publishers: New York, NY, USA, 2011. [Google Scholar]
- Muller, G.W.; Shire, M.G.; Wong, L.M.; Corral, L.G.; Patterson, R.T.; Chen, Y.; Stirling, D.I. Thalidomide analogs and PDE4 inhibition. Bioorg. Med. Chem. Lett. 1998, 8, 2669–2674. [Google Scholar] [CrossRef]
- Muller, G.W.; Corral, L.G.; Shire, M.G.; Wang, H.; Moreira, A.; Kaplan, G.; Stirling, D.I. Structural modifications of thalidomide produce analogs with enhanced tumor necrosis factor inhibitory activity. J. Med. Chem. 1996, 39, 3238–3240. [Google Scholar] [CrossRef] [PubMed]
- Marriott, J.B.; Westby, M.; Cookson, S.; Guckian, M.; Goodbourn, S.; Muller, G.; Shire, M.G.; Stirling, D.; Dalgleish, A.G. CC-3052: A water-soluble analog of thalidomide and potent inhibitor of activation-induced TNF-α production. J. Immunol. 1998, 161, 4236–4243. [Google Scholar] [PubMed]
- Corral, L.G.; Haslett, P.A.; Muller, G.W.; Chen, R.; Wong, L.M.; Ocampo, C.J.; Patterson, R.T.; Stirling, D.I.; Kaplan, G. Differential cytokine modulation and t cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-α. J. Immunol. 1999, 163, 380–386. [Google Scholar] [PubMed]
- Man, H.W.; Schafer, P.; Wong, L.M.; Patterson, R.T.; Corral, L.G.; Raymon, H.; Blease, K.; Leisten, J.; Shirley, M.A.; Tang, Y.; et al. Discovery of (S)-N-[2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1h-isoindol-4-yl] acetamide (apremilast), a potent and orally active phosphodiesterase 4 and tumor necrosis factor-alpha inhibitor. J. Med. Chem. 2009, 52, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Shmygol, A.; Blanks, A.M.; Bru-Mercier, G.; Gullam, J.E.; Thornton, S. Control of uterine Ca2+ by membrane voltage: Toward understanding the excitation-contraction coupling in human myometrium. Ann. N. Y. Acad. Sci. 2007, 1101, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Yousif, F.B.; Triggle, D.J. Inhibitory actions of a series of Ca2+ channel antagonists against agonist and K+ depolarization induced responses in smooth muscle: An assessment of selectivity of action. Can. J. Physiol. Pharmacol. 1986, 64, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.I.; Ponce-Monter, H.A.; Mora-Rodriguez, J.A.; Barragan-Ramirez, G.; Barron-Guerrero, B.S. Synergistic relaxing effect of the paracetamol and pyrilamine combination in isolated human myometrium. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 157, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Granger, S.E.; Hollingsworth, M.; Weston, A.H. Effects of calcium entry blockers on tension development and calcium influx in rat uterus. Br. J. Pharmacol. 1986, 87, 147–156. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, K.F.; Wallace, D.A.; Hill, E.V.; Anthony, D.F.; Henderson, D.J.; Houslay, D.M.; Arthur, J.S.; Baillie, G.S.; Houslay, M.D. Phosphorylation of cAMP-specific PDE4A5 (phosphodiesterase-4A5) by MK2 (MAPKAPK2) attenuates its activation through protein kinase a phosphorylation. Biochem. J. 2011, 435, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Mahdian, D.; Shafiee-Nick, R.; Mousavi, S.H. Different effects of adenylyl cyclase activators and phosphodiesterases inhibitors on cervical cancer (HeLa) and breast cancer (MCF-7) cells proliferation. Toxicol. Mech. Methods 2014, 24, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Mehats, C.; Tanguy, G.; Dallot, E.; Robert, B.; Rebourcet, R.; Ferre, F.; Leroy, M.J. Selective up-regulation of phosphodiesterase-4 cyclic adenosine 3′,5′-monophosphate (cAMP)-specific phosphodiesterase variants by elevated camp content in human myometrial cells in culture. Endocrinology 1999, 140, 3228–3237. [Google Scholar] [CrossRef] [PubMed]
- Dousa, T.; Rychlik, I. Adenyl cyclase and adenosine 3′,5′-cyclic phosphate phosphodiesterase in the receptor tissues of neurohypophysial hormones. Life Sci. 1968, 7, 1039–1044. [Google Scholar] [CrossRef]
- Ferre, F.; De Pariente, D.; Breuiller, M.; Cedard, L. Inhibition of human cyclic AMP phosphodiesterase by uterine relaxant drugs. Biochem. Pharmacol. 1978, 27, 1292–1294. [Google Scholar] [CrossRef]
- Mehats, C.; Tanguy, G.; Paris, B.; Robert, B.; Pernin, N.; Ferre, F.; Leroy, M.J. Pregnancy induces a modulation of the camp phosphodiesterase 4-conformers ratio in human myometrium: Consequences for the utero-relaxant effect of PDE4-selective inhibitors. J. Pharmacol. Exp. Ther. 2000, 292, 817–823. [Google Scholar] [PubMed]
- Bardou, M.; Cortijo, J.; Loustalot, C.; Taylor, S.; Perales-Marin, A.; Mercier, F.J.; Dumas, M.; Deneux-Tharaux, C.; Frydman, R.; Morcillo, E.J.; et al. Pharmacological and biochemical study on the effects of selective phosphodiesterase inhibitors on human term myometrium. Naunyn Schmiedebergs Arch. Pharmacol. 1999, 360, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Croci, T.; Cecchi, R.; Marini, P.; Rouget, C.; Viviani, N.; Germain, G.; Guagnini, F.; Fradin, Y.; Descamps, L.; Pascal, M.; et al. In vitro and in vivo pharmacological characterization of ethyl-4-[trans-4-[((2S)-2-hydroxy-3-[4-hydroxy-3[(methylsulfonyl)amino]-phenoxy]propyl)amino]cyclohexyl]benzoate hydrochloride (SAR150640), a new potent and selective human beta3-adrenoceptor agonist for the treatment of preterm labor. J. Pharmacol. Exp. Ther. 2007, 321, 1118–1126. [Google Scholar] [PubMed]
- Franova, S.; Janicek, F.; Visnovsky, J.; Dokus, K.; Zubor, P.; Sutovska, M.; Nosalova, G. Utero-relaxant effect of PDE4-selective inhibitor alone and in simultaneous administration with beta2-mimetic on oxytocin-induced contractions in pregnant myometrium. J. Obstet. Gynaecol. Res. 2009, 35, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Verli, J.; Klukovits, A.; Kormanyos, Z.; Hajagos-Toth, J.; Ducza, E.; Seres, A.B.; Falkay, G.; Gaspar, R. Uterus-relaxing effect of β2-agonists in combination with phosphodiesterase inhibitors: Studies on pregnant rat in vivo and on pregnant human myometrium in vitro. J. Obstet. Gynaecol. Res. 2013, 39, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Oger, S.; Mehats, C.; Barnette, M.S.; Ferre, F.; Cabrol, D.; Leroy, M.J. Anti-inflammatory and utero-relaxant effects in human myometrium of new generation phosphodiesterase 4 inhibitors. Biol. Reprod. 2004, 70, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Herve, R.; Schmitz, T.; Evain-Brion, D.; Cabrol, D.; Leroy, M.J.; Mehats, C. The PDE4 inhibitor rolipram prevents NF-κB binding activity and proinflammatory cytokine release in human chorionic cells. J. Immunol. 2008, 181, 2196–2202. [Google Scholar] [CrossRef] [PubMed]
- Leroy, M.J.; Cedrin, I.; Breuiller, M.; Giovagrandi, Y.; Ferre, F. Correlation between selective inhibition of the cyclic nucleotide phosphodiesterases and the contractile activity in human pregnant myometrium near term. Biochem. Pharmacol. 1989, 38, 9–15. [Google Scholar] [CrossRef]
- Klukovits, A.; Verli, J.; Falkay, G.; Gaspar, R. Improving the relaxing effect of terbutaline with phosphodiesterase inhibitors: Studies on pregnant rat uteri in vitro. Life Sci. 2010, 87, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.A.; Heesom, K.J.; Lopez Bernal, A. Phasic contractions of isolated human myometrium are associated with Rho-kinase (rock)-dependent phosphorylation of myosin phosphatase-targeting subunit (MYPT1). Mol. Hum. Reprod. 2012, 18, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, H.N.; Mitchell, B.F. Physiological pathways and molecular mechanisms regulating uterine contractility. Hum. Reprod. Update 2010, 16, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Webb, R.C. Smooth muscle contraction and relaxation. Adv. Physiol. Educ. 2003, 27, 201–206. [Google Scholar] [PubMed]
- Aguilar, H.N.; Tracey, C.N.; Zielnik, B.; Mitchell, B.F. Rho-kinase mediates diphosphorylation of myosin regulatory light chain in cultured uterine, but not vascular smooth muscle cells. J. Cell. Mol. Med. 2012, 16, 2978–2989. [Google Scholar] [CrossRef] [PubMed]
- Julian, L.; Olson, M.F. Rho-associated coiled-coil containing kinases (ROCK): Structure, regulation, and functions. Small GTPases 2014, 5, e29846. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Karginov, A.V. Phosphorylation-mediated regulation of GEFS for RhoA. Cell Adhes. Migr. 2014, 8, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E.; Palmer, T.M. Protein kinase A-mediated phosphorylation of RhoA on serine 188 triggers the rapid induction of a neuroendocrine-like phenotype in prostate cancer epithelial cells. Cell Signal. 2012, 24, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, J.A.; Weiner, C.P. Mechanisms underlying myometrial quiescence during pregnancy. Fetal Matern. Med. Rev. 2012, 14, 209–237. [Google Scholar] [CrossRef]
- Woodrum, D.A.; Brophy, C.M. The paradox of smooth muscle physiology. Mol. Cell. Endocrinol. 2001, 177, 135–143. [Google Scholar] [CrossRef]
- Shojo, H.; Kaneko, Y. Oxytocin-induced phosphorylation of myosin light chain is mediated by extracellular calcium influx in pregnant rat myometrium. J. Mol. Recognit. 2001, 14, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Poondra, R.R.; Nallamelli, R.V.; Meda, C.L.; Srinivas, B.N.; Grover, A.; Muttabathula, J.; Voleti, S.R.; Sridhar, B.; Pal, M.; Parsa, K.V. Discovery of novel 1,4-dihydropyridine-based PDE4 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Azam, M.A.; Tripuraneni, N.S. Selective phosphodiesterase 4b inhibitors: A review. Sci. Pharm. 2014, 82, 453–481. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Handa, H. Teratogenic effects of thalidomide: Molecular mechanisms. Cell. Mol. Life Sci. 2011, 68, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Schafer, P.H.; Parton, A.; Capone, L.; Cedzik, D.; Brady, H.; Evans, J.F.; Man, H.W.; Muller, G.W.; Stirling, D.I.; Chopra, R. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell Signal. 2014, 26, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Richter, W.; Mehats, C.; Livera, G.; Park, J.Y.; Jin, C. Cyclic AMP-specific PDE4 phosphodiesterases as critical components of cyclic AMP signaling. J. Biol. Chem. 2003, 278, 5493–5496. [Google Scholar] [CrossRef] [PubMed]
- GeneCards. Genecards the Human Database. Weizmann Institute of Science: Israel. Available online: http://www.genecards.org/cgi-bin/carddisp.pl?gene=MYL9&keywords=MYL (accessed on 25 July 2015).
- Aguilar, H.N.; Zielnik, B.; Tracey, C.N.; Mitchell, B.F. Quantification of rapid myosin regulatory light chain phosphorylation using high-throughput in-cell western assays: Comparison to western immunoblots. PLoS ONE 2010, 5, e9965. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.B.; Lutz, S.; Steffens, F.; Korth, M.; Wieland, T. Oxytocin receptors differentially signal via Gq and Gi proteins in pregnant and nonpregnant rat uterine myocytes: Implications for myometrial contractility. Mol. Endocrinol. 2007, 21, 740–752. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.; Baker, P.N.; Tribe, R.M.; Taggart, M.J. Expression of scaffolding, signalling and contractile-filament proteins in human myometria: Effects of pregnancy and labour. J. Cell. Mol. Med. 2005, 9, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.L.; Bushnik, T.; Lan, L.; Conti, M. Subcellular localization of rolipram-sensitive, cAMP-specific phosphodiesterases. Differential targeting and activation of the splicing variants derived from the PDE4d gene. J. Biol. Chem. 1998, 273, 19672–19678. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Mika, D.; Blanchard, E.; Day, P.; Conti, M. Beta1-adrenergic receptor antagonists signal via PDE4 translocation. EMBO Rep. 2013, 14, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Shealy, Y.F.; Opliger, C.E.; Montgomery, J.A. Synthesis of d- and l-thalidomide and related studies. J. Pharm. Sci. 1968, 57, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Kalvin, D.M.; Woodard, R.W. Synthesis of (4R)-d,l-[4-2H]- and (4S)-d,l-[4-2H] homoserine lactones. J. Org. Chem. 1985, 50, 2259–2263. [Google Scholar] [CrossRef]
- Fernández-Martínez, E.; Morales-Rios, M.S.; Perez-Alvarez, V.; Muriel, P. Effects of thalidomide and 3-phthalimido-3-(3,4-dimethoxyphenyl)-propanamide on bile duct obstruction-induced cirrhosis in the rat. Drug Dev. Res. 2001, 54, 209–218. [Google Scholar] [CrossRef]
- Fernández-Martínez, E.; Morales-Rios, M.S.; Perez-Alvarez, V.; Muriel, P. Immunomodulatory effects of thalidomide analogs on LPS-induced plasma and hepatic cytokines in the rat. Biochem. Pharmacol. 2004, 68, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Osorio-Espinoza, A.; Escamilla-Sanchez, J.; Aquino-Jarquin, G.; Arias-Montaño, J.A. Homologous desensitization of human histamine H(3) receptors expressed in CHO-K1 cells. Neuropharmacology 2014, 77, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the thalidomide analogs are not available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contraction | Spontaneous | Tonic KCl-Induced | ||
|---|---|---|---|---|
| Compounds | IC50 (µM) | Emax (%) | IC50 (µM) | Emax (%) |
| Rolipram | 22.74 ± 2.32 | 94.13 ± 7.53 | 98.45 ± 8.86 | 97.10 ± 8.73 |
| 4APDPMe | 62.17 ± 5.61 a | 88.16 ± 7.93 | 125 ± 13.72 | 88.65 ± 7.09 |
| 4NO2PDPMe | 128.30 ± 14.56 a,b | 67.99 ± 8.15 | 203.45 ± 18.31 a,b | 70.61 ± 6.78 |
| Ca2+-Induced Contractile Response (%) | ||||
|---|---|---|---|---|
| [Ca2+] (mM) | 1 | 2 | 3 | 5 |
| Control | 34.21 ± 3.53 | 68.14 ± 4.45 | 81.09 ± 5.15 | 93.03 ± 5.06 |
| 4NO2PDPMe | 23.32 ± 2.32 | 50.15 ± 3.19 a | 61.37 ± 3.32 a | 70.29 ± 3.54 a |
| 4APDPMe | 13.23 ± 2.16 a | 30.32 ± 3.32 a,b | 47.18 ± 3.50 a | 55.34 ± 3.59 a |
| Rolipram | 5.35 ± 2.51 a,b | 18.12 ± 3.32 a,b | 28.58 ± 3.35 a,b,c | 35.28 ± 3.54 a,b,c |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Martínez, E.; Ponce-Monter, H.; Soria-Jasso, L.E.; Ortiz, M.I.; Arias-Montaño, J.-A.; Barragán-Ramírez, G.; Mayén-García, C. Inhibition of Uterine Contractility by Thalidomide Analogs via Phosphodiesterase-4 Inhibition and Calcium Entry Blockade. Molecules 2016, 21, 1332. https://doi.org/10.3390/molecules21101332
Fernández-Martínez E, Ponce-Monter H, Soria-Jasso LE, Ortiz MI, Arias-Montaño J-A, Barragán-Ramírez G, Mayén-García C. Inhibition of Uterine Contractility by Thalidomide Analogs via Phosphodiesterase-4 Inhibition and Calcium Entry Blockade. Molecules. 2016; 21(10):1332. https://doi.org/10.3390/molecules21101332
Chicago/Turabian StyleFernández-Martínez, Eduardo, Héctor Ponce-Monter, Luis E. Soria-Jasso, Mario I. Ortiz, José-Antonio Arias-Montaño, Guillermo Barragán-Ramírez, and Cynthia Mayén-García. 2016. "Inhibition of Uterine Contractility by Thalidomide Analogs via Phosphodiesterase-4 Inhibition and Calcium Entry Blockade" Molecules 21, no. 10: 1332. https://doi.org/10.3390/molecules21101332