2. Results
Compound
1 was obtained as a white amorphous powder. The molecular composition of
1, C
20H
28O
5, was deduced from the positive ion peak at
m/
z 349.2001 [M + H]
+ (calcd. 349.2010) in its HR-ESI-MS, with seven degrees of unsaturation. The IR spectrum exhibited the absorptions at 3430, 2932, 1696, 1616, and 1418 cm
−1, indicating the presence of hydroxyl, an aromatic ring, and carboxyl functionalities. The
1H-NMR spectrum of
1 exhibited the signals of three methyl groups (δ
H 1.13, s; 1.22, d,
J = 6.8 Hz; 1.24, s), one hydroxyl methyl group (δ
H 3.48, dd,
J = 10.2, 7.4 Hz; 3.69, dd,
J = 10.2, 5.7 Hz), one oxymethine group (δ
H 3.52, dd,
J = 11.7, 4.9 Hz), and two aromatic protons (δ
H 6.36, s; 8.05, s) (
Table 1 and
Supplementary Figure S2). The
13C-NMR spectrum of
1 displayed 20 carbons signals attributed to one carbonyl carbon (δ
C 181.2), six aromatic carbons (δ
C 153.7, 140.0, 136.1, 130.0, 128.9, and 115.5), two quaternary carbons (δ
C 45.0 and 44.3), three methine carbons (δ
C 79.1, 53.9, and 37.6), five methylene carbons (δ
C 68.4, 37.0, 33.5, 31.5, and 22.4), and three methyl carbons (δ
C 29.1, 18.2, and 17.3) (
Table 1 and
Supplementary Figure S3). The
1H- and
13C-NMR data of
1 were quite similar to those of the aglycone of 19-
O-
d-glucopyranoside of 16-hydroxylambertic acid except that a methylene group in ring A of the aglycone of 19-
O-
d-glucopyranoside of 16-hydroxylambertic acid was replaced by an oxymethine group in compound
1, as well as the down-field shift of an aromatic acid (8.05 in
1; 6.54 in 19-
O-
d-glucopyranoside of 16-hydroxylambertic acid) [
13].
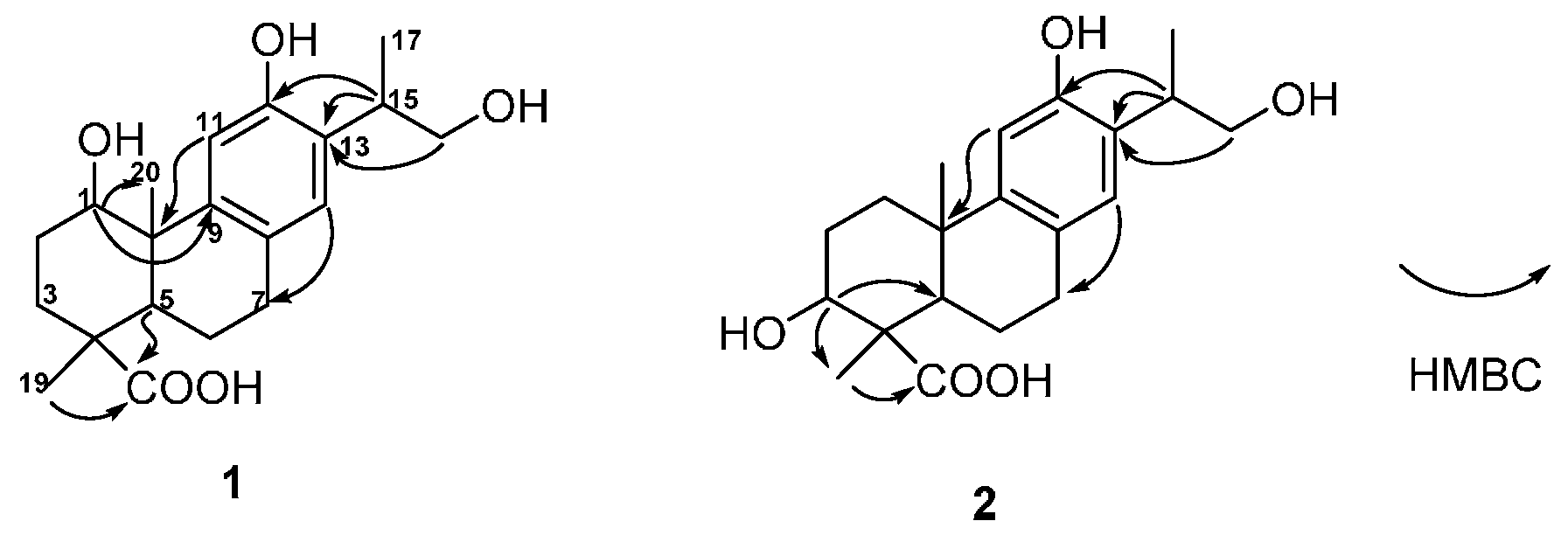
The structure of
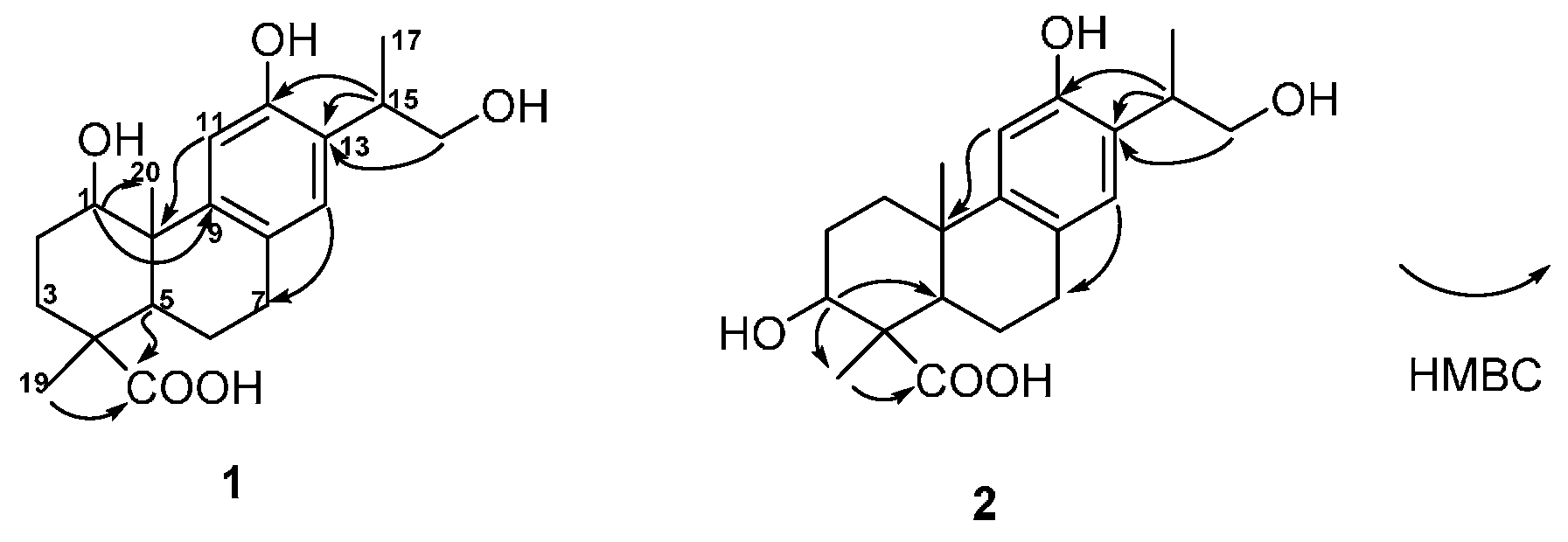
1 was constructed by the detailed analysis of the HSQC and HMBC spectra (
Figure 2 and
Supplementary Figure S4 and S5). The correlations from H-11 to C-10, from H-14 to C-7, from H-15 to C-12, C-13, and from H
2-16 to C-13 manifested a hydroxyl group on C-12, as well as a hydroxyl-substituted isopropyl substituent on C-13. Furthermore, the HMBC cross-peaks from H-1 to C-9 and C-20 suggested a hydroxyl group was substituted on C-1. The down-field shift of H-11 also supported a hydroxyl group was substituted on C-1 [
14,
15]. In addition, the correlated signals of H
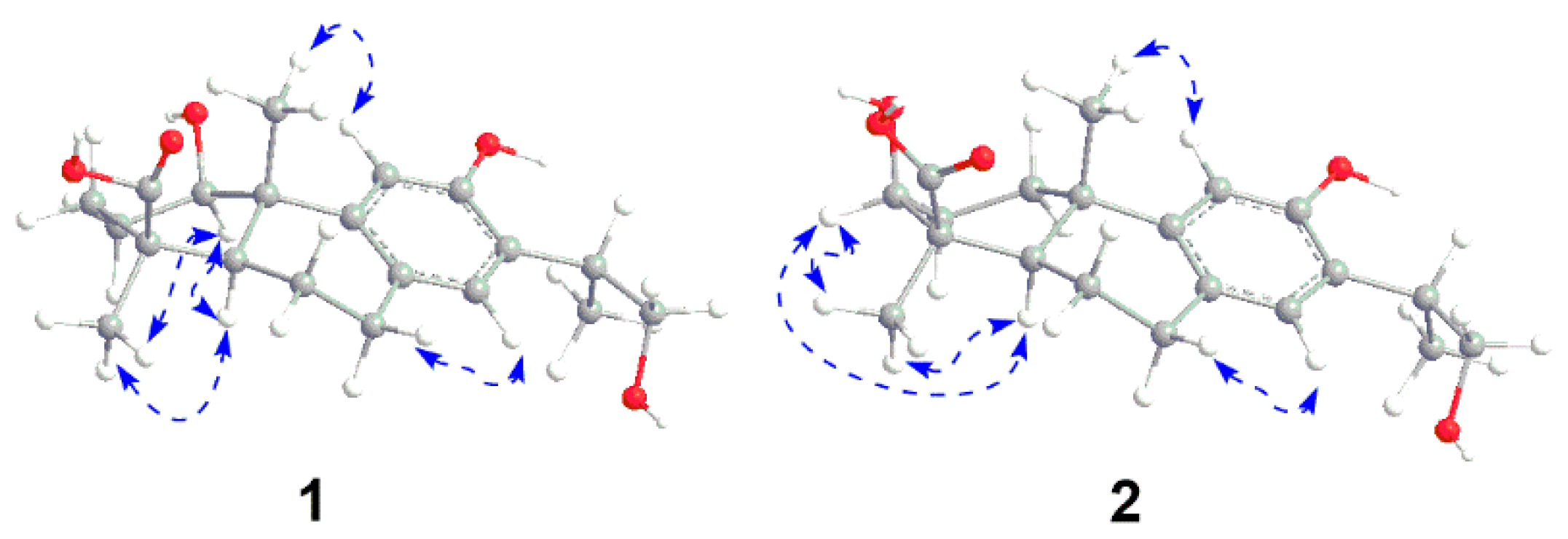
3-18 and H-5 to C-19 confirmed the carboxyl group on C-19. The relative configuration of
1 was subsequently deduced from the NOESY experiment. The hydroxyl group on C-1 was assigned as
β-oriented based on the correlations from H
3-18 to H-1 and H-5, as well as H-5 to H-1 (
Figure 3 and
Supplementary Figure S6). The absolute configuration of compound
1 was determined by CD spectrum. The Cotton effect of compound
1 (Δε
211 4.14, Δε
227 3.91, Δε
265 −2.99) was consistent with that of ferruginol (Δε
211 4.11, Δε
227 3.97, and Δε
265 −2.95), indicating a 10
S configuration [
16]. Thus, the structure of
1 was established as shown in
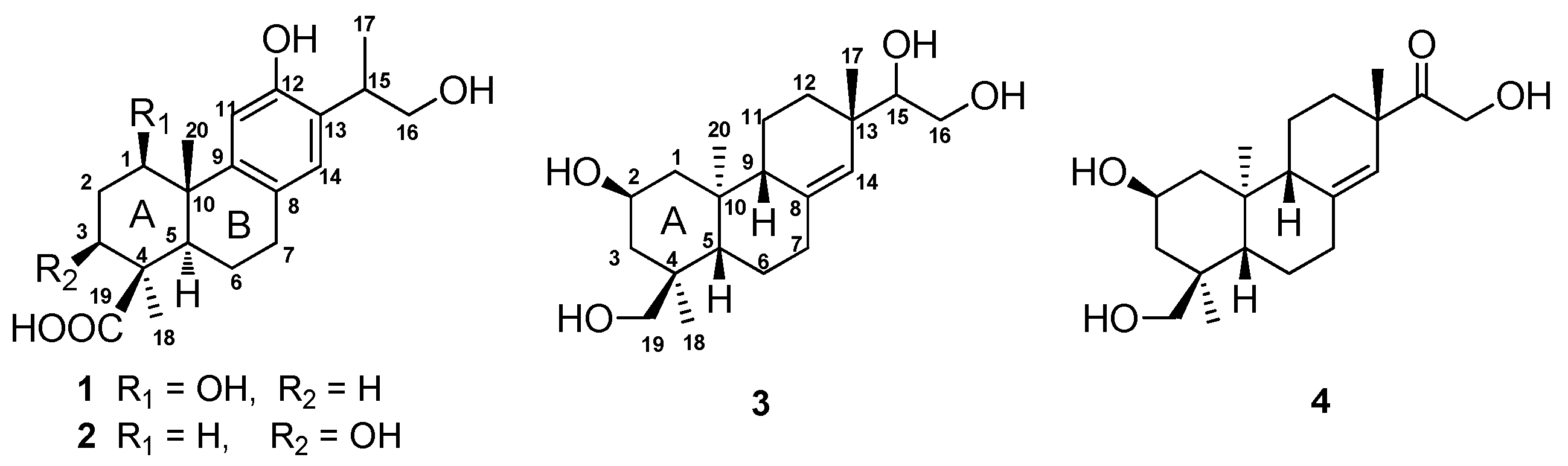
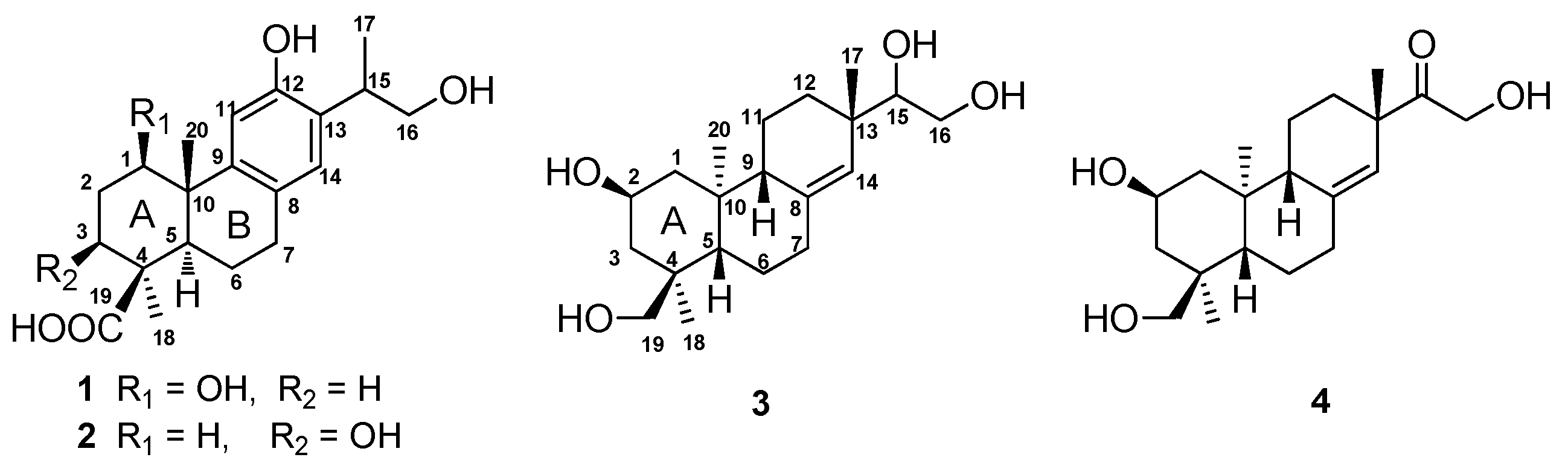
Figure 1, and it was named 1β,16-dihydroxylambertic acid.
Compound
2 was isolated as a white amorphous powder. The molecular formula was determined to be C
20H
28O
5 from the ion peak at
m/z 349.2004 [M + H]
+ (calcd. 349.2010) in its HR-ESI-MS. The IR spectrum of
2 displayed the absorptions for hydroxyl groups (3530 and 3385 cm
−1), an aromatic ring (1616 and 1383 cm
−1), and a carboxyl group (1688 cm
−1). The
1H- and
13C-NMR spectra of
2 were quite similar to those of the aglycone of the 19-
O-
d-glucopyranoside of 16-hydroxylambertic acid except that a methylene group in ring A of the aglycone of the 19-
O-
d-glucopyranoside of 16-hydroxylambertic acid was replaced by an oxymethine group in compound
2, which inferred a hydroxyl group might substitute at ring A [
13]. The hydroxyl group was assigned on C-3 based on the HMBC correlations from H-3 to C-5 and C-18 (
Figure 2). The NOE cross-peaks from H
3-18 to H-3 and H-5, and H-5 to H-3 indicated the hydroxyl group was β-oriented (
Figure 3). Moreover, the identical Cotton effect of compounds
2 and
1 suggested they shared the same absolute configuration at C-10. Thus, the structure of
2 was determined as shown in
Figure 1, and it was named 3β,16-dihydroxylambertic acid.
Compound
3 was obtained as a white amorphous powder. The molecular formula of
3, C
20H
34O
4, was deduced by the ion peak at
m/
z 383.2438 [M + HCOO]
− (calcd. 383.2439) in its HR-ESI-MS. The existence of hydroxyl group and double bond was deduced by the IR absorptions at 3424 and 1647 cm
−1, respectively. The
1H-NMR spectrum of
3 showed the signals of three methyl groups (δ
H 0.85, s; 0.90, s; 0.99, s), two oxygenated methylenes (δ
H 3.05, d,
J = 11.0 Hz, 3.36, m; 3.38, m, 3.74, dd,
J = 11.1, 2.4 Hz), two oxygenated methines (δ
H 3.34, m; 3.85, m), and an olefinic proton (δ
H 5.37, s) (
Table 2). The
13C-NMR spectrum revealed 20 carbon signals attributed to three methyl carbons, eight methylene carbons, four
sp3 methine carbons, three
sp3 quaternary carbons, one
sp2 methine carbon, and one
sp2 quaternary carbon (
Table 2). The
1H- and
13C-NMR data of
3 quite resembled to those of kirenol except for the chemical shifts of H
3-18, C-18, and C-19 [
17,
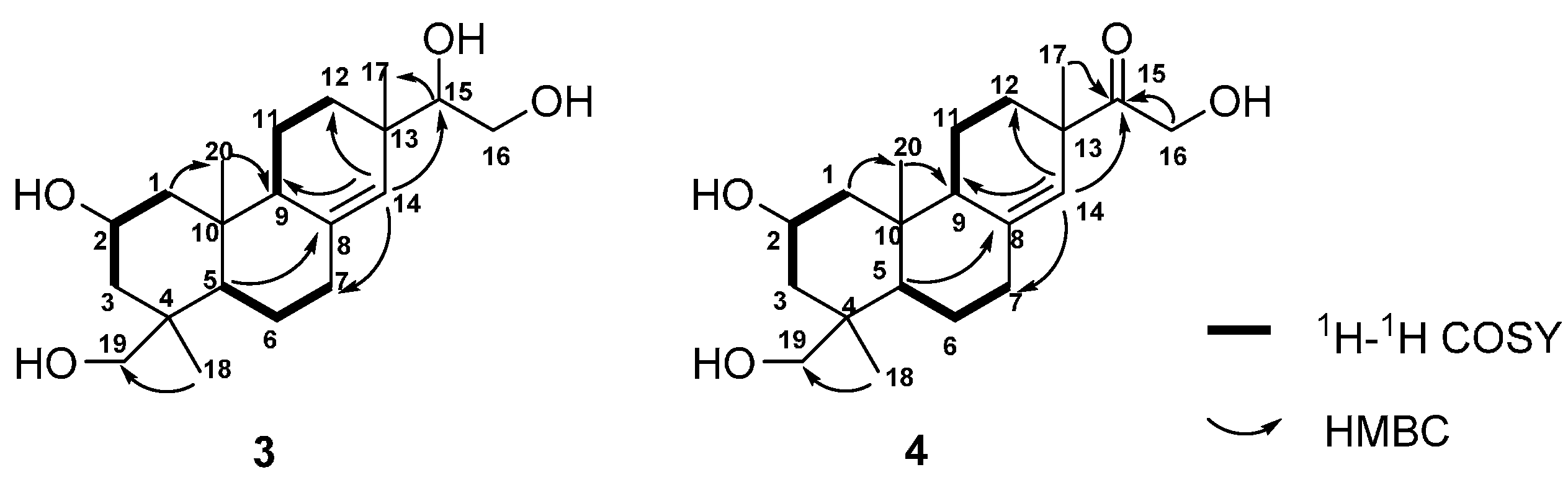
18]. Detailed analysis of the
1H-
1H COSY, HSQC, and HMBC spectra resulted in the construction of planar structure of
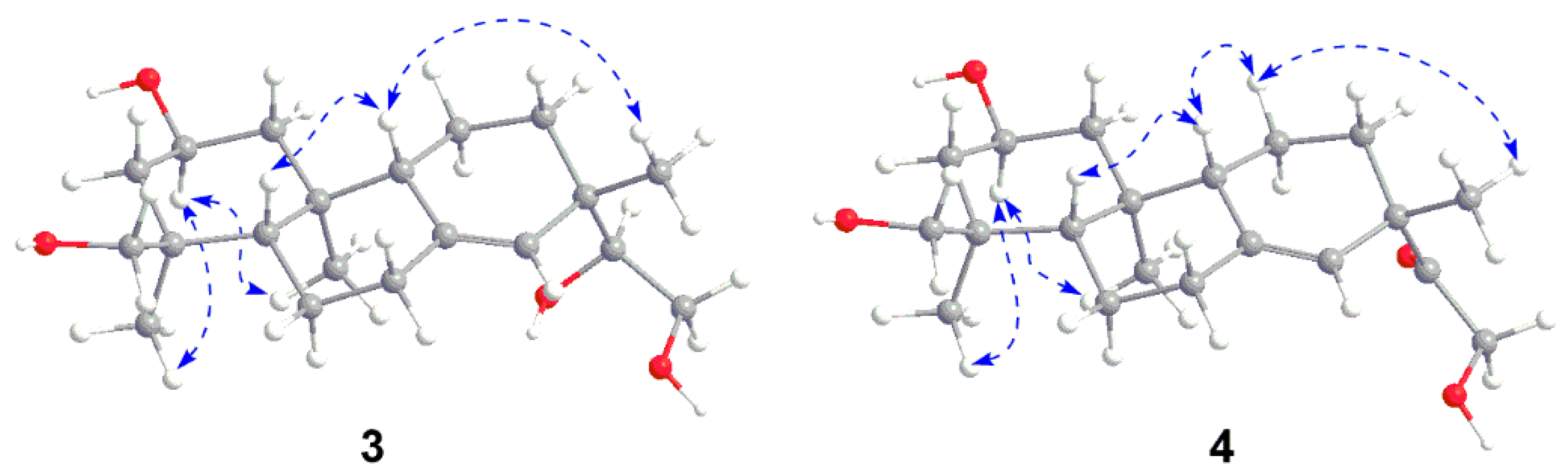
3 (
Figure 4). Subsequently, 2-hydroxyl group and H
3-18 were assigned as β-oriented and α-oriented, respectively, by the correlations from H
3-20 to H-2, and H-2 to H
3-18 in the NOESY spectrum (
Figure 5). The correlations from H-5 to H-9, together with H-9 to H
3-17 indeed confirmed the two protons and H
3-17 were
β-oriented. Thus, the structure of
3 was elucidated as
ent-2β,15,16,18-tetrahydroxypimar-8(14)-ene (
Figure 1).
Compound
4 was obtained as a white amorphous powder. The molecular formula was determined to be C
20H
28O
5 from the ion peak at
m/
z 337.2374 [M + H]
+ (calcd. 337.2373) in its HR-ESI-MS, suggesting the presence of five degrees of unsaturation. The IR spectrum showed absorptions for a double bond at 1618 cm
−1, a ketone carbonyl group at 1710 cm
−1 and hydroxyl groups at 3426 cm
−1. The
1H- and
13C-NMR spectra of
4 were quite similar to those of compound
3 (
Table 2). After careful comparison, the chemical shifts of C-15 (80.8 ppm in
3; 215.3 ppm in
4) and H
2-16 (3.38 and 3.74 ppm in
3; 4.36 and 4.32 ppm in
4) were obviously downfield shifted, implying that a ketone group in
4 might replace the hydroxyl group on C-15 in
3. The HMBC correlations from H-14, H-16, and H
3-17 to the carbonyl carbon assigned it as C-15 (
Figure 4). The NOESY experiment further confirmed the relative configuration of
4 (
Figure 5). Thus, the structure of
4 was established as
ent-15-oxo-2β,16,18-trihydroxypimar-8(14)-ene as shown in
Figure 1.
All the new compounds were tested for their cytotoxic activity on human cervical cancer Hela cells, human lung cancer A549 cells, and human breast cancer MCT-7 cells. The MTT assay results showed compounds 3 and 4 significantly inhibited proliferation on three cell lines at the concentration of 10 μM. The inhibitory rates for compounds 3 and 4 were 60.7% ± 1.9% and 62.6% ± 1.6% on Hela cells, 26.4% ± 4.1% and 29.0% ± 1.1% on A549 cells, and 41.2% ± 1.4% and 44.7% ± 1.9% on MCF-7 cells, respectively. However, compounds 1 and 2 did not show obvious cytotoxicity at 20 μM against the three cell lines. Herein, paclitaxel was used as the positive control, and the inhibitory rate of paclitaxel (200 nM) on Hela, A549, and MCF-7 cells were 56.2% ± 5.4%, 66.7% ± 8.8% and 43.3% ± 8.1%, respectively.
Additionally, the new compounds were evaluated for their inhibitory effect on NO production in LPS-stimulated RAW264.7 cells. The results showed compounds 2 and 4 significantly reduced NO production, with IC50 values of 26.5 ± 6.1 and 17.1 ± 1.5 μM, respectively, which were comparable with that of the positive control indomethacin (IC50 4.5 ± 0.2 μM). The other two compounds did not show any inhibitory effect up to 100 μM. On the other hand, compounds 1–4 did not show obvious cytotoxicity at 100 μM against RAW264.7 cells.
3. Materials and Methods
3.1. General Experiments
TLC was performed on pre-coated silica gel GF254 plates (Merck Chemical Co. Ltd., Shanghai, China). MCI (Polystyrene) gel (CHP20P, 75−150 μm, Mitsubishi Chemical Industries, Tokyo, Japan), silica gel (Qingdao Marine Chemical Industrials, Qingdao, Shandong, China), macro porous resin AB-8 (Shandong Lu Kang Chemical Industrials, Jinan, Shandong, China), and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden) were used for column chromatography (CC). Analytical HPLC was applied on a Waters 2695 instrument (Milford, MD, USA) coupled with a 2998 PDA, a Waters 2424 ELSD, and a Waters 3100 MS detector. Preparative HPLC was performed on a Varian PrepStar pumps with an Alltech 3300 ELSD (Columbia, MD, USA) using a Waters Sunfire RP C18, 5 μm, 30 × 150 mm column. Optical rotations were measured on a Rudolph Autopol VI Automatic polarimeter (Hackettstown, NJ, USA). IR data were recorded on a Nicolet Magna FTIR-750 spectrophotometer (Waltham, MA, USA). NMR spectra were recorded on a Bruker Avance III (Bruker, Zurich, Switzerland) for 500M and 600M NMR spectrometer and a Varian MR-400 (Varian, Palo Alto, CA, USA) for 400M NMR spectrometer with TMS as the internal standard. CD spectra were measured on a Jasco J-180 spectrophotometer (Mitsubishi Chemical Industries, Tokyo, Japan). HR-ESI-MS were measured on a Waters Xevo Q-Tof mass detector and an Agilent G6520 Q-TOF mass detector (Santa Clara, CA, USA). All solvents used for CC and HPLC were of analytical grade (Shanghai Chemical Reagents Co. Ltd., Shanghai, China) and gradient grade (Merck KGaA, Darmstadt, Germany), respectively.
3.2. Plant Material
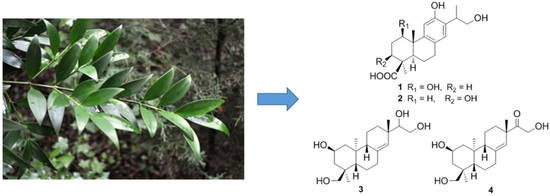
The twigs of P. nagi were collected in Ledong County, Hainan Province, China, and identified by Professor Chang-Qiang Ke, Shanghai Institute of Materia Medica. A voucher specimen (No. 20140611) was deposited at the herbarium of Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
3.3. Extraction, Isolation, and Characterization
Air-dried twigs of P. nagi (39.3 kg) were grounded and extracted with 95% EtOH (3 × 35 L) at room temperature (each 72 h). The concentrated extract (1.1 kg) was suspended in water and then partitioned with petroleum ether and EtOAc successively. The EtOAc extract (85 g) was separated into five fractions (Fr.1A‒Fr.1E) by MCI gel CC eluted with EtOH/H2O (from 20:80 to 95:5). The 40% ethanol fraction (Fr.1B, 17 g) was subjected to silica gel CC eluted with CH2Cl2/MeOH (from 80:1 to 10:1) to yield 10 fractions (Fr.1B1–Fr.1B10). Fr.1B5 was purified by CC over Sephadex LH-20 gel eluted with CHCl3/MeOH (1:1, v/v) to obtain 1 (8 mg). Fr.1B10 was subjected to Sephadex LH-20 gel eluted with MeOH to yield Fr.1B10A and Fr.1B10B, which were further purified by preparative HPLC (MeCN/H2O, 5%-35%, 0−110 min, 25.0 mL/min) to obtain 4 (6 mg) and 2 (10 mg), respectively. The water fraction was separated into two fractions (Fr.2B and Fr.2C) by macro porous resin AB-8 gel CC with EtOH/H2O (40:60 and 70:30). Fr.2C was subjected to silica gel CC eluted with CH2Cl2/MeOH (from 40:1 to 20:1) to yield two fractions (Fr.2C1 and Fr.2C2). Fr.2C2 was purified by Sephadex LH-20 gel eluted with CHCl3/MeOH (1:1, v/v) to obtain 3 (190 mg).
1β,16-Dihydroxylambertic acid (
1): white powder; [α]
+119.8 (
c 1.0, MeOH); UV (MeOH) λ
max(log ε), nm: 210 (4.84), 227 (4.62), 265 (3.66); CD (MeOH) λ
max (Δε), nm: 211 (4.11), 227 (3.97), 265 (−2.95); IR (KBr, cm
−1): ν
max 3430, 2961, 1695, 1616, 1506, 1418, 1384, 1251;
1H- and
13C-NMR data, see
Table 1; ESI-MS
m/
z 349.2 [M + H]
+ ; HR-ESI-MS
m/
z 349.2001 [M + H]
+ (calcd. for C
20H
29O
5, 349.2010).
3β,16-Dihydroxylambertic acid (
2): white powder; [α]
+25.7 (
c 1.0, MeOH); UV (MeOH) λ
max(log ε), nm: 210 (4.64), 227 (4.49), 265 (3.31); CD (MeOH) λ
max (Δε), nm: 211 (4.14), 227 (3.79), 265 (−2.99); IR (KBr, cm
−1): ν
max 3531, 3385, 2953, 2926, 1687, 1616,1470, 1422, 1383, 1200;
1H- and
13C-NMR data, see
Table 1; ESI-MS
m/
z 349.2 [M + H]
+ ; HR-ESI-MS
m/
z 349.2003 [M + H]
+ (calcd. for C
20H
29O
5, 349.2010).
ent-2β,15,16,18-Tetrahydroxypimar-8(14)-ene (
3): white powder; [α]
+8.8 (
c 1.0, MeOH); IR (KBr, cm
−1): ν
max 3424, 2932, 2872, 1769, 1720, 1646, 1459, 1383;
1H- and
13C-NMR data, see
Table 2; ESI-MS
m/
z 383.2 [M + HCOO]
−; HR-ESI-MS
m/
z 383.2438 [M + HCOO]
– (calcd. for C
21H
35O
6, 383.2439).
ent-15-oxo-2β,16,18-Trihydroxypimar-8(14)-ene (
4): white powder; [α]
+3.8 (
c 1.0, MeOH); IR (KBr, cm
–1): ν
max 3426, 2929, 2870, 1710, 1618, 1459, 1385;
1H- and
13C-NMR data, see
Table 2; ESI-MS
m/
z 337.2 [M + H]
+; HR-ESI-MS
m/
z 337.2374 [M + H]
+(calcd. for C
20H
33O
4, 337.2373).
3.4. Reagents
The compounds were dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO, USA) as a stock solution and stored at −20 °C. Dulbecco′s modified Eagle′s medium (DMEM), RPMI 1640 medium, 0.25% trypsin-EDTA, fetal bovine serum (FBS), Penicillin-Streptomycin (10,000 units/mL of penicillin and 10,000 μg/mL of streptomycin), and phosphate-buffered saline (PBS) were purchased from Gibco (Carlsbad, CA, USA). Paclitaxel, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT), indomethacin, and LPS were purchased from Sigma-Aldrich (St. Louis, MO, USA).
3.5. Cell Culture
The human cervical cancer Hela cells were acquired from American Type Culture Collection (Rockville, MD, USA) and cultured in DMEM medium supplemented with 10% (v/v) FBS and 1% (v/v) Penicillin-Streptomycin. The human non-small cell lung cancer A549 cells were obtained from American Type Culture Collection (Rockville, MD, USA) and cultured in RPMI 1640 medium containing 10% (v/v) FBS and 1% (v/v) Penicillin-Streptomycin. The human breast cancer MCF-7 cells were acquired from KeyGEN Biotech (Nanjing, Jiangsu, China) and cultured in DMEM medium supplemented with 10% (v/v) FBS and 1% (v/v) Penicillin-Streptomycin. The murine macrophage RAW264.7 cells were obtained from American Type Culture Collection (Rockville, MD, USA) and maintained in DMEM supplemented with 10% FBS. Cells were grown in a standard humidified incubator with 5% CO2 at 37 °C.
3.6. MTT Assay
Viability of the cells after treatment with the compounds was determined by MTT assay as described previously [
19]. Exponentially growing Hela, A549, MCF-7, and RAW264.7 cells were seeded into 96-well plates. Upon approximately 70%–80%, the cells were treated with series concentrations of different compounds (48 h for Hela, A549 and MCF-7 cells; 24 h for RAW264.7 cells). Paclitaxel was used as the positive control, and DMSO was used as the vehicle control. Then, a 1-mg/mL MTT solution was added to each well and the 96-well plates were further incubated for 4 h at 37 °C. A 100-μL portion of DMSO was added to each well to dissolve the formazan crystals. Absorbance at 570 nm was measured by a microplate reader (Perkin Elmer, 1420 Multilabel Counter Victor 3, Wellesley, MA, USA).
3.7. NO Production Assay
RAW264.7 cells were seeded into 24-well plates (1 × 10
5 cells/well). After 24 h, the cells were treated with different concentrations of compounds or vehicle (DMSO) for 1 h. Then, the cells were stimulated with 1 μg/mL LPS and further incubated for 18 h. Indomethacin was used as the positive control. To measure the NO content, the culture supernatant was collected and assayed using Griess reagent, as described previously [
20].
3.8. Statistical Analysis
Data were expressed as mean values and standard deviation. All the experiments were repeated under the same conditions at least three times (n = 9). Statistical significances were analyzed by one-way analysis of variance using SPSS 17 software (Statistical Package for the Social Sciences, SPSS Inc., Chicago, IL, USA). p < 0.05 was considered as the significant difference.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}