
Comparative Incorporation of PNA into DNA Nanostructures

Abstract
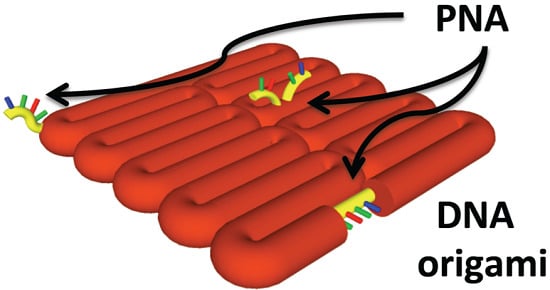
:
1. Introduction
2. Results and Discussion
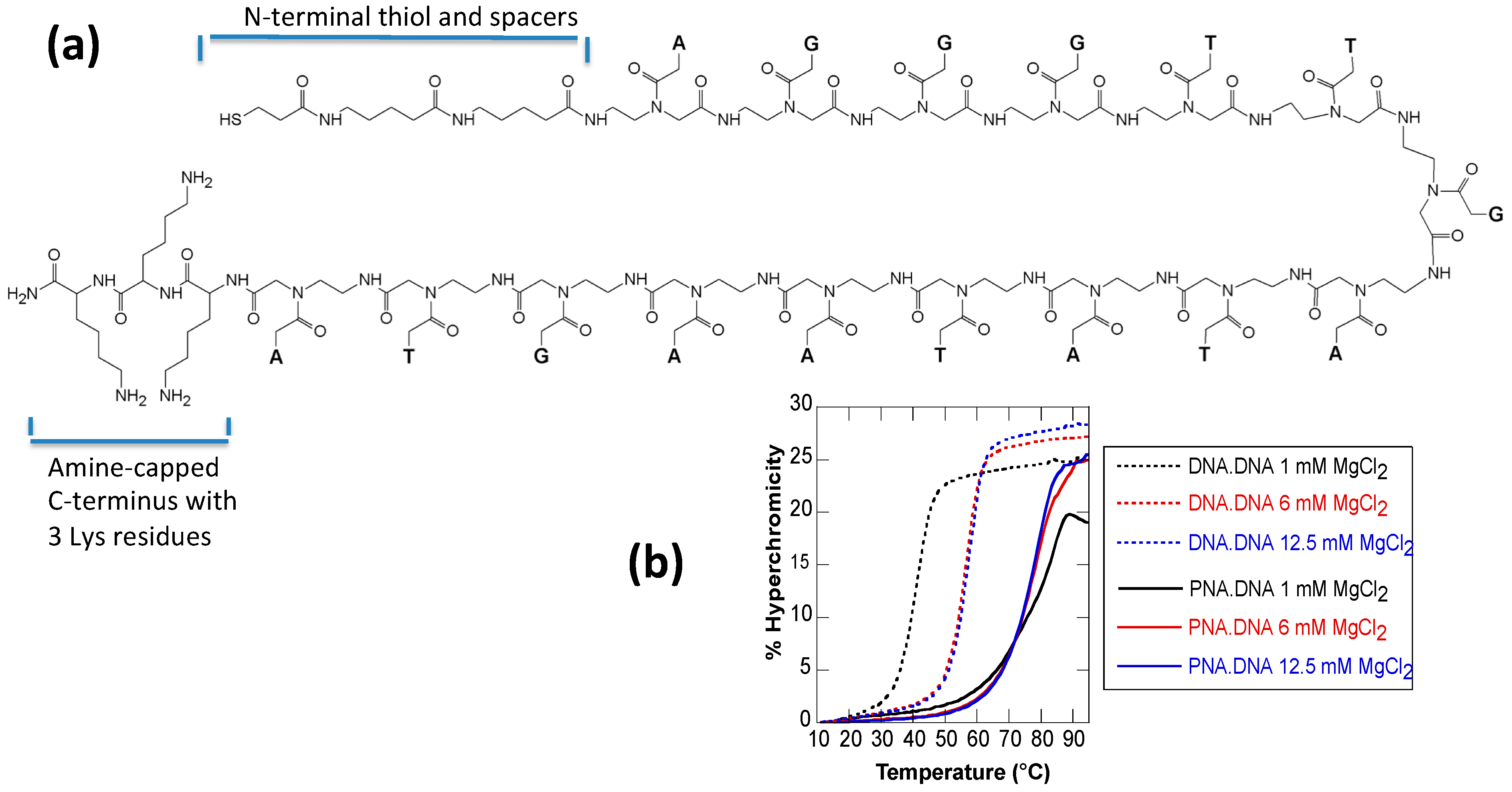
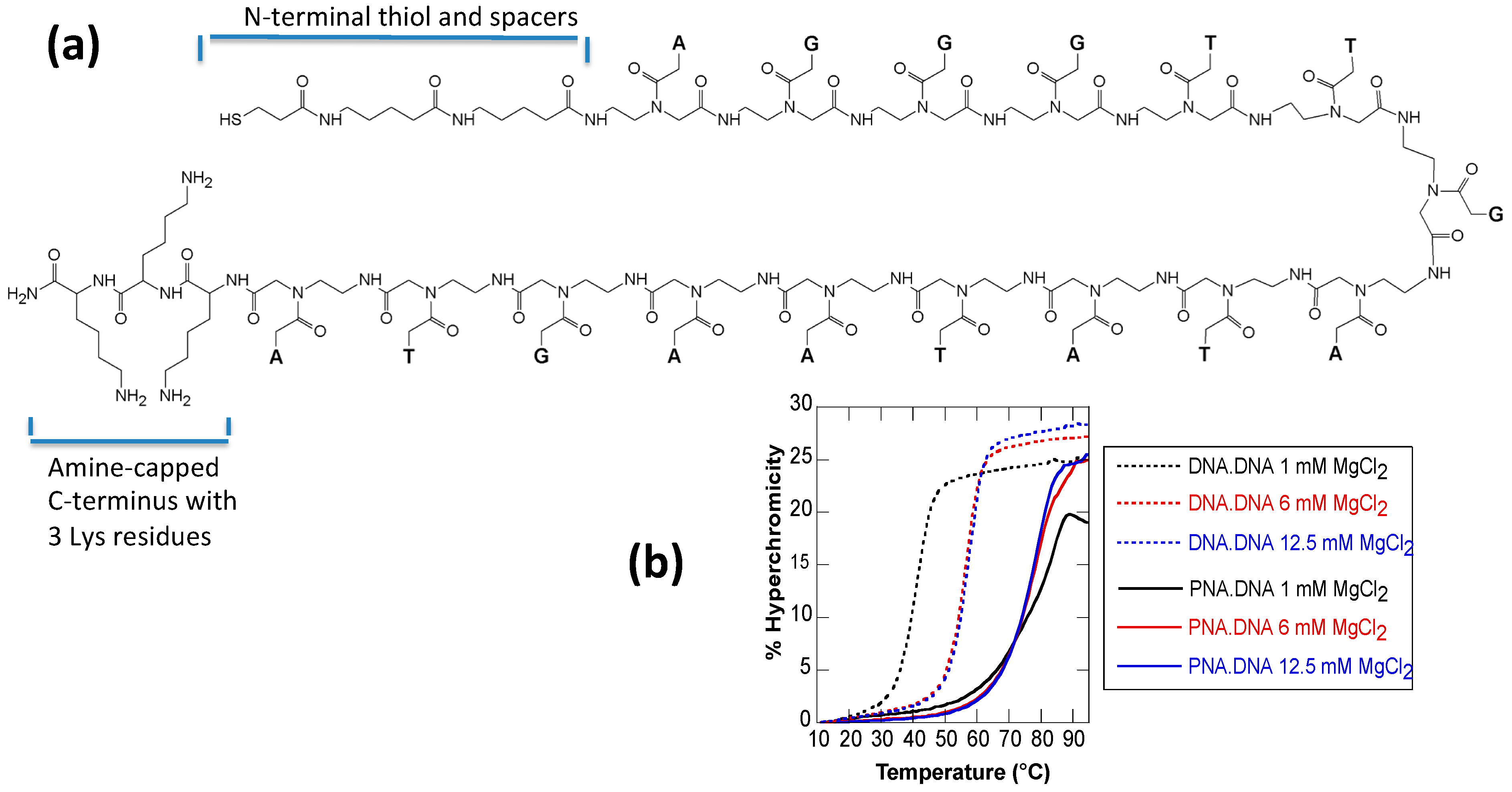
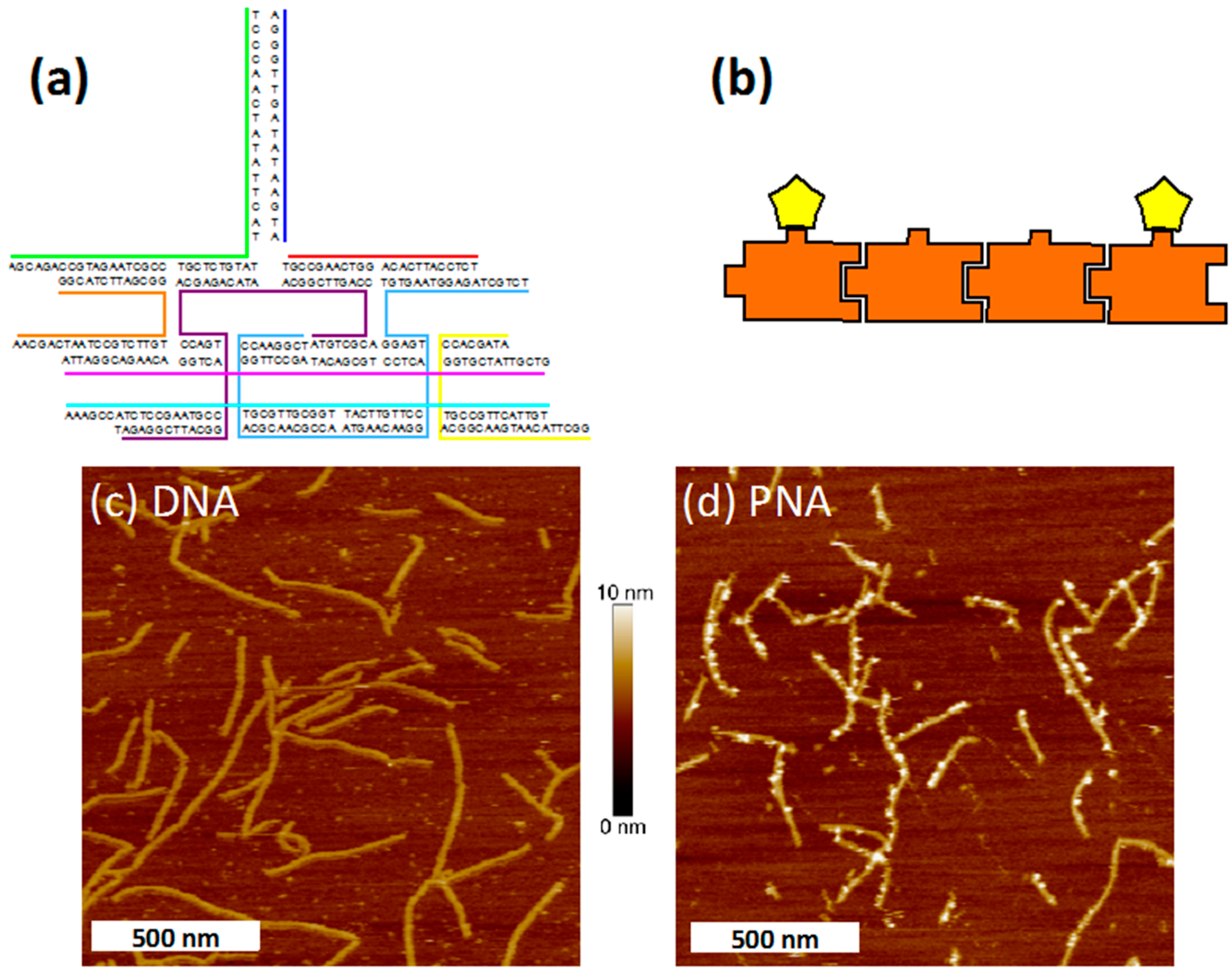
2.1. Melting Curves

{kind=link}
{kind=link}
{kind=link}
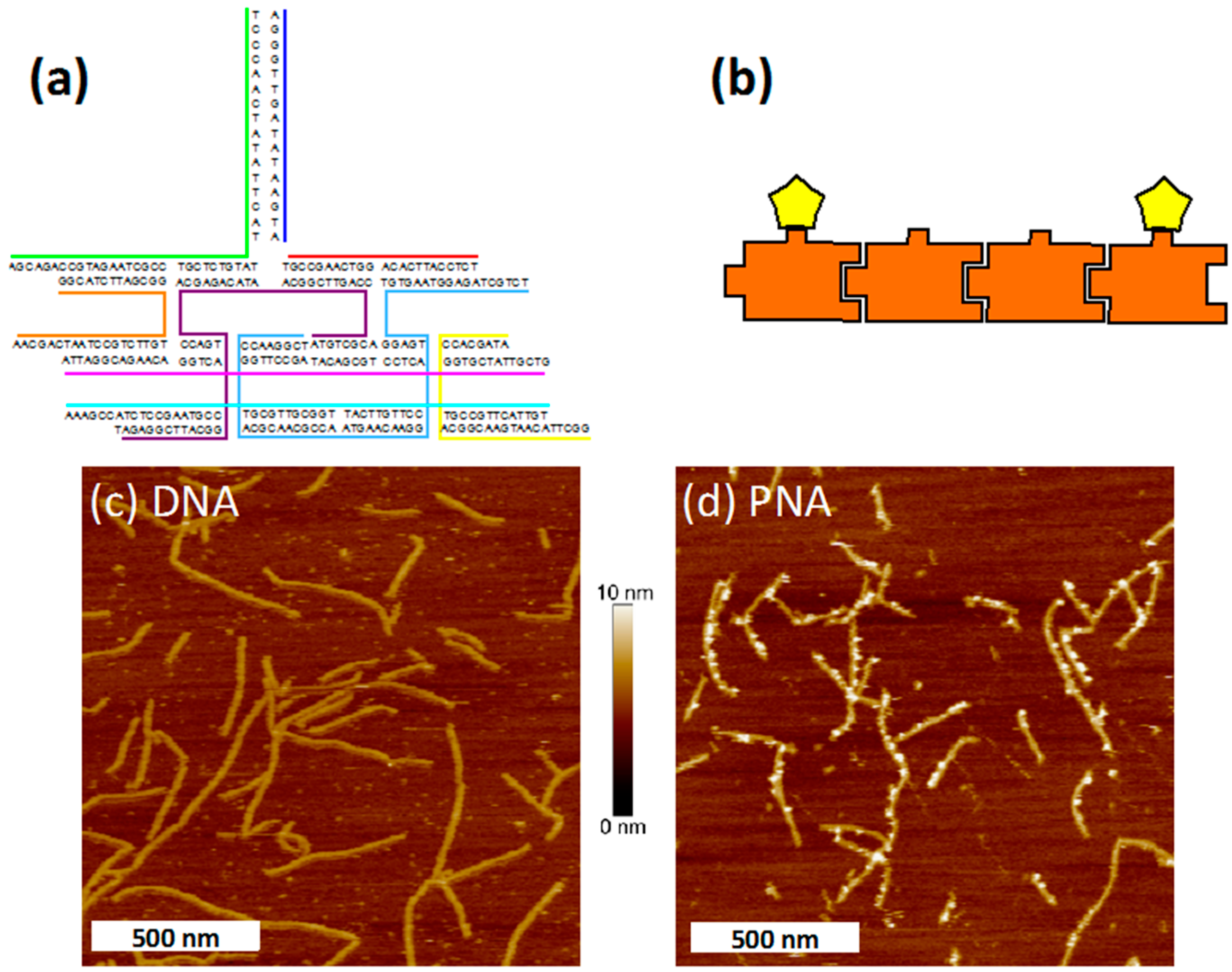{kind=link}
{kind=link}
| MgCl2 Concentration | DNA·DNA | PNA3K·DNA | ΔTm |
|---|---|---|---|
| 1 mM | 40 | 78 | 38 |
| 6 mM | 56 | 77 | 21 |
| 12.5 mM | 57 | 76 | 19 |
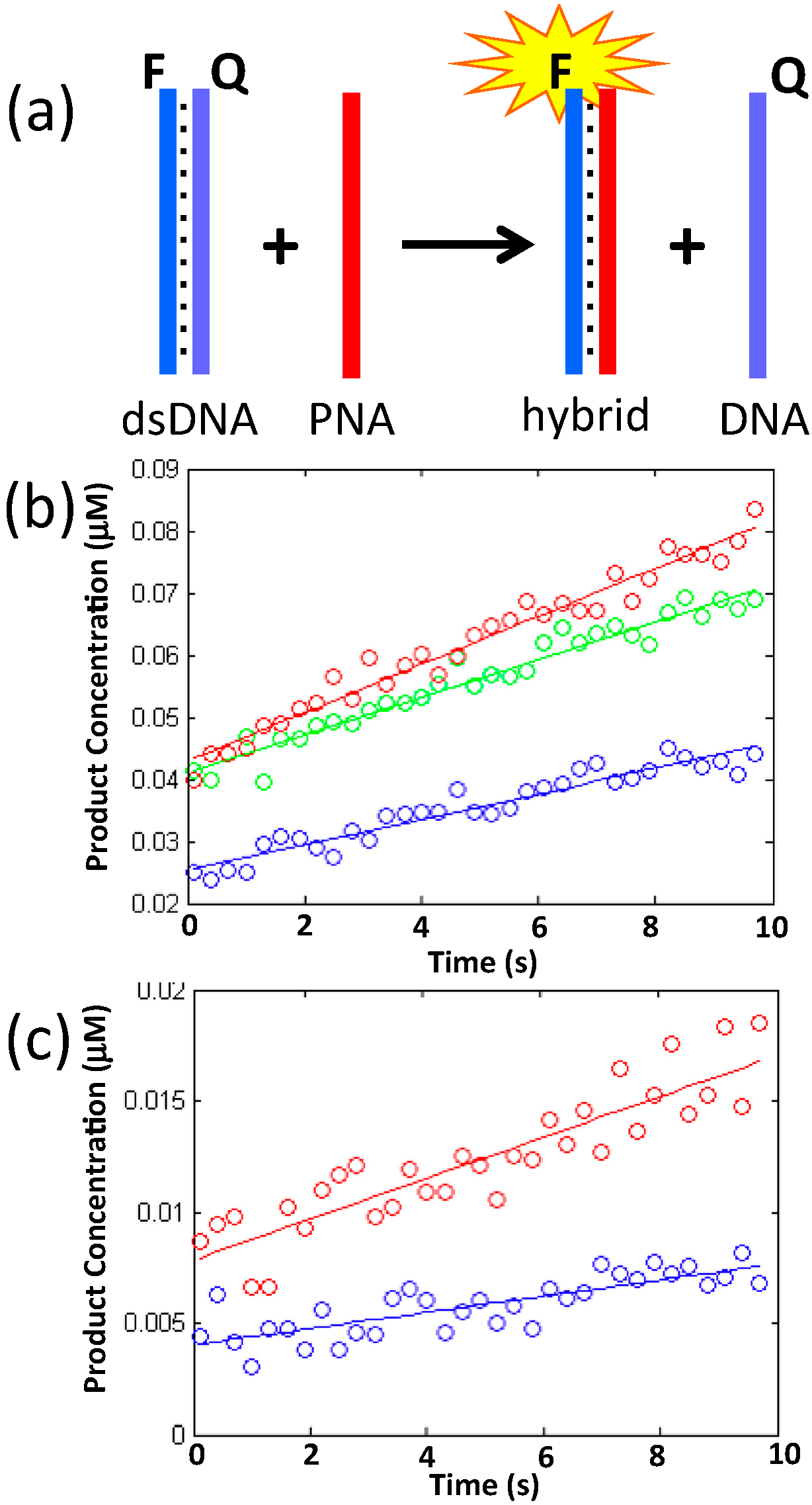
2.2. Duplex Invasion
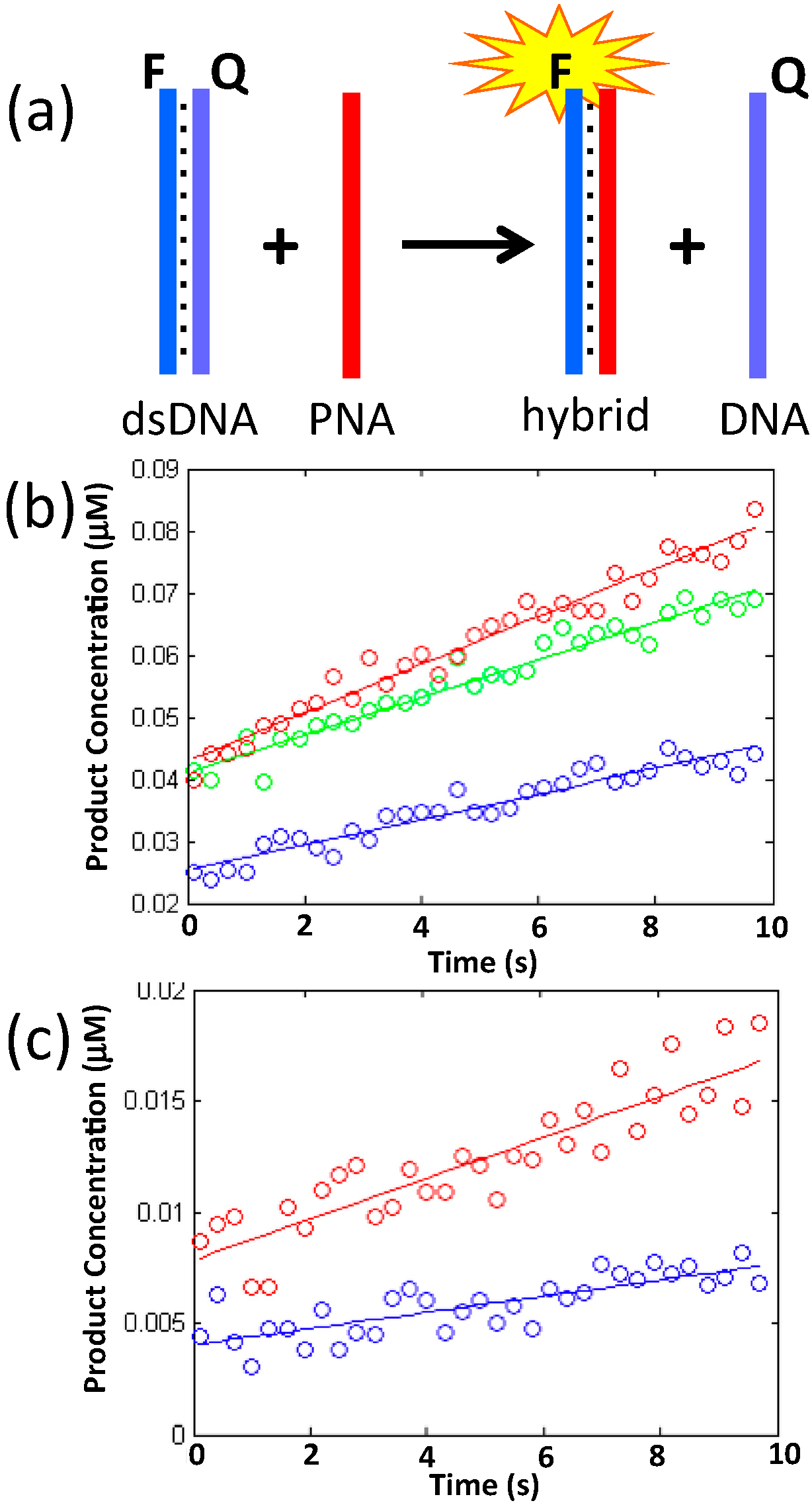
| 1 mM MgCl2 | 6 mM MgCl2 | 12.5 mM MgCl2 | |
|---|---|---|---|
| k (s−1∙M−1) | 0.0025 | 0.00072 | 0.00034 |
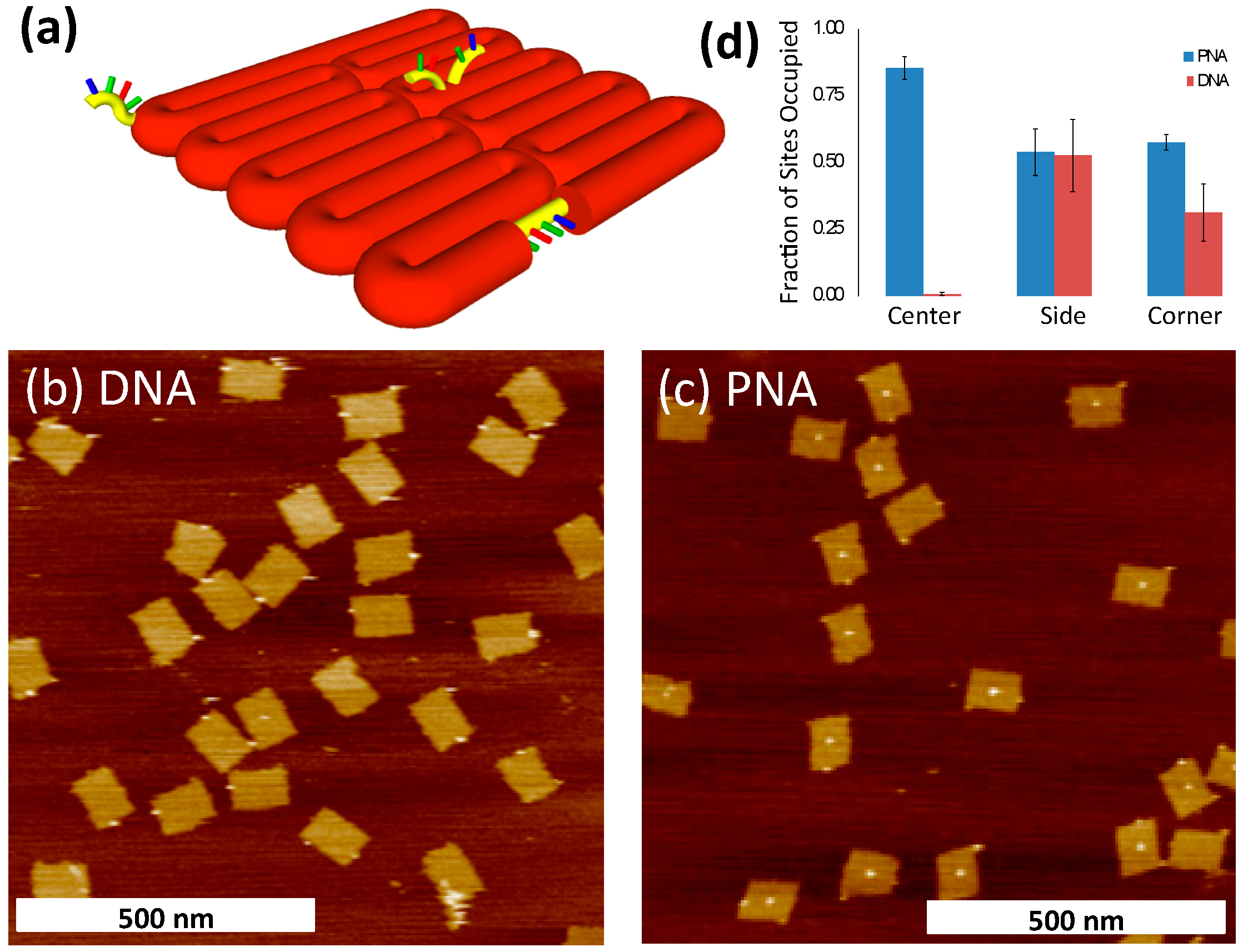
2.3. Binding of PNA to DNA Origami
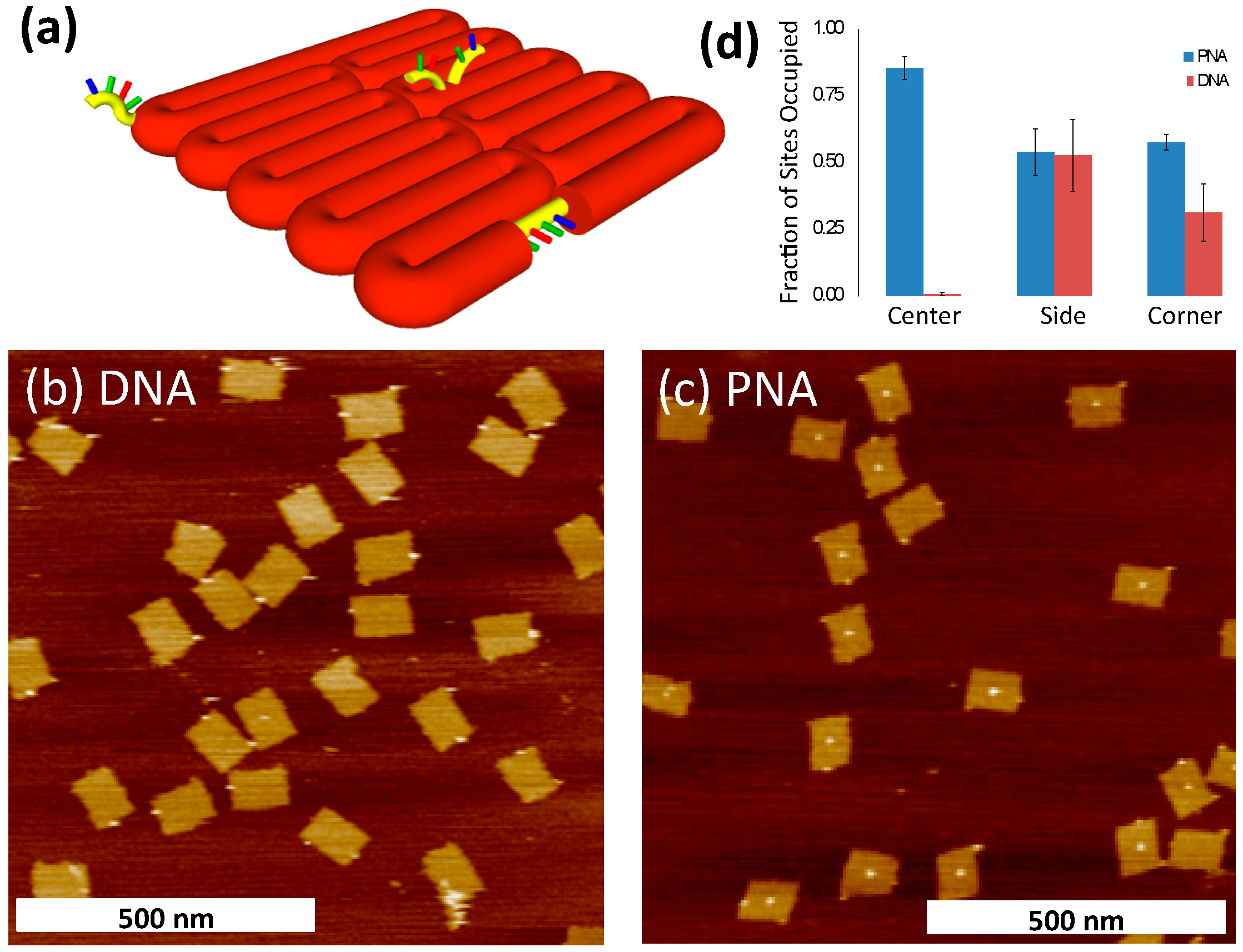
| bPNA3K | DNA | |
|---|---|---|
| Origami | 596 | 883 |
| Center site | 510 | 5 |
| Side site | 322 | 458 |
| Corner site | 344 | 276 |
3. Experimental Section
3.1. PNA Synthesis
3.2. UV-Vis Melting Curves
3.3. Nanostructure Assembly
3.4. Atomic Force Microscopy
3.5. Fluorescence
4. Conclusions
Supplementary Material
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seeman, N.C. Nanomaterials based on DNA. Ann. Rev. Biochem. 2010, 79, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Rangnekar, A.; LaBean, T.H. Building DNA nanostructures for molecular computation, templated assembly, and biological applications. Acc. Chem. Res. 2014, 47, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, A.V.; Han, D.; Shih, W.M.; Yan, H. Challenges and opportunities for structural DNA nanotechnology. Nat. Nanotech. 2011, 6, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Saaem, I.; LaBean, T.H. Overview of DNA origami for molecular self-assembly. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Yang, Y.; Sugiyama, H. DNA origami technology for biomaterials applications. Biomed. Sci. 2013, 1, 347–360. [Google Scholar] [CrossRef]
- Li, H.; LaBean, T.H.; Leong, K.W. Nucleic acid-based nanoengineering: Novel structures for biomedical applications. Interface Focus 2011, 1, 702–724. [Google Scholar] [CrossRef] [PubMed]
- Aldaye, F.A.; Senapedis, W.T.; Silver, P.A.; Way, J.C. A structurally tunable DNA-based extracellular matrix. J. Am. Chem. Soc. 2010, 132, 14727–14729. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, R.O.; Loboa, E.G.; LaBean, T.H. Sensitization of transforming growth factor-β signaling by multiple peptides patterned on DNA nanostructures. Biomacromolecules 2013, 14, 4157–4160. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Yang, C.-S.; Huang, D.-M. Aptamer-conjugated DNA icosahedral nanoparticles as a carrier of doxorubicin for cancer therapy. ACS Nano 2011, 5, 6156–6163. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Song, C.; Nangreave, J.; Liu, X.; Lin, L.; Qiu, D.; Wang, Z.-G.; Zou, G.; Liang, X.; Yan, H.; et al. DNA origami as a carrier for circumvention of drug resistance. J. Am. Chem. Soc. 2012, 134, 13396–13403. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, Y.; Yu, T.; Clifford, C.; Liu, Y.; Yan, H.; Chang, Y. A DNA nanostructure platform for directed assembly of synthetic vaccines. Nano Lett. 2012, 12, 4254–4259. [Google Scholar] [CrossRef] [PubMed]
- Verena, J.; Schuller, S.H.; Sandholzer, N.; Nickels, P.C.; Suhartha, N.A.; Endres, S.; Bourquin, C.; Liedl, T. Cellular immunostimulation by CpG-sequence-coated DNA origami structures. ACS Nano 2011, 5, 9696–9702. [Google Scholar]
- Li, J.; Pei, H.; Zhu, B.; Liang, L.; Wei, M.; He, Y.; Chen, N.; Li, D.; Huang, Q.; Fan, C. Self-assembled multivalent DNA nanostructures for noninvasive intracellular delivery of immunostimulatory CpG oligonucleotides. ACS Nano 2011, 5, 8783–8789. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, D.; Surana, S.; Chakraborty, S.; Koushika, S.P.; Krishnan, Y. A synthetic icosahedral DNA-based host-cargo complex for functional in vivo imaging. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Chhabra, R.; Andersen, C.S.; Gothelf, K.V.; Yan, H.; Liu, Y. Toward reliable gold nanoparticle patterning on self-assembled DNA nanoscaffold. J. Am. Chem. Soc. 2008, 130, 7820–7821. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Deng, Z.; Yan, H.; Cabrini, S.; Zuckermann, R.N.; Bokor, J. Gold nanoparticle self-similar chain structure organized by DNA origami. J. Am. Chem. Soc. 2010, 132, 3248–3249. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.M.; Micheel, C.M.; Bozano, L.D.; Osterbur, L.W.; Wallraff, G.M.; Cha, J.N. Large-area spatially ordered arrays of gold nanoparticles directed by lithographically confined DNA origami. Nat. Nanotechnol. 2010, 5, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Pilo-Pais, M.; Goldberg, S.; Samano, E.; LaBean, T.H.; Finkelstein, G. Connecting the nanodots: Programmable nanofabrication of fused metal shapes on DNA templates. Nano Lett. 2011, 11, 3489–3492. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lan, X.; Wang, Q. DNA origami directed large-scale fabrication of nanostructures resembling room temperature single-electron transistors. Small 2013, 3567–3571. [Google Scholar] [CrossRef] [PubMed]
- Samano, E.C.; Pilo-Pais, M.; Goldberg, S.; Vogen, B.N.; Finkelstein, G.; LaBean, T.H. Self-assembling DNA templates for programmed artificial biomineralization. Soft Matter. 2011, 7, 3240–3245. [Google Scholar] [CrossRef]
- Voigt, N.V.; Torring, T.; Rotaru, A.; Jacobsen, M.F.; Ravnsbaek, J.B.; Subramani, R.; Mamdouh, W.; Kjems, J.; Mokhir, A.; Besenbacher, F.; et al. Single-molecule chemical reactions on DNA origami. Nat. Nanotechnol. 2010, 5, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Jahn, K.; Tørring, T.; Voigt, N.V.; Sørensen, R.S.; Bank Kodal, A.L.; Andersen, E.S.; Gothelf, K.V.; Kjems, J. Functional patterning of DNA origami by parallel enzymatic modification. Bioconjugate Chem. 2011, 22, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Yoshidome, T.; Endo, M.; Kashiwazaki, G.; Hidaka, K.; Bando, T.; Sugiyama, H. Sequence-selective single-molecule alkylation with a pyrrole-imidazole polyamide visualized in a DNA nanoscaffold. J. Am. Chem. Soc. 2012, 134, 4654–4660. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Tian, Y.; Ribbe, A.E.; Mao, C. Antibody nanoarrays with a pitch of ~20 nanometers. J. Am. Chem. Soc. 2006, 128, 12664–12665. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; LaBean, T.H.; Kenan, D.J. Single-chain antibodies against DNA aptamers for use as adapter molecules on DNA tile arrays in nanoscale materials organization. Org. Biomol. Chem. 2006, 4, 3420–3426. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, R.; Sharma, J.; Ke, Y.; Liu, Y.; Rinker, S.; Lindsay, S.; Yan, H. Spatially addressable multiprotein nanoarrays templated by aptamer-tagged DNA nanoarchitectures. J. Am. Chem. Soc. 2007, 129, 10304–10305. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.A.R.; Lund, K.; Liu, Y.; Yan, H.; Chaput, J.C. Self-assembled peptide nanoarrays: An approach to studying protein-protein interactions. Angew. Chem. Int. Ed. 2007, 46, 3051–3054. [Google Scholar] [CrossRef] [PubMed]
- Saccà, B.; Meyer, R.; Erkelenz, M.; Kiko, K.; Arndt, A.; Schroeder, H.; Rabe, K.S.; Niemeyer, C.M. Orthogonal protein decoration of DNA origami. Angew. Chem. Int. Ed. 2010, 49, 9378–9383. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Niemeyer, C.M. Orthogonal protein decoration of DNA nanostructures. Small 2011, 7, 3211–3218. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.D.; LaBean, T.H. Organization of inorganic nanomaterials via programmable DNA self-assembly and peptide molecular recognition. ACS Nano 2011, 5, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Nakata, E.; Liew, F.F.; Uwatoko, C.; Kiyonaka, S.; Mori, Y.; Katsuda, Y.; Endo, M.; Sugiyama, H.; Morii, T. Zinc-finger proteins for site-specific protein positioning on DNA-origami structures. Angew. Chem. Int. Ed. 2012, 51, 2421–2424. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M. An introduction to peptide nucleic acid. Curr. Issues Mol. Biol. 1999, 1, 89–104. [Google Scholar] [PubMed]
- De Costa, N.T.S.; Heemstra, J.M. Evaluating the effect of ionic strength on duplex stability for PNA having negatively or positively charged side chains. PLoS ONE 2013, 8, e58670. [Google Scholar] [CrossRef] [PubMed]
- Flory, J.D.; Shinde, S.; Lin, S.; Liu, Y.; Yan, H.; Ghirlanda, G.; Fromme, P. PNA-peptide assembly in a 3D DNA nanocage at room temperature. J. Am. Chem. Soc. 2013, 135, 6985–6993. [Google Scholar] [CrossRef] [PubMed]
- Lukeman, P.S.; Mittal, A.C.; Seeman, N.C. Two dimensional PNA/DNA arrays: Estimating the helicity of unusual nucleic acid polymers. Chem. Commun. 2004, 1694–1695. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Klibanov, A.M. Nanocrystals modified with peptide nucleic acids (PNAs) for selective self-assembly and DNA detection. J. Am. Chem. Soc. 2003, 125, 12531–12540. [Google Scholar] [CrossRef] [PubMed]
- Xiaodi, S.; Kanjanawarut, R. Control of metal nanoparticles aggregation and dispersion by PNA and detection. ACS Nano 2009, 3, 2751–2759. [Google Scholar]
- Stadler, A.L.; Sun, D.; Maye, M.M.; Lelie, D.V.D.; Gang, O. Site-selective binding of nanoparticles to double-stranded DNA via peptide nucleic acid “invasion”. ACS Nano 2011, 5, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Stadler, A.L.; Gurevich, M.; Palma, E.; Stach, E.; van der Lelie, D.; Gang, O. Heterogeneous nanoclusters assembled by PNA-templated double-stranded DNA. Nanoscale 2012, 4, 6722–6725. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, D.; Famulok, M. Pseudo-complementary PNA actuators as reversible switches in dynamic DNA nanotechnology. Nucleic Acids Res. 2013, 41, 4729–4739. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Aiba, Y.; Yasuda, K.; Sakai, Y.; Yamanaka, Y.; Kuzuya, A.; Ohya, Y.; Komiyama, M. Clear-cut observation of PNA invasion using nanomechanical DNA origami devices. Chem. Commun. 2012, 48, 11361–11363. [Google Scholar] [CrossRef] [PubMed]
- Wittung, P.; Nielsen, P.; Norde, B. Direct observation of strand invasion by peptide nucleic acid (PNA) into double-stranded DNA. J. Am. Chem. Soc. 1996, 118, 7049–7054. [Google Scholar] [CrossRef]
- LaBean, T.H.; Yan, H.; Kopatsch, J.; Liu, F.; Winfree, E.; Reif, J.H.; Seeman, N.C. Construction, analysis, ligation, and self-assembly of DNA triple crossover complexes. J. Am. Chem. Soc. 2000, 122, 1848–1860. [Google Scholar] [CrossRef]
- Li, H.; Park, S.H.; Reif, J.H.; LaBean, T.H.; Yan, H. DNA-templated self-assembly of protein and nanoparticle linear arrays. J. Am. Chem. Soc. 2004, 126, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Lindsay, S.; Chang, Y.; Liu, Y.; Yan, H. Self-assembled water-soluble nucleic acid probe tiles for label-free RNA hybridization assays. Science 2008, 319, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Jungmann, R.; Steinhauer, C.; Scheible, M.; Kuzyk, A.; Tinnefeld, P.; Simmel, F.C. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett. 2010, 10, 4756–4761. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Buck, A.; Nangreave, J.; Jiang, S.; Yan, H.; Walter, N.G. Multifactorial modulation of binding and dissociation kinetics on two-dimensional DNA nanostructures. Nano Lett. 2013, 13, 2754–2759. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Hogberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2009, 459, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Douglas, S.M.; Liu, M.; Sharma, J.; Cheng, A.; Leung, A.; Liu, Y.; Shih, W.M.; Yan, H. Multilayer DNA origami packed on a square lattice. J. Am. Chem. Soc. 2009, 131, 15903–15908. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E. Peptide Nucleic Acids: Protocols and Applications, 2nd ed.; Horizon Bioscience: Norfolk, UK, 2004. [Google Scholar]
- Sample Availability: Samples of the materials not available but can be constructed by the methods described in the manuscript.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedersen, R.O.; Kong, J.; Achim, C.; LaBean, T.H. Comparative Incorporation of PNA into DNA Nanostructures. Molecules 2015, 20, 17645-17658. https://doi.org/10.3390/molecules200917645
Pedersen RO, Kong J, Achim C, LaBean TH. Comparative Incorporation of PNA into DNA Nanostructures. Molecules. 2015; 20(9):17645-17658. https://doi.org/10.3390/molecules200917645
Chicago/Turabian StylePedersen, Ronnie O., Jing Kong, Catalina Achim, and Thomas H. LaBean. 2015. "Comparative Incorporation of PNA into DNA Nanostructures" Molecules 20, no. 9: 17645-17658. https://doi.org/10.3390/molecules200917645