
Enantioselective Cycloaddition Reactions Catalyzed by BINOL-Derived Phosphoric Acids and N-Triflyl Phosphoramides: Recent Advances



Abstract
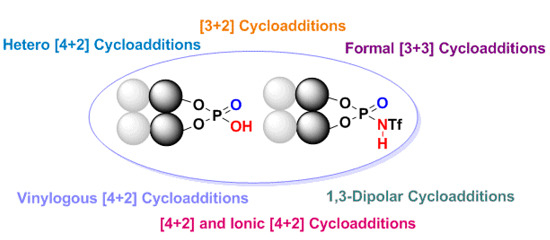
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Cycloadditions Catalyzed by Chiral BINOL-Derived Phosphoric Acids
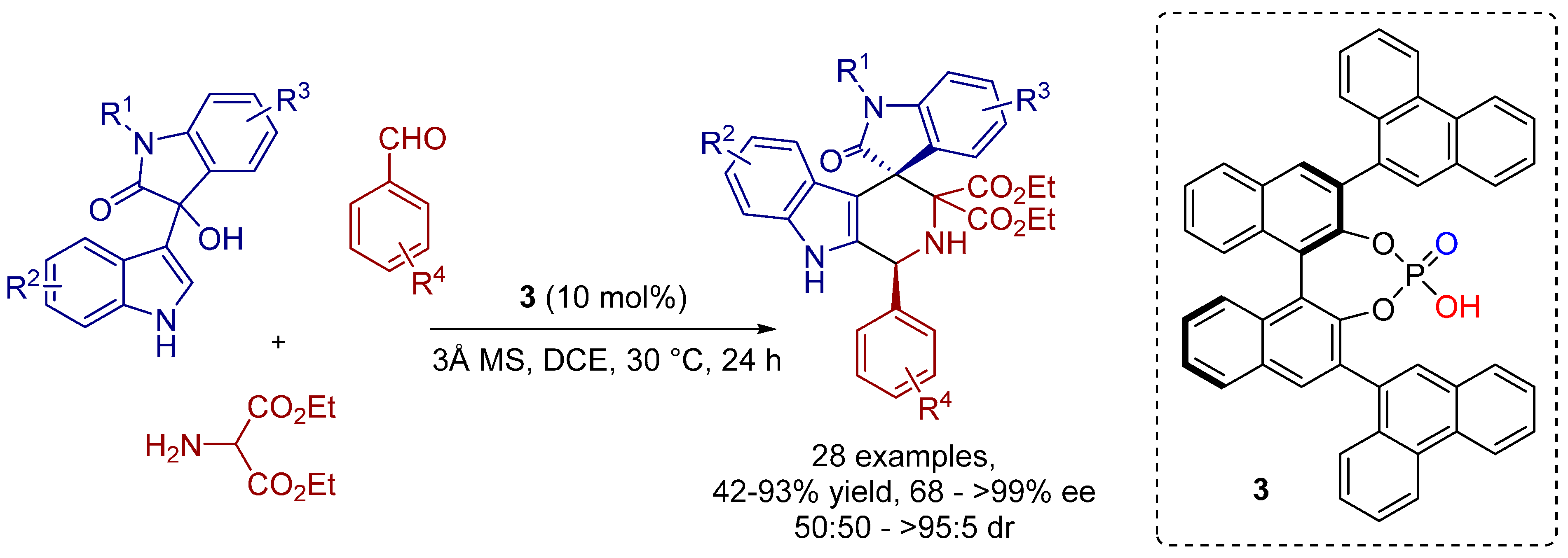
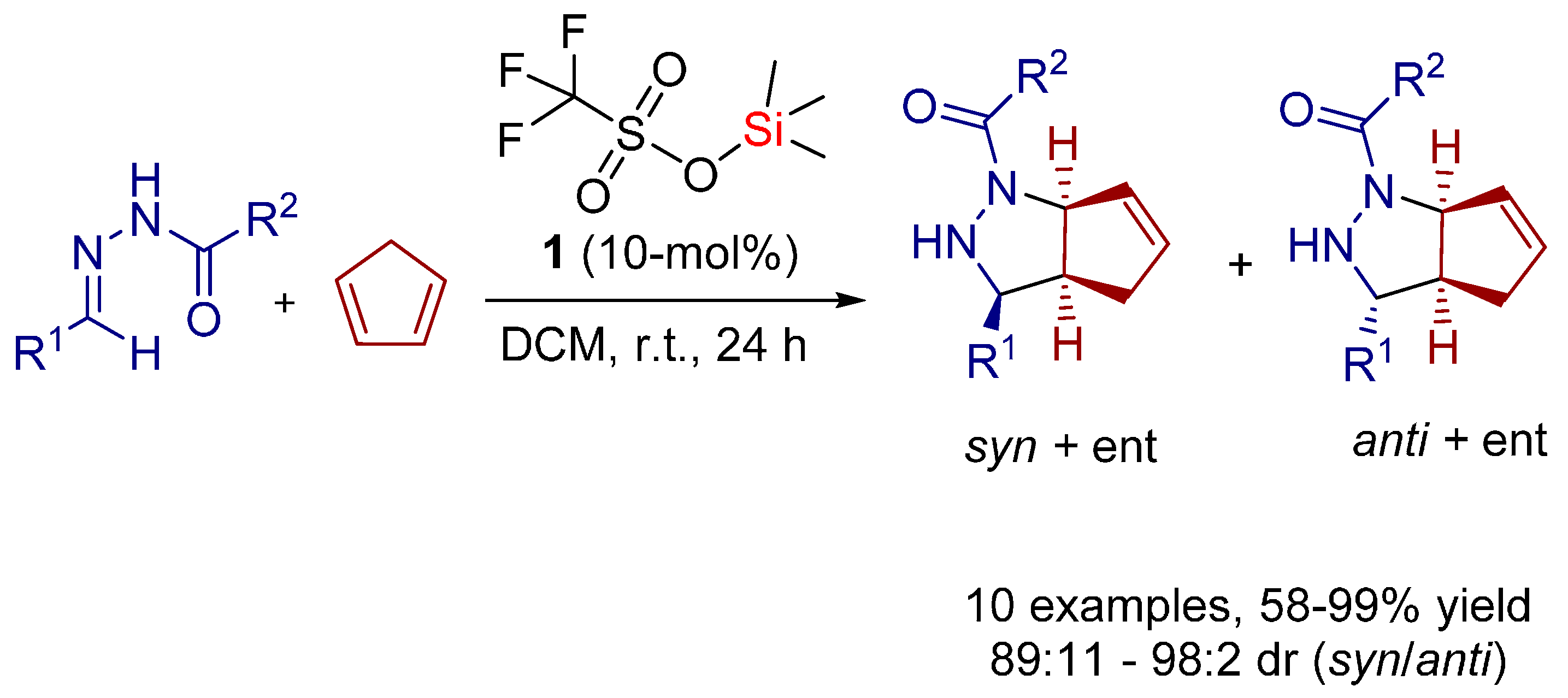
2.1. [3+2] Cycloadditions

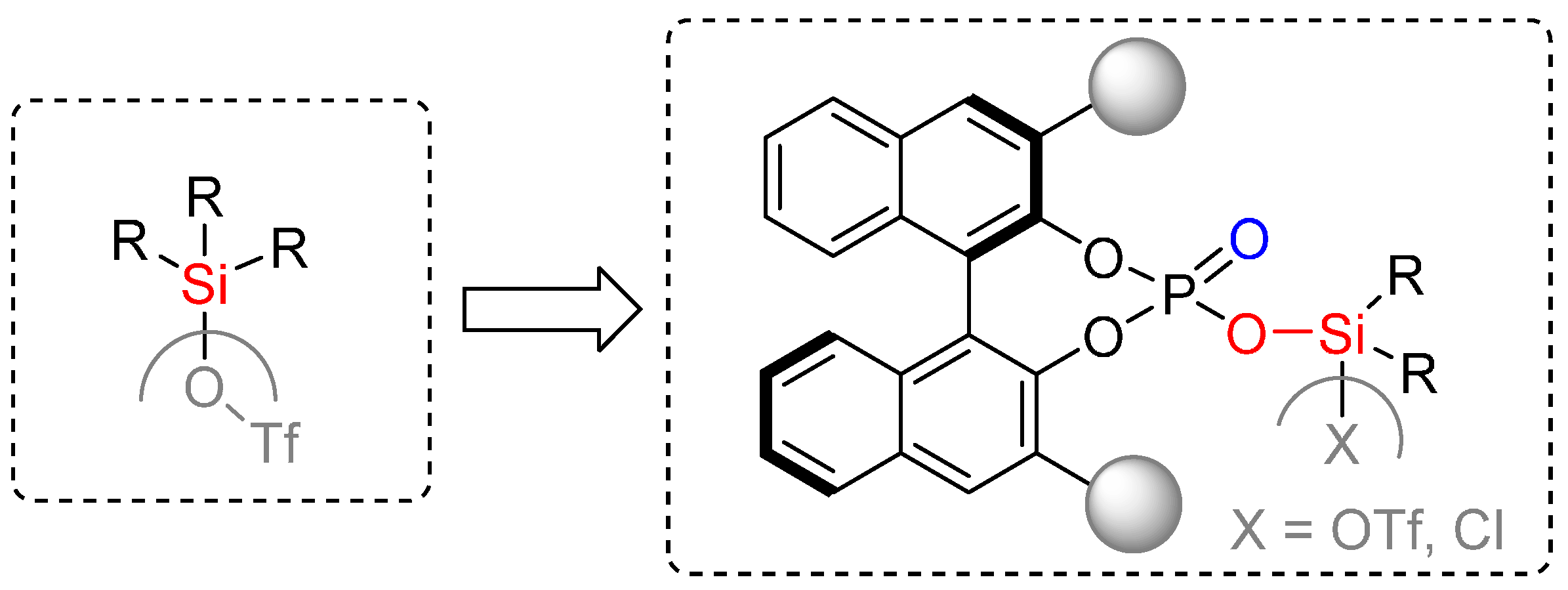
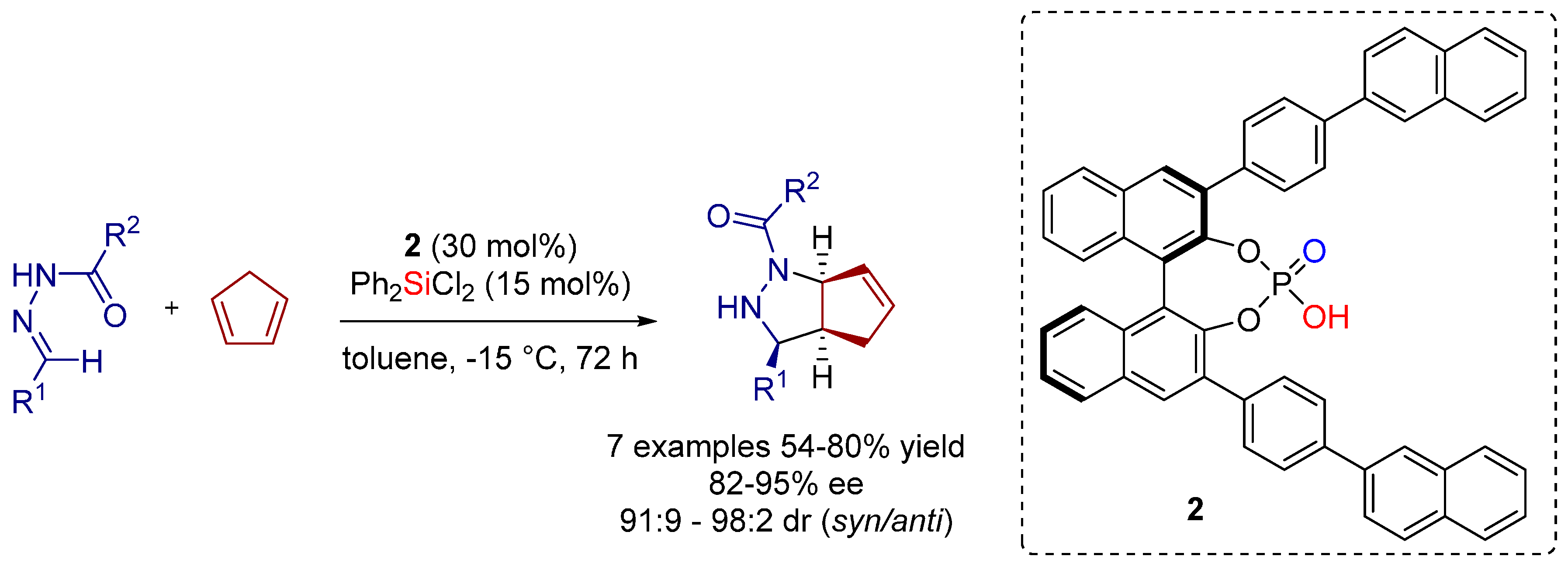
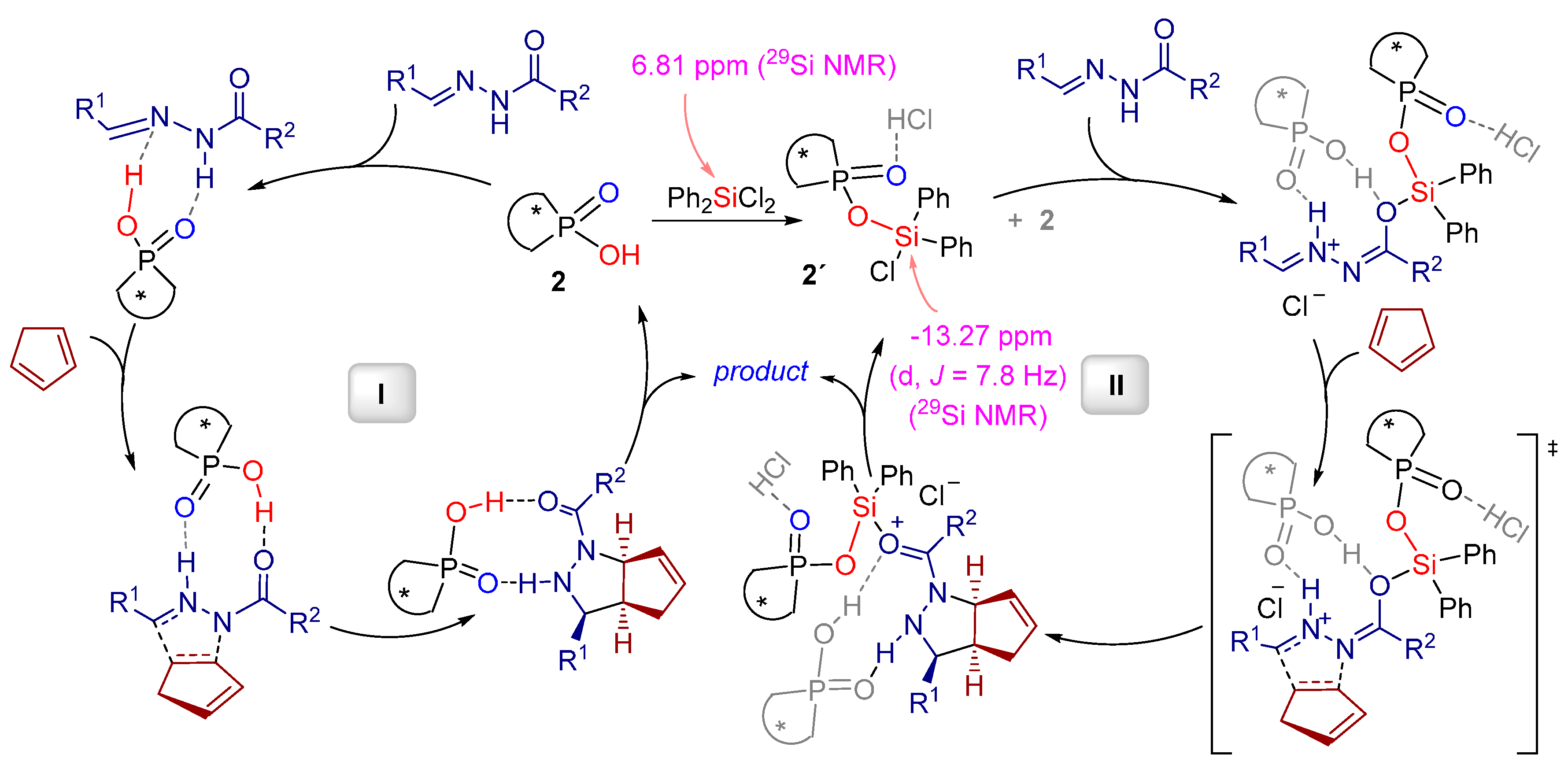
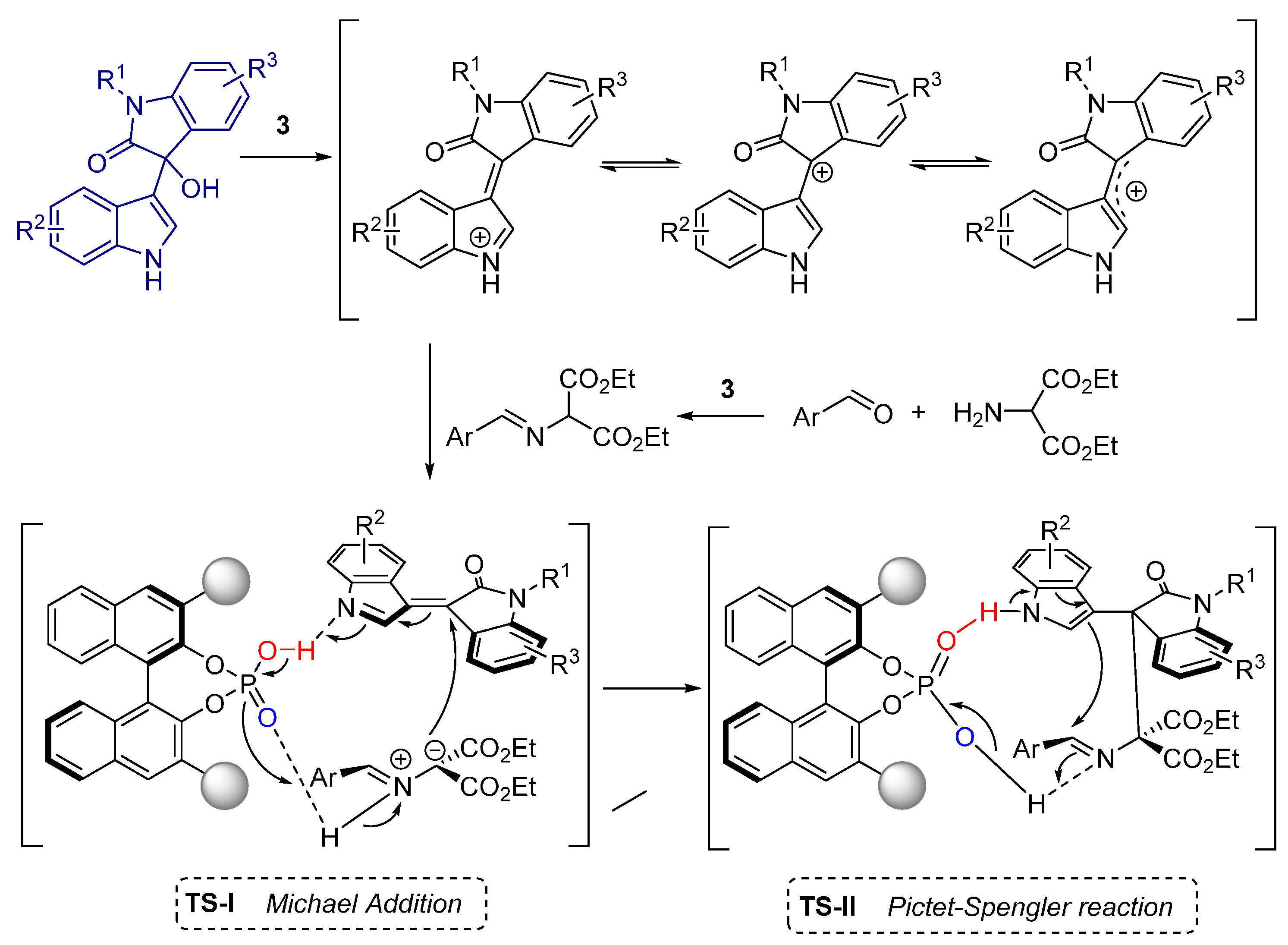
2.2. Formal [3+3] Cycloadditions

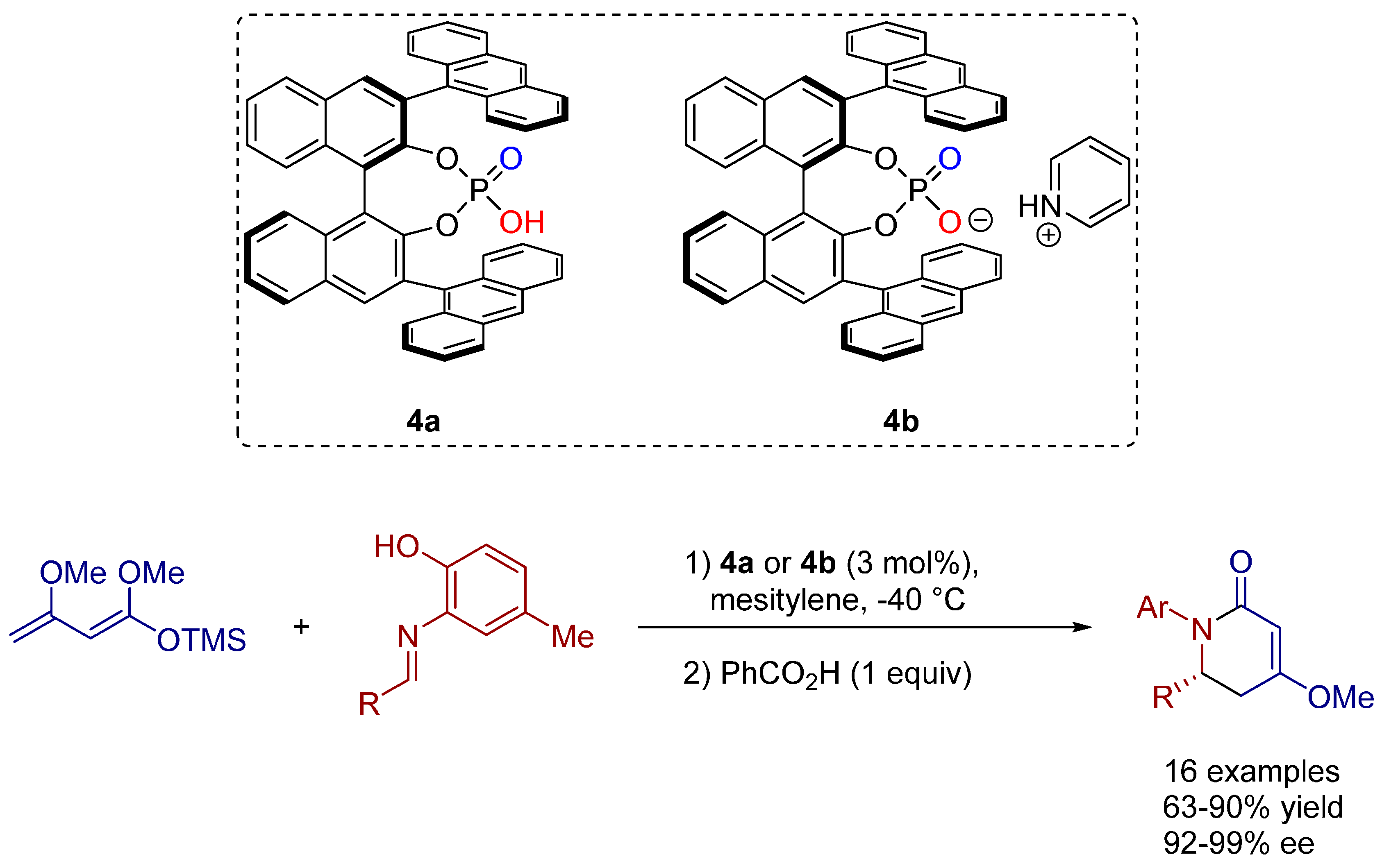
2.3. [4+2] and Vinylogous [4+2] Cycloadditions
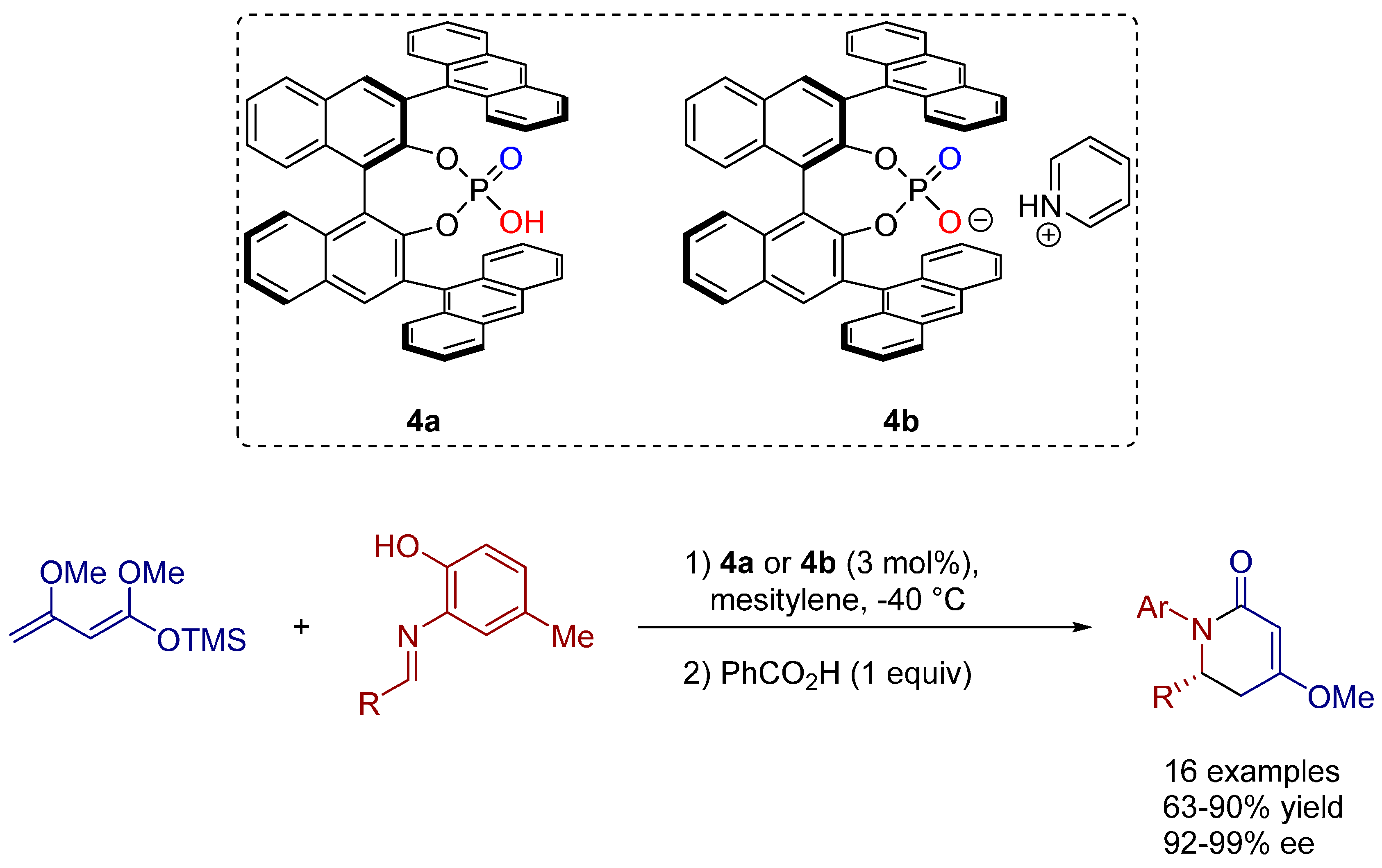
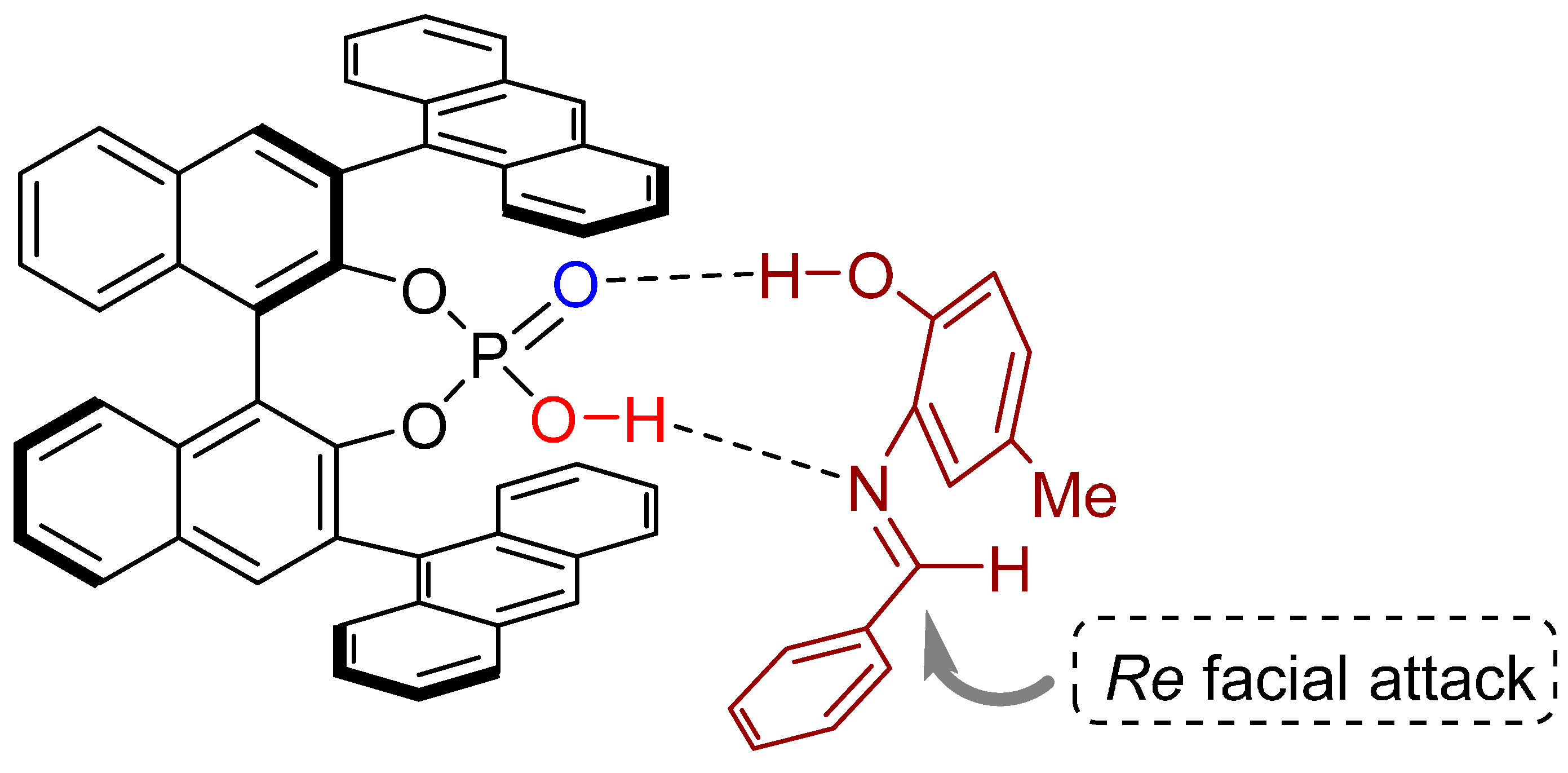

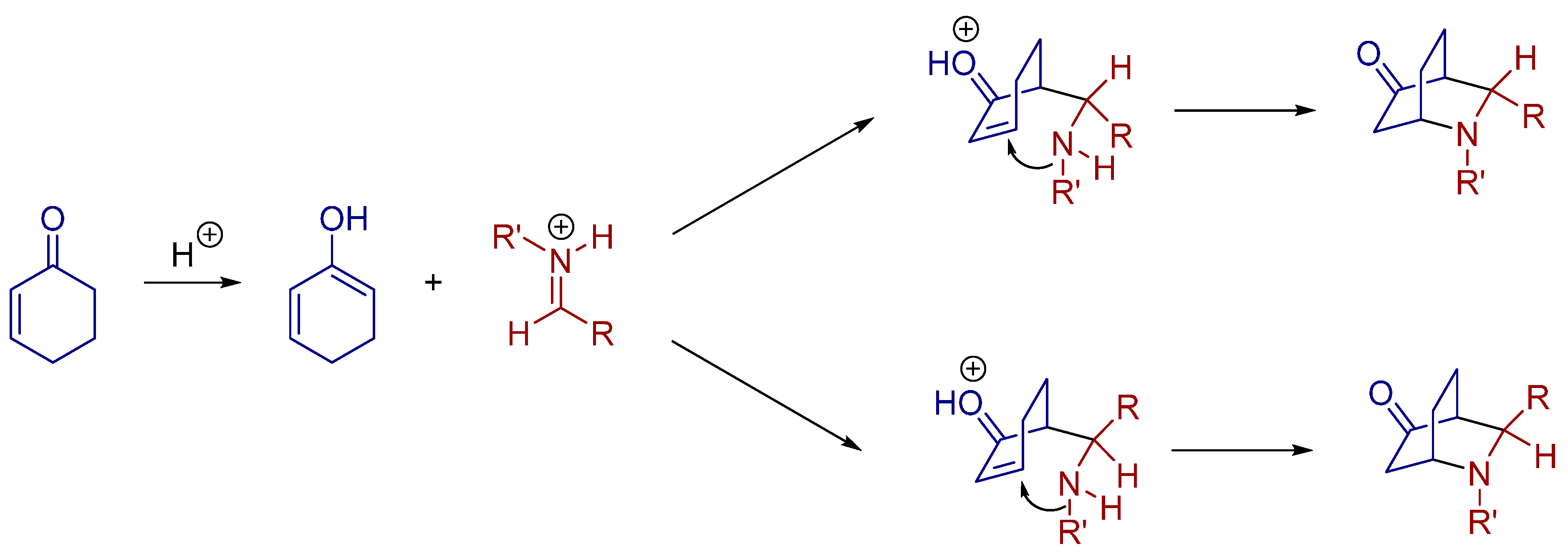
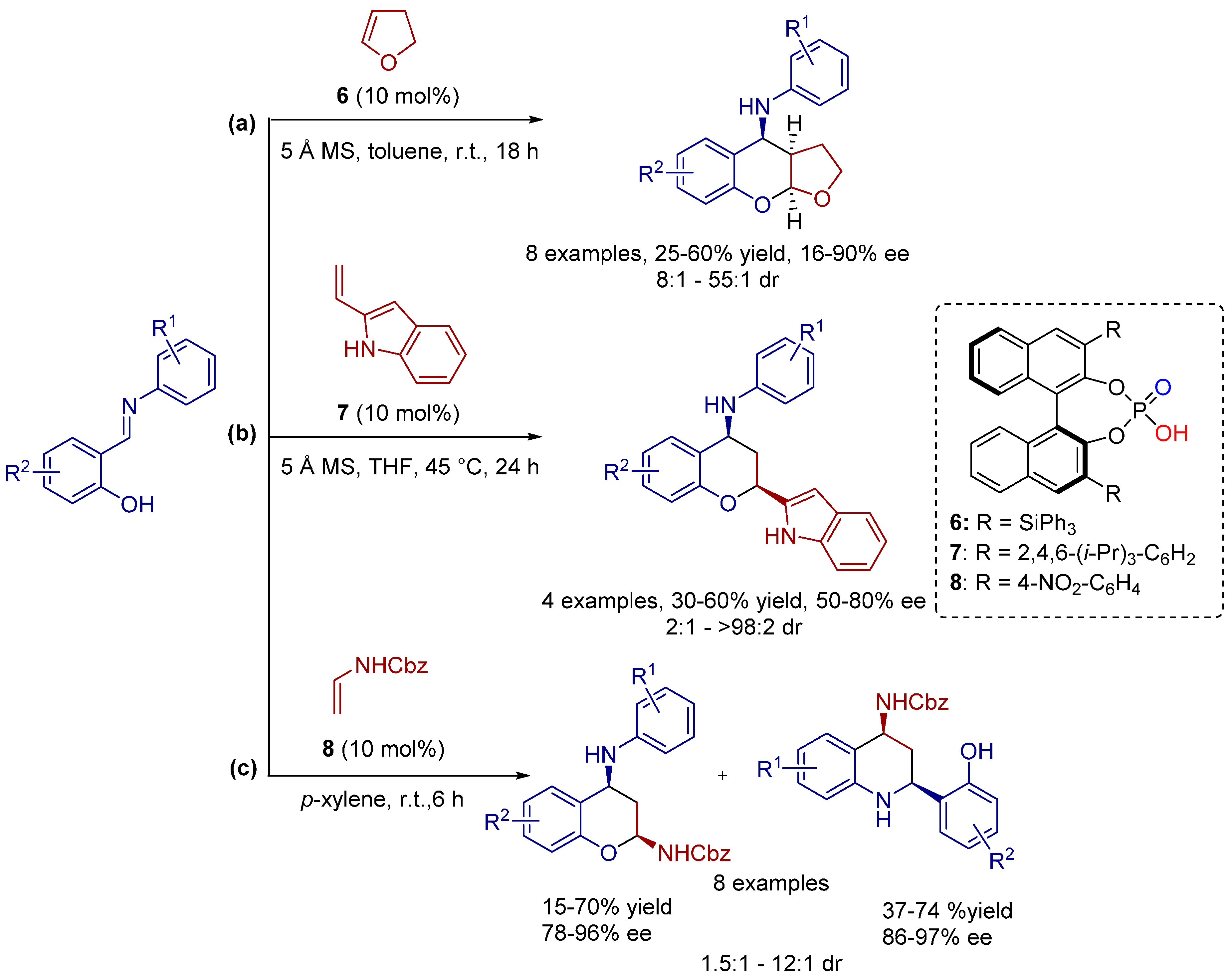
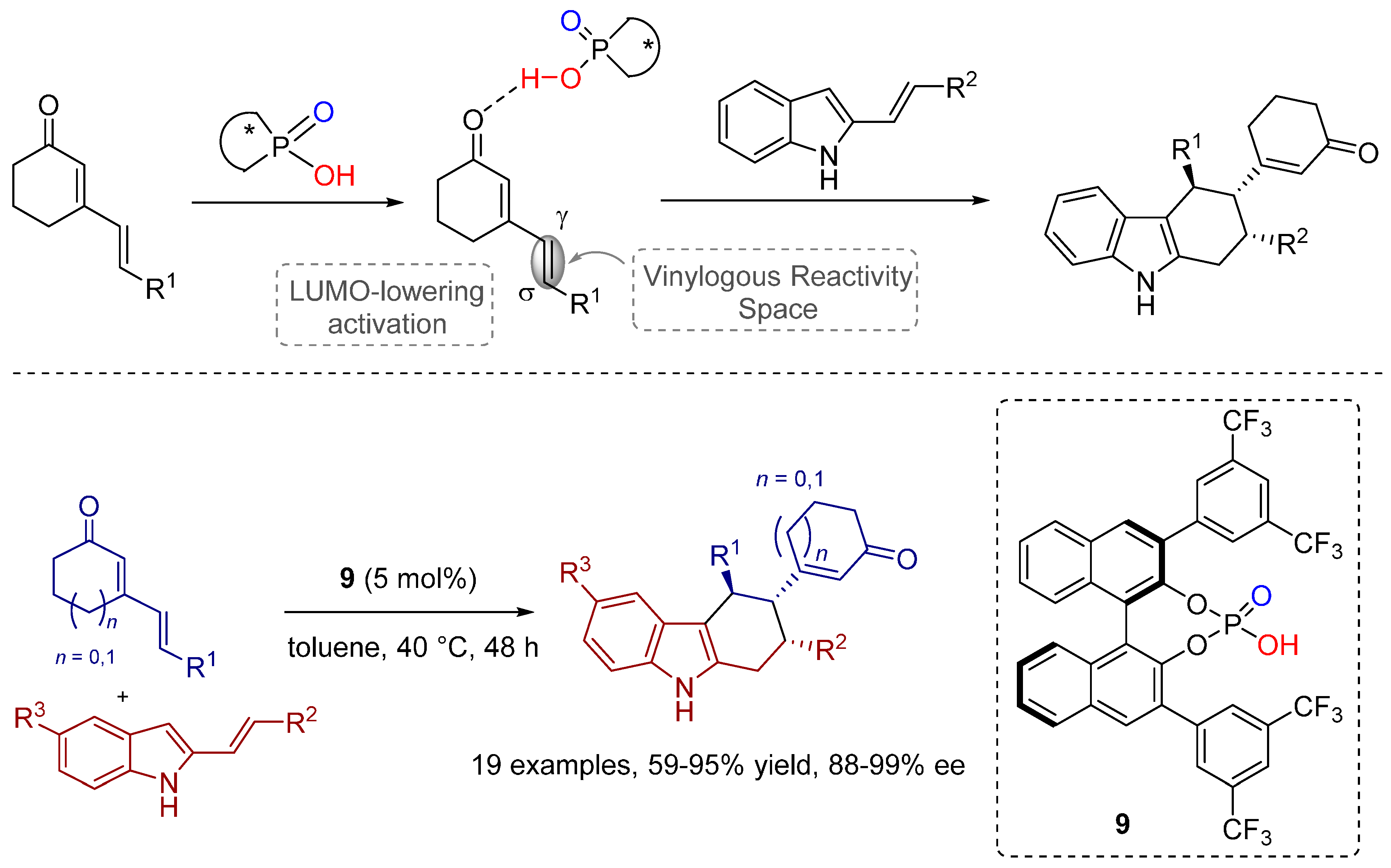

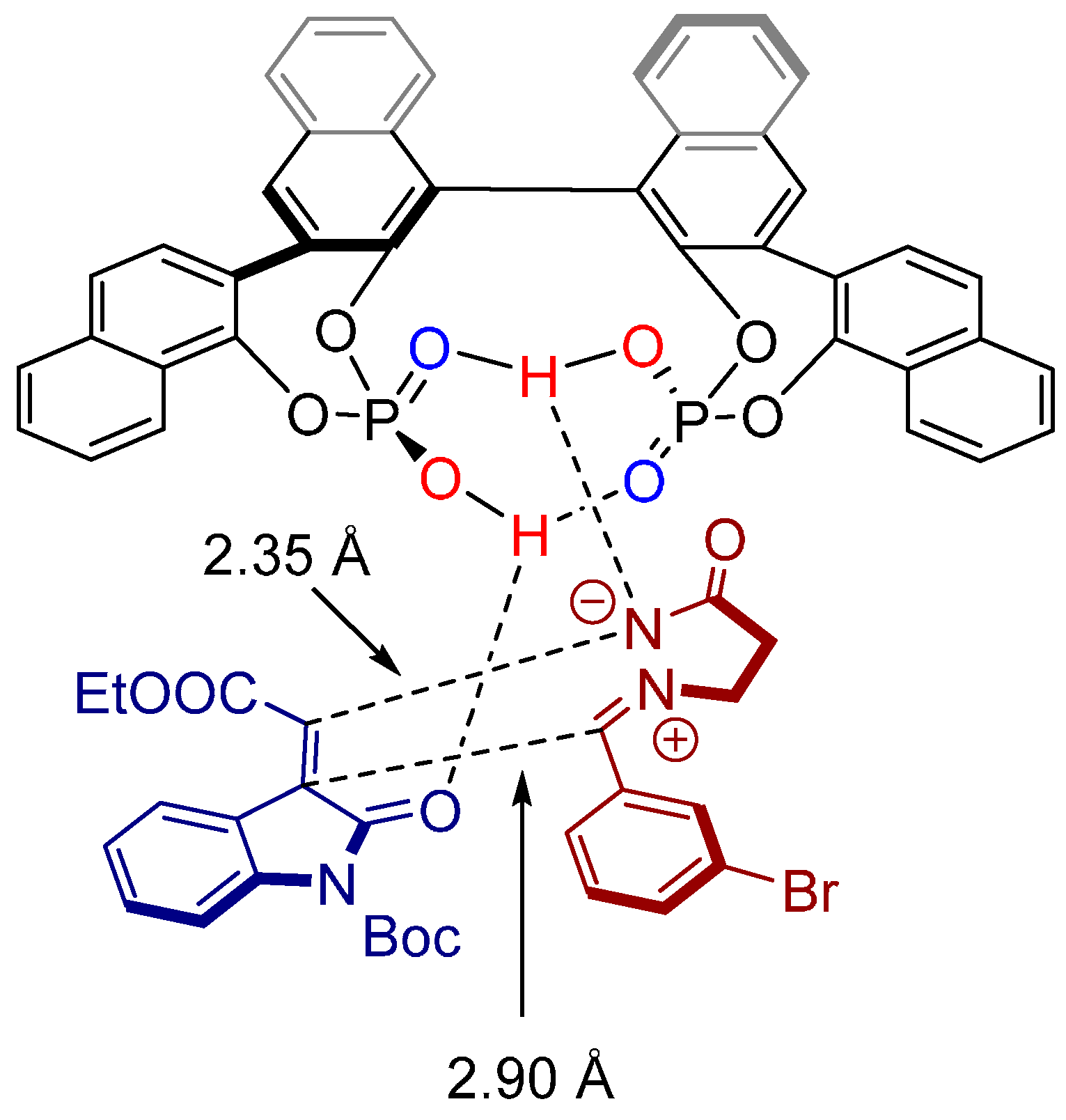
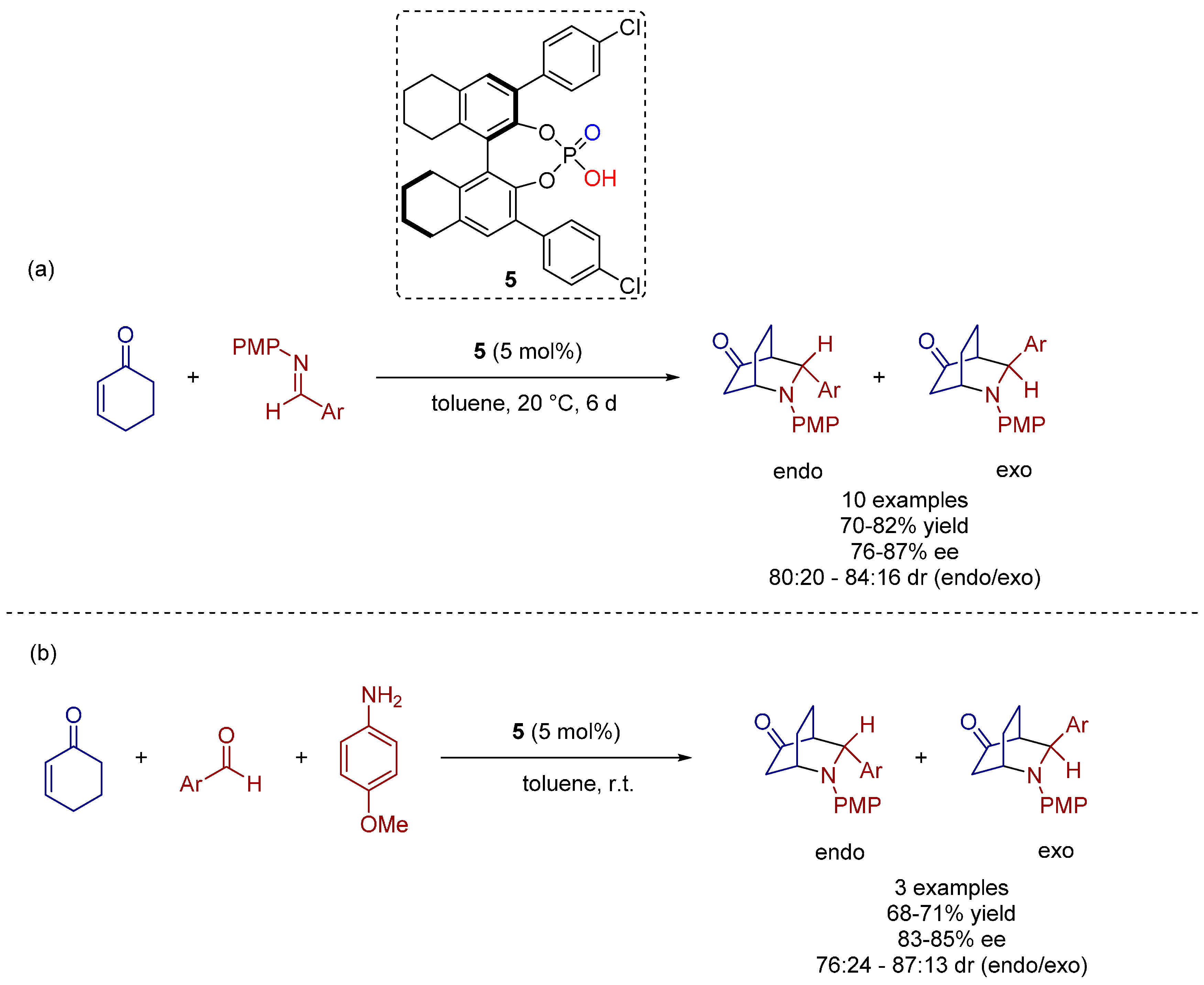
2.4. 1,3-Dipolar Cycloadditions

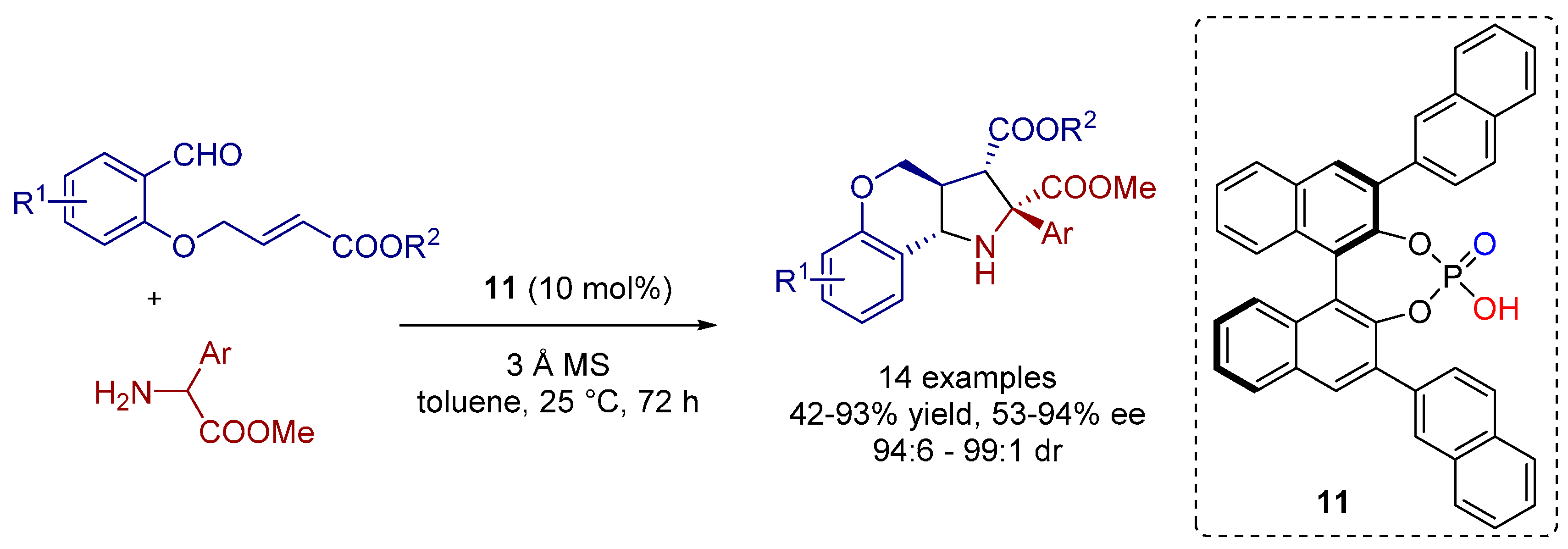


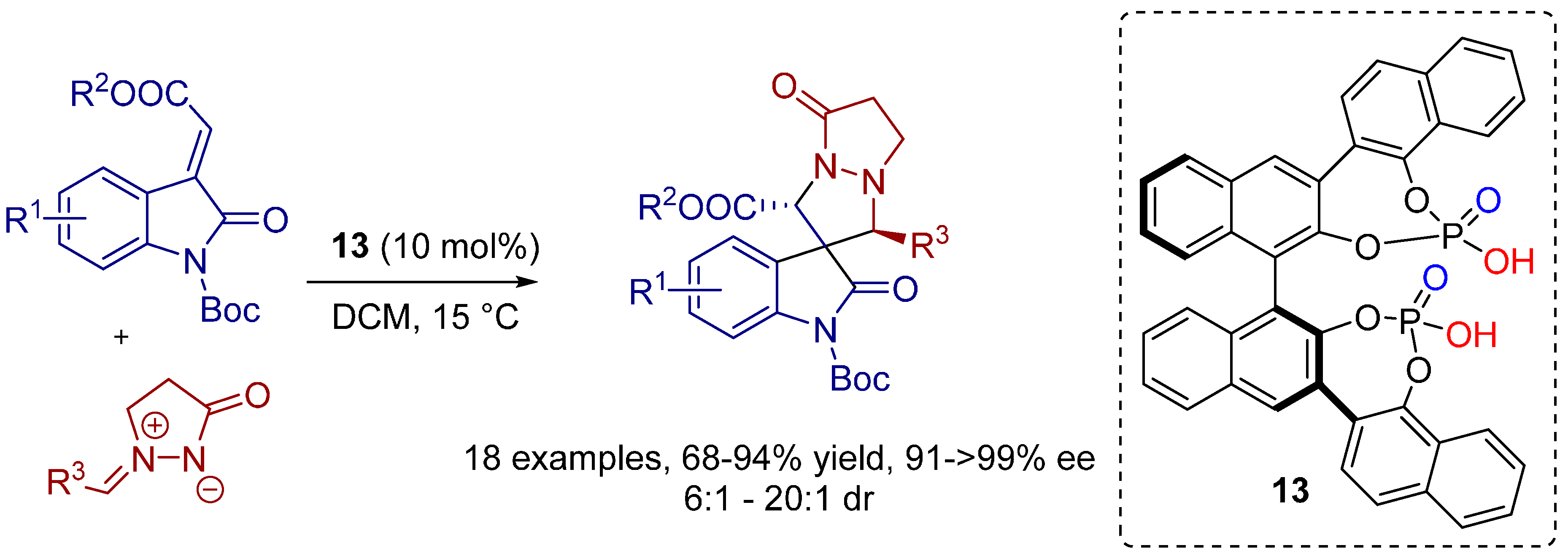
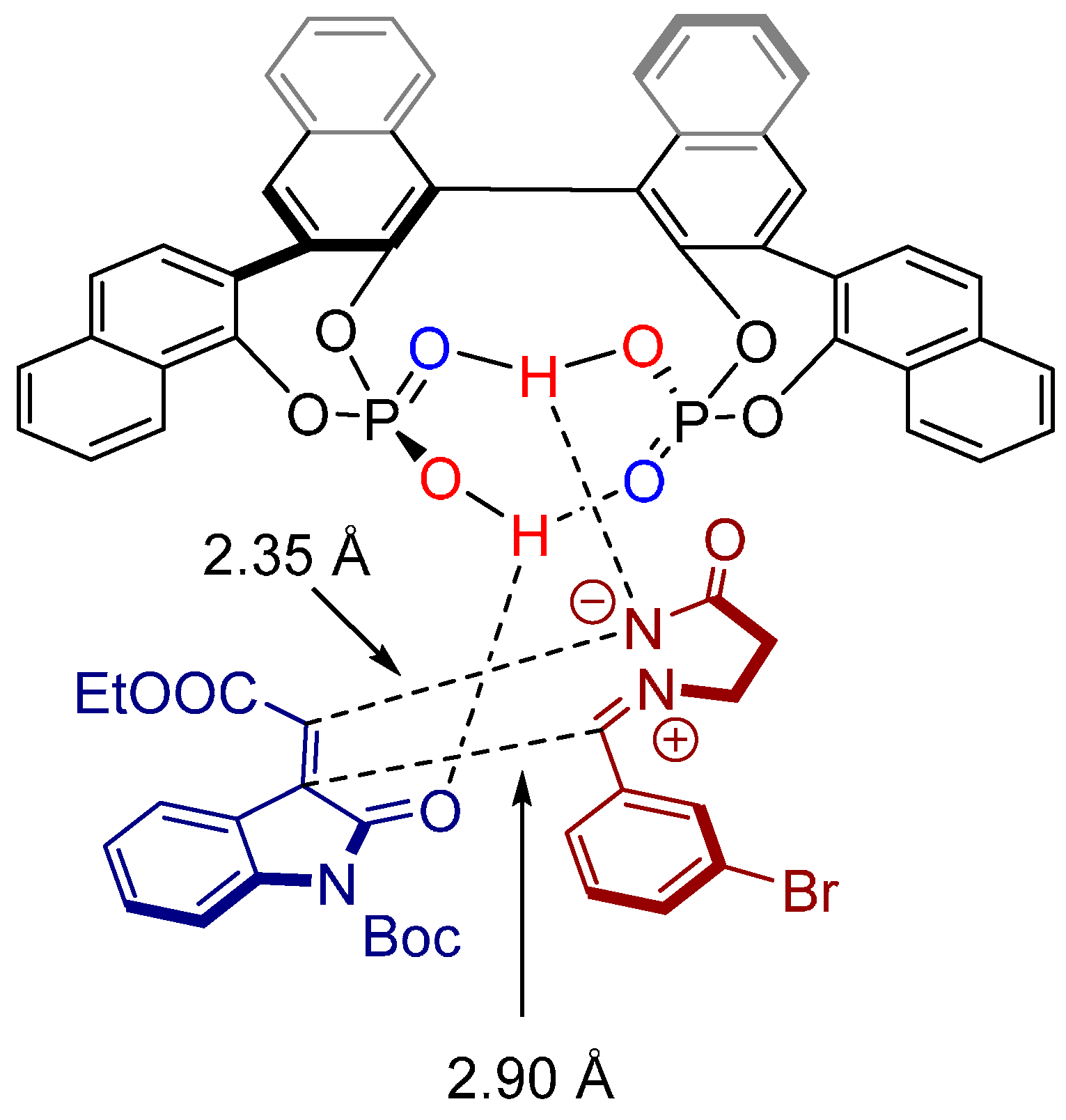
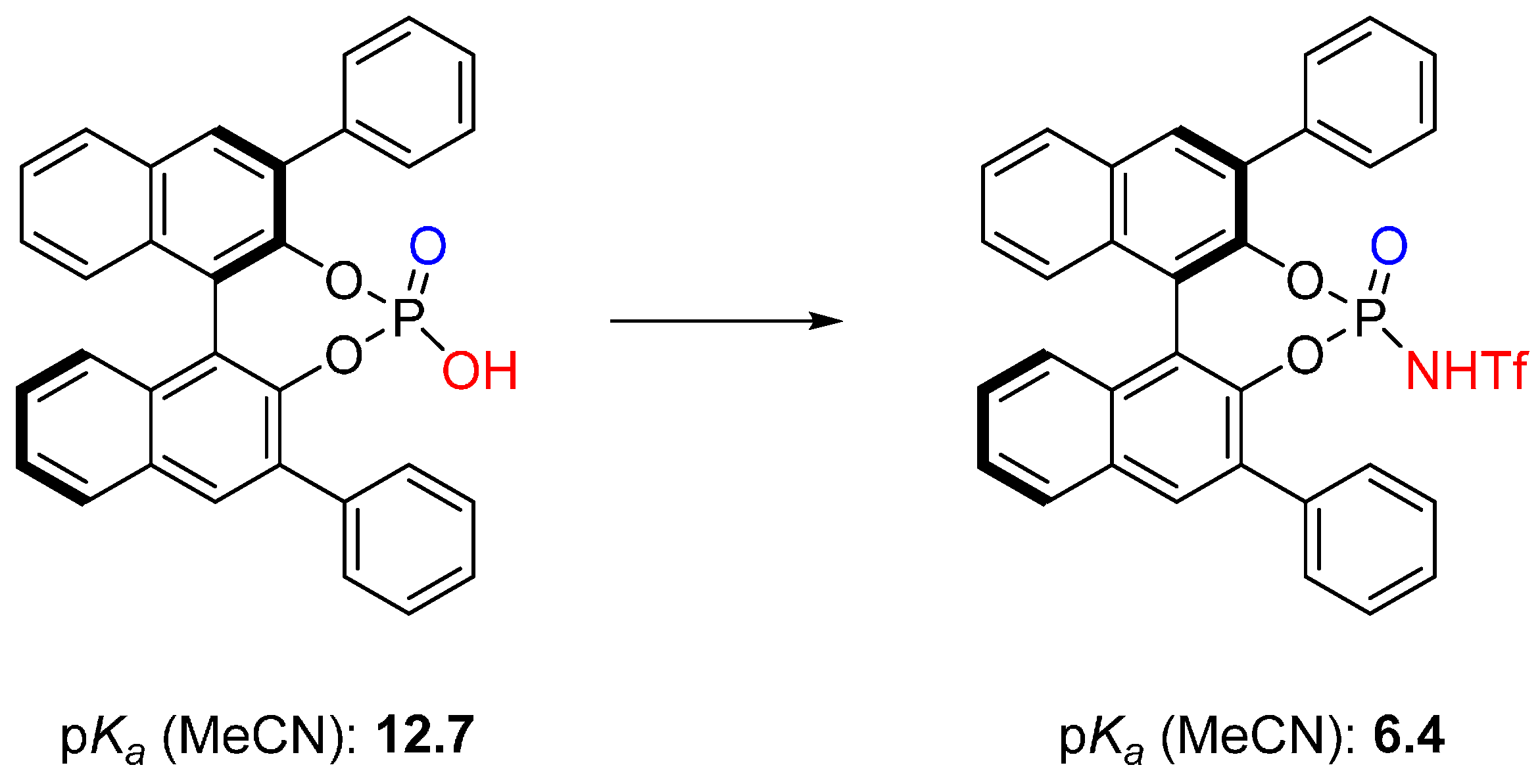
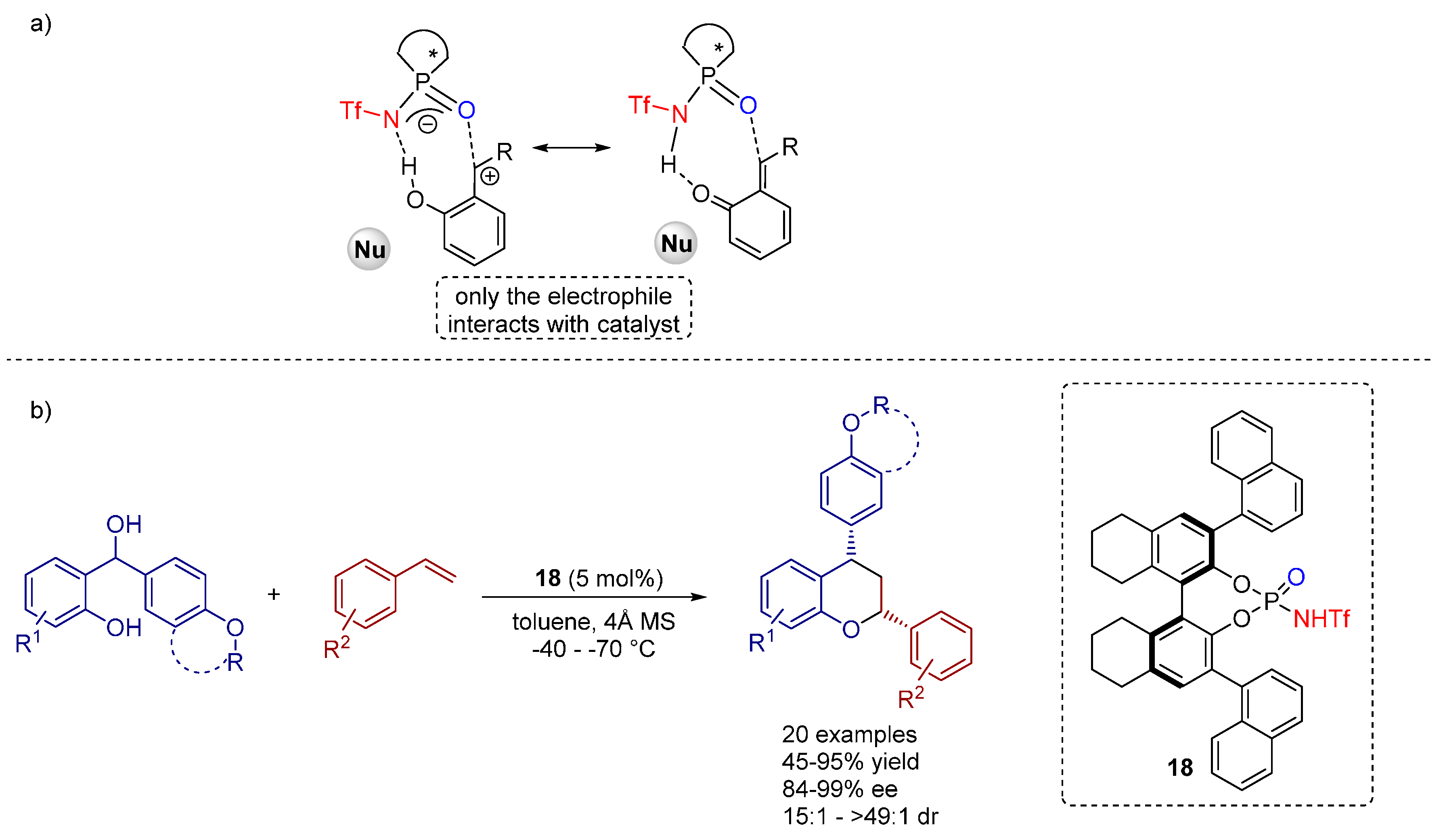
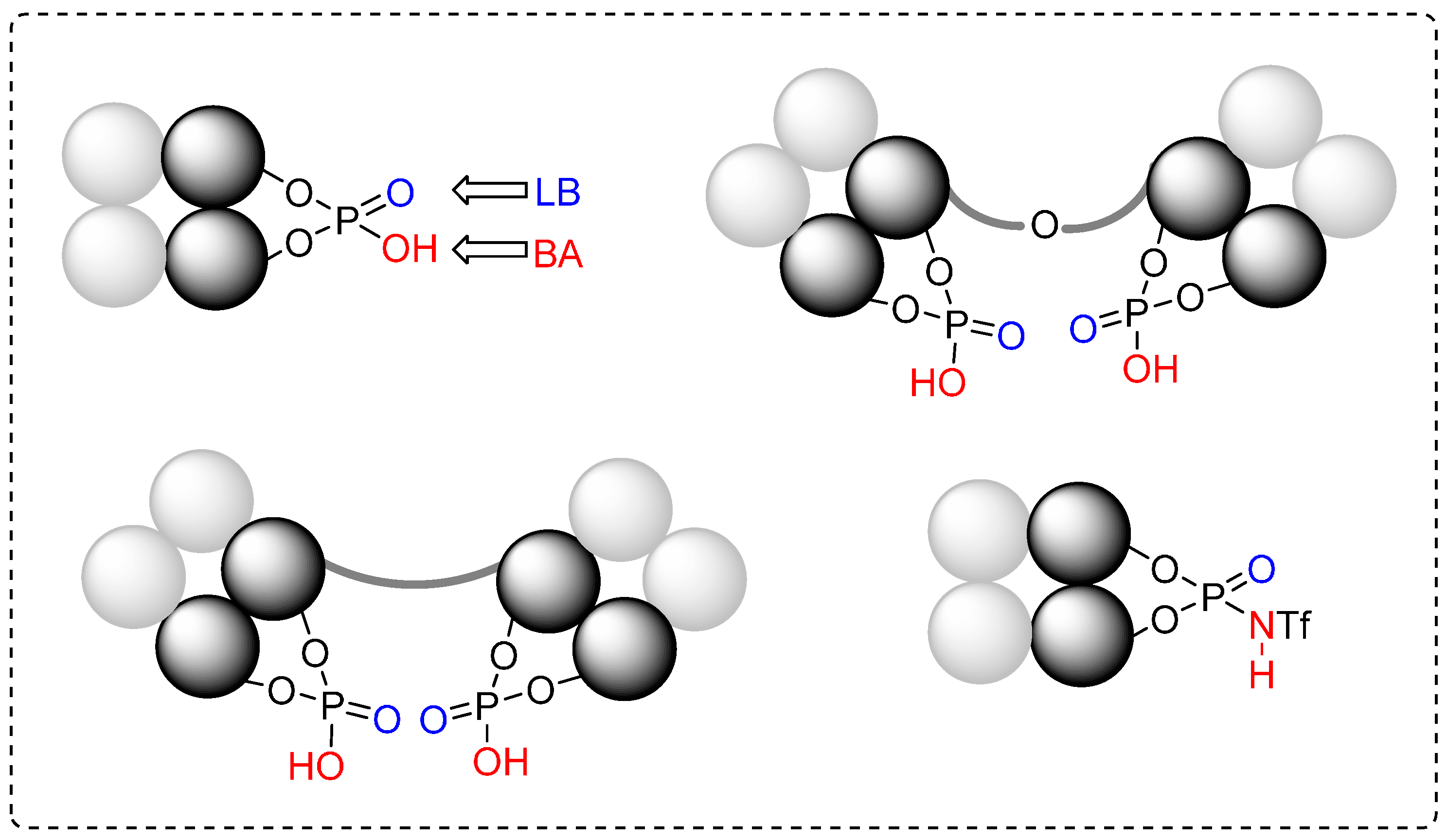
3. Cycloadditions Catalyzed by BINOL-(/SPINOL-)Derived N-Triflyl Phosphoramides
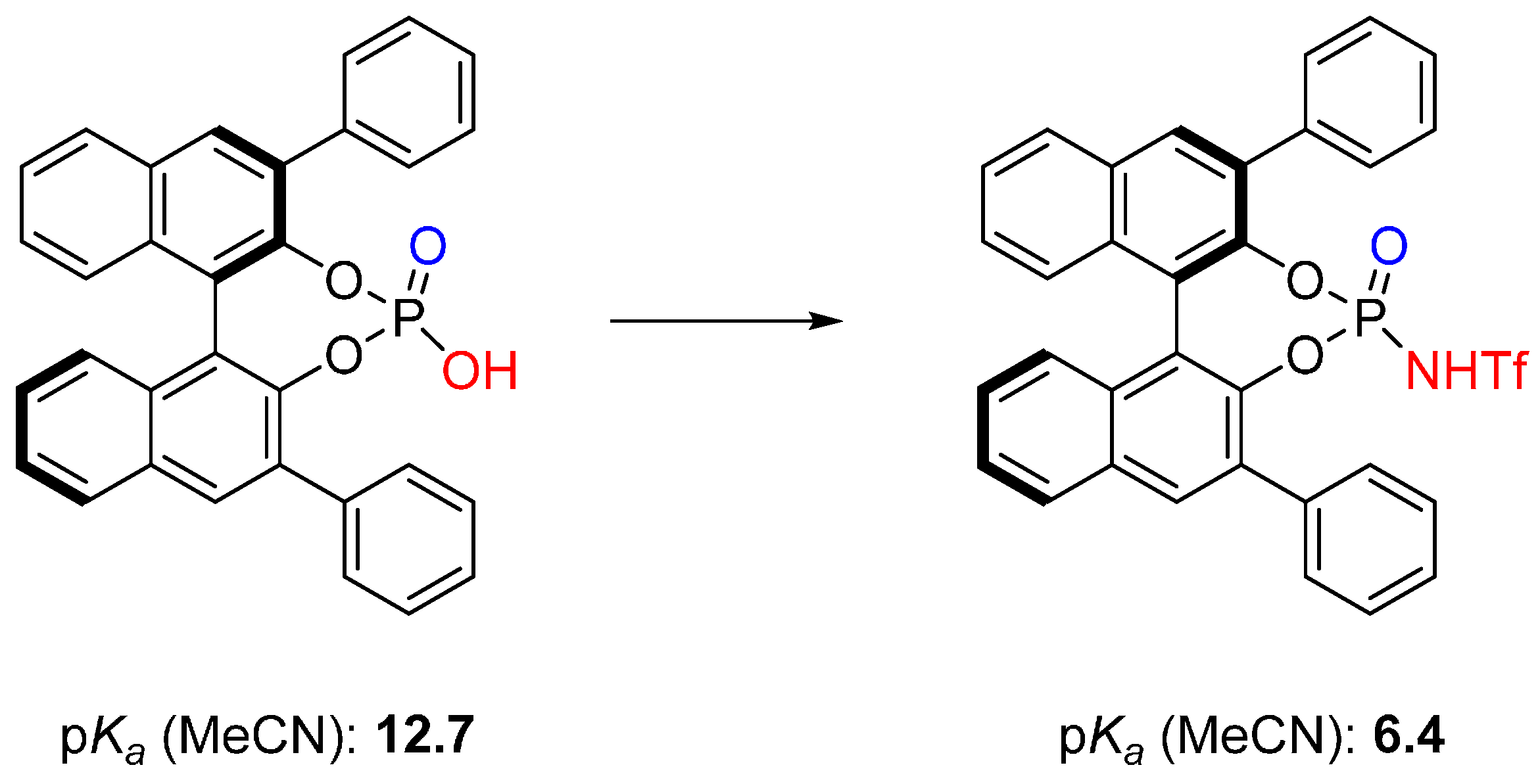
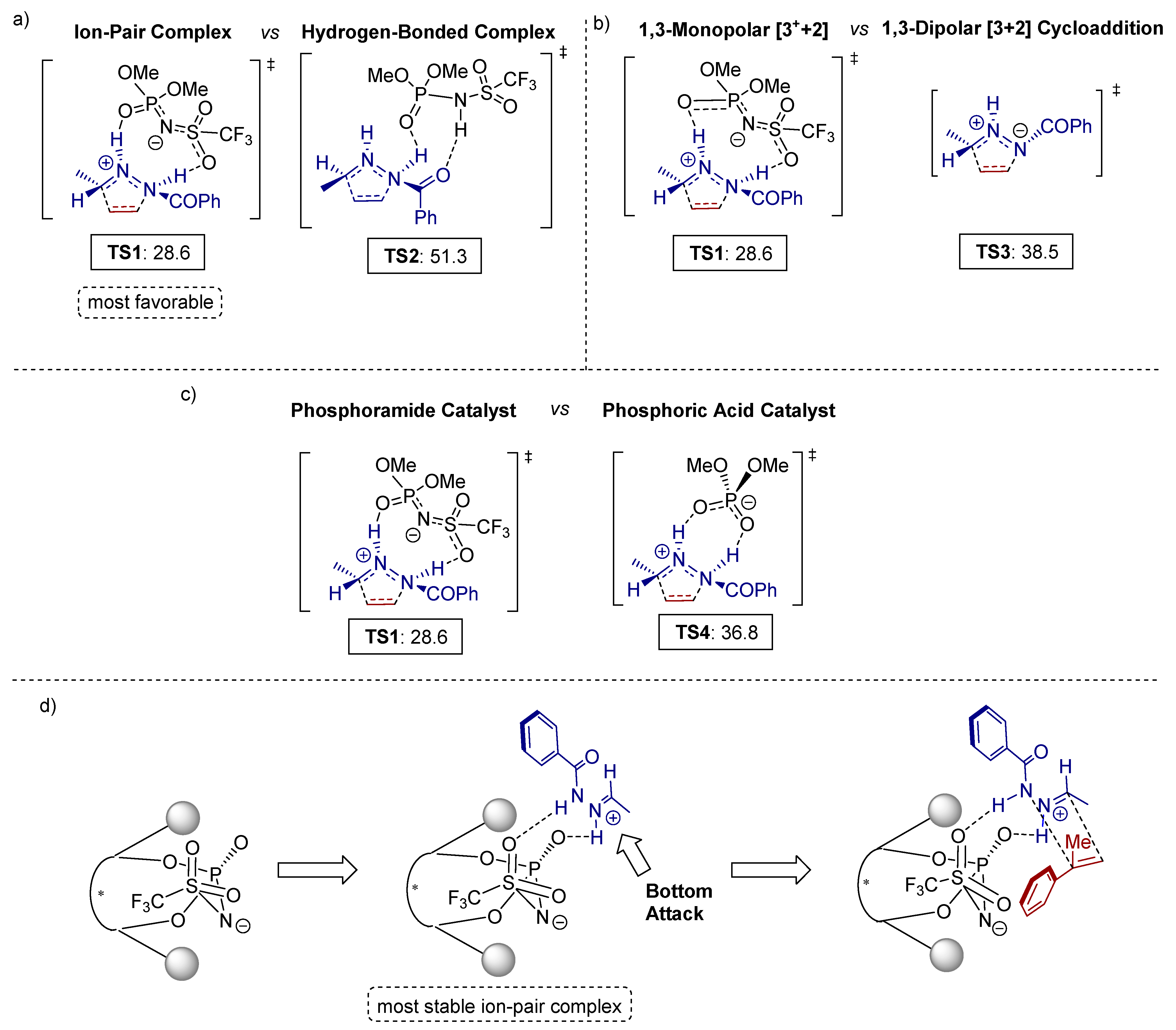
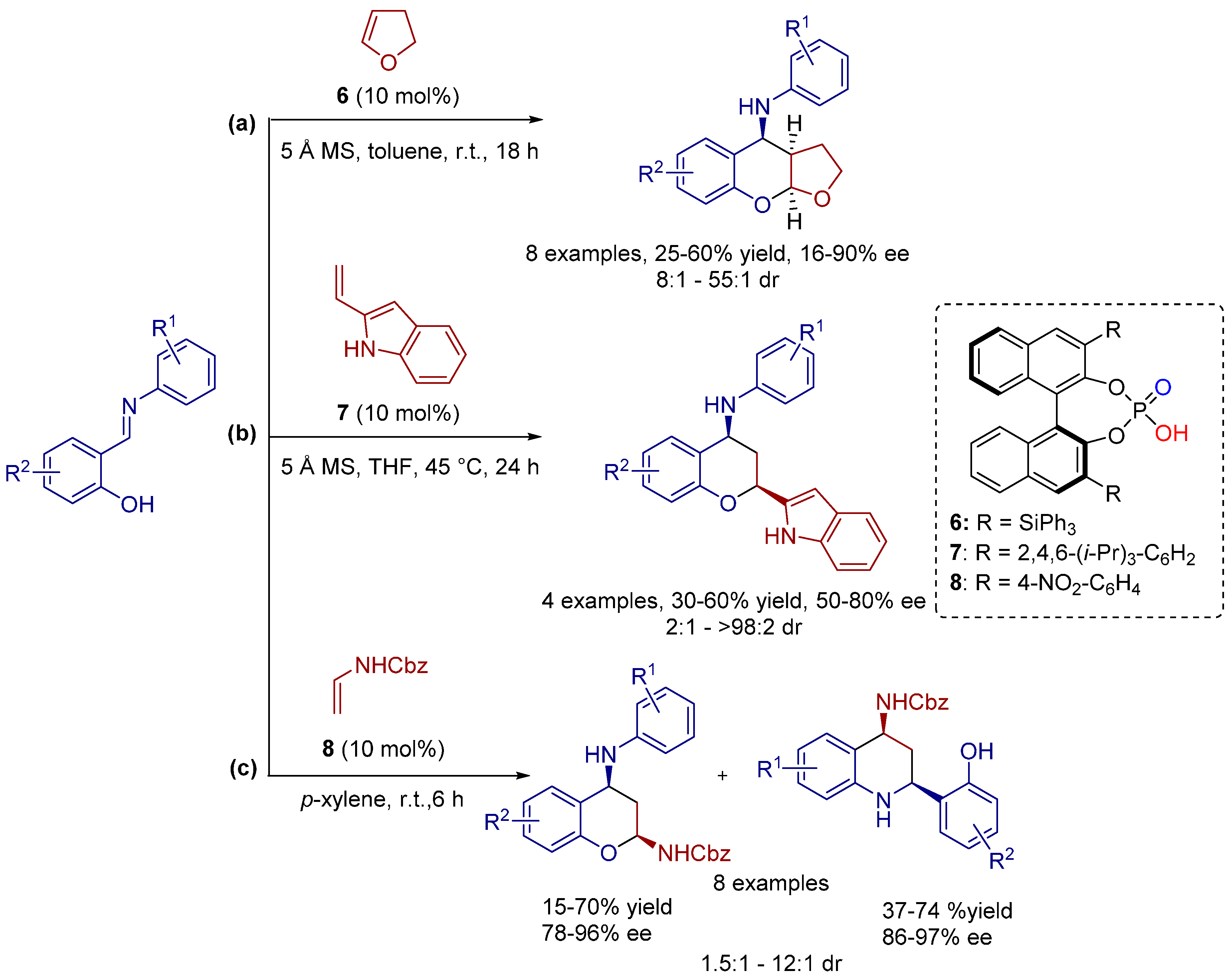
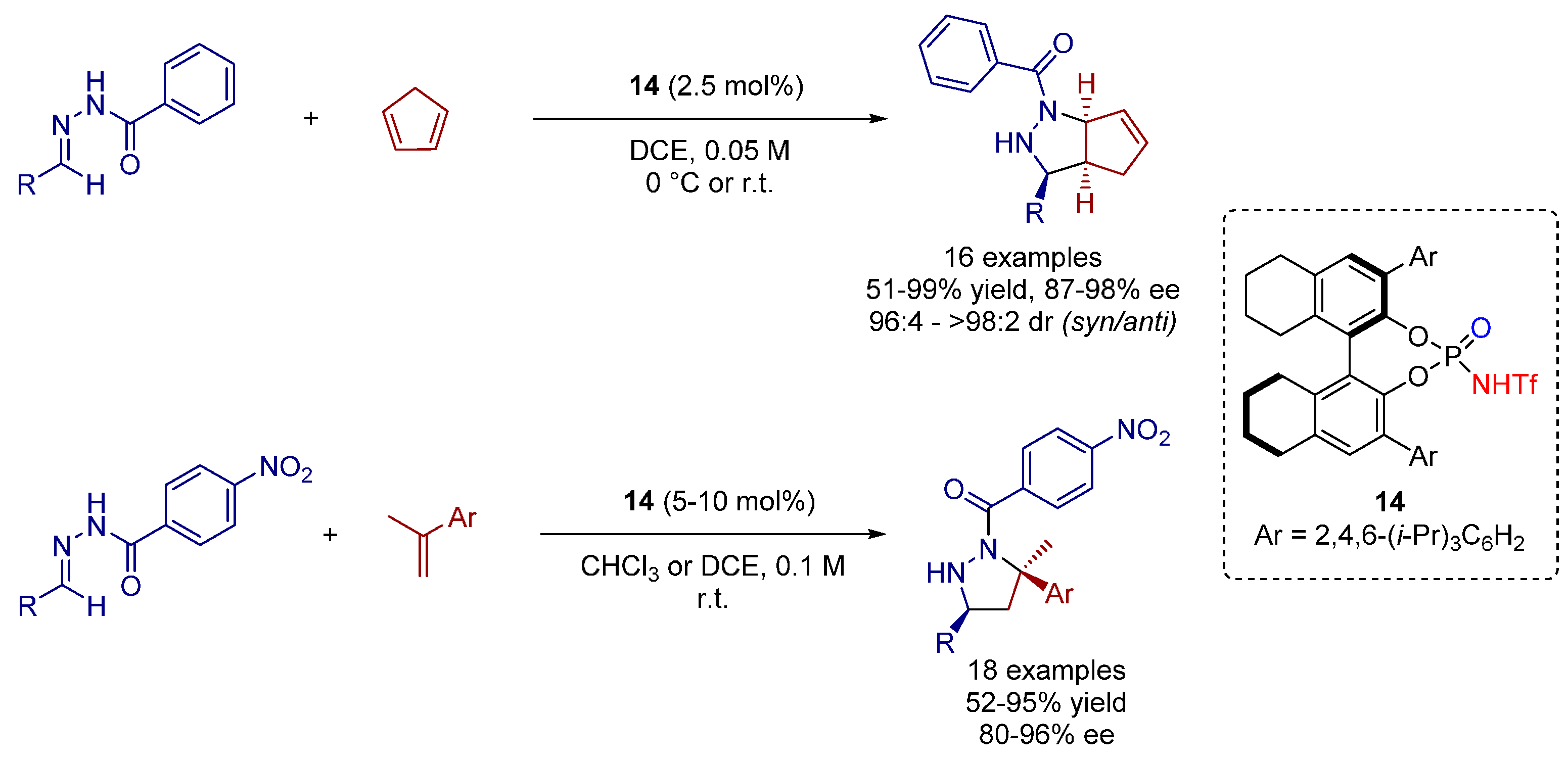
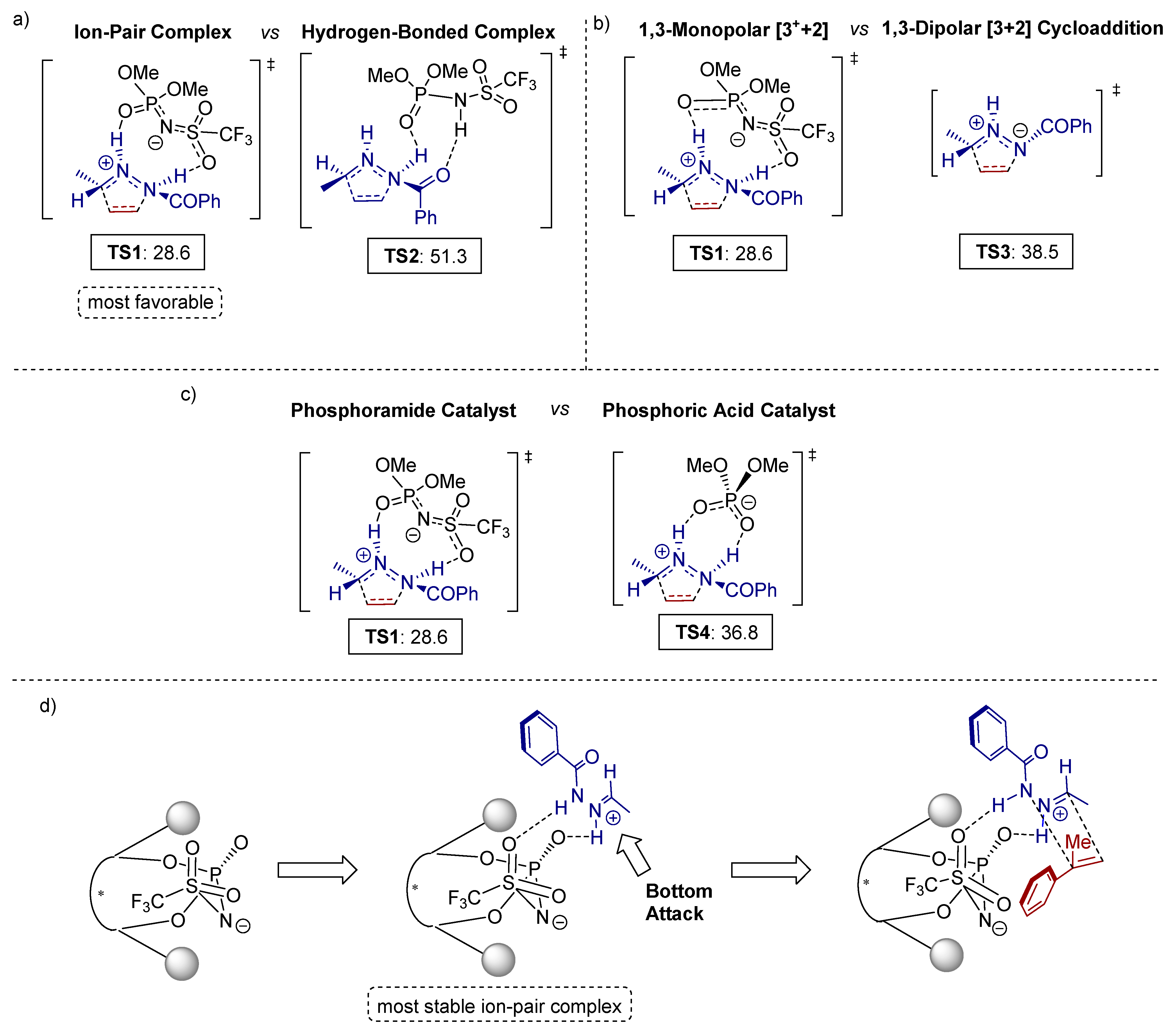
3.1. [3+2] Cycloadditions
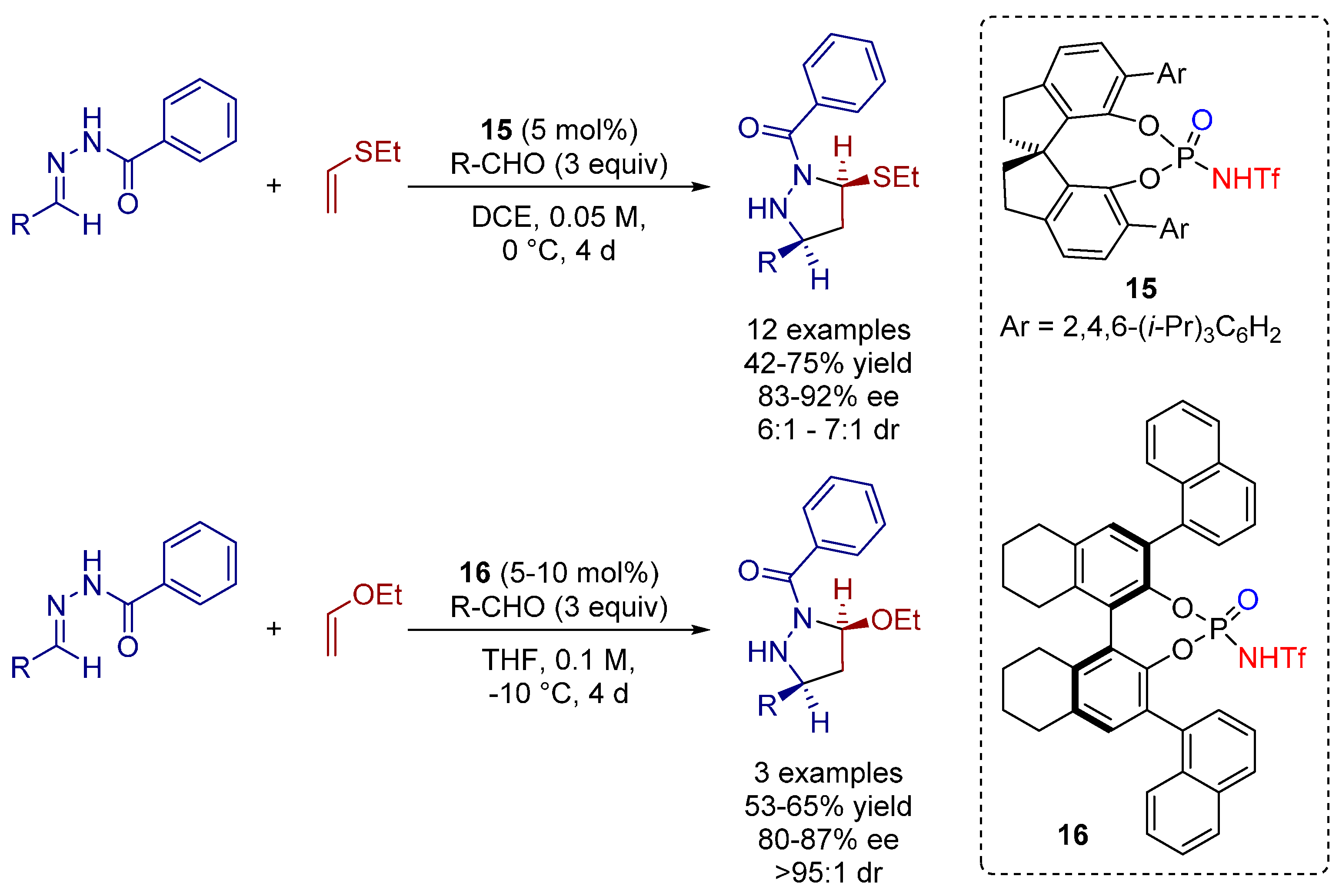
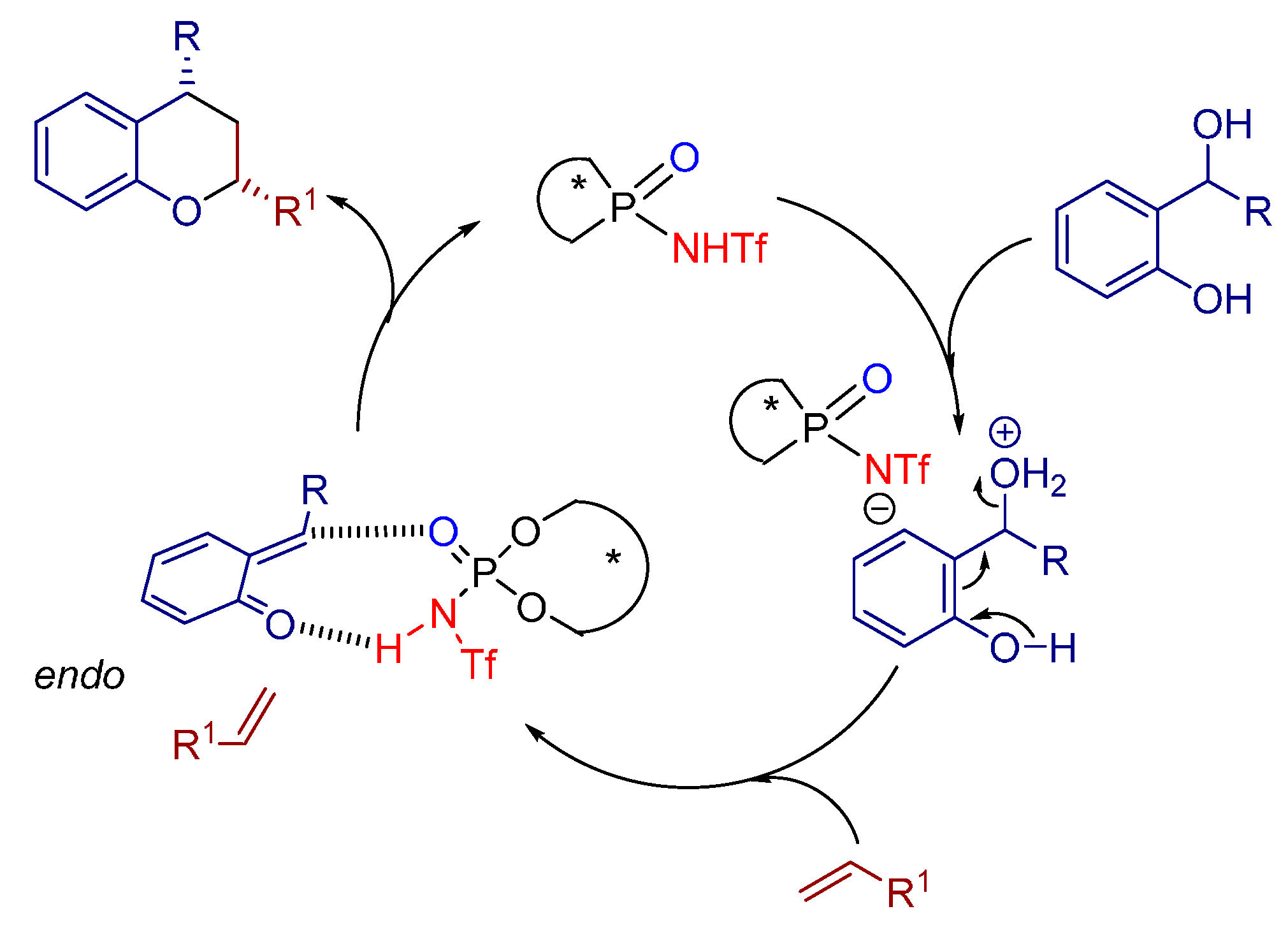
3.2. [4+2], Ionic [4+2] and Hetero [4+2] Cycloadditions
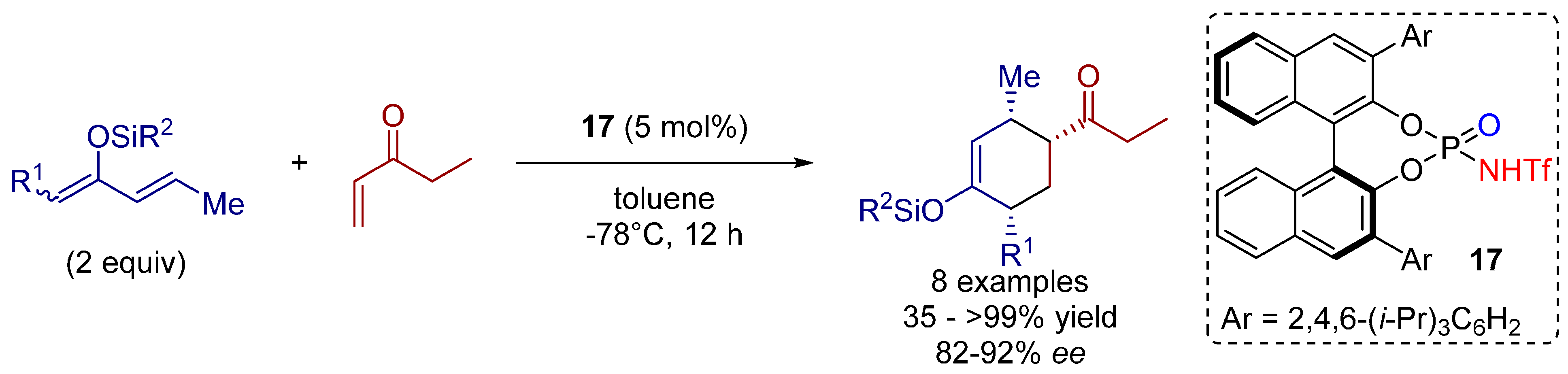
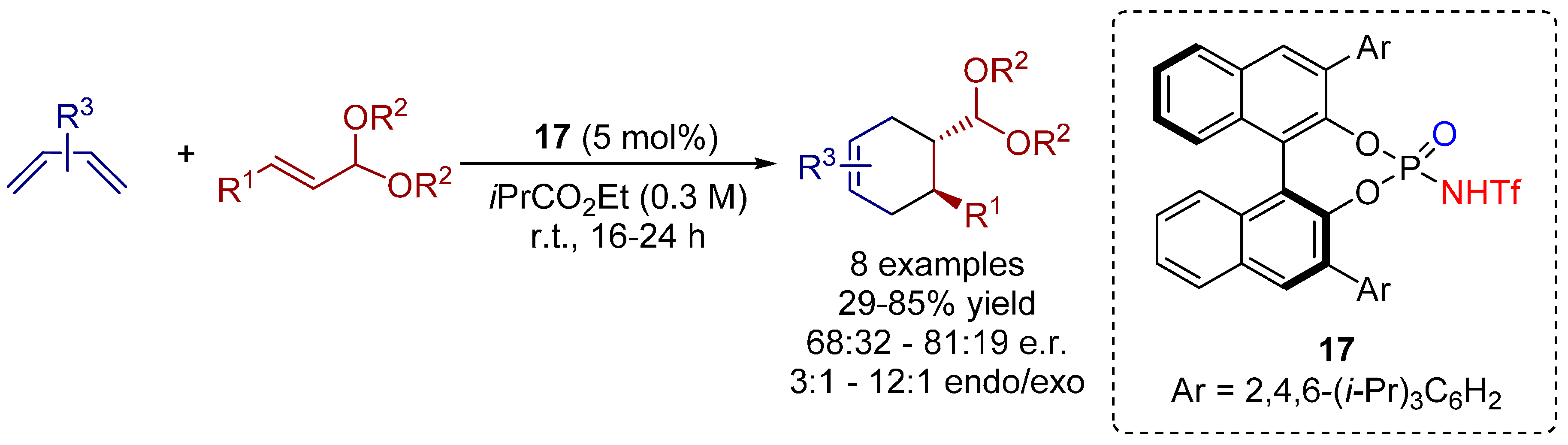
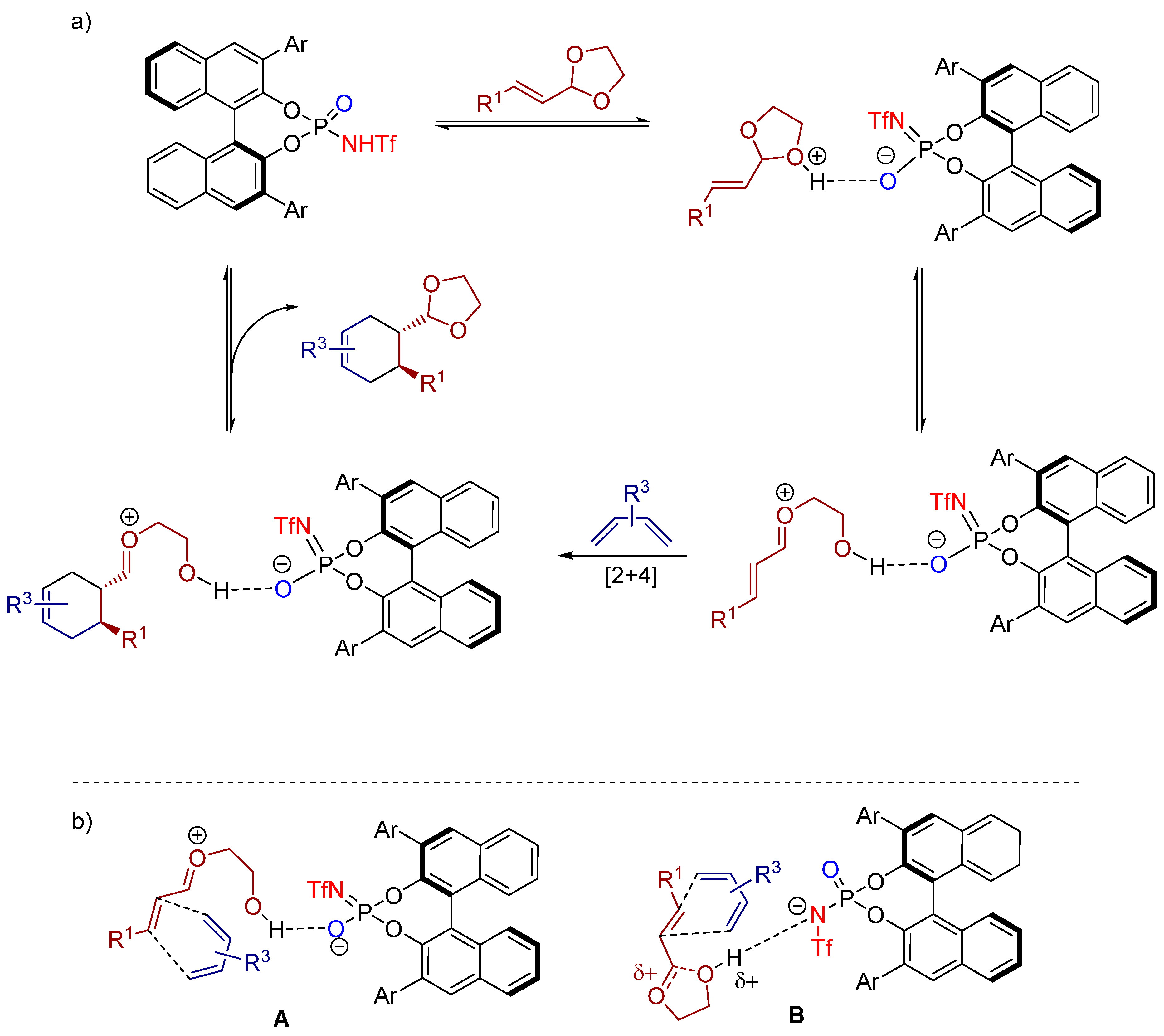
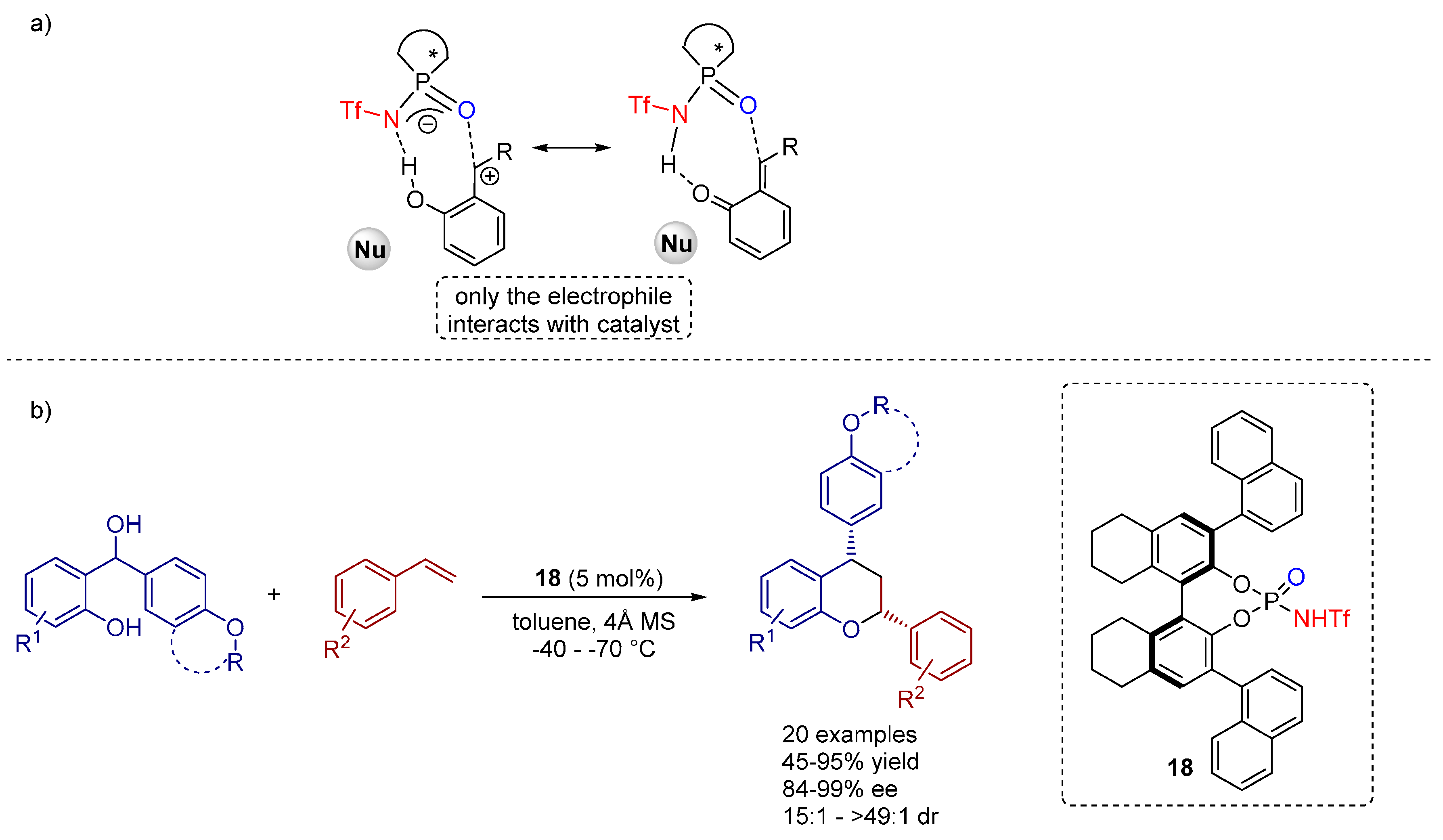
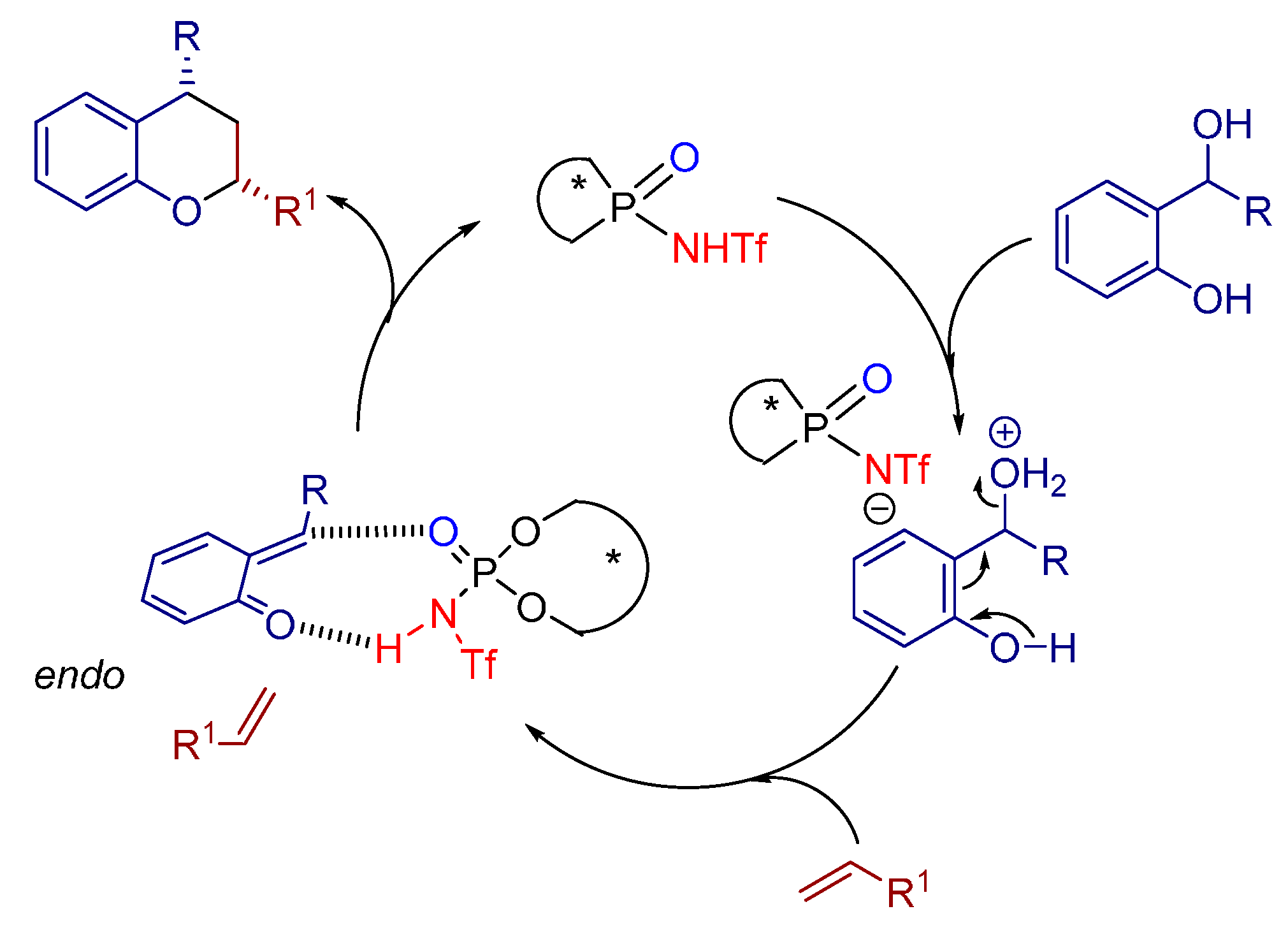
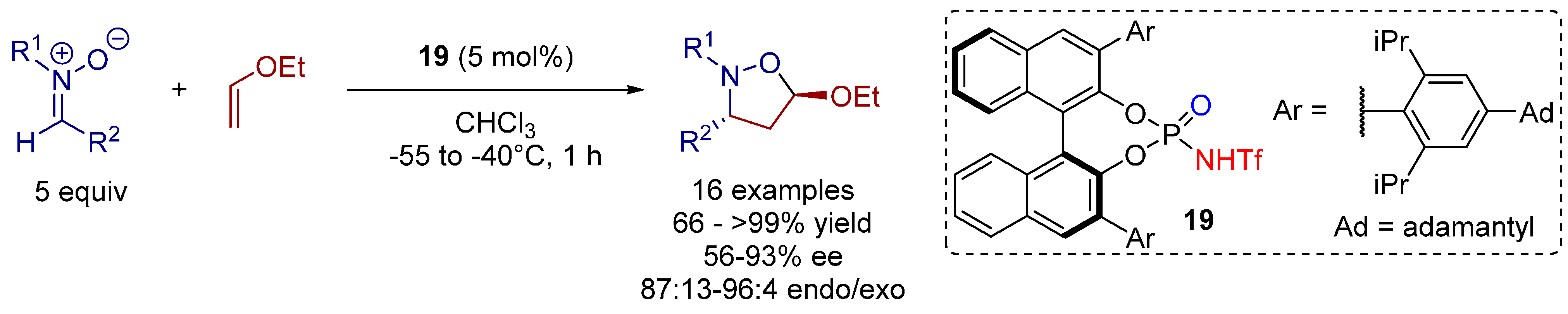
3.3. 1,3-Dipolar Cycloadditions
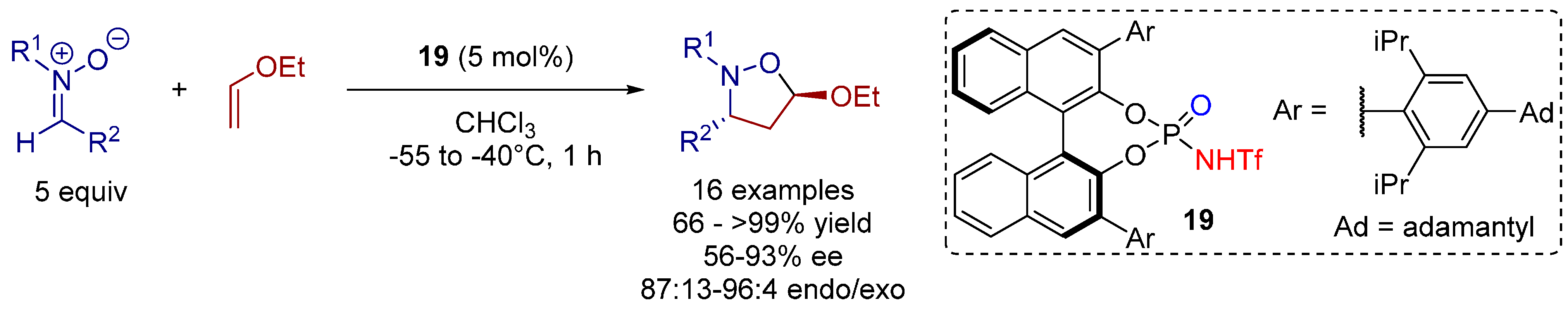

4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Evans, D.A.; Johnson, J.S. Catalytic enantioselective Diels-Alder reactions. In Comprehensive Asymmetric Catalysis; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: Berlin, Germany, 1999; Volume 3, pp. 1177–1235. [Google Scholar]
- Trost, M.; Jiang, C. Catalytic enantioselective construction of all-carbon quaternary stereocenters. Synthesis 2006, 3, 369–396. [Google Scholar] [CrossRef]
- Moyano, A.; Rios, R. Asymmetric organocatalytic cyclization and cycloaddition reactions. Chem. Rev. 2011, 111, 4703–4832. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Enantioselective Mannich-type reaction catalyzed by a chiral brønsted acid. Angew. Chem. Int. Ed. 2004, 43, 1566–1568. [Google Scholar] [CrossRef] [PubMed]
- Uraguchi, D.; Terada, M. Chiral brønsted acid-catalyzed direct Mannich reactions via electrophilic activation. J. Am. Chem. Soc. 2004, 126, 5356–5357. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived brønsted acid and metal catalysis: History and classification by mode of activation; brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef] [PubMed]
- Terada, M. Binaphthol-derived phosphoric acid as a versatile catalyst for enantioselective carbon-carbon bond forming reactions. Chem. Commun. 2008, 35, 4097–4112. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T. Stronger brønsted acids. Chem. Rev. 2007, 107, 5744–5758. [Google Scholar] [CrossRef] [PubMed]
- Terada, M. Chiral phosphoric acids as versatile catalysts for enantioselective carbon-carbon bond forming reactions. Bull. Chem. Soc. Jpn. 2010, 83, 101–119. [Google Scholar] [CrossRef]
- Kampen, D.; Reisinger, C.M.; List, B. Chiral bronsted acids for asymmetric organocatalysis. Top. Curr. Chem. 2010, 291, 395–456. [Google Scholar] [PubMed]
- Zamfir, A.; Schenker, S.; Freund, M.; Tsogoeva, S.B. Chiral BINOL-derived phosphoric acids: Privileged brønsted acid organocatalysts for C-C bond formation reactions. Org. Biomol. Chem. 2010, 8, 5262–5276. [Google Scholar] [CrossRef] [PubMed]
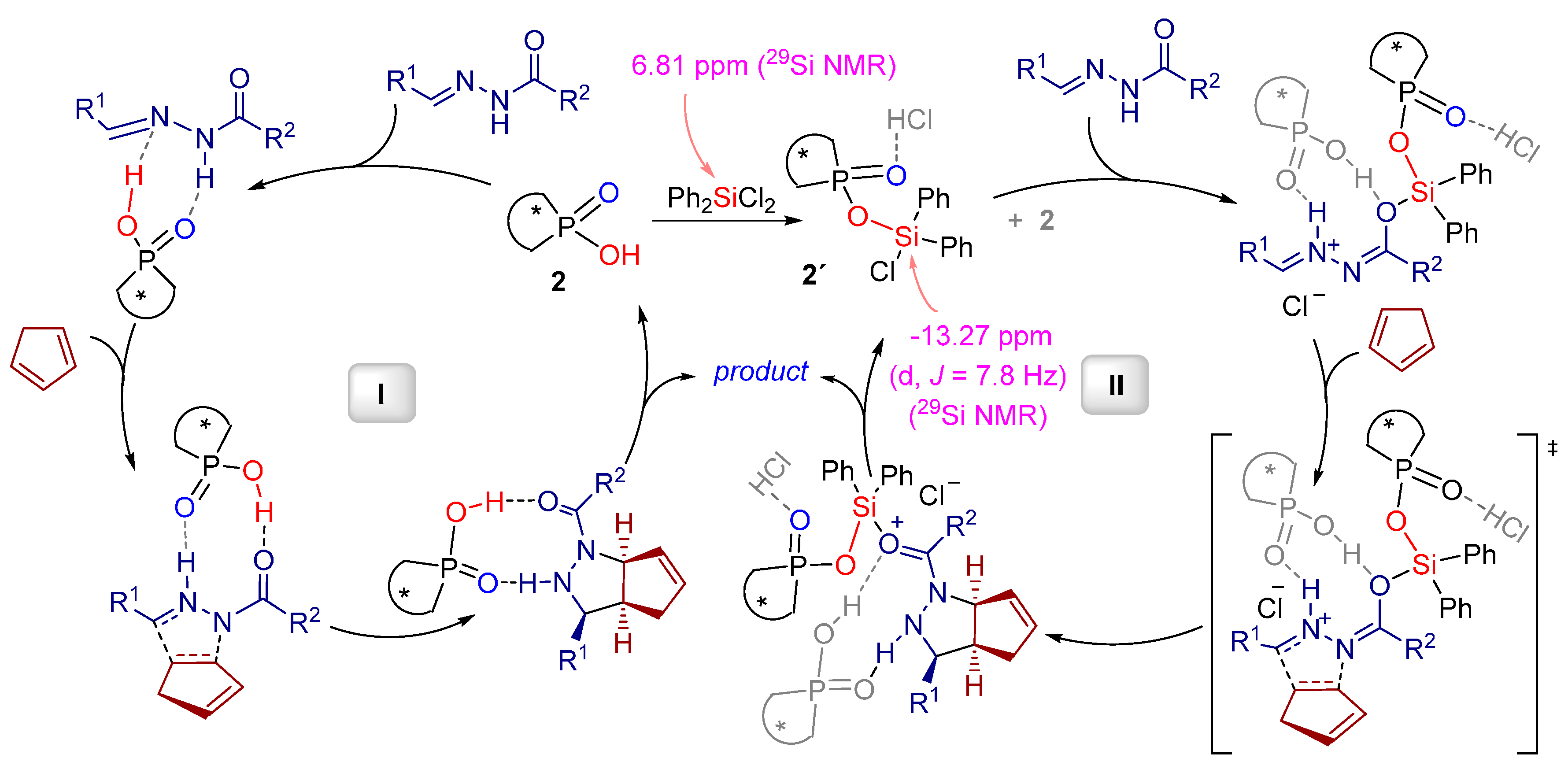
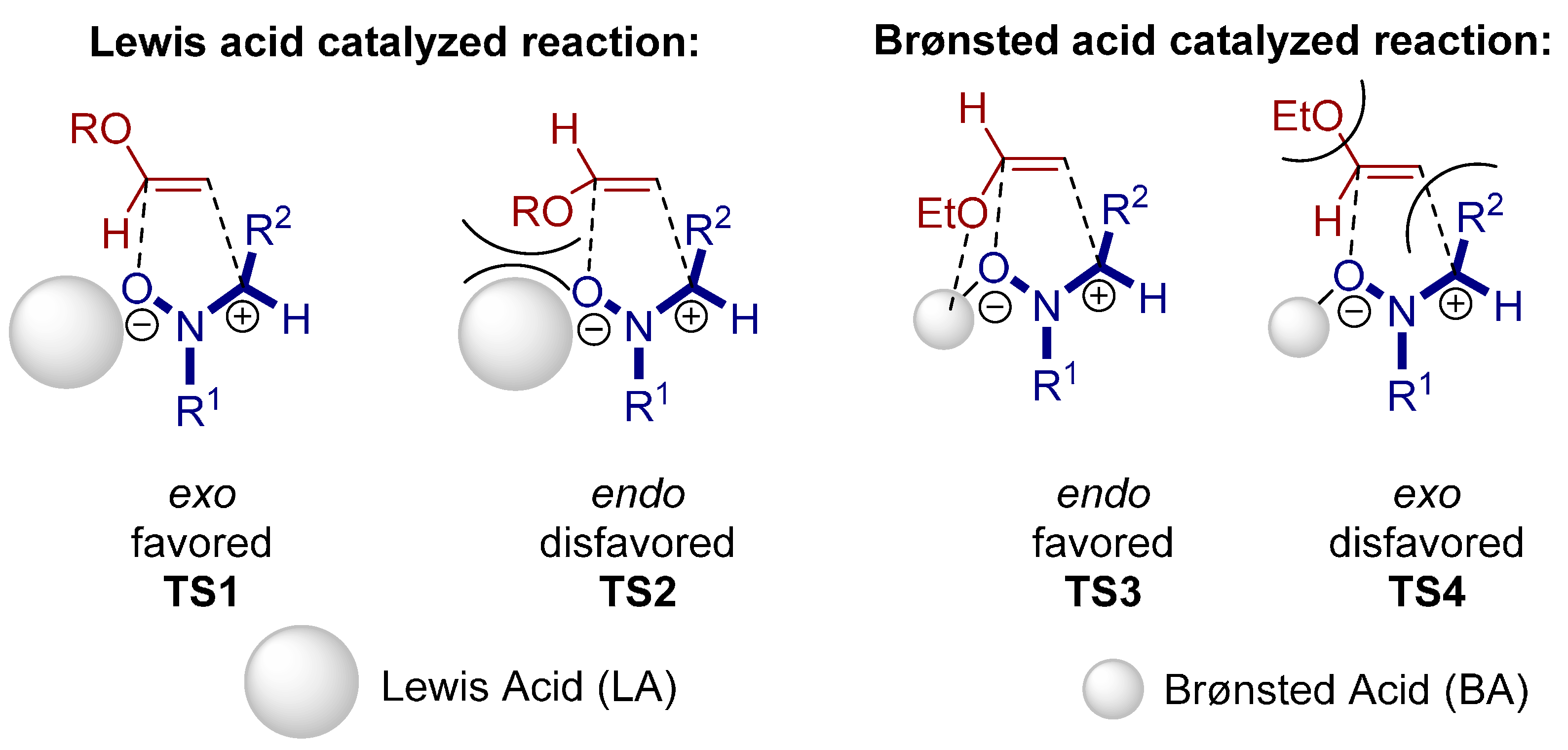
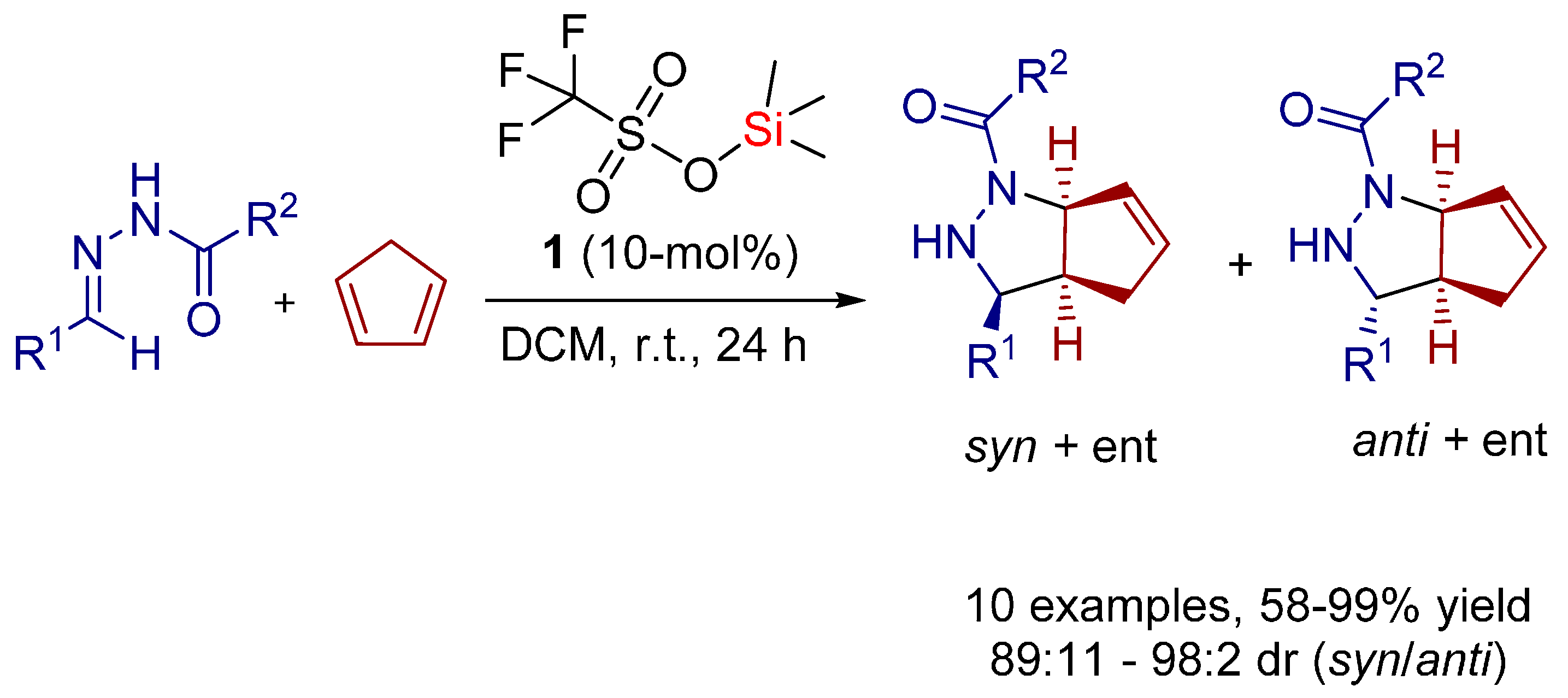
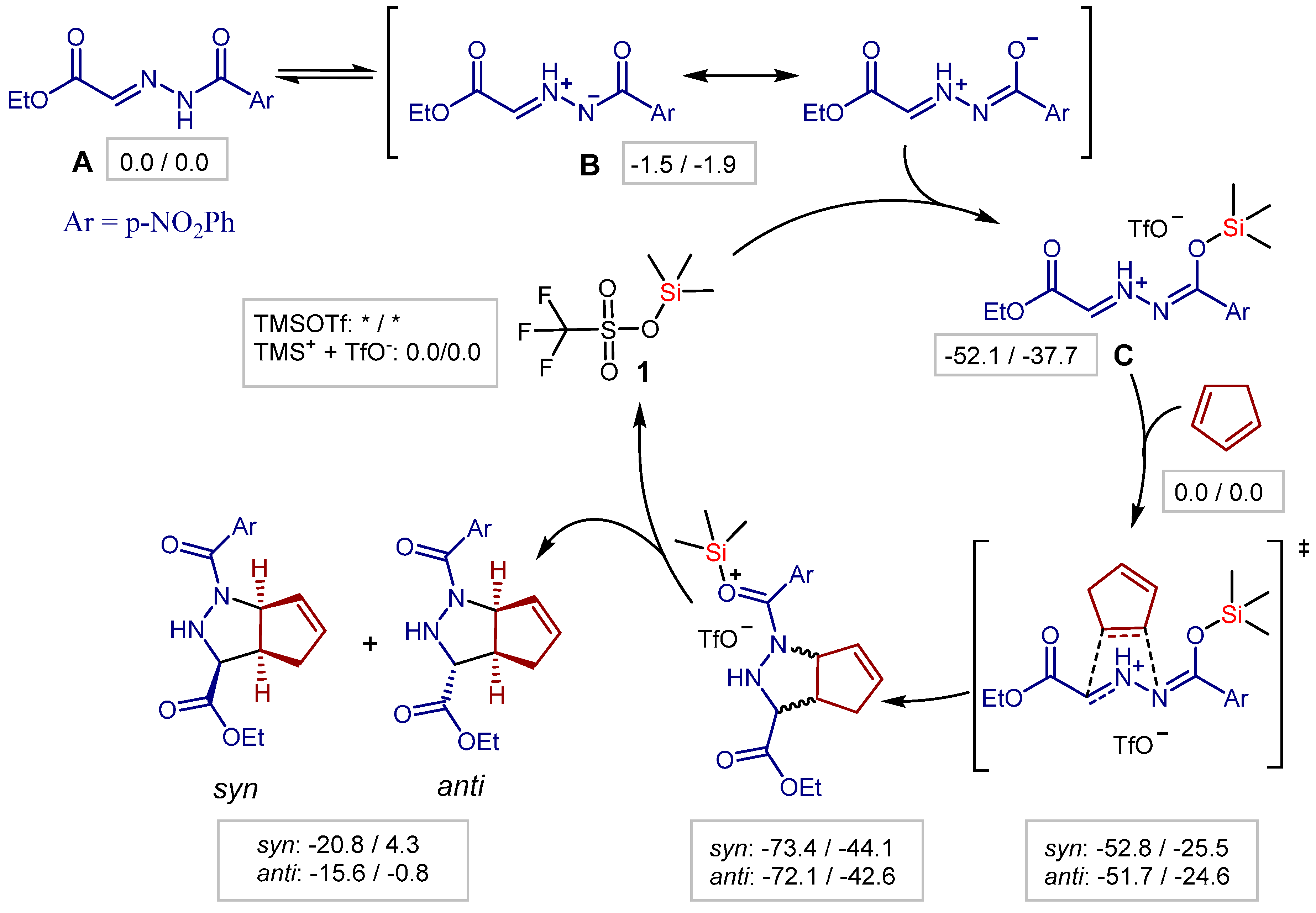
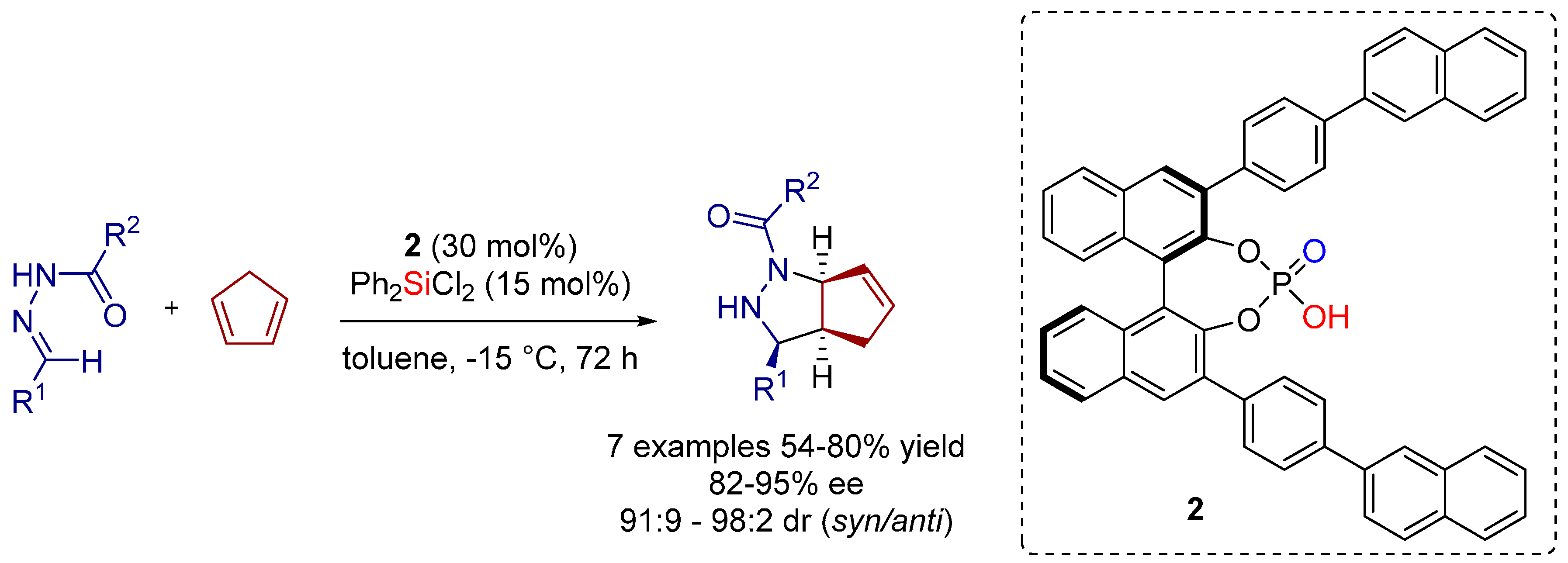
- Zamfir, A.; Schenker, S.; Bauer, W.; Clark, T.; Tsogoeva, S.B. Silicon lewis acid catalyzed [3+2] cycloaddition reactions of hydrazones/cyclopentadiene: Mild access to pyrazolidine derivatives. Eur. J. Org. Chem. 2011, 2011, 3706–3709. [Google Scholar] [CrossRef] [Green Version]
- Serdyuk, O.V.; Zamfir, A.; Hampel, F.; Tsogoeva, S.B. Combining in situ generated chiral silicon lewis acid and chiral brønsted acid catalysts for [3+2] cycloadditions: Cooperative catalysis as a convenient enantioselective route to pyrazolidines. Adv. Synth. Catal. 2012, 354, 3115–3121. [Google Scholar] [CrossRef]
- Zamfir, A.; Tsogoeva, S.B. Towards a catalytic asymmetric version of the [3+2] cycloaddition between hydrazones and cyclopentadiene. Synthesis 2011, 12, 1988–1992. [Google Scholar]
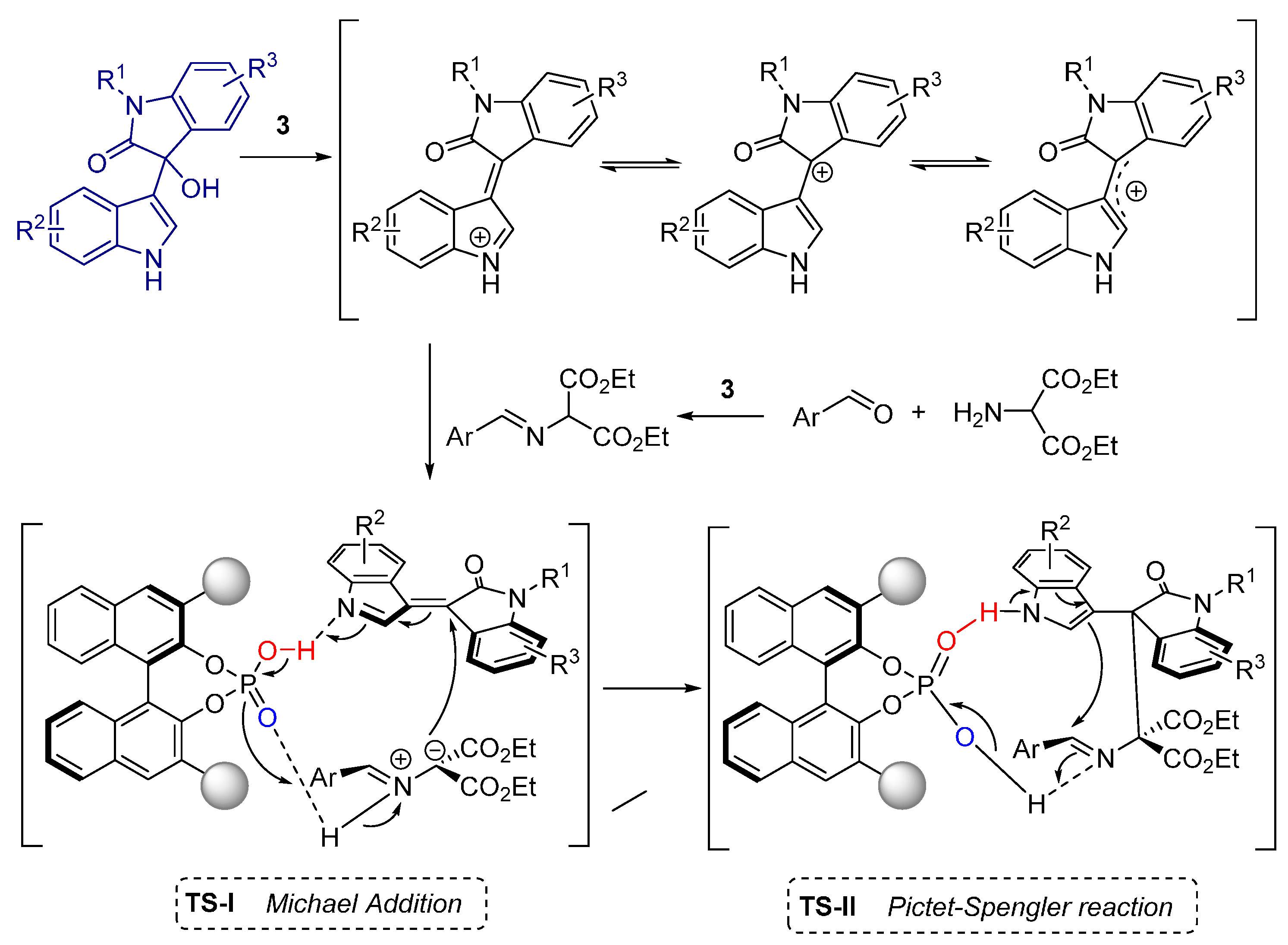
- Shi, F.; Zhu, R.-Y.; Dai, W.; Wang, C.-S.; Tu, S.-J. Catalytic asymmetric formal [3+3] cycloaddition of an azomethine ylide with 3-indolylmethanol: Enantioselective construction of a six-membered piperidine framework. Chem. Eur. J. 2014, 20, 2597–2604. [Google Scholar] [CrossRef] [PubMed]
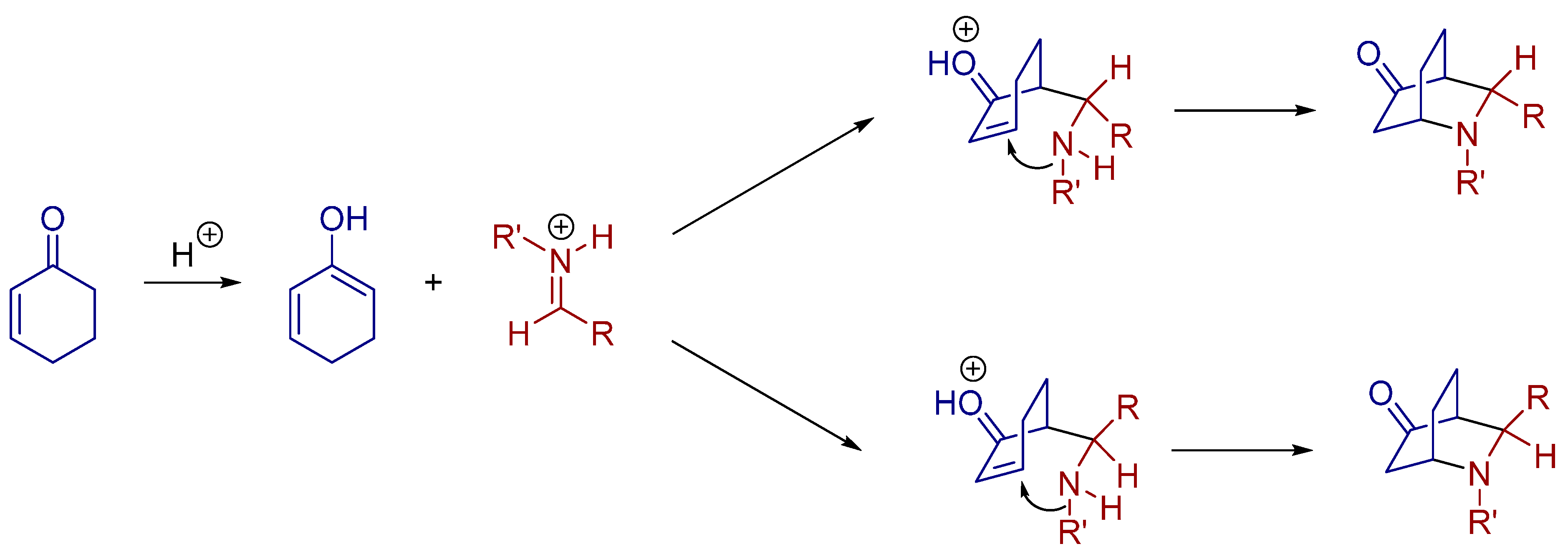
- Bernardi, L.; Comes-Franchini, M.; Fochi, M.; Leo, V.; Mazzanti, A.; Ricci, A. Catalytic asymmetric inverse-electron-demand (IED) [4+2] cycloaddition of salicylaldimines: Preparation of optically active 4-aminobenzopyran derivatives. Adv. Synth. Catal. 2010, 352, 3399–3406. [Google Scholar] [CrossRef]
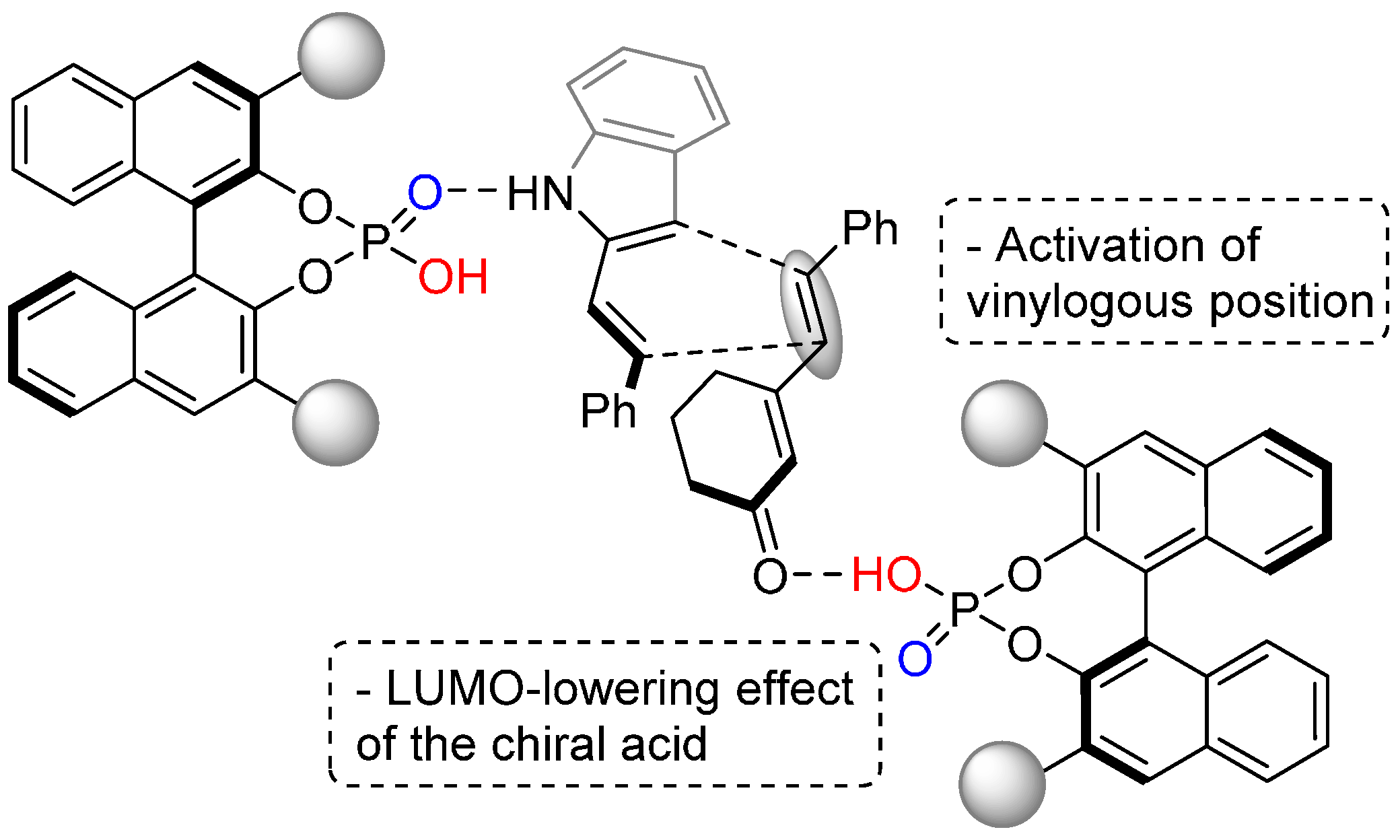
- Tian, X.; Hofman, N.; Melchiorre, P. Asymmetric vinylogous Diels-Alder reactions catalyzed by a chiral phosphoric acid. Angew. Chem. Int. Ed. 2014, 53, 2997–3000. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, W.-J.; Gong, L.-Z. Kinetic resolution of racemic 2,3-allenoates by organocatalytic asymmetric 1,3-dipolar cycloaddition. Org. Lett. 2010, 12, 4050–4053. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Song, J.; Tu, X.-F.; Liu, B.; Chen, X.H.; Gong, L.-Z. Organocatalytic asymmetric intramolecular [3+2] cycloaddition: A straightforward approach to access multiply substituted hexahydrochromeno[4,3-b]pyrrolidine derivatives in high optical purity. Org. Biomol. Chem. 2010, 8, 2016–2019. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Zhu, R.-Y.; Liang, X.; Tu, S.-J. Catalytic asymmetric 1,3-dipolar cycloadditions of alkynes with isatin-derived azomethine ylides: Enantioselective synthesis of spiro[indoline-3,2′-pyrrole] derivatives. Adv. Synth. Catal. 2013, 355, 2447–2458. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Wang, X.; Zhao, Y.-L.; Zhu, F.; Zeng, X.-P.; Chen, L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. One-pot tandem approach to spirocyclic oxindoles featuring adjacent spiro-stereocenters. Angew. Chem. Int. Ed. 2013, 52, 13735–13739. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Kai, M.; Wu, C.; Sun, W.; Zhu, G.; Li, G.; Yaoa, X.; Wang, R. Enantioselective 1,3-dipolar cycloaddition of methyleneindolinones and N,N′-cyclic azomethine imines. Chem. Commun. 2013, 49, 6713–6715. [Google Scholar] [CrossRef] [PubMed]
- Weiß, K.; Wei, S.; Tsogoeva, S.B. Novel one-pot process for the synthesis of 1,3-thiazoles via organocatalysed epoxidation of nitro-olefins. Org. Biomol. Chem. 2011, 9, 3457–3461. [Google Scholar] [CrossRef] [PubMed]
- Yalalov, D.A.; Tsogoeva, S.B.; Shubina, T.E.; Martynova, I.M.; Clark, T. Evidence for an enol mechanism in a highly enantioselective Mannich-type reaction catalyzed by primary amine-thiourea. Angew. Chem. Int. Ed. 2008, 47, 6624–6628. [Google Scholar] [CrossRef] [PubMed]
- Tsogoeva, S.B.; Wei, S.; Freund, M.; Mauksch, M. Generation of highly enantioenriched crystalline products in reversible asymmetric reactions with racemic or achiral catalysts. Angew. Chem. Int. Ed. 2009, 48, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Zamfir, A.; Tsogoeva, S.B. Asymmetric hydrocyanation of hydrazones catalyzed by in situ formed O-silylated binol-phosphate: A convenient access to versatile α-hydrazino acids. Org. Lett. 2009, 12, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Itoh, J.; Fuchibe, K.; Akiyama, T. Chiral brønsted acid catalyzed enantioselective aza-Diels-Alder reaction of brassard’s diene with imines. Angew. Chem. Int. Ed. 2006, 45, 4796–4798. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Cun, L.-F.; Mi, A.-Q.; Jiang, Y.-Z.; Gong, L.-Z. Enantioselective direct aza hetero-Diels-Alder reaction catalyzed by chiral brønsted acids. Org. Lett. 2006, 8, 6023–6026. [Google Scholar] [CrossRef] [PubMed]
- Koppel, I.A.; Koppel, J.; Leito, I.; Koppel, I.; Mishima, M.; Yagupolskii, L.M. The enormous acidifying effect of the supersubstituent [double bond, length half m-dash]NSO2CF3 on the acidity of derivatives of benzenesulfonamide and toluene-p-sulfonamide in the gas phase and in dimethyl sulfoxide. J. Chem. Soc., Perkin Trans. 2001, 2, 229–232. [Google Scholar] [CrossRef]
- Leito, I.; Kaljurand, I.; Koppel, I.A.; Yagupolskii, L.M.; Vlasov, V.M. Spectrophotometric acidity scale of strong neutral brønsted acids in acetonitrile. J. Org. Chem. 1998, 63, 7868–7874. [Google Scholar] [CrossRef]
- Nakashima, D.; Yamamoto, H. Design of chiral N-triflyl phosphoramide as a strong chiral brønsted acid and its application to asymmetric Diels-Alder reaction. J. Am. Chem. Soc. 2006, 128, 9626–9627. [Google Scholar] [CrossRef] [PubMed]
- Kaupmees, K.; Tolstoluzhsky, N.; Raja, S.; Rueping, M.; Leito, I. On the acidity and reactivity of highly effective chiral brønsted acid catalysts: Establishment of an acidity scale. Angew. Chem. Int. Ed. 2013, 52, 11569–11572. [Google Scholar] [CrossRef] [PubMed]
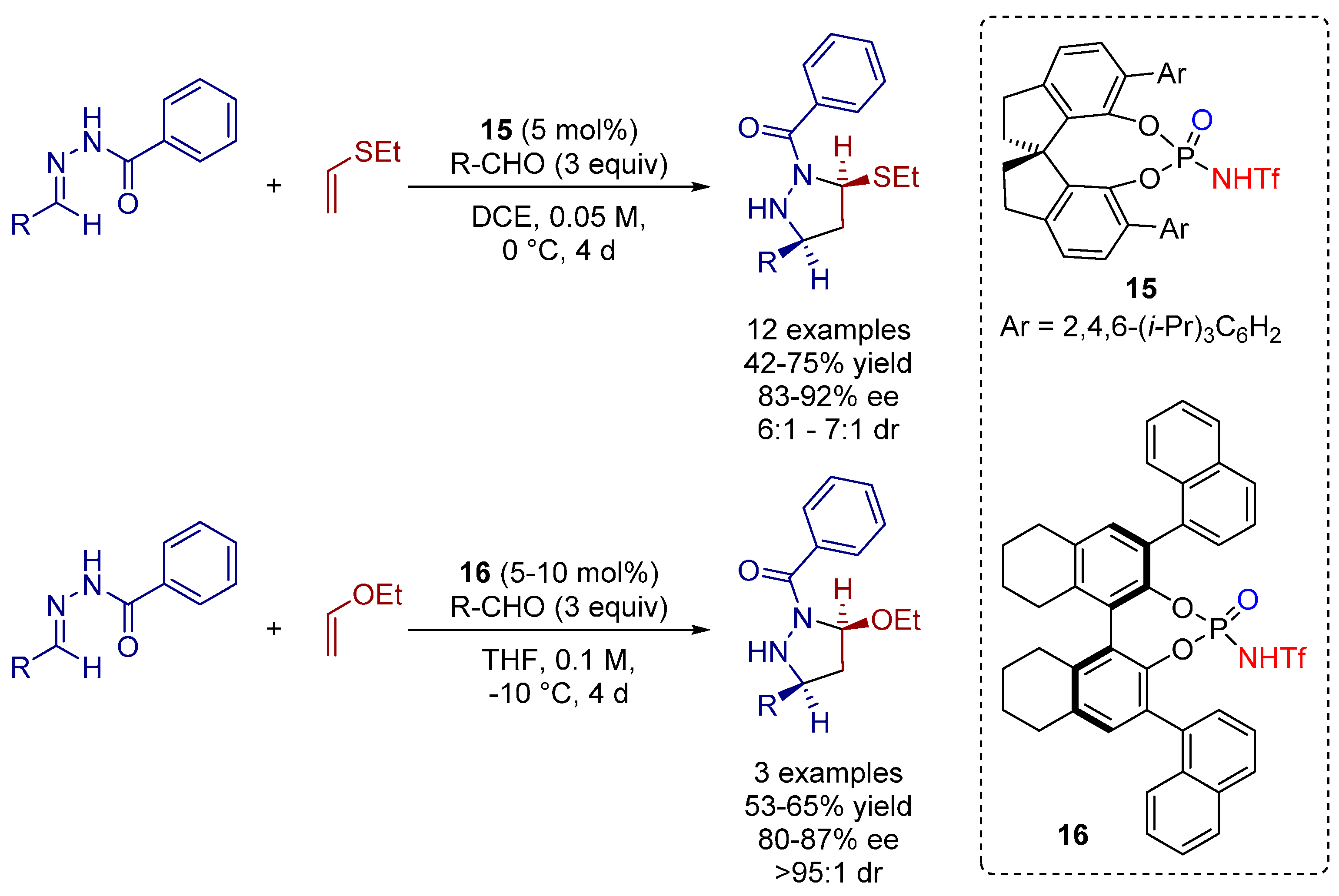
- Rueping, M.; Maji, M.S.; Küçük, H.B.; Atodiresei, I. Asymmetric brønsted acid catalyzed cycloadditions—Efficient enantioselective synthesis of pyrazolidines, pyrazolines, and 1,3-diamines from N-acyl hyrazones and alkenes. Angew. Chem. Int. Ed. 2012, 51, 12864–12868. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Küçük, H.B.; Maji, M.S.; Yang, Y.-F.; Rueping, M.; Houk, K.N. Mechanism and selectivity of N-triflylphosphoramide catalyzed (3++2) cycloaddition between hydrazones and alkenes. J. Am. Chem. Soc. 2014, 136, 13769–13780. [Google Scholar] [CrossRef] [PubMed]
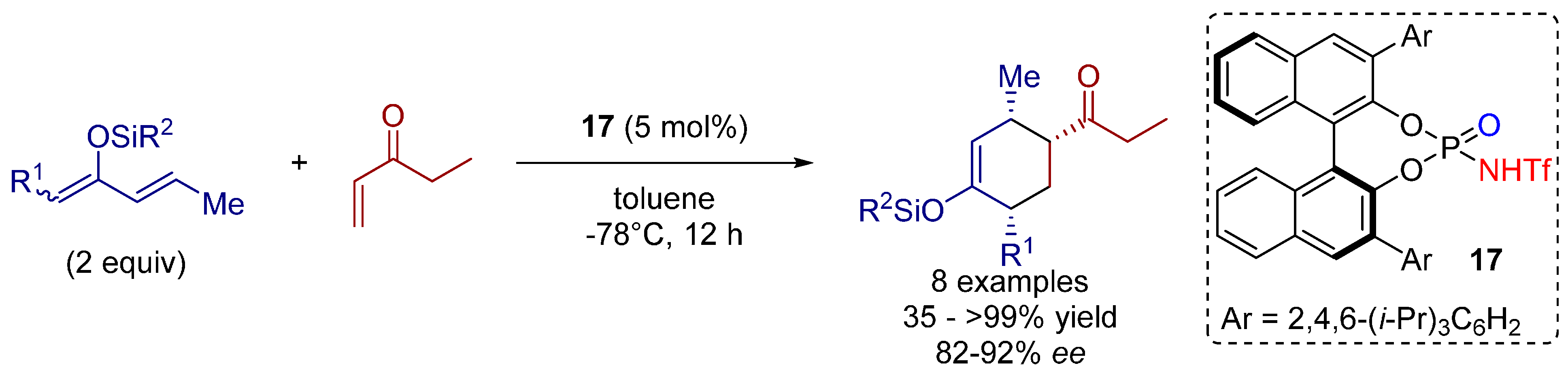
- Borovika, A.; Nagorny, P. Chiral brønsted acid-catalyzed enantioselective ionic [2+4] cycloadditions. Tetrahedron 2013, 69, 5719–5725. [Google Scholar] [CrossRef]
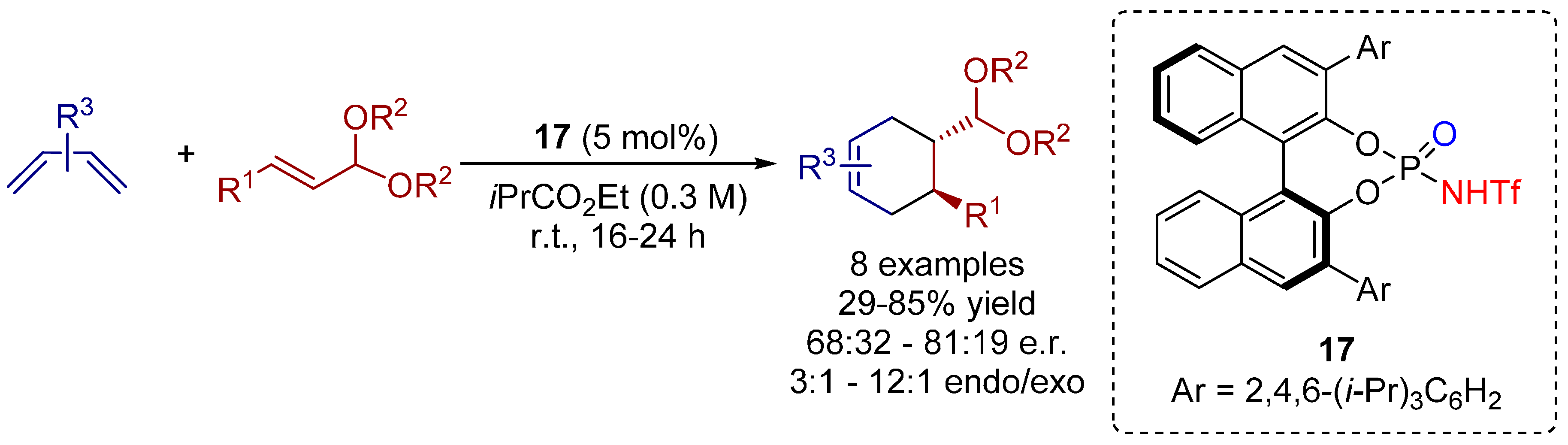
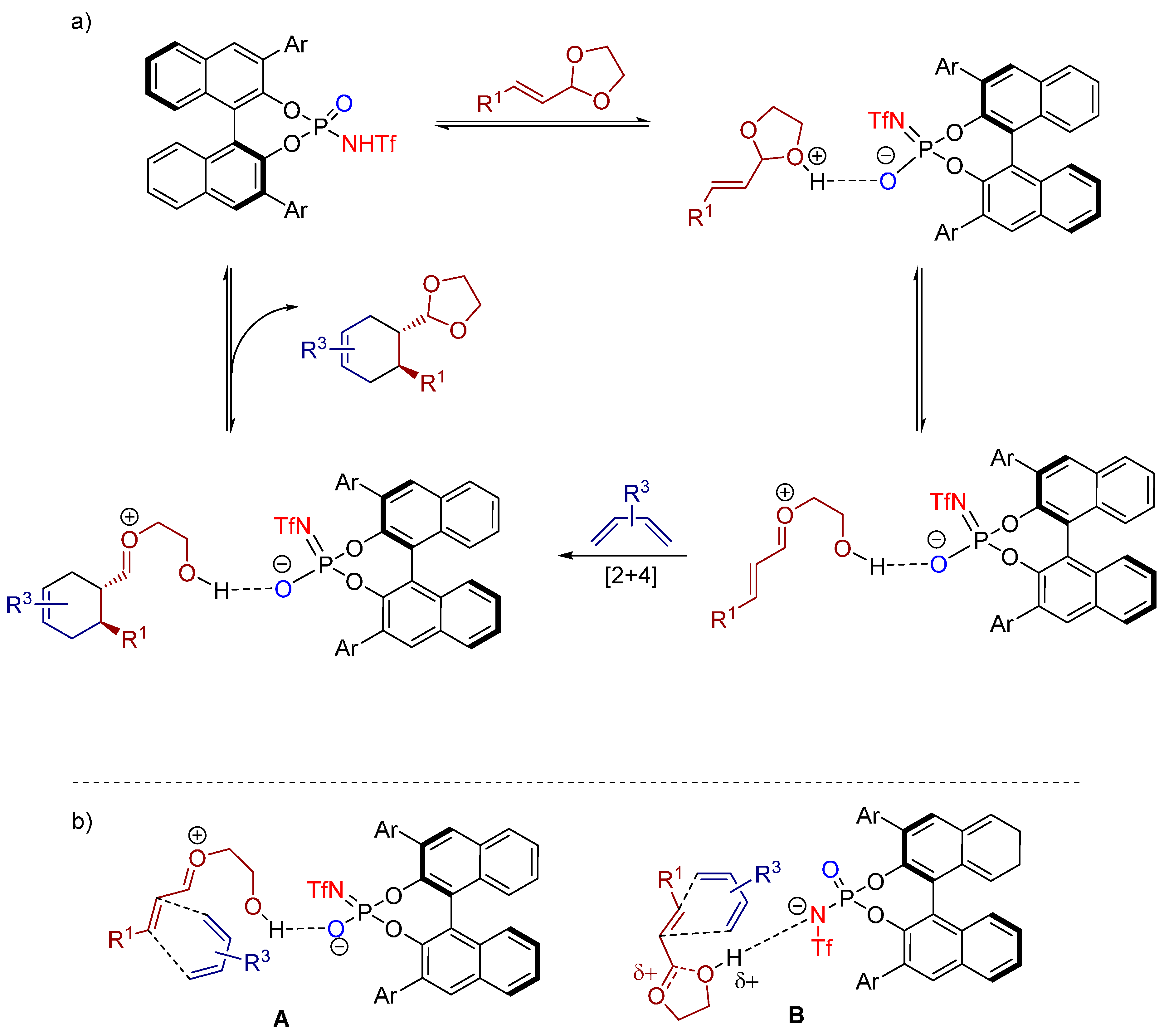
- Hsiao, C.-C.; Raja, S.; Liao, H.-H.; Atodiresei, I.; Rueping, M. Ortho-quinone methides as reactive intermediates in asymmetric brønsted acid catalyzed cycloadditions with unactivated alkenes by exclusive activation of the electrophile. Angew. Chem. Int. Ed. 2015, 54, 5762–5765. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Nakashima, D.; Yamamoto, H. Enantioselective 1,3-dipolar cycloaddition of nitrones with ethyl vinyl ether: The difference between brønsted and lewis acid catalysis. Angew. Chem. Int. Ed. 2008, 47, 2411–2413. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Held, F.E.; Grau, D.; Tsogoeva, S.B. Enantioselective Cycloaddition Reactions Catalyzed by BINOL-Derived Phosphoric Acids and N-Triflyl Phosphoramides: Recent Advances. Molecules 2015, 20, 16103-16126. https://doi.org/10.3390/molecules200916103
Held FE, Grau D, Tsogoeva SB. Enantioselective Cycloaddition Reactions Catalyzed by BINOL-Derived Phosphoric Acids and N-Triflyl Phosphoramides: Recent Advances. Molecules. 2015; 20(9):16103-16126. https://doi.org/10.3390/molecules200916103
Chicago/Turabian StyleHeld, Felix E., Dominik Grau, and Svetlana B. Tsogoeva. 2015. "Enantioselective Cycloaddition Reactions Catalyzed by BINOL-Derived Phosphoric Acids and N-Triflyl Phosphoramides: Recent Advances" Molecules 20, no. 9: 16103-16126. https://doi.org/10.3390/molecules200916103