
Nanostructures for the Inhibition of Viral Infections



Abstract
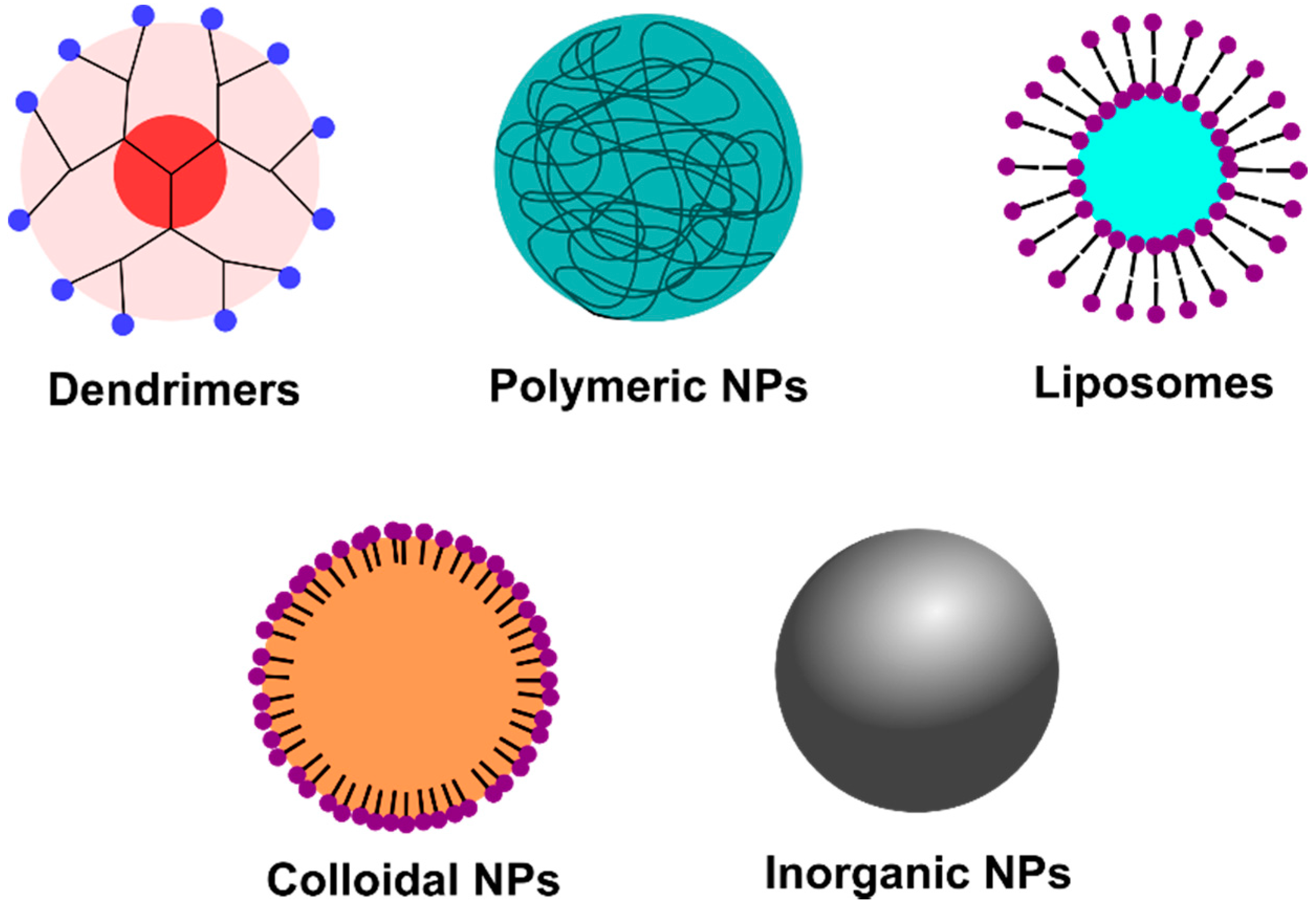
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
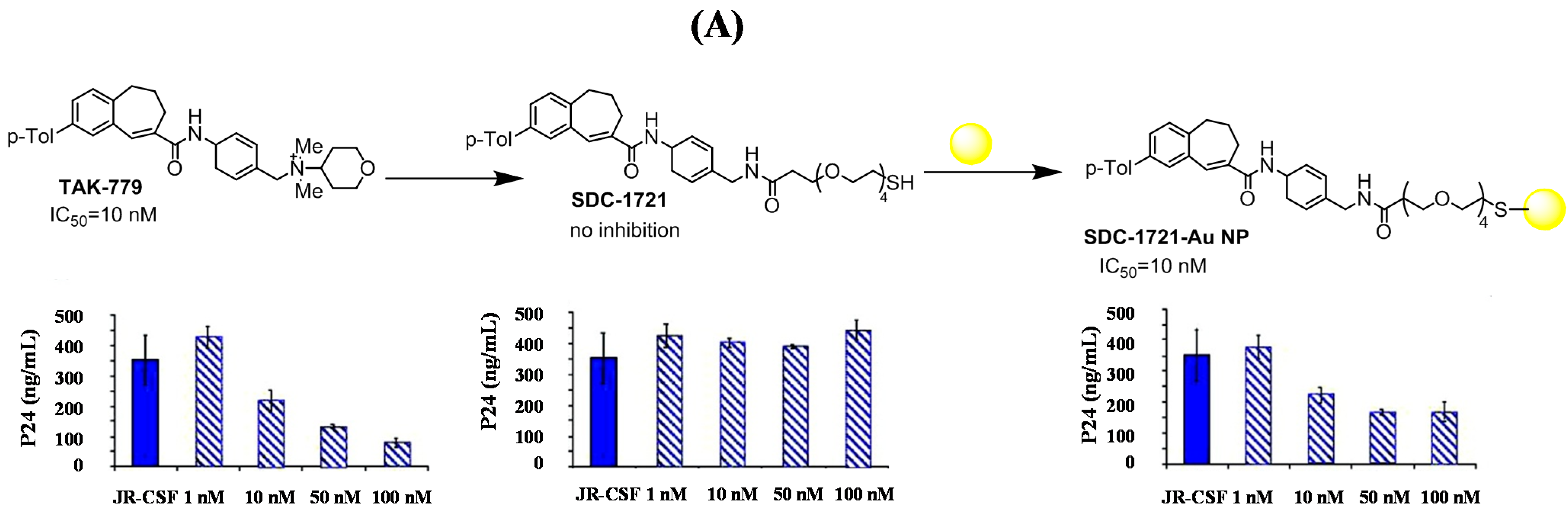{kind=link}
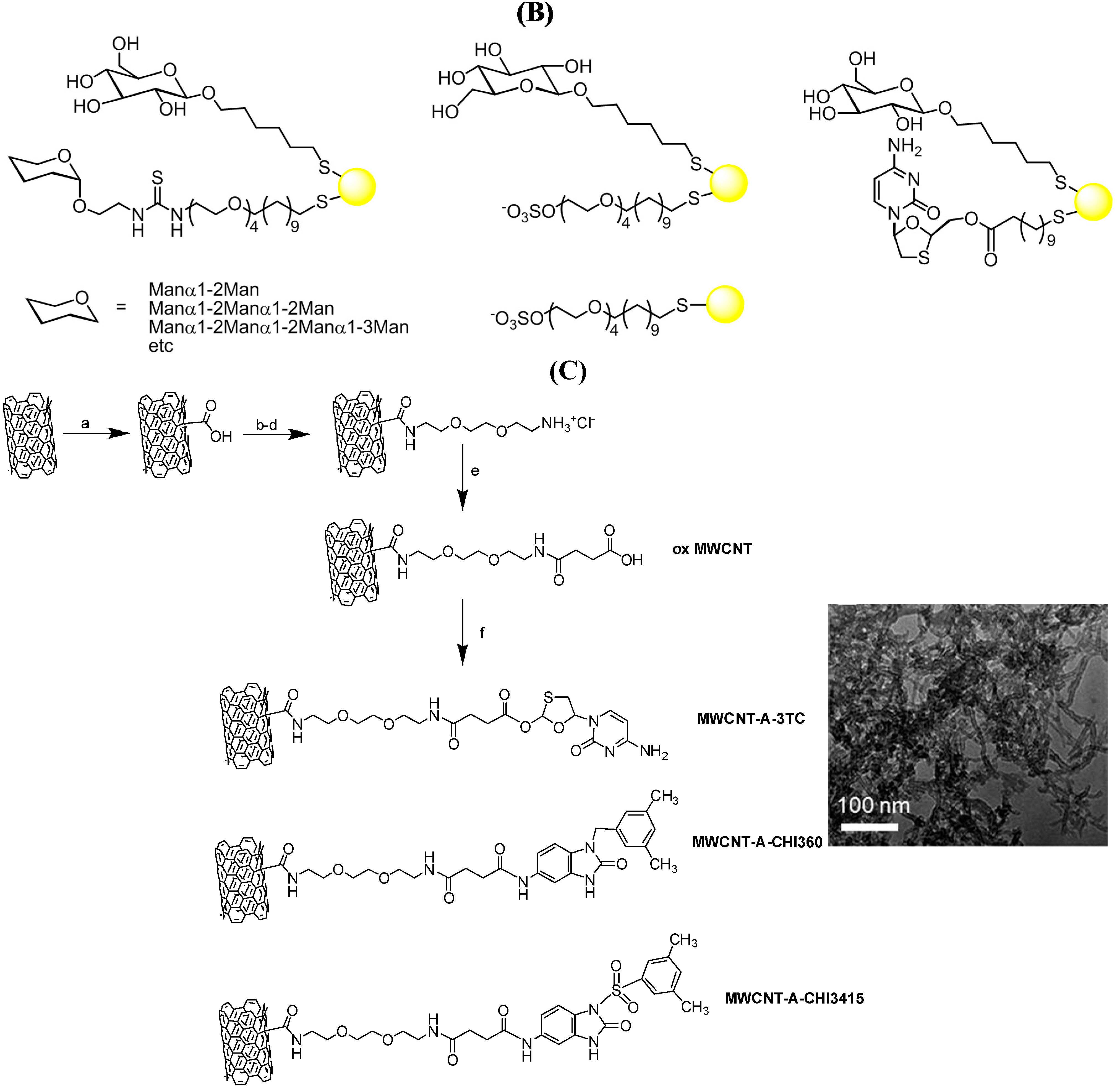{kind=link}
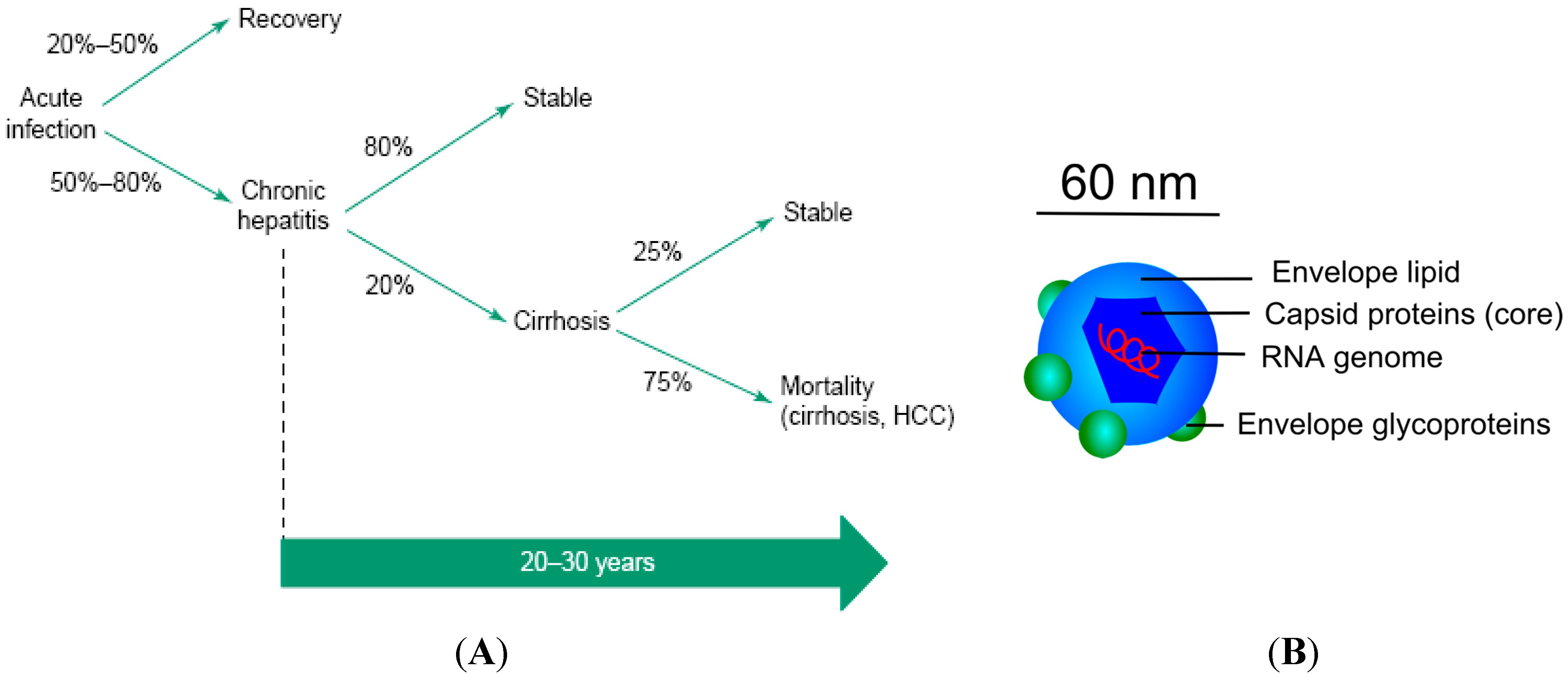{kind=link}
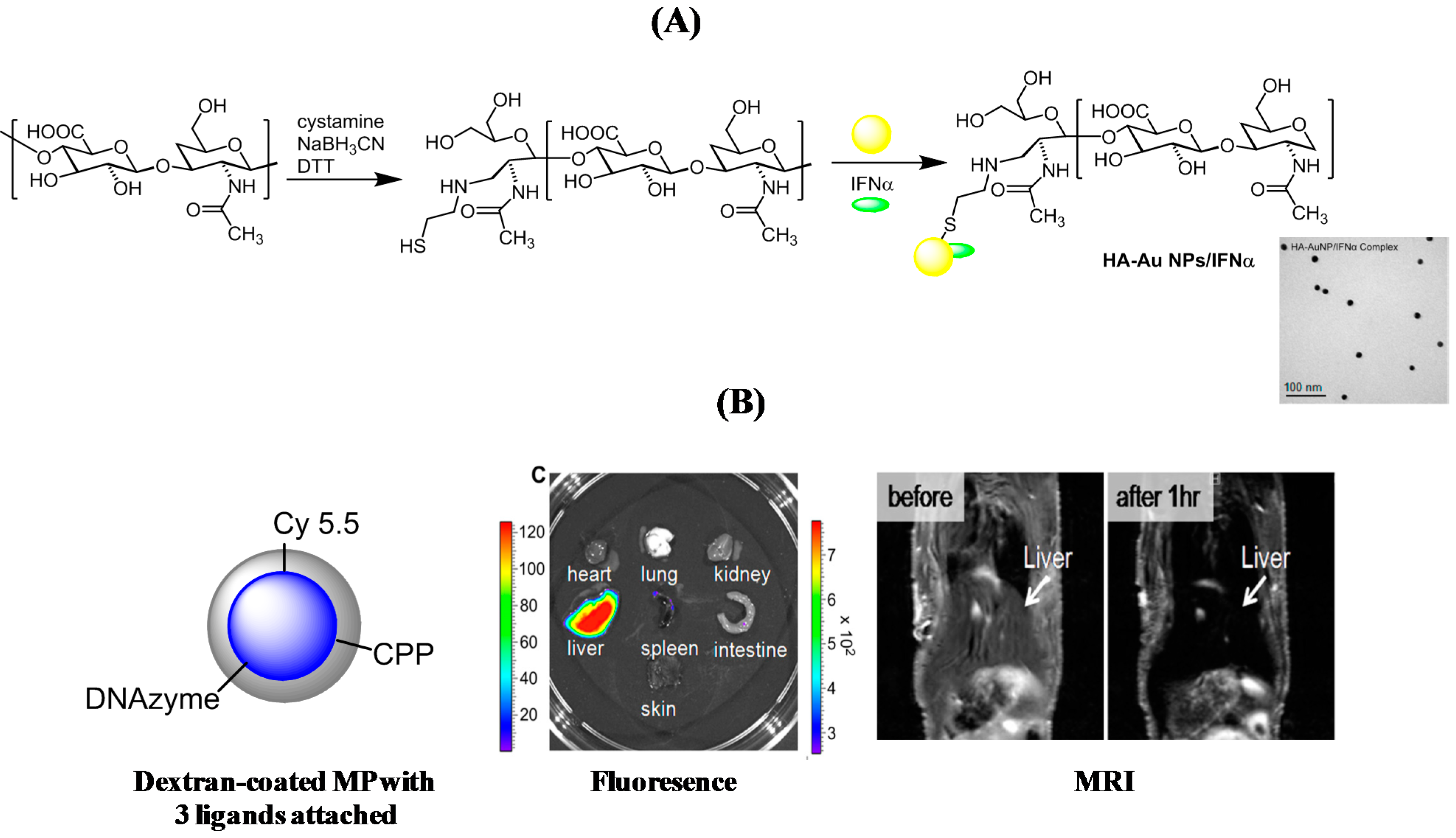{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
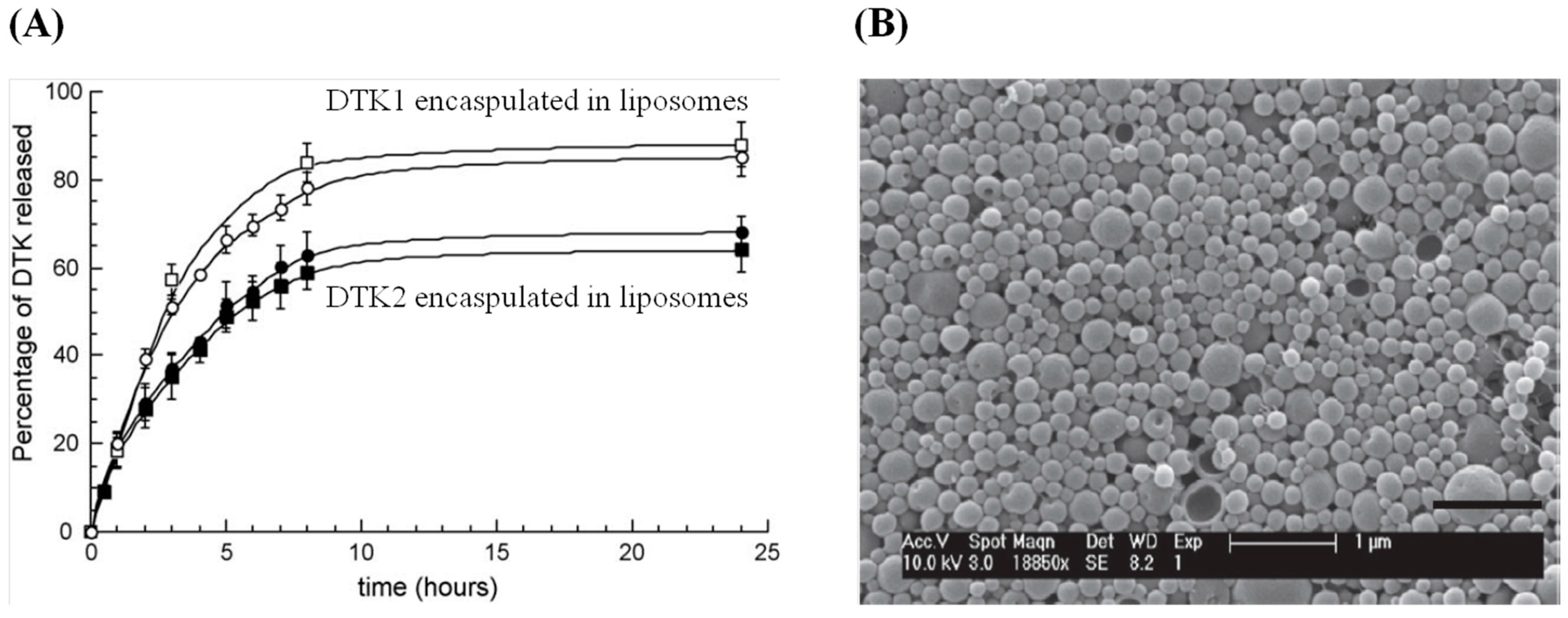{kind=link}
| Virus | Antiviral Agent | Administration | Viral Target |
|---|---|---|---|
| HIV | Efavirenz, Nevirapine, Rilpivirine | Oral | Non-nucleoside reverse transcriptase inhibitor |
| Indinavir | Recombinant protease inhibition | ||
| Raltegravir | Integrase strand transfer inhibitor | ||
| Maraviroc | Antagonists of CCR5 receptor | ||
| Tenofovir | Nucleotide reverse transcriptase inhibitor | ||
| Atazanavir, Ritonavir, Lopinavirb, Ritonavirc, Saquinavir, Tipranavir, Darunavir, Indinavir | Protease inhibitors | ||
| Abacavir, Didanosine, Emtricitabine, Lamivudine | Nucleoside reverse transcriptase inhibitors | ||
| Dapivirine | Intravaginal rings | Non-nucleoside reverse transcriptase inhibitor | |
| Enfuvirtide | Subcutaneous injection | Inhibitor of gp 41 | |
| Zidovudine | Oral + intravenous | Reverse transcriptase inhibitor and stop DNA elongation | |
| HCV | Ribavarin | Oral | Nucleoside analogue |
| Pegylated interfereon-α | Subcutaneous | Major histocompatibility complex stimulator | |
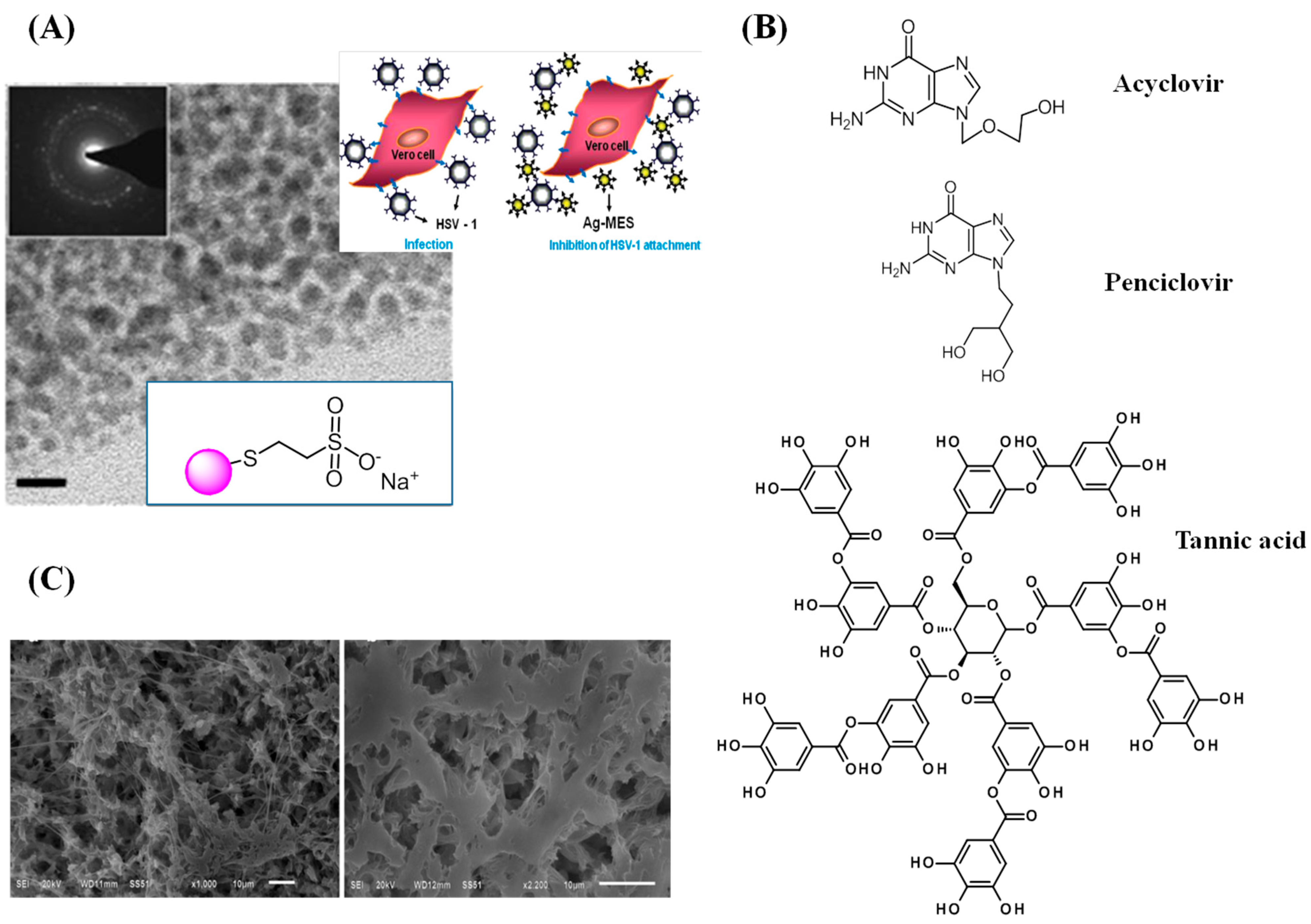
| HSV | Acyclovir | Oral, topical, intravenous | Inhibitor DNA syntheses |
| Penciclovir | Topical | DNA elongation inhibitor | |
| Famciclovir, Brivudin, Valaciclovir | Oral | DNA elongation inhibitor | |
| Iodoxuridine | Intravenous | DNA elongation inhibitor | |
| Trifluridine | Eye drops | DNA elongation inhibitor |
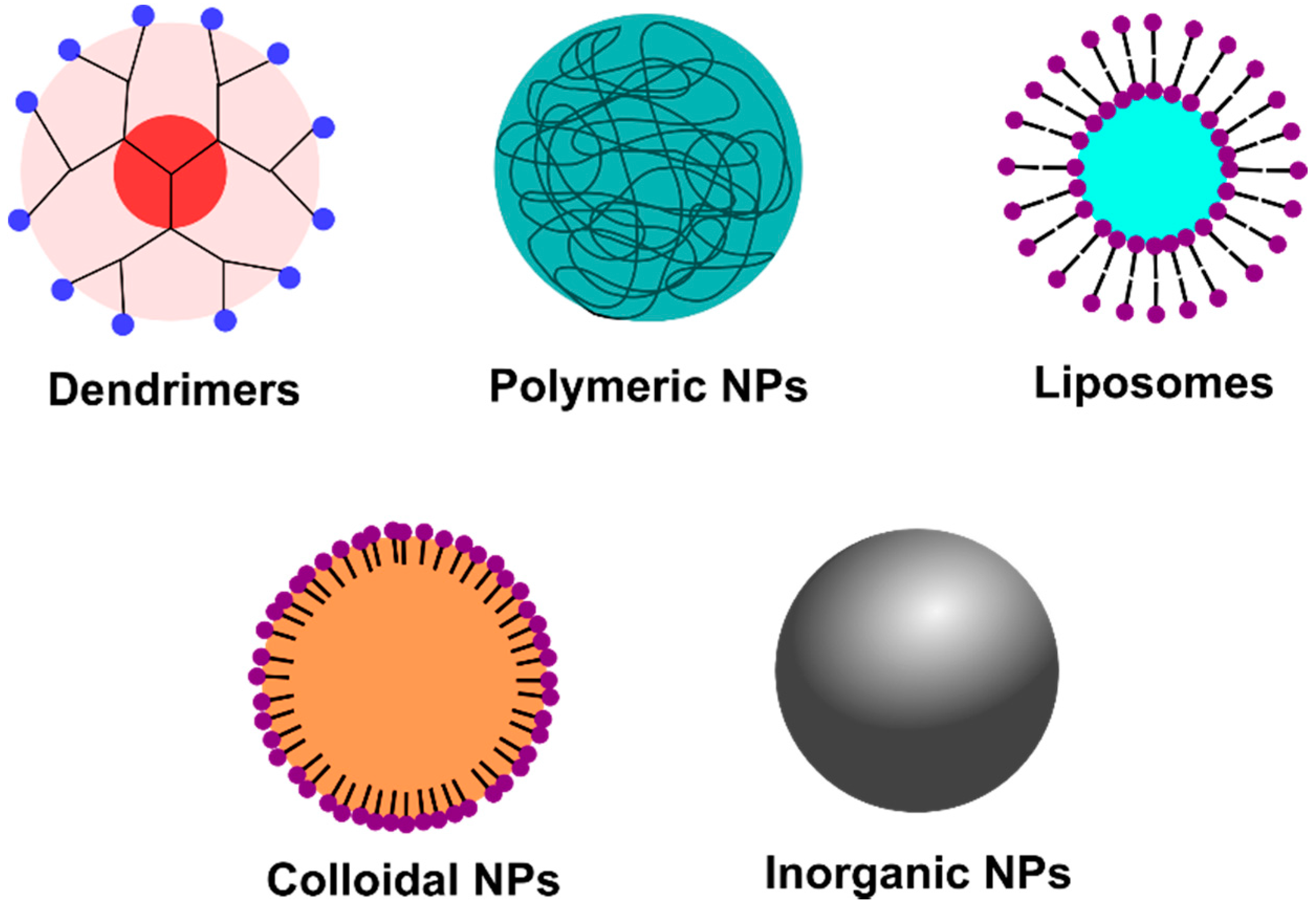
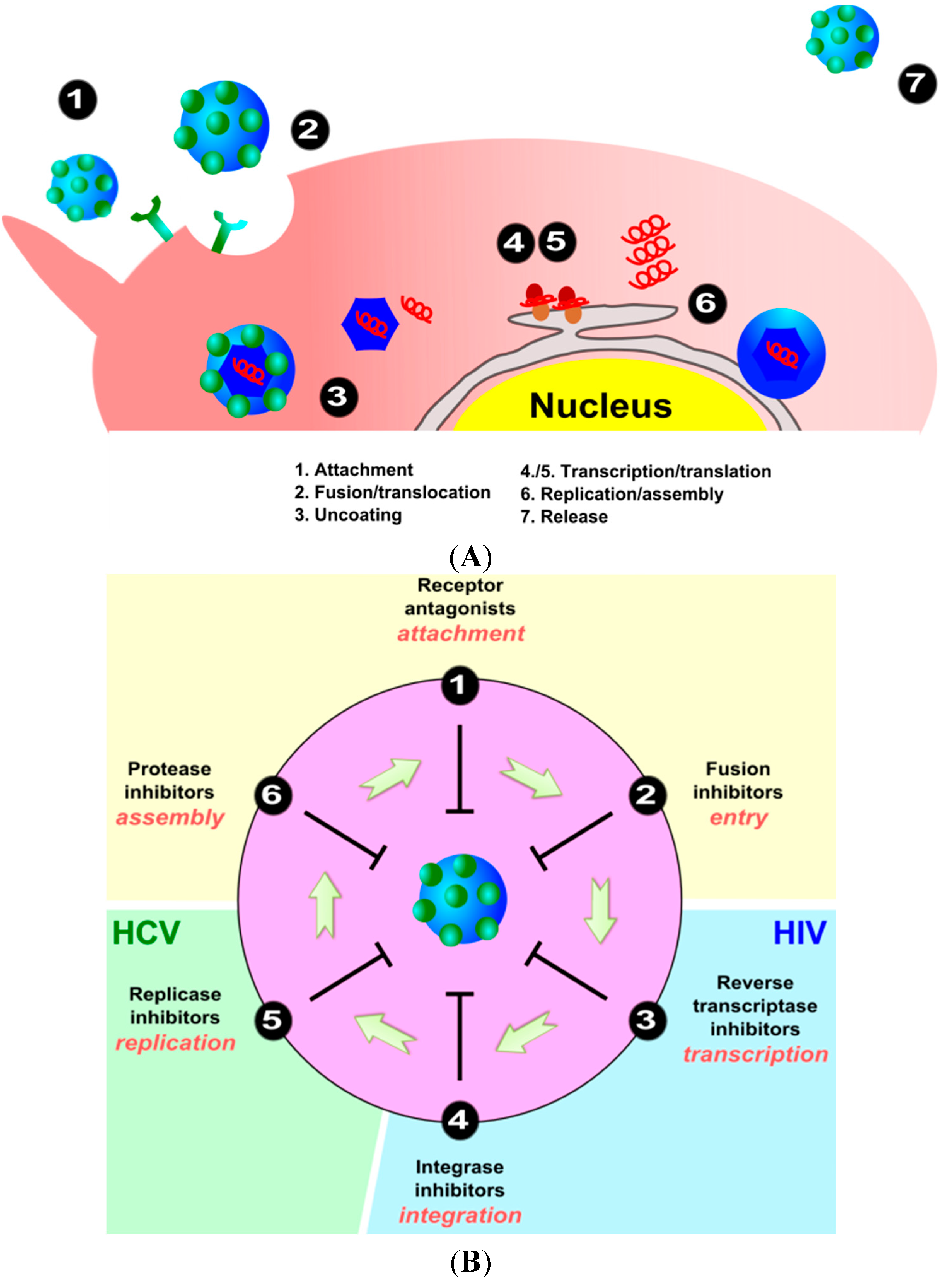
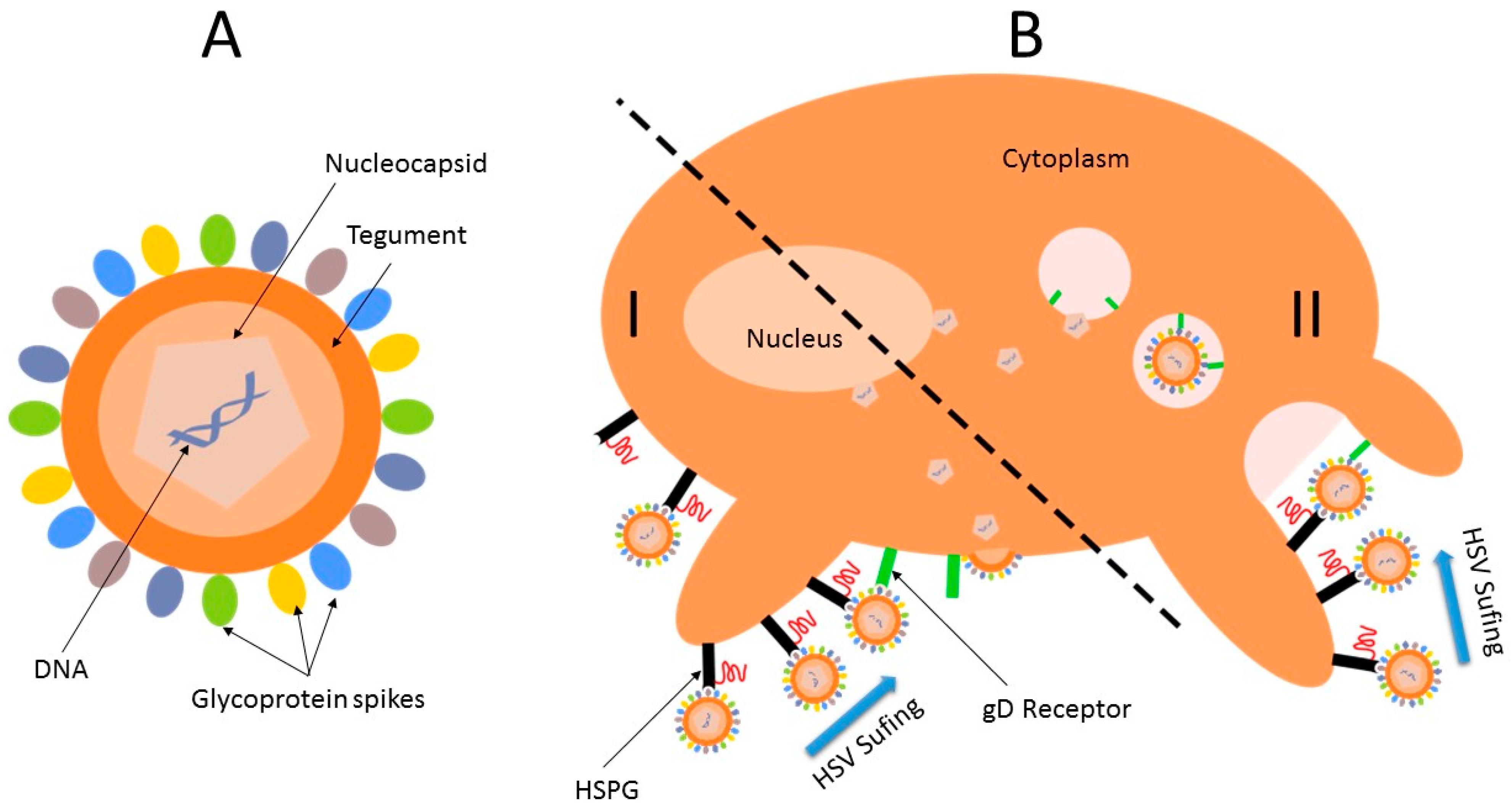
2. Viral Replication Cycle
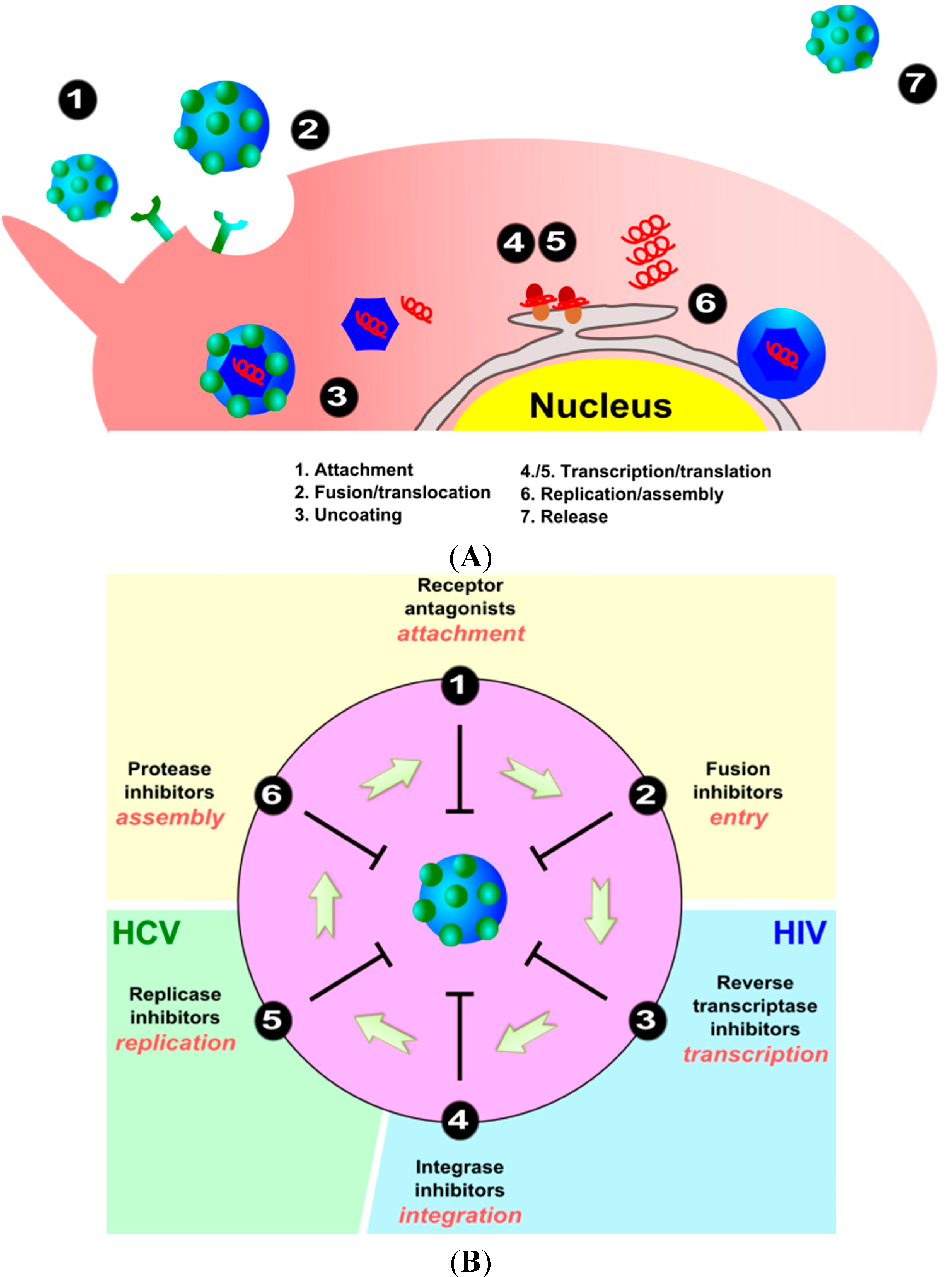
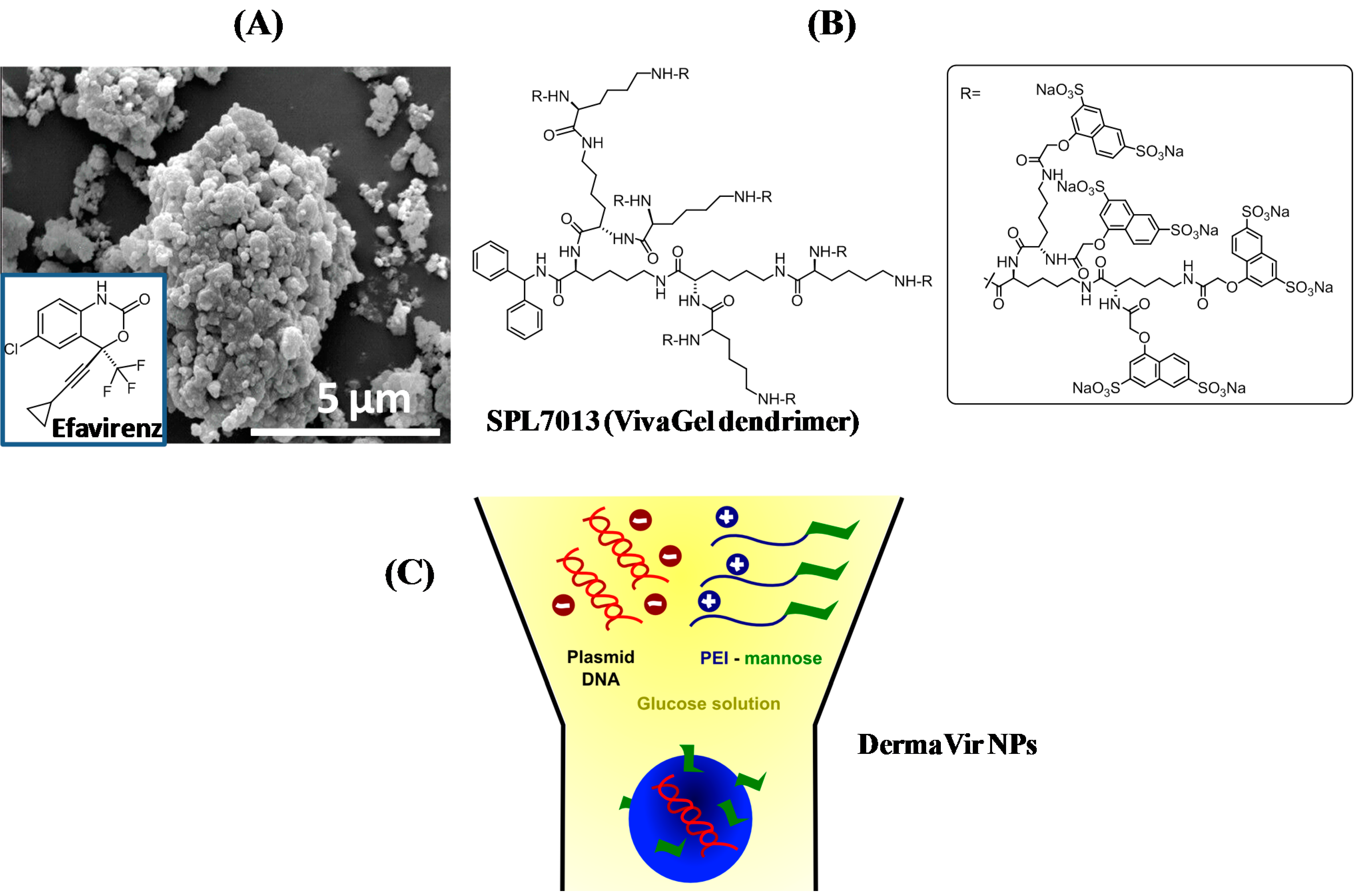
3. Interaction and Treatment of Different Enveloped Viruses with Nanocarriers
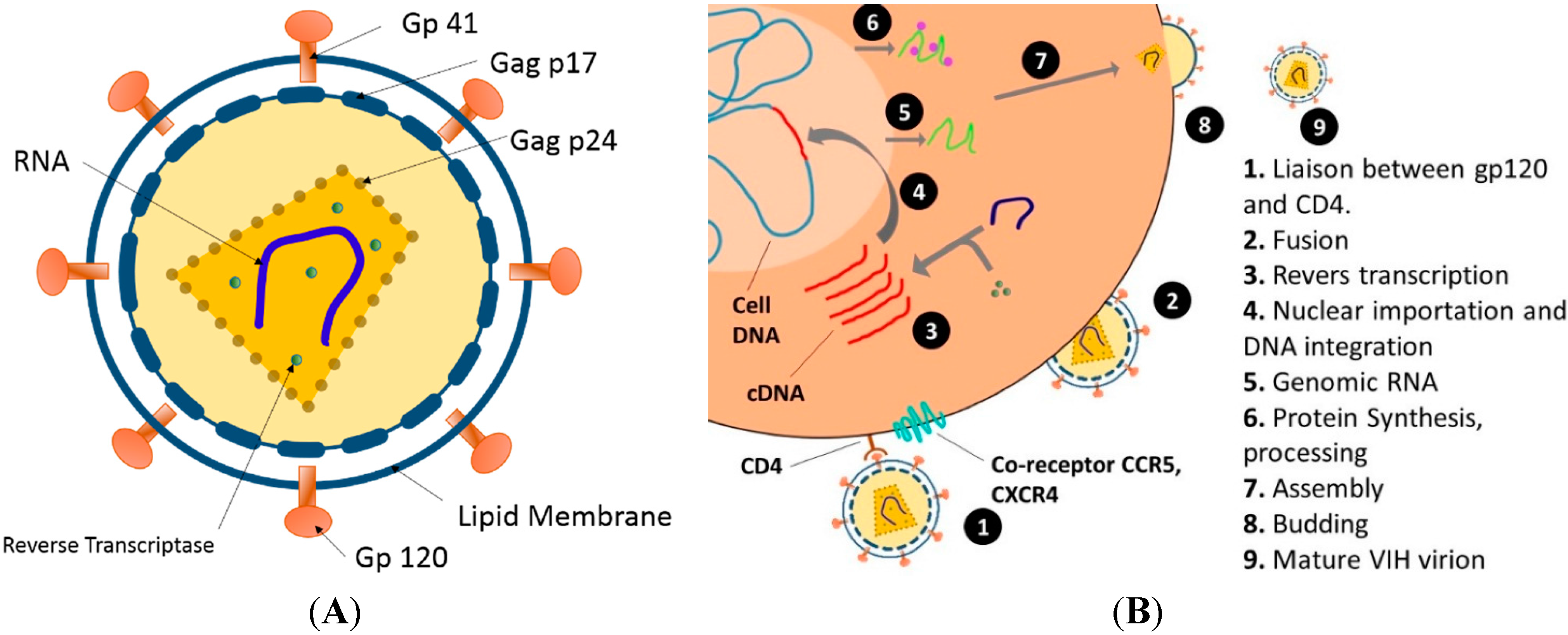
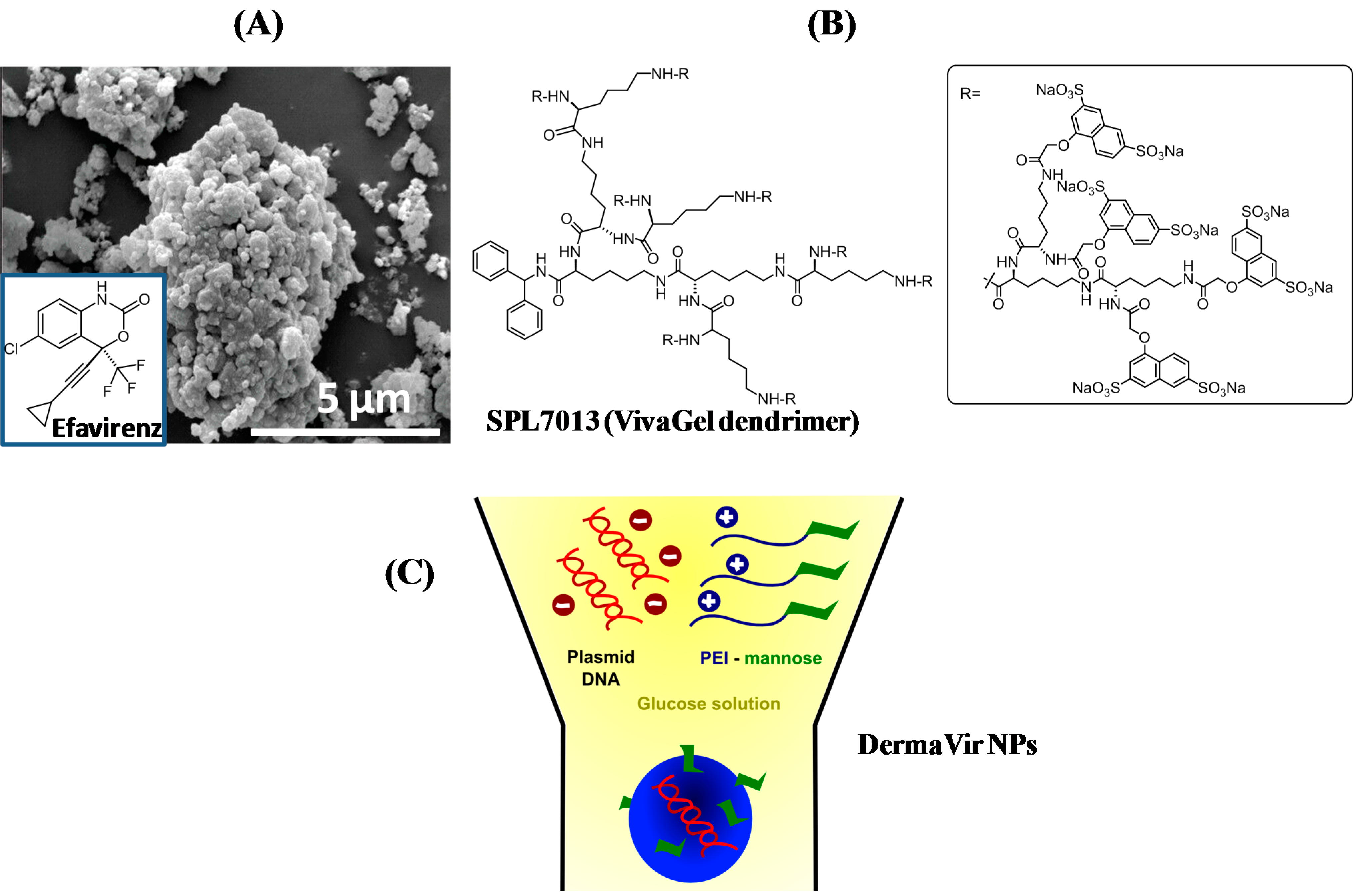
3.1. Human Immunodeficiency Virus (HIV)

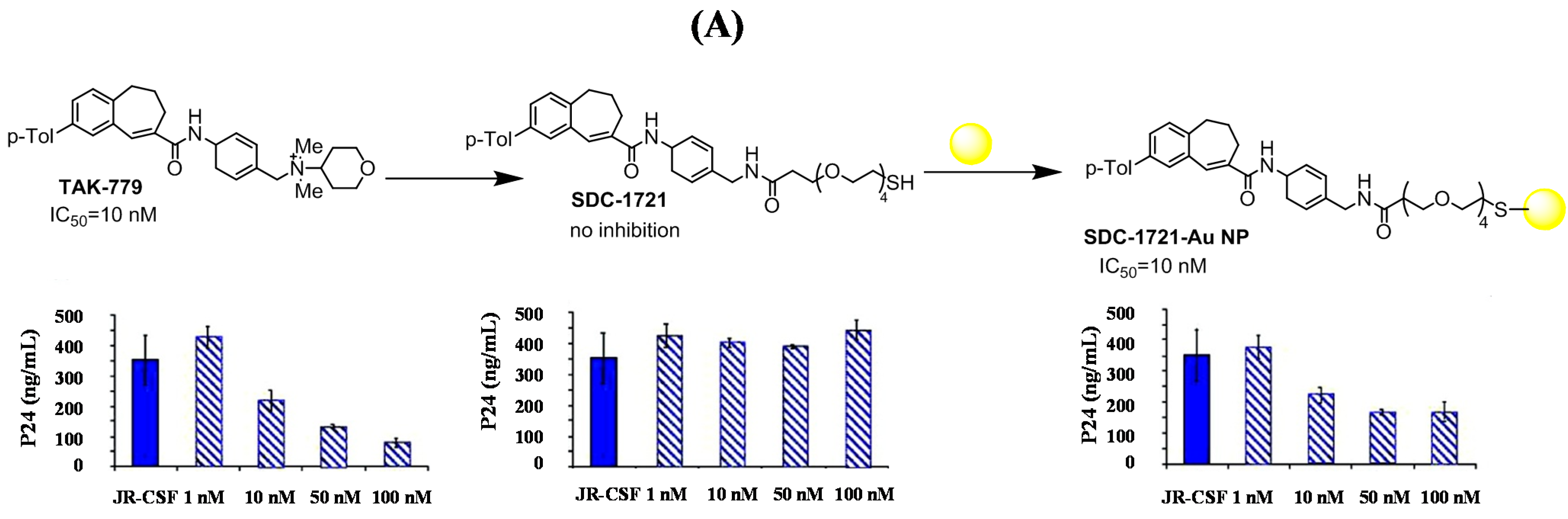
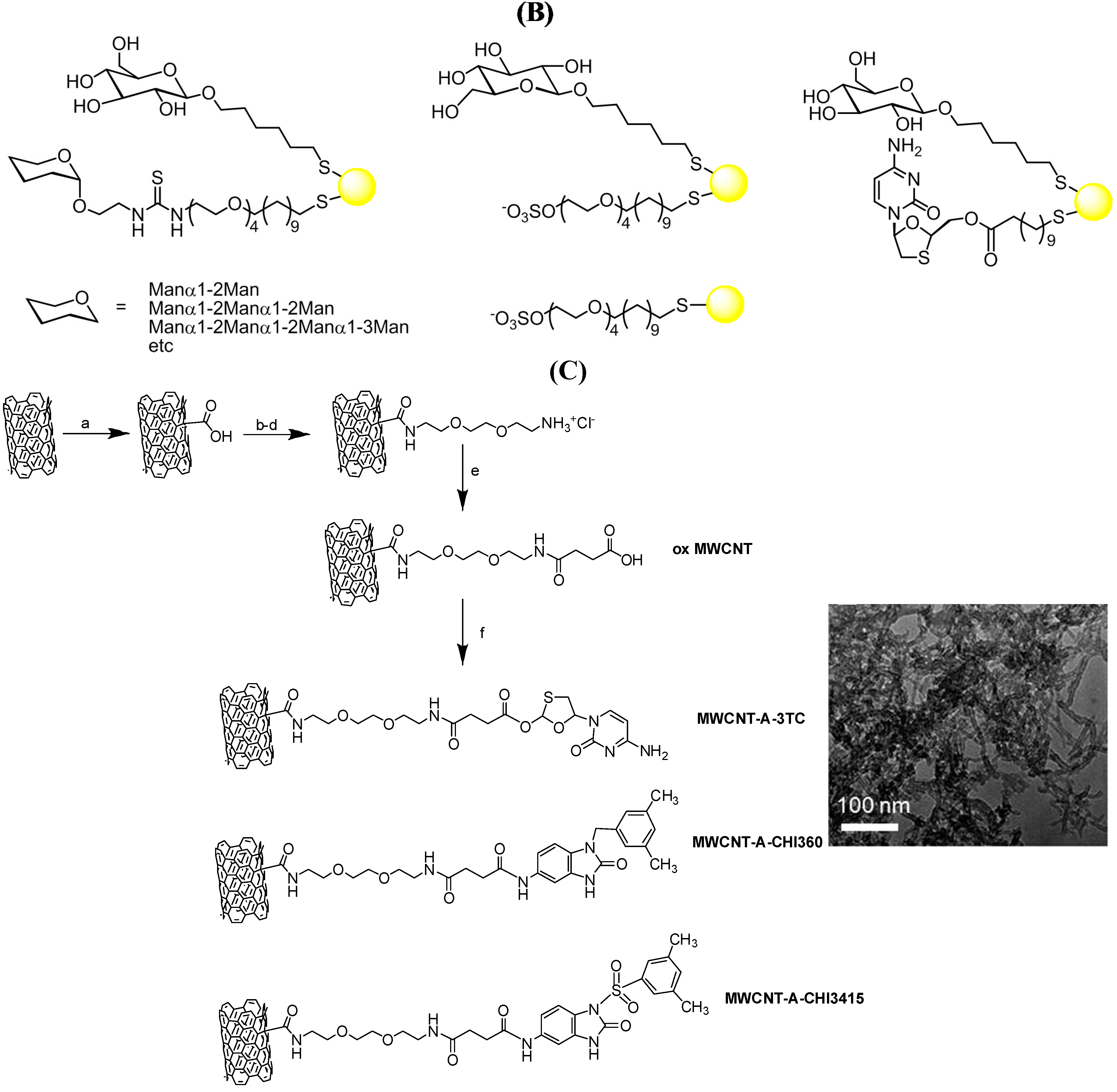
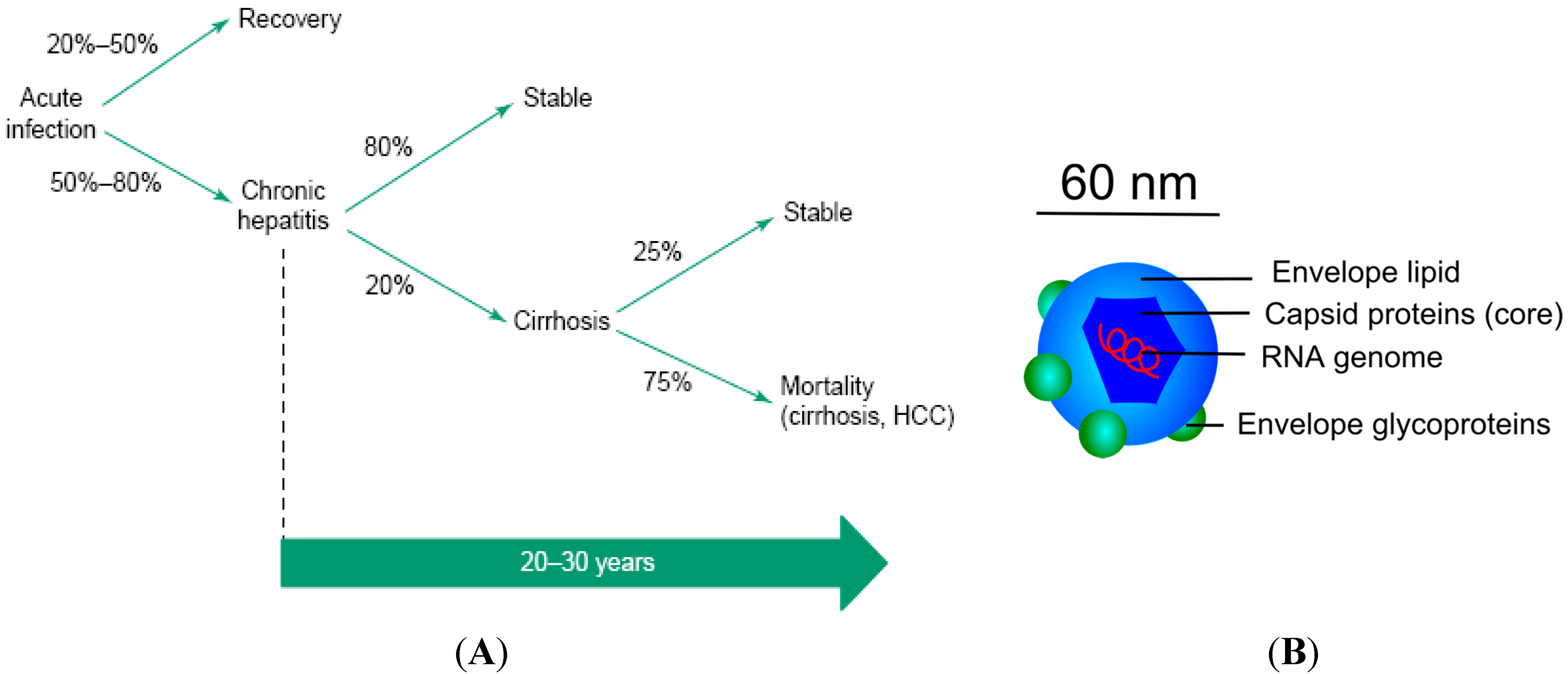
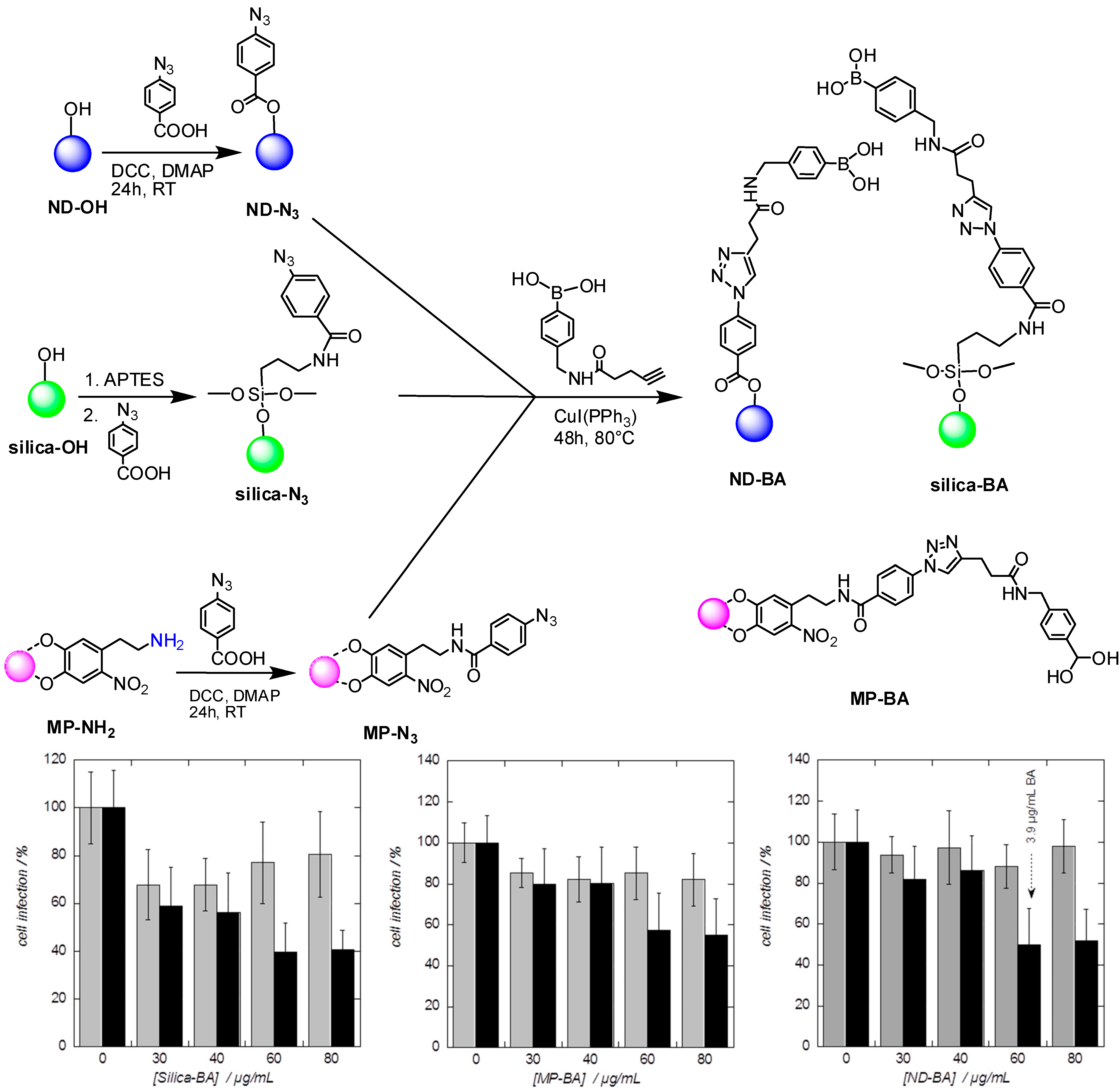
3.2. Hepatitis C Virus (HCV)
| Target | Examples of Compounds | Stage of Development |
|---|---|---|
| HCV E1E2 | Neutralizing antibodies | |
| Polyclonal HCV IgG (civacir) | Phase II | |
| HCV-AbXTL68 | Phase II | |
| Human mAB AR3 | Mouse model | |
| CBH and HC antibodies | Cell culture | |
| IGH antibodies | Cell culture | |
| AP33 | Cell culture | |
| 3/11 | Cell culture | |
| Fab e137 | Cell culture | |
| mAbs 1:7 and A8 | Cell culture | |
| HCV1 and HCV 95-2 | Cell culture | |
| Heparin + HS analogues | Cell culture | |
| Lectins | Cell culture | |
| HCV particle | Anti-apoEmAb | Cell culture |
| SR-BI | Anti-SR-BI pAb and mAb | Cell culture |
| ITX5061 | Cell culture | |
| Serum amyloid A | Cell culture | |
| CD81 | Anti-CD81 mAb | Cell culture, mouse model |
| Imidazole-based compounds | Cell culture | |
| CLDN1 | Anti-CLDN1 pAb and mAb | Cell culture |
| Internalization/fusion | PS-ON | Mouse model |
| Arbidol | Cell culture | |
| Chloroquine | Cell culture | |
| Silymarin | Cell culture |

3.3. Herpes Simplex Virus

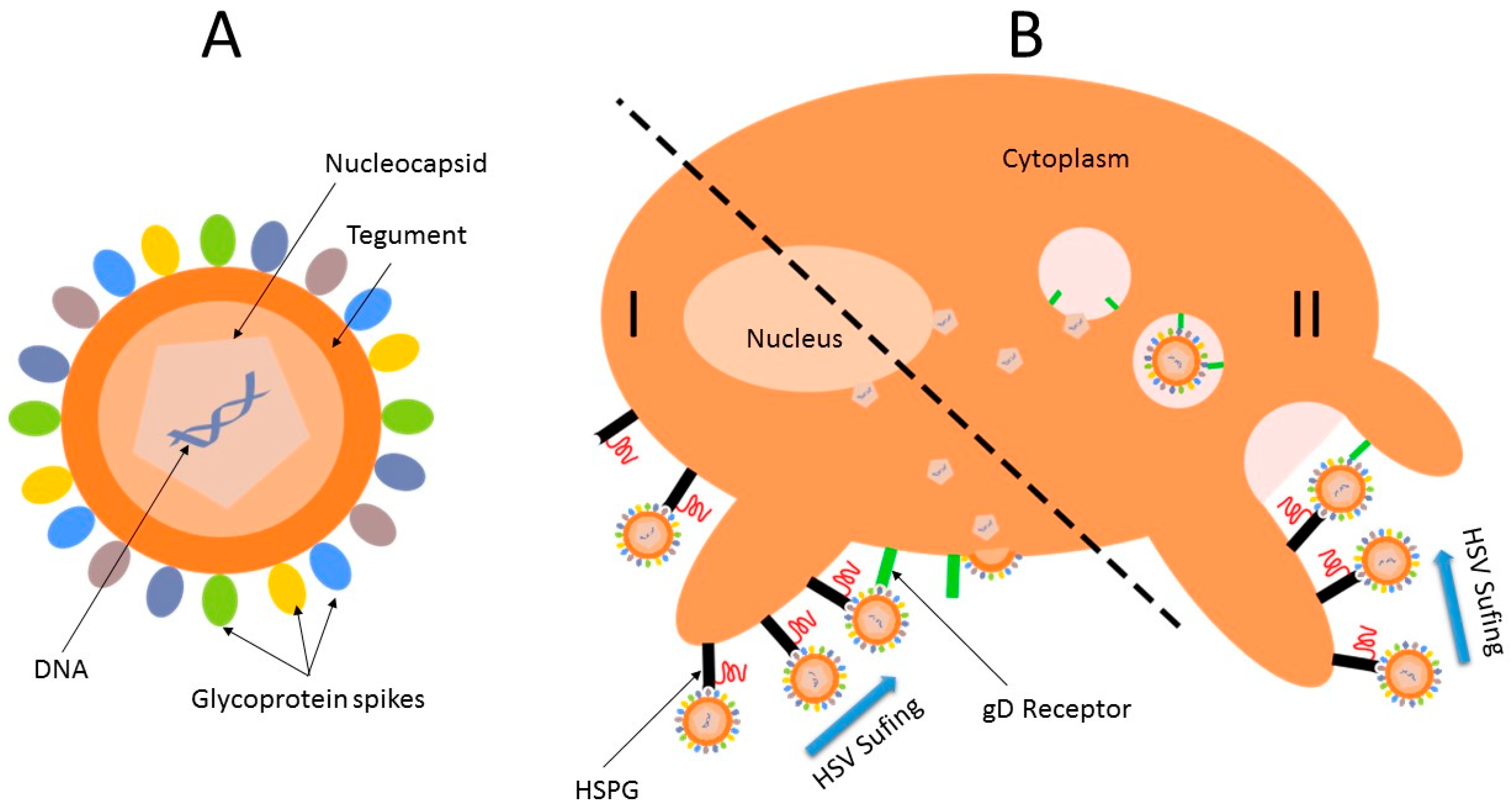
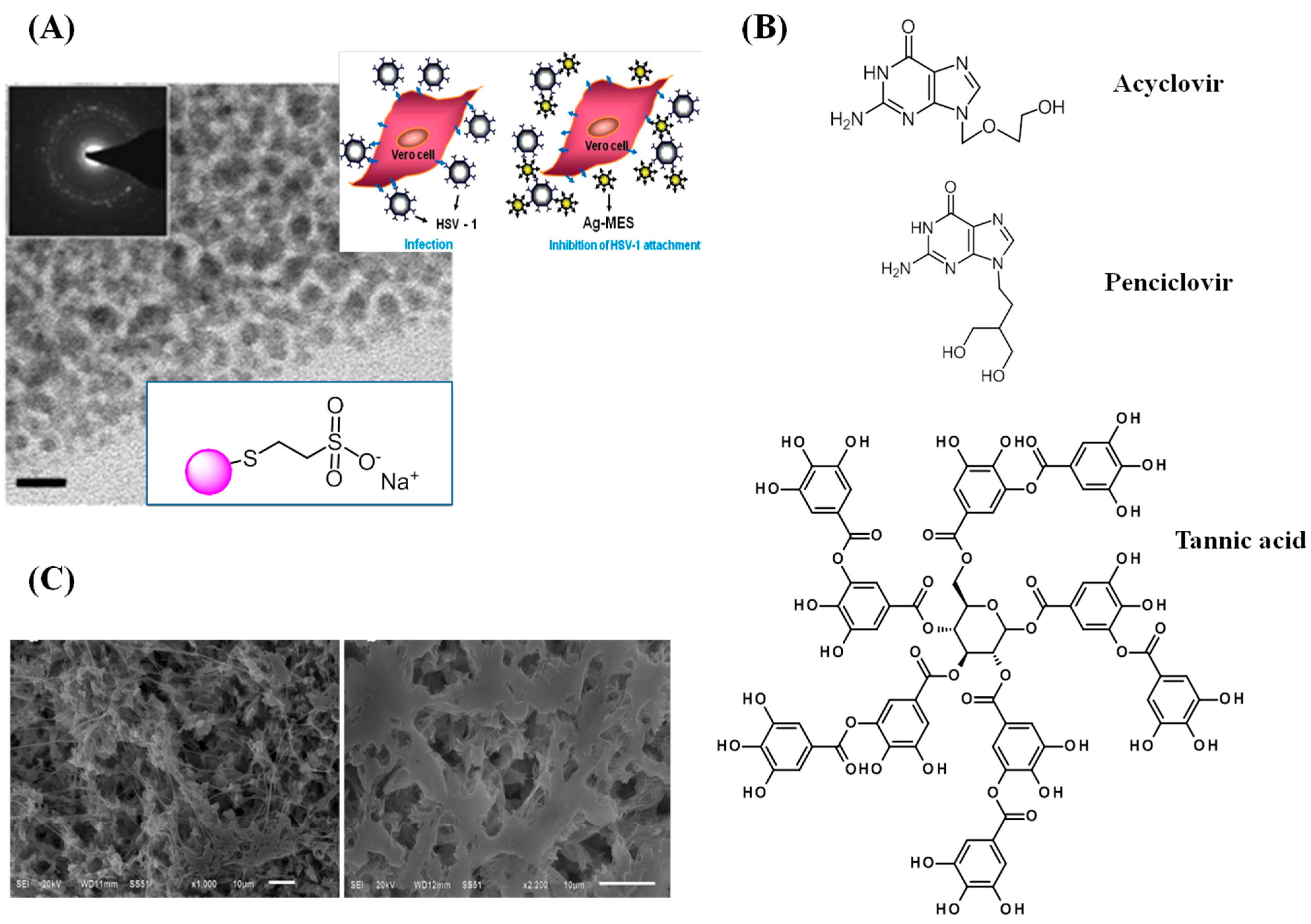
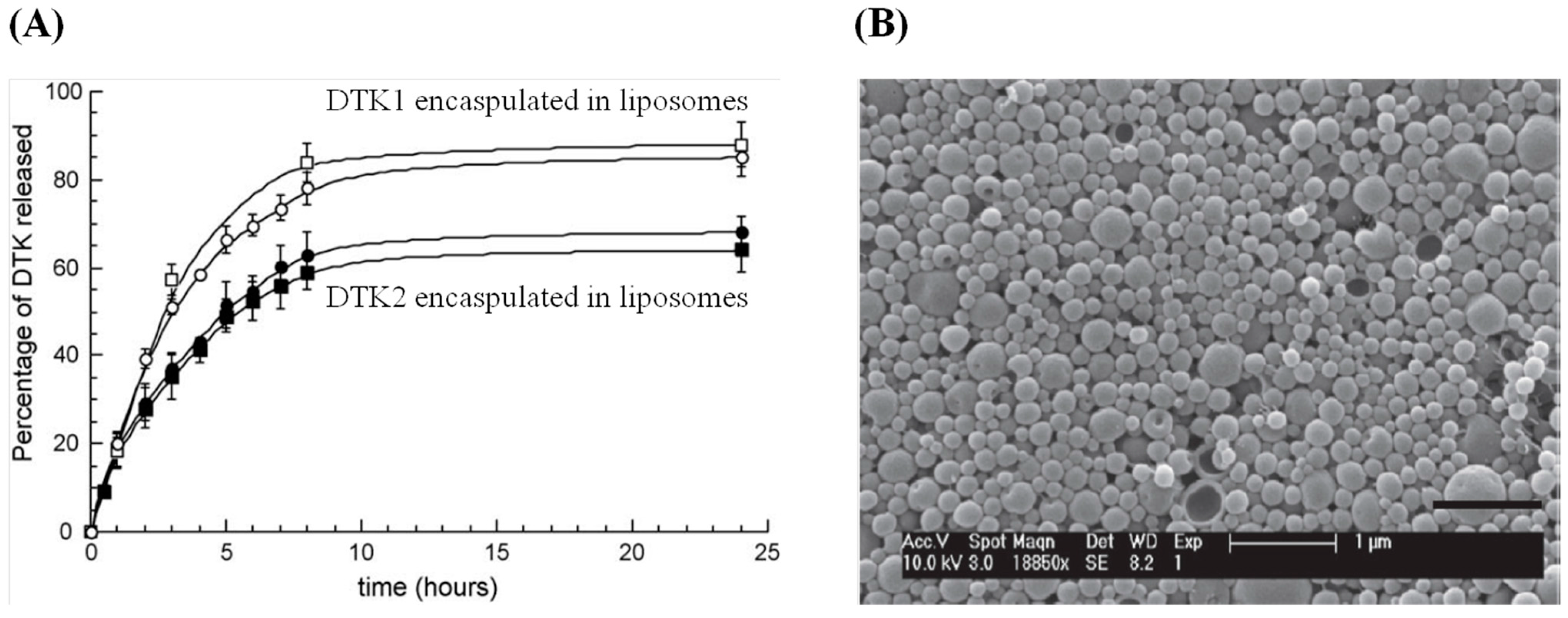
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Milroy, D.; Featherstone, J. Antiviral market overview. Nat. Rev. Drug. Discov. 2002, 1, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Lembro, D.; Cavalli, R. Nanoparticulate delivery systems for antiviral drugs. Antivir. Chem. Chemotherap. 2010, 21, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Tyring, S.K.; Baker, D.; Snowden, W. Valacyclovir for herpes simplex virus infection: Long-term safety and sustained efficacy after 20 years’ experience with acyclovir. J. Infect. Dis. 2002, 186 (Suppl. S1), S40–S46. [Google Scholar] [CrossRef] [PubMed]
- Hearnden, V.; Sankar, V.; Hull, K.; Juras, D.V.; Greenberg, M.; Kerr, A.R.; Lockhart, P.B.; Patton, L.L.; Porter, S.; Thornhill, M.H. New developments and opportunities in oral mucosal drug delivery for local and systemic disease. Adv. Drug Deliv. Rev. 2012, 64, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Resistance of herpes simplex viruses to nucleoside analogues: Mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 2012, 55, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Kim, J.S.; Mitchell, S.; Kish, P.; Kijek, P.; Hilfinger, J. 5′-O-d-valyl ara A, a potential prodrug for improving oral bioavailability of the antiviral agent vidarabine. Nucleic Acids 2009, 28, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, Y.; Wen, X.; Ma, H. Current prodrug strategies for improving oral absorption of nucleoside analogues. Asian J. Pharm. Sci. 2014, 9, 65–74. [Google Scholar] [CrossRef]
- Johnson, T.P.; Frey, R.; Modugno, M.; Brennan, T.P.; Margulies, B.J. Development of an aciclovir implant for the effective long-term control of herpes simplex virus type-1 infection in Vero cells and in experimentally infected SKH-1 mice. Int. J. Antimicrob. Agents 2007, 30, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; MacKay, J.A.; Frechet, J.M.; Szoka, F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005, 23, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Rupp, R.; Rosenthal, S.L.; Stanberry, L.R. VivaGel (SPL7013 Gel): A candidate dendrimer—Microbicide for the prevention of HIV and HSV infection. Int. J. Nanomed. 2007, 2, 561–566. [Google Scholar]
- Ham, A.S.; Cost, M.R.; Sassi, A.B.; Dezzutti, C.S.; Rohan, L.C. Targeted Delivery of PSC-RANTES for HIV-1 Prevention using Biodegradable Nanoparticles. Pharm. Res. 2009, 26, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Woodrow, K.A.; Cu, Y.; Booth, C.J.; Saucier-Sawyer, J.K.; Wood, M.J.; Saltzman, W.M. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat. Mater. 2009, 8, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Jederstrom, G.; Andersson, A.; Grasjo, J.; Sjoholm, I. Formulating insulin for oral administration: Preparation of hyaluronan-insulin complex. Pharm. Res. 2004, 21, 2040–2047. [Google Scholar] [CrossRef] [PubMed]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Rajaonarivony, M.; Vauthier, C.; Couarraze, G.; Puisieux, F.; Couvreur, P. Development of a new drug carrier made from alginate. J. Pharm. Sci. 1993, 82, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, C.; Bouchemal, K. Methods for the preparation and manufacture of polymeric nanoparticles. Pharm. Res. 2009, 26, 1025–1058. [Google Scholar] [CrossRef] [PubMed]
- Rawat, M.; Singh, D.; Saraf, S.; Saraf, S. Nanocarriers: Promising vehicle for bioactive drugs. Biol. Pharm. Bull. 2006, 29, 1790–1798. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Martins, S.; Sarmento, B.; Ferreira, D.C.; Souto, E.B. Lipid-based colloidal carriers for peptide and protein delivery—Liposomes versus lipid nanoparticles. Int. J. Nanomed. 2007, 2, 595–607. [Google Scholar]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar] [PubMed]
- Mahajan, S.D.; Aalinkeel, R.; Law, W.C.; Reynolds, J.L.; Nair, B.B.; Sykes, D.E.; Yong, K.T.; Roy, I.; Prasad, P.N.; Schwartz, S.A. Anti-HIV-1 nanotherapeutics: Promises and challenges for the future. Int. J. Nanomed. 2012, 7, 5301–5314. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, J.; Rodríguez-Gascón, A.; Solinís, M.Á.; del Pozo-Rodríguez, A. Lipid nanoparticles as carriers for RNAi against viral infections: Current status and future perspectives. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.J.; Rees, G.D. Microemulsion-based media as novel drug delivery systems. Adv. Drug Deliv. Rev. 2000, 45, 9–121. [Google Scholar] [CrossRef]
- Talegaonkar, S.; Azeem, A.; Ahmad, F.J.; Khar, R.K.; Pathan, S.A.; Khan, Z.I. Microemulsions: A novel approach to enhanced drug delivery. Recent Pat. Drug Deliv. Formul. 2008, 2, 238–257. [Google Scholar] [CrossRef] [PubMed]
- Shishu; Rajan, S.; Kamalpreet. Development of novel microemulsion-based topical formulations of acyclovir for the treatment of cutaneous herpetic infections. AAPS PharmSciTech 2009, 10, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Valetti, S.; Simona Mura, S.; Stella, B.; Couvreur, P. Rational design for multifunctional non-liposomal lipid-based nanocarriers for cancer management: Theory to practice. J. Nanobiotechnol. 2013, 11. [Google Scholar] [CrossRef] [PubMed]
- Mehnert, W.; Mader, K. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev. 2001, 47, 165–196. [Google Scholar] [CrossRef]
- Heurtault, B.; Saulnier, P.; Pech, B.; Proust, J.E.; Benoit, J.P. A novel phase inversion-based process for the preparation of lipid nanocarriers. Pharm. Res. 2002, 19, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, R.; Ravani, L.; Menegatti, E.; Drechsler, M.; Esposito, E. Colloidal dispersions for the delivery of acyclovir: A comparative study. Indian J. Pharm. Sci. 2011, 73, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Elechiguerra, J.L.; Burt, J.L.; Morones, J.R.; Camacho-Bragado, A.; Gao, X.; Lara, H.H.; Yacaman, M.J. Interaction of silver nanoparticles with HIV-1. J. Nanobiotechnol. 2005, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joe Grove, J.; Marsh, M. Host-pathogen interactions: The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Mannan, M.; Choi, S.K.; Whitesides, G.M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Mu ltivalent Ligands and Inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2745–2794. [Google Scholar]
- Wang, Z.; Liu, H.; Yang, S.H.; Wang, T.; Liu, C.; Cao, Y.C. Nanoparticle-based artificial RNA silencing machinery for antiviral therapy. Proc. Natl. Acad. Sci. USA 2012, 109, 12387–12392. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Singh, I.; Helenius, A. Mechanisms of virus uncoating. Trends Microbiol. 1994, 2, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Church, G.A.; Wilson, D.W. Study of herpes simplex virus maturation during a synchronous wave of assembly. J. Virol. 1997, 71, 3603–3612. [Google Scholar] [PubMed]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. The design of drugs for HIV and HCV. Nat. Rev. Drug. Discov. 2007, 6, 1001–1018. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.H.; Pedersen, F.S.; Kjems, J. Molecular strategies to inhibit HIV-1 replication. Retrovirology 2005, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enjuanes, L.; Sola, I.; Izeta, A.; Sanchez-Morgado, J.M.; Gonzalez, J.M.; Alonso, S.; Escors, D.; Sanchez, C.M. Interference with virus and bacteria replication by the tissue specific expression of antibodies and interfering molecules. Adv. Exp. Med. Biol. 1999, 473, 31–45. [Google Scholar] [PubMed]
- Yan, L.; Zhang, J.; Guo, H.; Yan, S.; Chen, Q.; Zhang, F.; Fang, Q. Aquareovirus NS80 Initiates Efficient Viral Replication by Retaining Core Proteins within Replication-Associated Viral Inclusion Bodies. PLoS ONE 2015, 10, e0126127. [Google Scholar] [CrossRef] [PubMed]
- Badley, A. D.; Sainski, A.; Wightman, F.; Lewin, S.R. Altering cell death pathways as an approach to cure HIV infection. Cell Death Dis. 2013, 4, e718. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.C.; Lacey, C.J. Microbicides and HIV prevention: Lessons from the past, looking to the future. Curr. Opin. Infect. Dis. 2010, 23, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Date, A.A.; Destache, C.J. A review of nanotechnological approaches for the prophylaxis of HIV/AIDS. Biomaterials 2013, 34, 6202–6228. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.H.; Taylor, K.A. AIDS virus envelope spike structure. Curr. Opin. Struct. Biol. 2007, 17, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Lisziewicz, J.; Toke, E.R. Nanomedicine applications towards the cure of HIV. Nanomedicine 2013, 9, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.; Kon, K.; Ingle, A.; Duran, N.; Galdiero, S.; Galdiero, M. Broad-spectrum bioactivities of silver nanoparticles: The emerging trends and future prospects. Appl. Microbiol. Biotechnol. 2014, 98, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.B.; Patterson, B.K.; Naus, G.J.; Landers, D.V.; Gupta, P. Development of an in vitro organ culture model to study transmission of HIV-1 in the female genital tract. Nat. Med. 2000, 6, 475–479. [Google Scholar] [PubMed]
- Caron, M.; Besson, G.; Lekana-Douki Etenna, S.; Mintsa-Ndong, A.; Mourtas, S.; Radaelli, A.; de Giuli Morghen, C.; Loddo, R.; la Colla, P.; Antimisiaris, S.G.; et al. Protective properties of non-nucleoside reverse transcriptase inhibitor (MC1220) incorporated into liposome against intravaginal challenge of Rhesus Macaques with RT-SHIV. Virology 2010, 405, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Mamo, T.; Moseman, E.A.; Kolishetti, N.; Salvador-Morales, C.; Shi, J.; Kuritzkes, D.R.; Langer, R.; von Andrian, U.; Farokhzad, O.C. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine (Lond.) 2010, 5, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Neff, C.P.; Liu, X.; Zhang, J.; Li, H.; Smith, D.D.; Swiderski, P.; Aboellail, T.; Huang, Y.; Du, Q.; et al. Systemic administration of combinatorial dsiRNAs via nanoparticles efficiently suppresses HIV-1 infection in humanized mice. Mol. Ther. 2011, 19, 2228–2238. [Google Scholar] [CrossRef] [PubMed]
- Gaur, P.K.; Mishra, S.; Bajpai, M.; Mishra, A. Enhanced oral bioavailability of efavirenz by solid lipid nanoparticles: In vitro drug release and pharmacokinetics studies. BioMed Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Feeling, J.P.; Koehn, J.; Shu, C.; Sun, J.; Ho, R.J. Anti-HIV drug-combination nanoparticles enhance plasma drug exposure duration as well as triple-drug combination levels in cells within lymph nodes and blood in primates. AIDS Res. Hum. Retrov. 2015, 31, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Chaowanachan, T.; Krogstad, E.; Ball, C.; Woodrow, K.A. Drug synergy of tenofovir and nanoparticle-based antiretrovirals for HIV prophylaxis. PLoS ONE 2013, 8, e61416. [Google Scholar] [CrossRef] [PubMed]
- Destache, C.J.; Belgum, T.; Christensen, K.; Shibata, A.; Sharma, A.; Dash, A. Combination antiretroviral drugs in PLGA nanoparticle for HIV-1. BMC Infect. Dis. 2009, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- De Jaeghere, F.; Allémann, E.; Kubel, F.; Galli, B.; Cozens, R.; Doelker, E.; Gurny, R. Oral bioavailability of a poorly water soluble HIV-1 protease inhibitor incorporated into pH-sensitive particles: Effect of the particle size and nutritional state. J. Control. Release 2000, 68, 291–298. [Google Scholar] [CrossRef]
- Parboosing, R.; Maguire, G.E.; Govender, P.; Kruger, H.G. Nanotechnology and the treatment of HIV infection. Viruses 2012, 4, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Jenita, J.L.; Chocalingam, V.; Wilson, B. Albumin nanoparticles coated with polysorbate 80 as a novel drug carrier for the delivery of antiretroviral drug-Efavirenz. Int. J. Pharm. Investig. 2014, 4, 142–148. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.; Singh, M.; Ugozzoli, M.; Wild, C.; Barnett, S.; Chen, M.; Schaefer, M.; Doe, B.; Otten, G.R.; Ulmer, J.B. Induction of potent immune responses by cationic microparticles with adsorbed human immunodeficiency virus DNA vaccines. J. Virol. 2001, 75, 9037–9043. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, T.D.; Karellas, P.; Henderson, S.A.; Giannis, M.; O'Keefe, D.F.; Heery, G.; Paull, J.R.; Matthews, B.R.; Holan, G. Dendrimers as Drugs: Discovery and Preclinical and Clinical Development of Dendrimer-Based Microbicides for HIV and STI Prevention. Mol. Pharm. 2005, 2, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, O.; Toke, E.R.; Somogyi, E.; Horkay, F.; Chandran, P.L.; Douglas, J.F.; Szebeni, J.; Lisziewicz, J. Structures and biologial activity of pathogen-like synthetic nanomedicines. Nanomedicines 2012, 8, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Katz, A.A. Cell Membranes Open “Doors” for Cationic Nanoparticles/Biomolecules: Insights into Uptake Kinetics. ACS Nano 2013, 7, 10799–10808. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.C.; Ballard, T.E.; Ackerson, C.J.; Feldheim, D.L.; Margolis, D.M.; Melander, C. Inhibition of HIV fusion with multivalent gold nanoparticles. J. Am. Chem. Soc. 2008, 130, 6896–6897. [Google Scholar] [CrossRef] [PubMed]
- Chiodo, F.; Marradi, M.; Calvo, J.; Yuste, E.; Penades, S. Glycosystems in nanotechnology: Gold glyconanoparticles as carrier for anti-HIV prodrugs. Beilstein J. Org. Chem. 2014, 10, 1339–1346. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, G.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. Highly active antiretroviral therapy: Current state of the art, new agents and their pharmacological interactions useful for improving therapeutic outcome. Curr. Pharm. Des. 2005, 11, 1805–1843. [Google Scholar] [CrossRef] [PubMed]
- Di Gianvincenzo, P.; Chiodo, F.; Marradi, M.; Penades, S. Gold manno-glyconanoparticles for intervening in HIV gp120 carbohydrate-mediated processes. Methods Enzymol. 2012, 509, 21–40. [Google Scholar] [PubMed]
- Martinez-Avila, O.; Bedoya, L.M.; Marradi, M.; Clavel, C.; Alcami, J.; Penades, S. Multivalent manno-glyconanoparticles inhibit DC-SIGN-mediated HIV-1 trans-infection of human T cells. ChemBioChem 2009, 10, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Di Gianvincenzo, P.; Marradi, M.; Martinez-Avila, O.M.; Bedoya, L.M.; Alcami, J.; Penades, S. Gold nanoparticles capped with sulfate-ended ligands as anti-HIV agents. Bioorg. Med. Chem. Lett. 2010, 20, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Marrardi, M.; Chiodo, F.; Garcia, I.; Penadés, S. Glyconanoparticles as multifunctinal and multimodal carbohydrate systems. Chem. Soc. Rev. 2013, 42, 4728–4745. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, Y.; Chen, Z.; Li, W.; Liu, Y.; Wang, L.; Liu, Y.; Wu, X.; Ji, Y.; Zhao, Y.; et al. Surface-engineered gold nanorods: Promising DNA vaccine adjuvant for HIV-1 treatment. Nano Lett. 2012, 12, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Iannazzo, D.; Pistone, A.; Galvagno, S.; Ferro, S.; de Luca, L.; Monforte, A.; da Ros, T.; Hadad, C.; Prato, M.; Pannecouque, C. Synthesis and anti-HIV activity of carboxylated and drug-conjugated multi-walled carbon nanotubes. Carbon 2015, 82, 548–561. [Google Scholar] [CrossRef]
- Marchesan, S.; da Ros, T.; Spalluto, G.; Balzarini, J.; Prato, M. Anti-HIV properties of cationic fullerene derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 3615–3618. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Pause, A.; Shi, Y.; Sonenberg, N. Hepatitis C therapeutics: Current status and emerging strategies. Nat. Rev. Drug Discov. 2002, 1, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Pathophysiology of hepatitis C virus infection and related liver disease. Trends Microbiol. 2004, 12, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Zeuzem, S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010, 138, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Fofana, I.; Fafi-Kremer, S.; Baumert, T.F. Hepatitis C virus entry into hepatocytes: Molecular mechanisms and targets for antiviral therapies. J. Hepatol. 2011, 54, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Hesselgesser, J.; Paulson, M.; Vanwolleghem, T.; Desombere, I.; Reiser, H.; Leroux-Roels, G. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 2008, 48, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Syder, A.J.; Lee, H.; Zeisel, M.B.; Grove, J.; Soulier, E.; Macdonald, J.; Chow, S.; Chang, J.; Baumert, T.F.; McKeating, J.A.; et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J. Hepatol. 2011, 54, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Baldick, C.J.; Wichroski, M.J.; Pendri, A.; Walsh, A.W.; Fang, J.; Mazzucco, C.E.; Pokornowski, K.A.; Rose, R.E.; Eggers, B.J.; Hsu, M.; et al. A novel small molecule inhibitor of hepatitis C virus entry. PLoS Pathog. 2010, 6, e1001086. [Google Scholar] [CrossRef] [PubMed]
- Helle, F.; Goffard, A.; Morel, V.; Duverlie, G.; McKeating, J.A.; Keck, Z.Y.; Foung, S.; Penin, F.; Dubuisson, J.; Voisset, C. The Neutralizing Activity of Anti-Hepatitis C Virus Antibodies Is Modulated by Specific Glycans on the E2 Envelope Protein. J. Virol. 2007, 81, 8101–8111. [Google Scholar] [CrossRef] [PubMed]
- Goffard, A.; Callens, N.; Bartosch, B.; Wychowski, C.; Cosset, F.L.; Montpellier, C.; Dubuisson, J. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 2005, 79, 8400–8409. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Catanese, M.T.; Verhoye, L.; Desombere, I.; Farhoudi, A.; Jones, C.T.; Sheahan, T.; Grzyb, K.; Cortese, R.; Rice, C.M.; et al. A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology 2012, 55, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Albecka, A.; Belouzard, S.; Vercauteren, K.; Verhoye, L.; Wychowski, C.; Leroux-Roels, G.; Palmer, K.E.; Dubuisson, J. Griffithsin has antiviral activity against hepatitis C virus. Antimicrob. Agents Chemother. 2011, 55, 5159–5167. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Umehara, T.; Yasui, F.; Nakagawa, S.; Yano, J.; Ohgi, T.; Sonoke, S.; Satoh, K.; Inoue, K.; Yoshiba, M.; et al. Liver target delivery of small interfering RNA to the HCV gene by lactosylated cationic liposome. J. Hepatol. 2007, 47, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Shin, D.; Lee, H.; Ahn, B.Y.; Yoon, Y.; Kim, M. Targeted delivery of siRNA against hepatitis C virus by apolipoprotein A-I-bound cationic liposomes. J. Hepatol. 2009, 50, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.K.; Kundu, A.K.; Hazari, S.; Chandra, S.; Bao, L.; Ooms, T.; Morris, G.F.; Wu, T.; Mandal, T.K.; Dash, S. Inhibition of hepatitis C virus replication by intracellular delivery of multiple siRNAs by nanosomes. Mol. Ther. 2012, 20, 1724–1736. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Yang, J.A.; Jung, H.S.; Beack, S.; Choi, J.E.; Hur, W.; Koo, H.; Kim, K.; Yoon, S.K.; Hahn, S.K. Hyaluronic acid-gold nanoparticle/interferon alpha complex for targeted treatment of hepatitis C virus infection. ACS Nano 2012, 6, 9522–9531. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, S.R.; Jang, H.; Kim, K.S.; Lee, B.; Kim, K.B.; Kim, Y.K.; Yeo, W.S.; Lee, Y.; Kim, D.E.; Min, D.H. Functional delivery of DNAzyme with iron oxide nanoparticles for hepatitis C virus gene knockdown. Biomaterials 2012, 33, 2754–2761. [Google Scholar] [CrossRef] [PubMed]
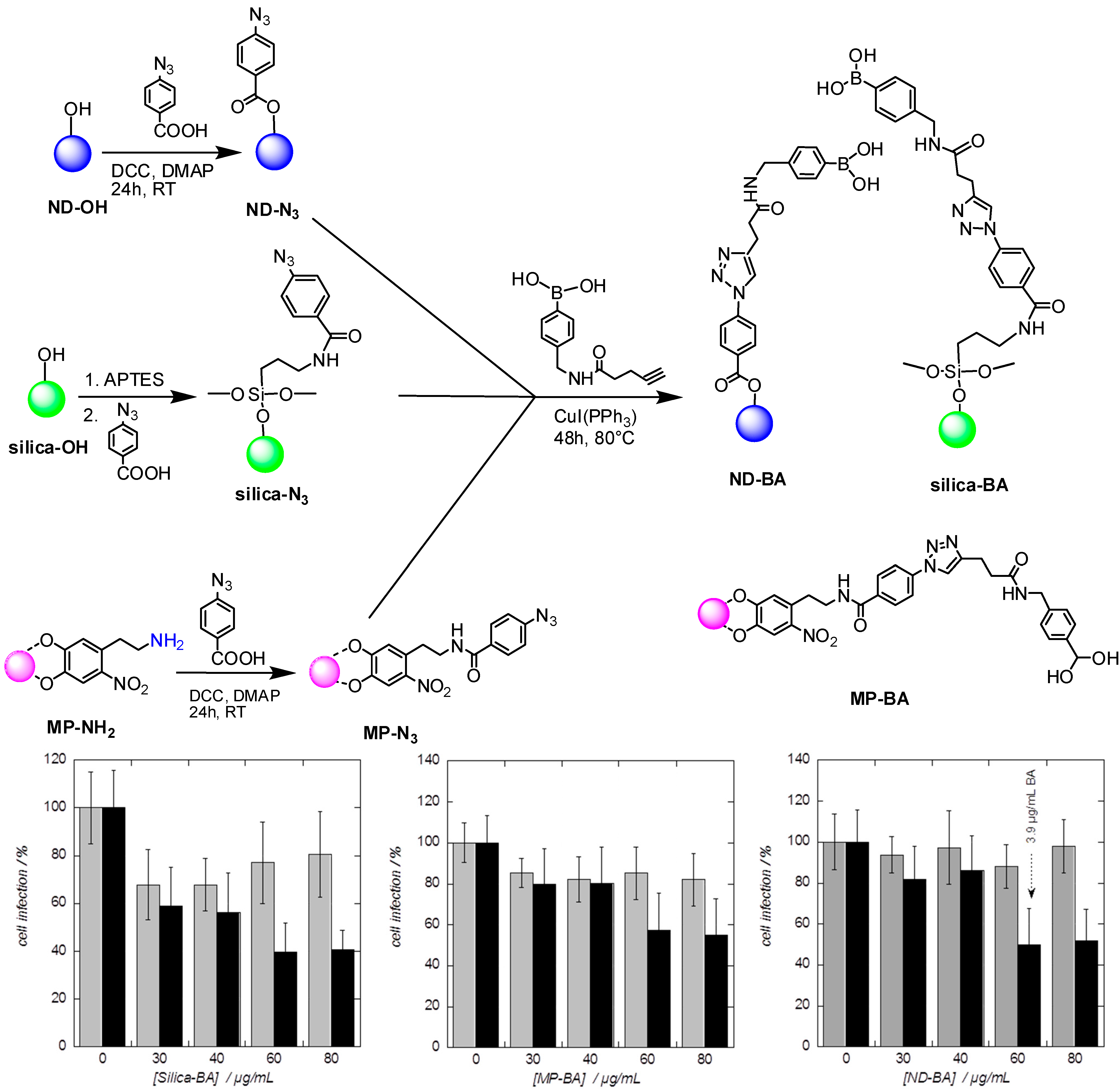
- Khanal, M.; Vausselin, T.; Barras, A.; Bande, O.; Turcheniuk, K.; Benazza, M.; Zaitsev, V.; Teodorescu, C.M.; Boukherroub, R.; Siriwardena, A.; et al. Phenylboronic-acid-modified nanoparticles: Potential antiviral therapeutics. ACS Appl. Mater. Interfaces 2013, 5, 12488–12498. [Google Scholar] [CrossRef] [PubMed]
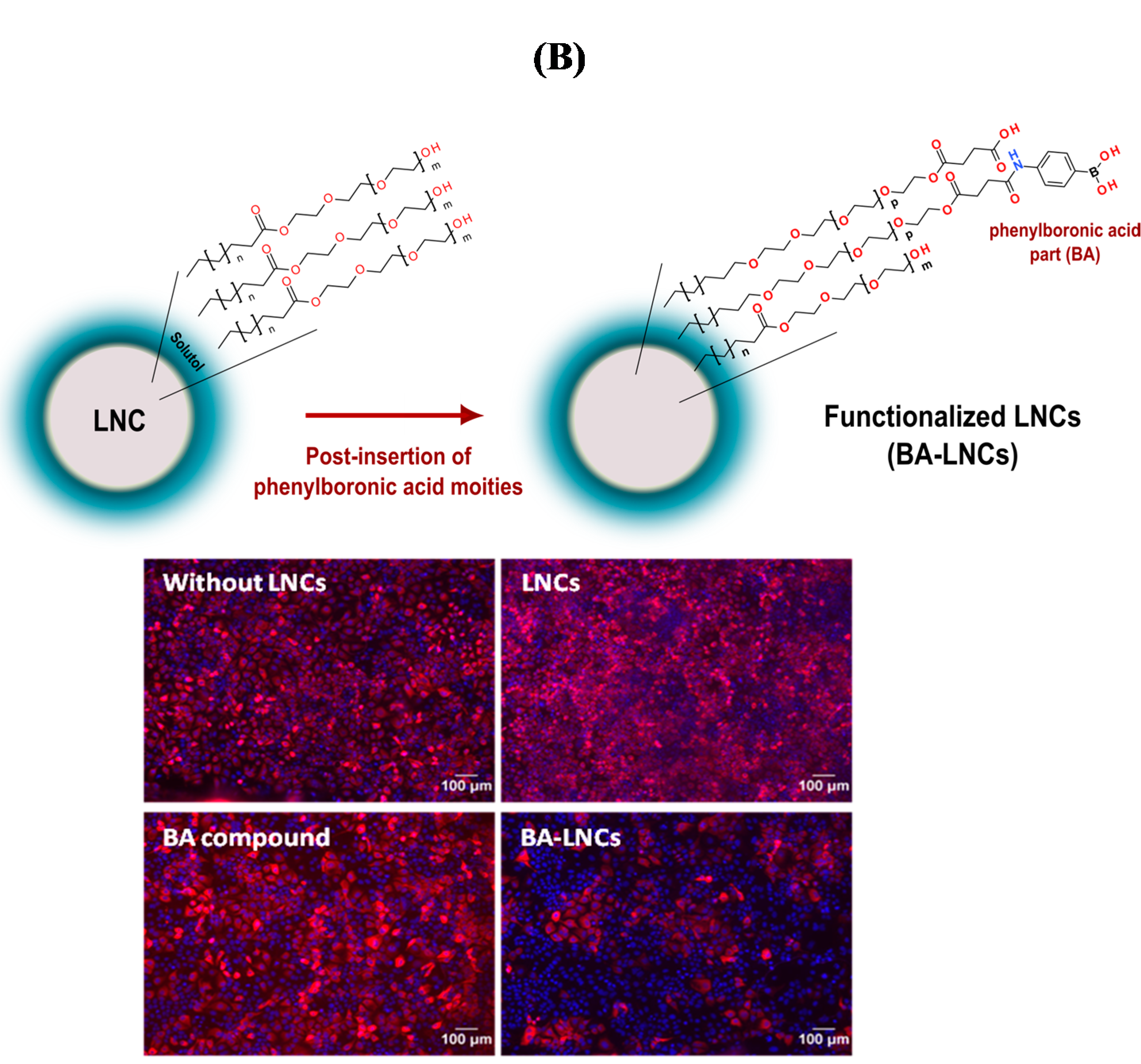
- Khanal, M.; Barras, A.; Vausselin, T.; Feneant, L.; Boukherroub, R.; Siriwardena, A.; Dubuisson, J.; Szunerits, S. Boronic acid-modified lipid nanocapsules: A novel platform for the highly efficient inhibition of hepatitis C viral entry. Nanoscale 2015, 7, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Dubuisson, J. New advances in the molecular biology of hepatitis C virus infection: Towards the identification of new treatment targets. Gut 2012, 61, i25–i35. [Google Scholar]
- Helle, F.; Wychowski, C.; Vu-Dac, N.; Gustafson, K.R.; Voiseet, C.; Dubuisson, J. Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J. Biol. Chem. 2006, 281, 25177–25183. [Google Scholar]
- Barwell, N.P.; Crump, M.D.; Davis, A.P. A Synthetic Lectin for β-Glucosyl. Angew. Chem. Int. Ed. 2009, 48, 7673–7676. [Google Scholar] [CrossRef] [PubMed]
- Pa, A.; Berube, M.; Hall, D.G. Design, Synthesis, and Screening of a Library of Peptidyl Bis(Boroxoles) as Oligosaccharide Receptors in Water: Identification of a Receptor for the Tumor Marker TF-Antigen Disaccharide. Angew. Chem. Int. Ed. 2010, 49, 1492–1495. [Google Scholar]
- Ke, C.; Destecrix, H.; Crump, M.P.; Davis, A.P. A simple and accessible synthetic lectin for glucose recognition and sensing. Nat. Chem. 2012, 4, 718–723. [Google Scholar]
- Bicker, K.L.; Sun, J.; Lavigne, J.J.; Thompson, P.R. Boronic Acid Functionalized Peptidyl Synthetic Lectins: Combinatorial Library Design, Peptide Sequencing, and Selective Glycoprotein Recognition. ACS Comb. Sci. 2011, 13, 232–243. [Google Scholar]
- Arnaud, J.; Audfray, A.; Imberty, A. Binding sugars: From natural lectins to synthetic receptors and engineered neolectins. Chem. Soc. Rev. 2013, 42, 4798–4813. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Fang, H.; Wang, B. Boronolectins and fluorescent boronolectins: An examination of the detailed chemistry issues important for the design. Med. Res. Rev. 2005, 25, 490–520. [Google Scholar] [CrossRef] [PubMed]
- Wiskur, S.L.; Lavigne, J.L.; Metzger, A.; Tobey, S.L.; Lynch, V.; Anslyn, E.V. Thermodynamic analysis of receptors based on guanidinium/boronic acid groups for the complexation of carboxylates, alpha92 hydroxycarboxylates, and diols: Driving force for binding and cooperativity. Chem. Eur. J. 2004, 10, 3792–3804. [Google Scholar] [CrossRef] [PubMed]
- Springsteen, G.; Wang, B.H. A detailed examination of boronic acid-diol complexation. Tetrahedron 2002, 58, 5291–5300. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 2000, 275, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Liu, J.; Valyi-Nagy, T.; Shukla, D. Anti-heparan Sulfate Peptides That Block Herpes Simplex Virus Infection in Vivo. J. Biol. Chem. 2011, 15, 25406–25415. [Google Scholar] [CrossRef] [PubMed]
- Baleux, F.; Loureiro-Morais, I.; Hersant, Y.; Clayette, P.; Arenzana-Seisdedos, F.; Bonnaffé, D.; Lortat-Jacob, H. A synthetic CD4–heparan sulfate glycoconjugate inhibits CCR5 and CXCR4 HIV-1 attachment and entry. Nat. Chem. Biol. 2009, 5, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Diaz, J.J.; Thouvenot, D.; Morfin, F. Novel targets for the development of anti-herpes compounds. Infect. Disord. Drug Targets 2007, 7, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Baram-Pinto, D.; Shukla, S.; Gedanken, A.; Sarid, R. Inhibition of HSV-1 attachment, entry, and cell-to-cell spread by functionalized multivalent gold nanoparticles. Small 2010, 6, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Baram-Pinto, D.; Shukla, S.; Perkas, N.; Gedanken, A.; Sarid, R. Inhibition of herpes simplex virus type 1 infection by silver nanoparticles capped with mercaptoethane sulfonate. Bioconjugate Chem. 2009, 20, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Mishra, Y.K.; Adelung, R.; Rohl, C.; Shukla, D.; Spors, F.; Tiwari, V. Virostatic potential of micro-nano filopodia-like ZnO structures against herpes simplex virus-1. Antivir. Res. 2011, 92, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Antoine, T.E.; Mishra, Y.K.; Trigilio, J.; Tiwari, V.; Adelung, R.; Shukla, D. Prophylactic, therapeutic and neutralizing effects of zinc oxide tetrapod structures against herpes simplex virus type-2 infection. Antiviral. Res. 2012, 96, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Asvadi, N.H.; Dang, N.T.; Davis-Poynter, N.; Coombes, A.G. Evaluation of microporous polycaprolactone matrices for controlled delivery of antiviral microbicides to the female genital tract. J. Mater. Sci. Mater. Med. 2013, 24, 2719–2727. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, P.; Tomaszewska, E.; Gniadek, M.; Baska, P.; Nowakowska, J.; Sokolowska, J.; Nowak, Z.; Donten, M.; Celichowski, G.; Grobelny, J.; et al. Tannic Acid Modified Silver Nanoparticles Show Antiviral Activity in Herpes Simplex Virus Type 2 Infection. PLoS ONE 2014, 9, e104113. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, B.; Verma, S. Preparation and evaluation of novel in situ gels containing acyclovir for the treatment of oral herpes simplex virus infections. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Guo, C.; Zhou, Y.; Cao, F.; Zhu, W.; Sun, M.; Zhai, G. Skin irritation and the inhibition effect on HSV-1 in vivo of penciclovir-loaded microemulsion. Int. Immunopharmacol. 2010, 10, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Yu, A.; Wang, W.; Dong, R.; Wu, J.; Zhai, G. Formulation design of microemulsion for dermal delivery of penciclovir. Int. J. Pharm. 2008, 360, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Lembo, D.; Swaminathan, S.; Donalisio, M.; Civra, A.; Pastero, L.; Aquilano, D.; Vavia, P.; Trotta, F.; Cavalli, R. Encapsulation of Acyclovir in new carboxylated cyclodextrin-based nanosponges improves the agent’s antiviral efficacy. Int. J. Pharm. 2013, 443, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Sasivimolphan, P.; Lipipun, V.; Ritthidej, G.; Chitphet, K.; Yoshida, Y.; Daikoku, T.; Sritularak, B.; Likhitwitayawuid, K.; Pramyothin, P.; Hattori, M.; et al. Microemulsion-based oxyresveratrol for topical treatment of herpes simplex virus (HSV) infection: Physicochemical properties and efficacy in cutaneous HSV-1 infection in mice. AAPS PharmSciTech 2012, 13, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, R.; Argnani, R.; Esposito, E.; Dalpiaz, A.; Scatturin, A.; Bortolotti, F.; Lufino, M.; Guerrini, R.; Cavicchioni, G.; Incorvaia, C.; et al. Cationic liposomes as potential carriers for ocular administration of peptides with anti-herpetic activity. Int. J. Pharm. 2006, 317, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Seth, A.K.; Misra, A.; Umrigar, D. Topical liposomal gel of idoxuridine for the treatment of herpes simplex: Pharmaceutical and clinical implications. Pharm. Dev. Technol. 2004, 9, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Palliser, D.; Chowdhury, D.; Wang, Q.Y.; Lee, S.J.; Bronson, R.T.; Knipe, D.M.; Lieberman, J. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 2006, 439, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Navarro, F.; Lal, A.; Basar, E.; Pandey, R.K.; Manoharan, M.; Feng, Y.; Lee, S.J.; Lieberman, J.; Palliser, D. Durable protection from Herpes Simplex Virus-2 transmission following intravaginal application of siRNAs targeting both a viral and host gene. Cell Host Microbe 2009, 5, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, J.M.; Weller, C.E.; Booth, C.J.; Saltzman, W.M. Polymer nanoparticles encapsulating siRNA for treatment of HSV-2 genital infection. J. Control. Release 2012, 162, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Chiarantini, L.; Argnani, R.; Zucchini, S.; Stevanato, L.; Zabardi, P.; Grossi, M.P.; Magnani, M.; Manservigi, R. Red blood cells as delivery system for recombinant HSV-1 glycoprotein B: Immunogenicity and protection in mice. Vaccine 1997, 15, 276–280. [Google Scholar] [CrossRef]
- Rossi, L.; Serafini, S.; Cappellacci, L.; Balestra, E.; Brandi, G.; Schiavano, G.F.; Franchetti, P.; Grifantini, M.; Perno, C.F.; Magnani, M. Erythrocyte-mediated delivery of a new homodinucleotide active against human immunodeficiency virus and herpes simplex virus. J. Antimicrob. Chemother. 2001, 47, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Westeerndorf, A.M.; Buer, J.; Uberla, K.; Epple, M. The potential of nanoparticles for the immunization against vital infections. J. Mater. Chem. B 2015, 3, 4767–4779. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szunerits, S.; Barras, A.; Khanal, M.; Pagneux, Q.; Boukherroub, R. Nanostructures for the Inhibition of Viral Infections. Molecules 2015, 20, 14051-14081. https://doi.org/10.3390/molecules200814051
Szunerits S, Barras A, Khanal M, Pagneux Q, Boukherroub R. Nanostructures for the Inhibition of Viral Infections. Molecules. 2015; 20(8):14051-14081. https://doi.org/10.3390/molecules200814051
Chicago/Turabian StyleSzunerits, Sabine, Alexandre Barras, Manakamana Khanal, Quentin Pagneux, and Rabah Boukherroub. 2015. "Nanostructures for the Inhibition of Viral Infections" Molecules 20, no. 8: 14051-14081. https://doi.org/10.3390/molecules200814051
APA StyleSzunerits, S., Barras, A., Khanal, M., Pagneux, Q., & Boukherroub, R. (2015). Nanostructures for the Inhibition of Viral Infections. Molecules, 20(8), 14051-14081. https://doi.org/10.3390/molecules200814051