
Synthesis and Enzymatic Incorporation of Modified Deoxyuridine Triphosphates

Abstract
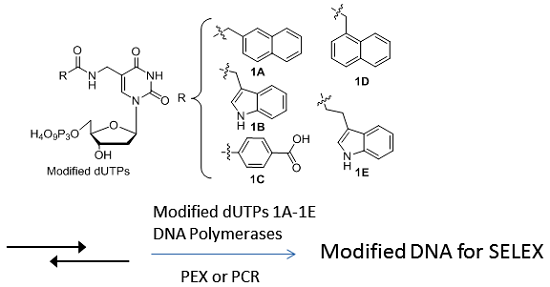
: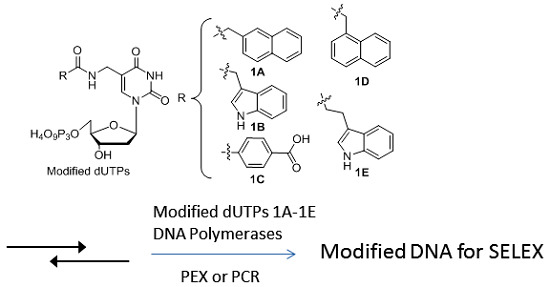
1. Introduction
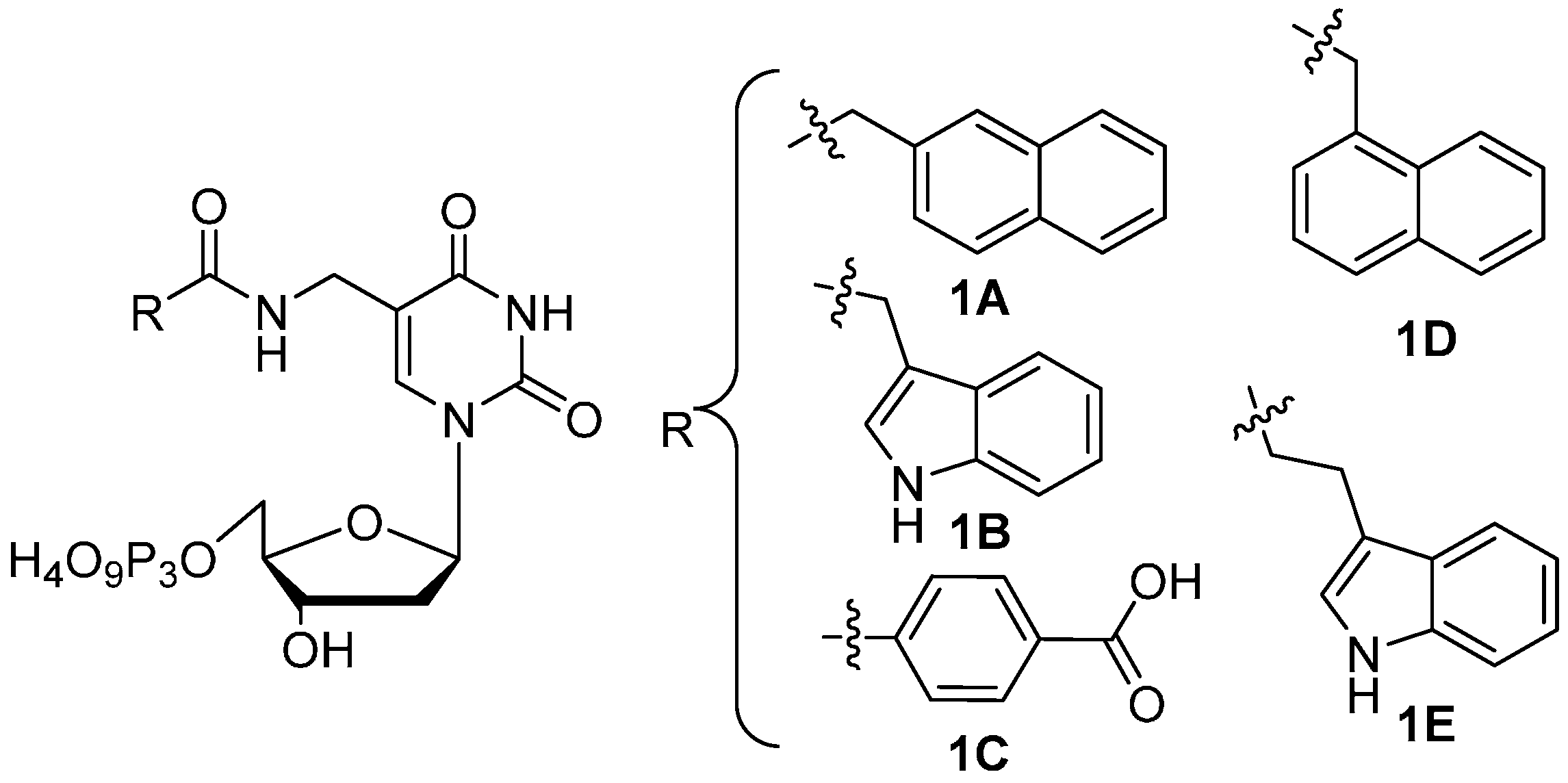
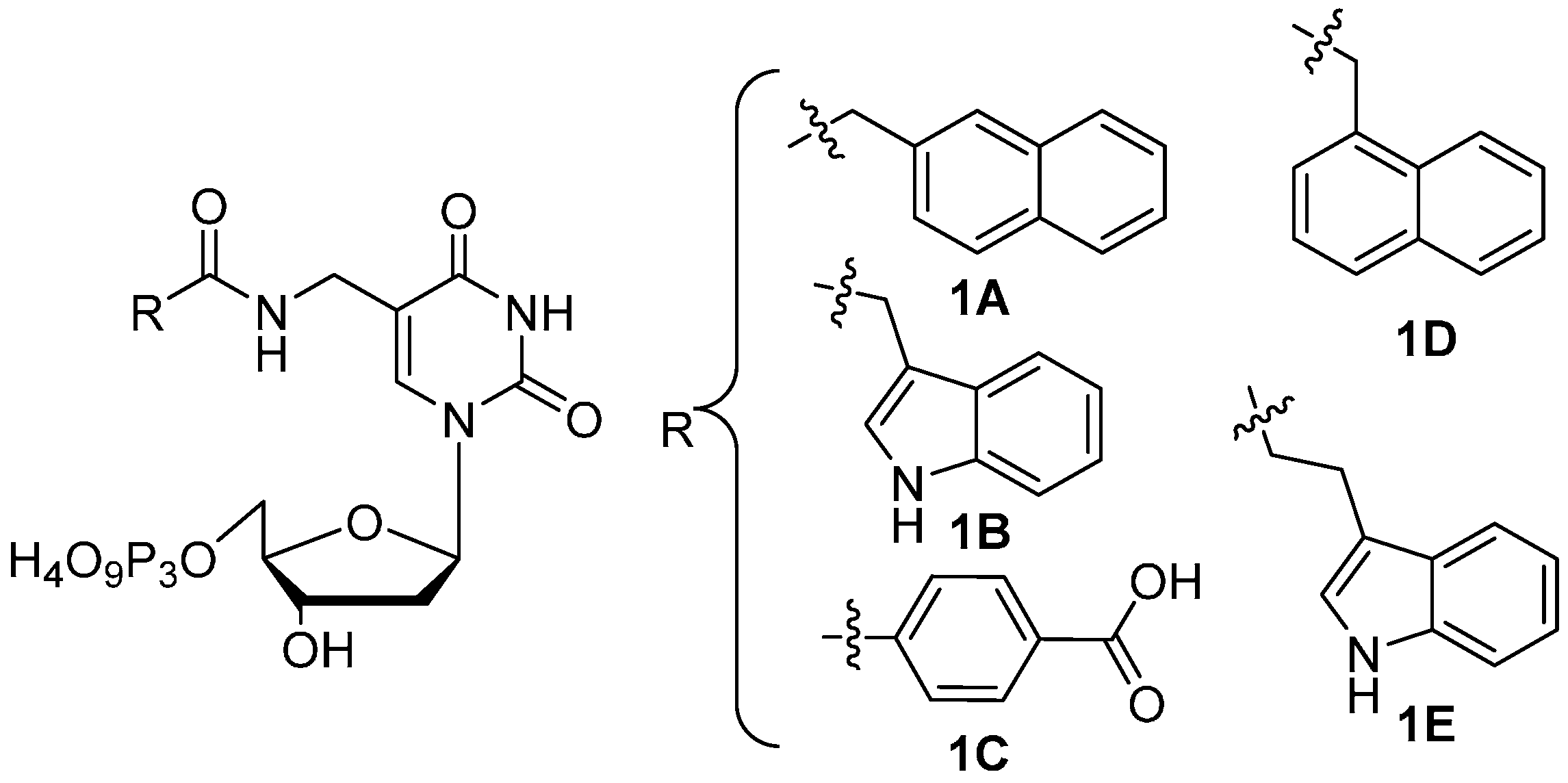
2. Results and Discussion
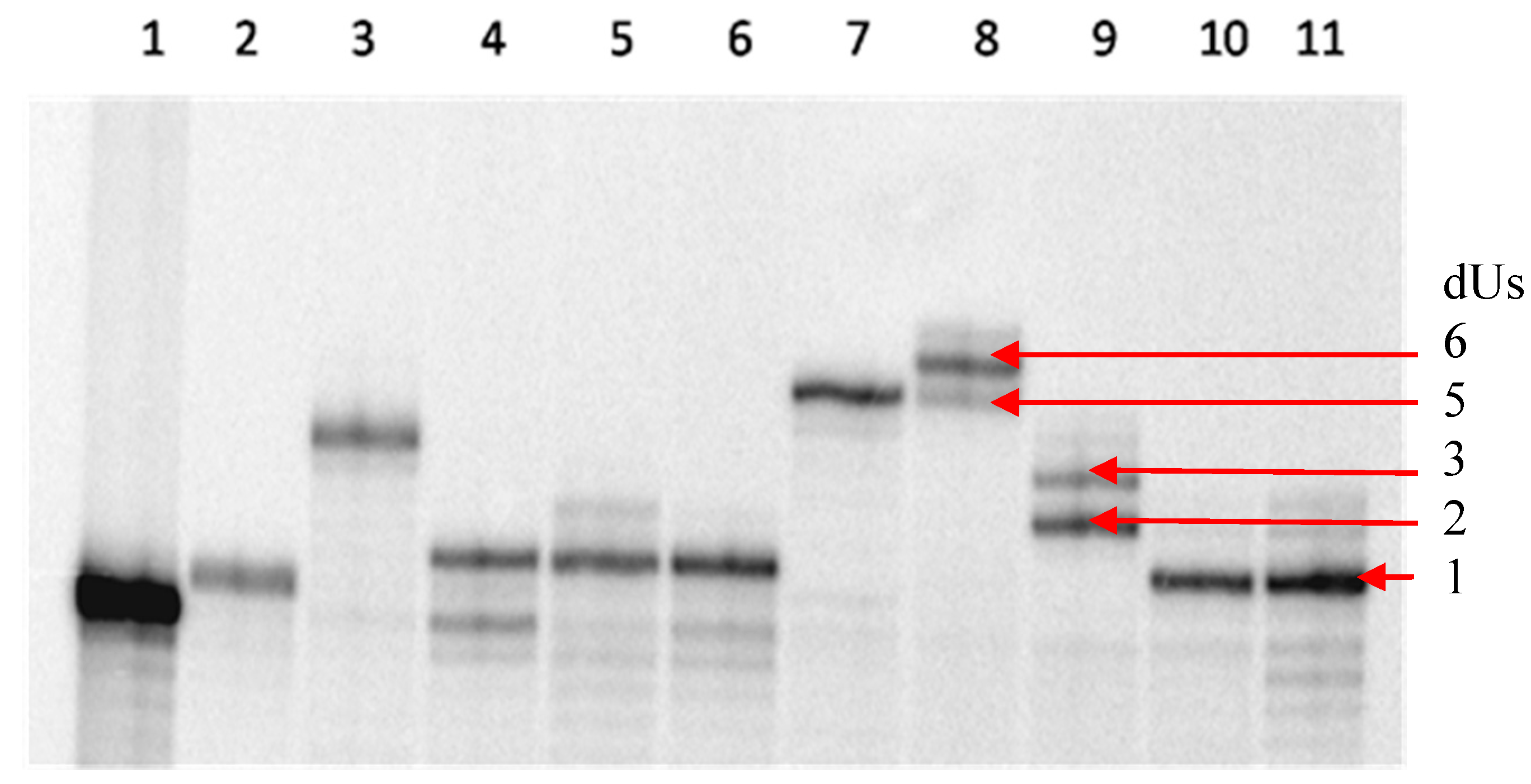
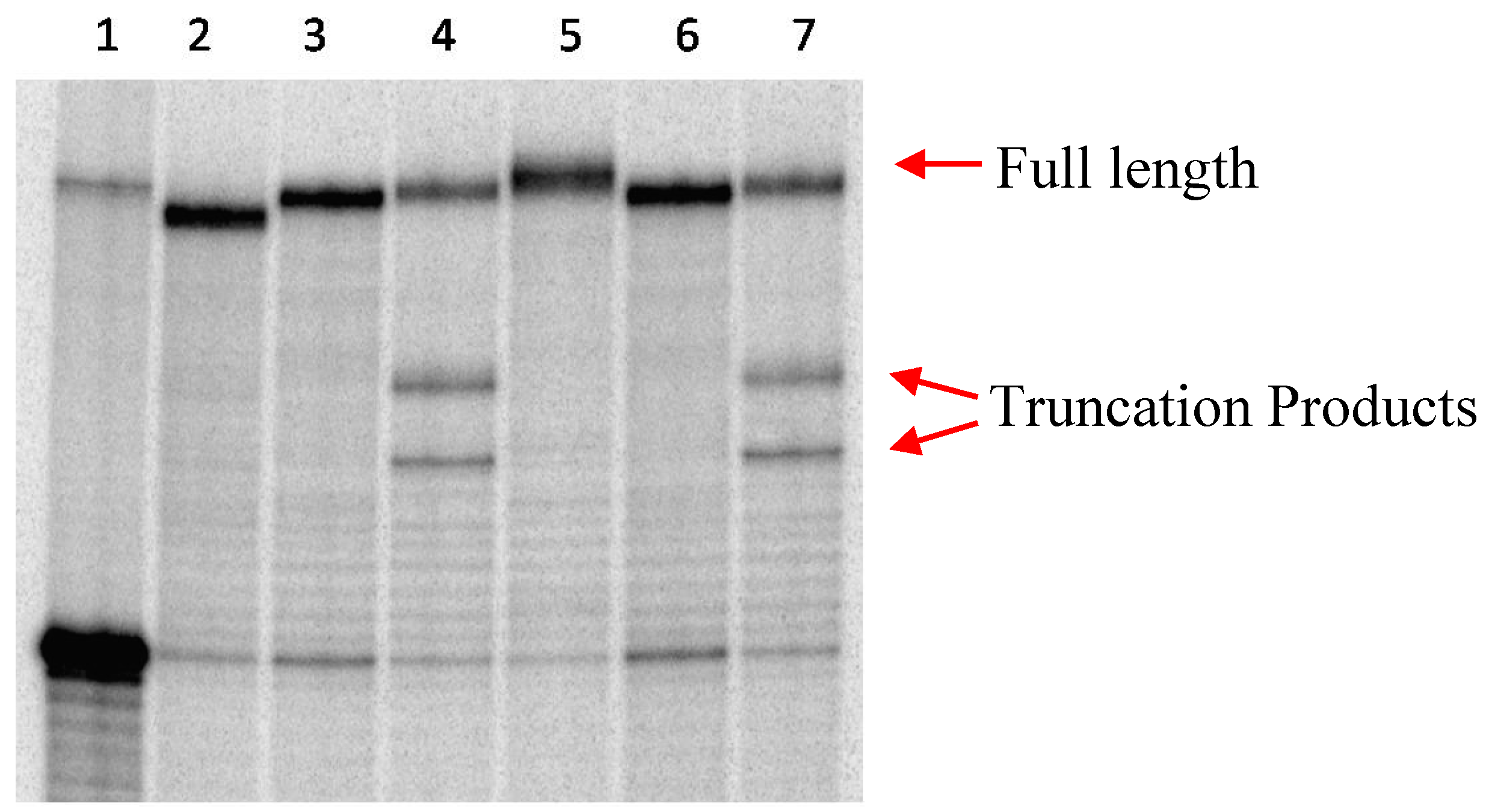
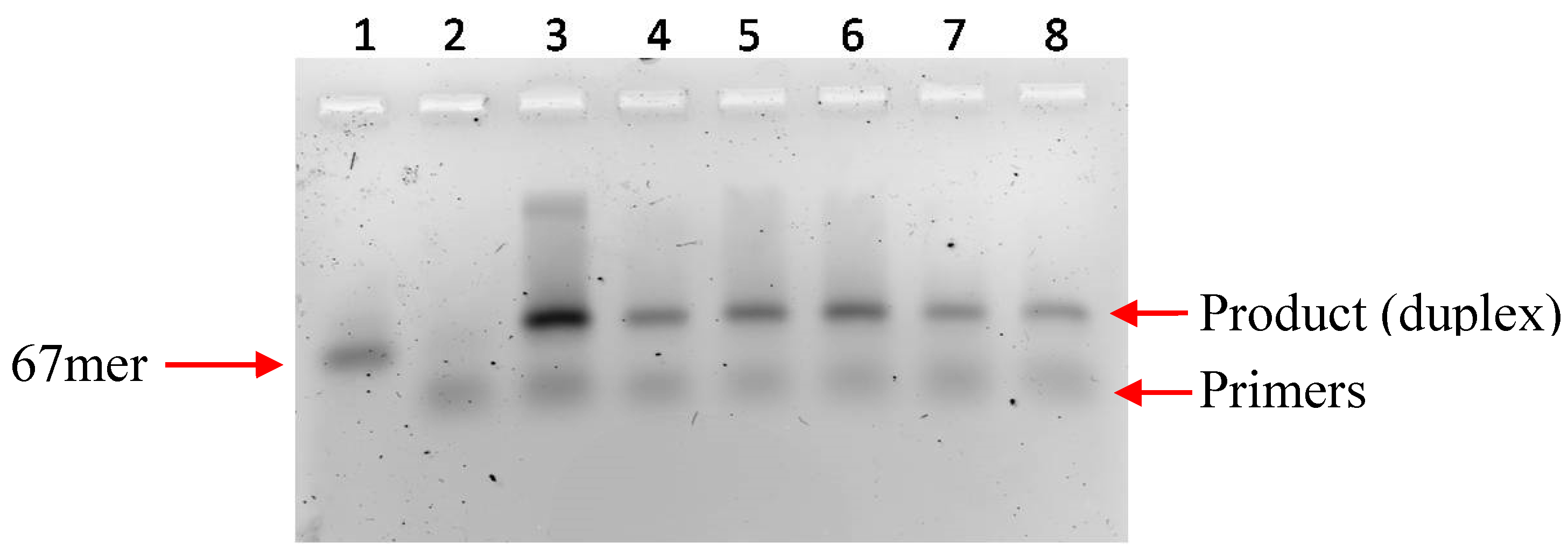
2.1 Primer Extension Assays

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequences Used for Primer Extension or PCR Amplification | |
|---|---|
| P1 | 5ʹ-TAATCGGGAAGGTCAGGGGGGAAAAGAAAA-3ʹ |
| T1 | 3ʹ-TTAGCCCTTCCAGTCCCCCCTTTTCTTTTGTAACTAAGATGGACAGCTCC-5ʹ |
| T2 | 3ʹ-TTAGCCCTTCCAGTCCCCCCTTTTCTTTTGGCCGTCTGTCTCGCTCC-5ʹ |
| Primers | 5ʹ-GCGCTCGCGCGCCGCG-3ʹ 3ʹ-CGGGGAGGCGTCCGGCTGCG-5ʹ |
| T3 | 5ʹ-GCGCTCGCGCGCCGCGCGCGCGCCCCCGCTCGGCGCCCCTCCGCAGGCCGACGC-3ʹ |
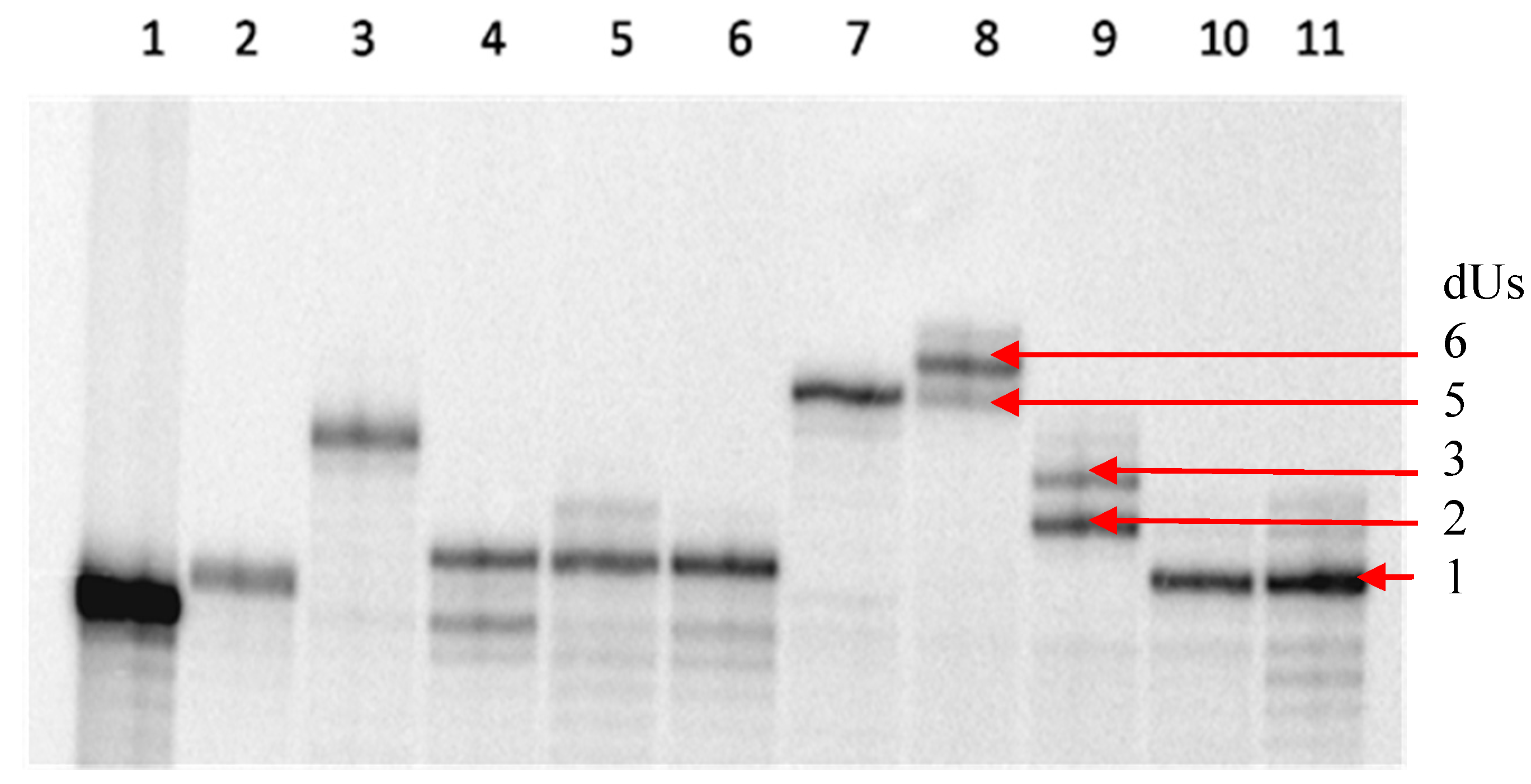
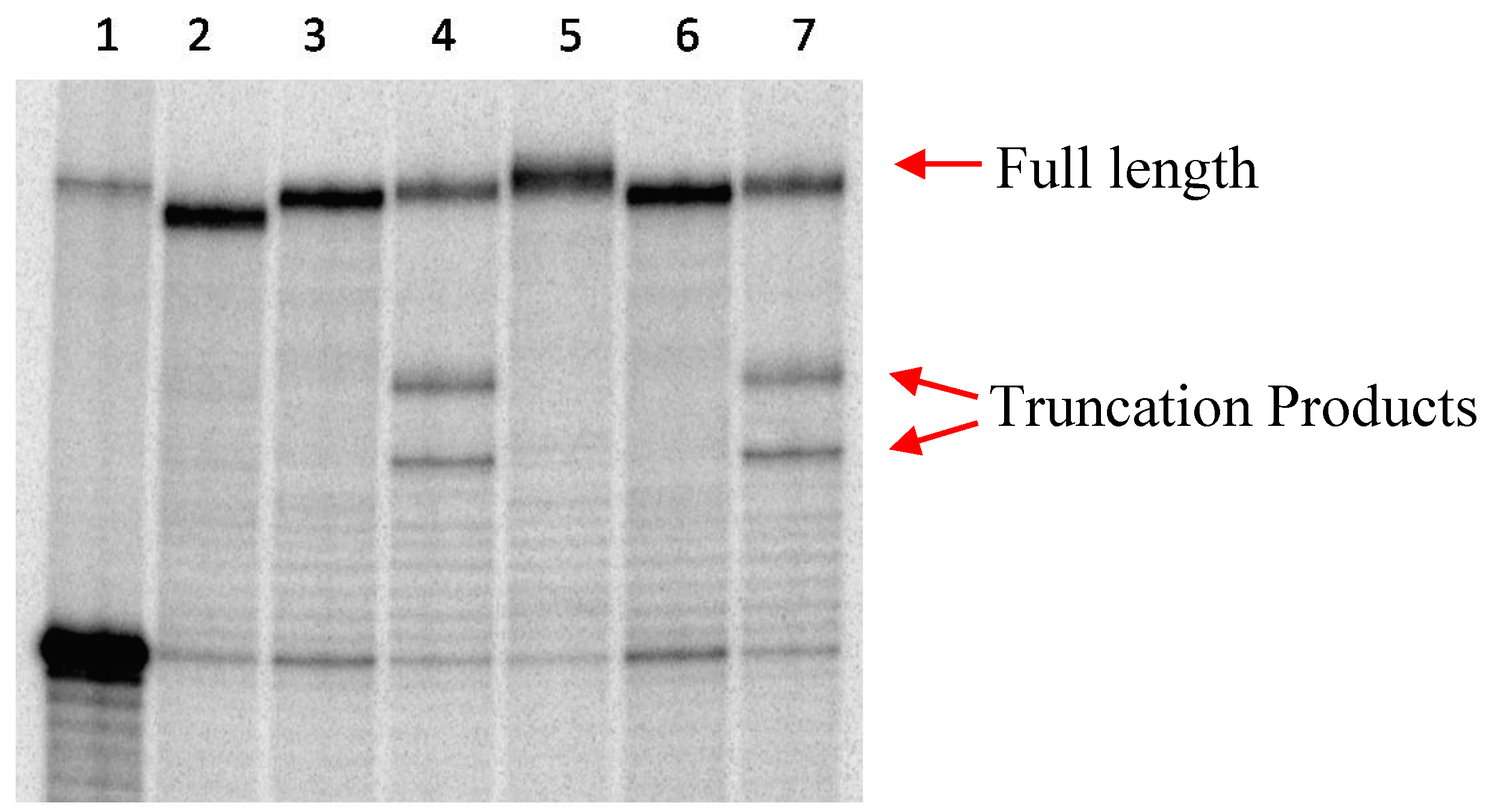
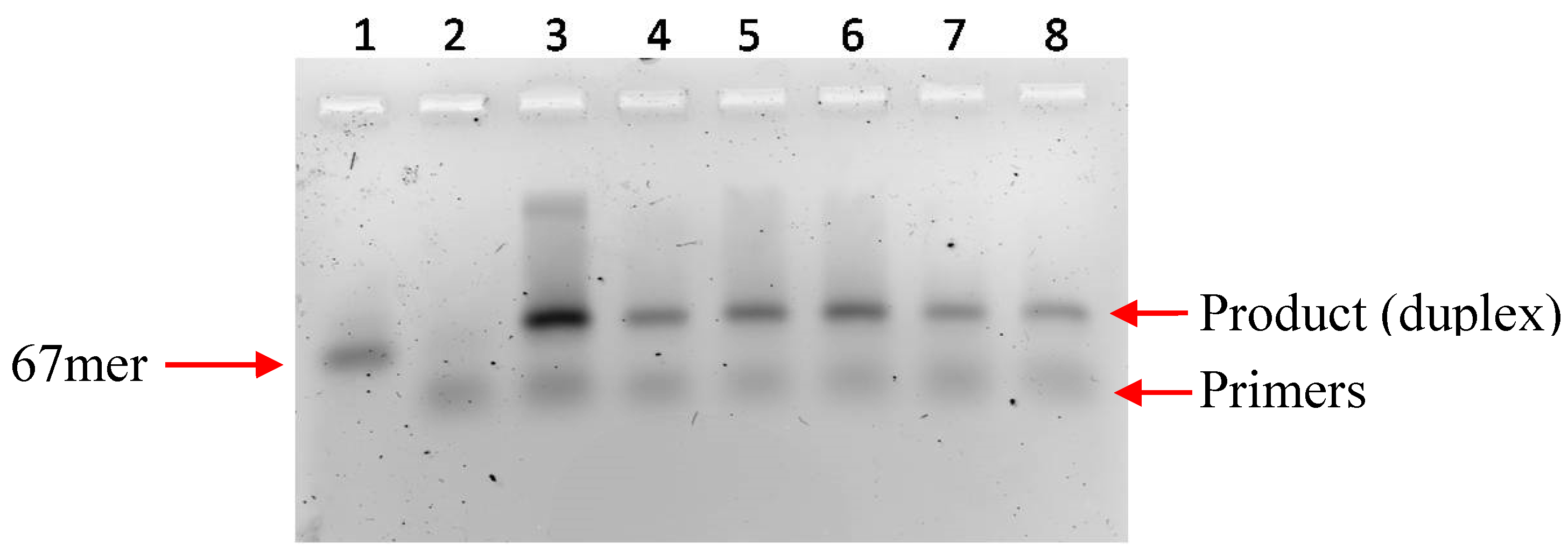
3. Experimental Section
3.1. General Information
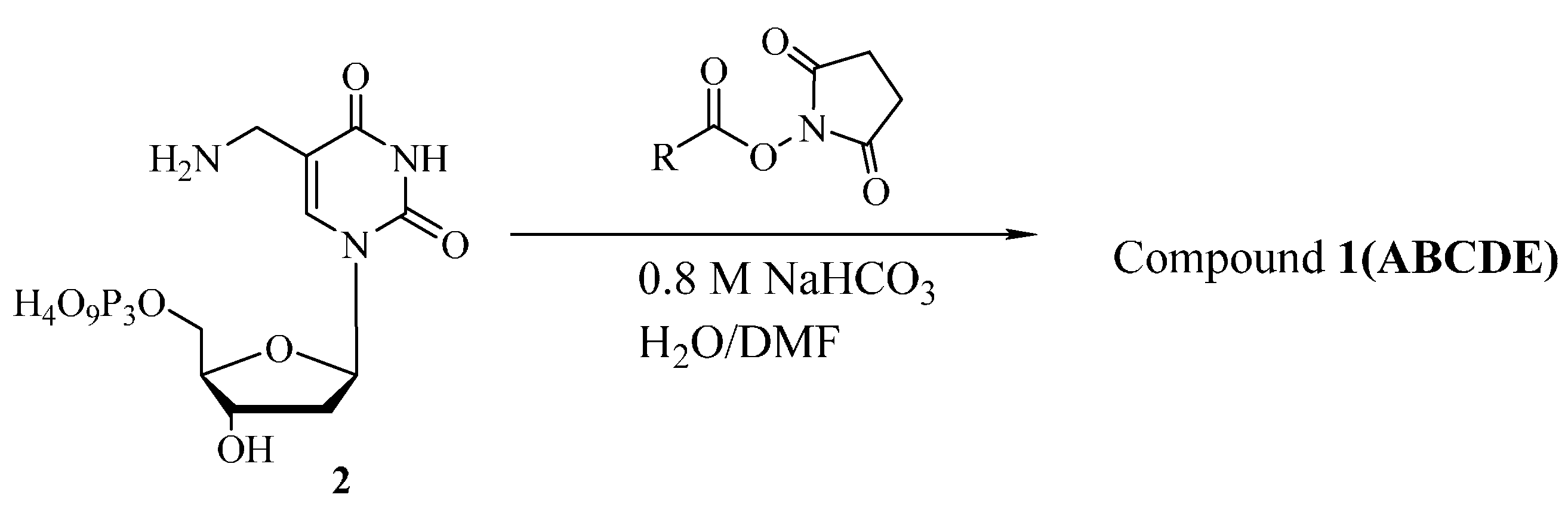
3.2. Synthesis
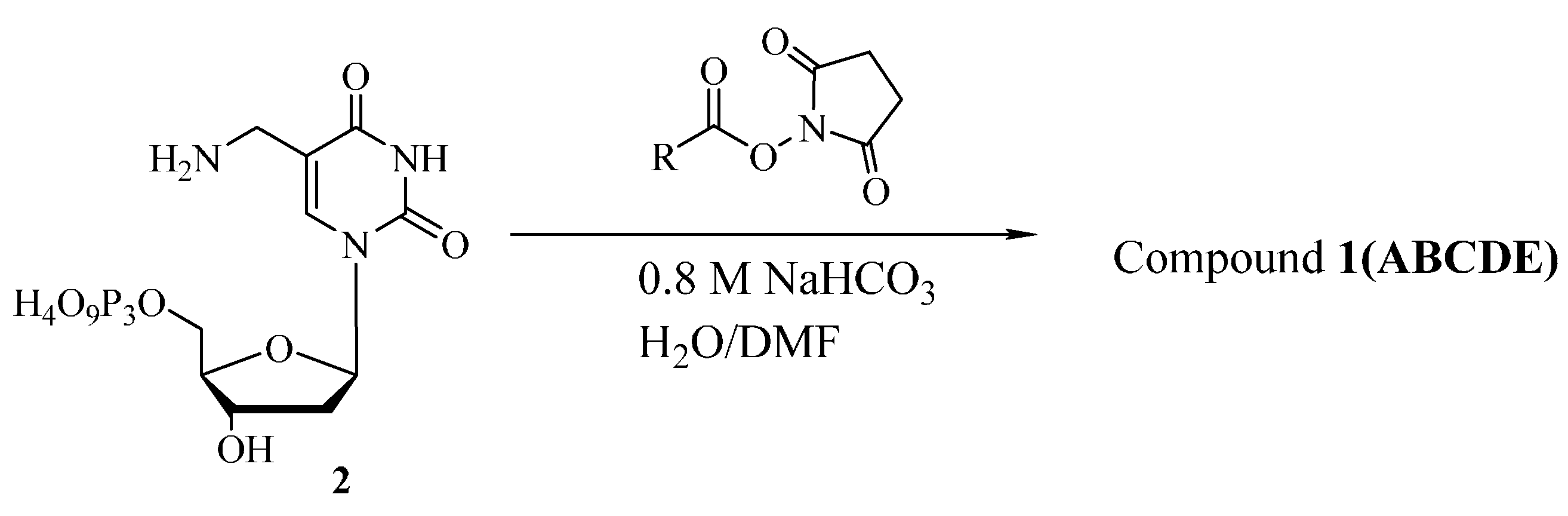
3.3. Enzymatic Incorporation
DNA Templates
3.4. Sequencing of the Modified DNA from PCR Amplification
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment-RNA ligands to bacteriophage-T4 DNA-polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R.; Joyce, G.F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223–229. [Google Scholar] [CrossRef]
- Santoro, S.W.; Joyce, G.F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 4262–4266. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.R.; Sen, D. Evidence for the metal-cofactor independence of an RNA phosphodiester-cleaving DNA enzyme. Chem. Biol. 1997, 4, 579–593. [Google Scholar] [CrossRef]
- Pradeepkumar, P.I.; Höbartner, C.; Baum, D.A.; Silverman, S.K. DNA-catalyzed formation of nucleopeptide linkages. Angew. Chem. Int. Ed. 2008, 47, 1753–1757. [Google Scholar] [CrossRef] [PubMed]
- Chandra, M.; Sachdeva, A.; Silverman, S.K. DNA-catalyzed sequence-specific hydrolysis of DNA. Nat. Chem. Biol. 2009, 5, 718–720. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, G.J.; Herschlag, D. Mechanistic aspects of enzymatic catalysis: Lessons from comparison of RNA and protein enzymes. Annu. Rev. Biochem. 1997, 66, 19–59. [Google Scholar] [CrossRef] [PubMed]
- McKeague, M.; Derosa, M.C. Challenges and opportunities for small molecule aptamer development. J. Nucleic Acids 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.W.S.; Khachigian, L.M. DNAzyme Delivery Approaches in Biological Settings. Curr. Med. Chem. 2013, 20, 3448–3455. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Ozaki, A.N.; Satoh, F.; Ohbayashi, T.; Masud, M.M.; Ozaki, H. Expansion of structural and functional diversities of DNA using new 5-substituted deoxyuridine derivatives by PCR with superthermophilic KOD Dash DNA polymerase. Chem. Commun. 2001, 24, 2604–2605. [Google Scholar] [CrossRef]
- Kuwahara, M.; Ohbayashi, T.; Hanawa, K.; Shoji, A.; Ozaki, A.N.; Ozaki, H.; Sawai, H. Enzymatic incorporation of chemically-modified nucleotides into DNAs. Nucleic Acids Res. Suppl. 2002, 2, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Takahata, Y.; Shoji, A.; Ozaki, A.N.; Ozaki, H.; Sawai, H. Substrate properties of C5-substituted pyrimidine 2ʹ-deoxynucleoside 5ʹ-triphosphates for thermostable DNA polymerases during PCR. Bioorg. Med. Chem. Lett. 2003, 13, 3735–3738. [Google Scholar] [CrossRef] [PubMed]
- Mehedi Masud, M.; Ozaki-Nakamura, A.; Kuwahara, M.; Ozaki, H.; Sawai, H. Modified DNA bearing 5(methoxycarbonylmethyl)-2ʹ-deoxyuridine: Preparation by PCR with thermophilic DNA polymerase and postsynthetic derivatization. Chem. Biol. Chem. 2003, 4, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Nagashima, J.; Kuwahara, M.; Kitagata, R.; Tamura, T.; Matsui, I. Differences in substrate specificity of C(5)-substituted or C(5)unsubstituted pyrimidine nucleotides by DNA Polymerases from thermophilic bacteria, archaea, and phages. Chem. Biodivers. 2007, 4, 1979–1995. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Tamura, T.; Kitagata, R.; Sawai, H.; Ozaki, H. Comparison study on PCR amplification of modified DNA by using various kinds of polymerase and modified nucleoside triphosphates. Nucleic Acids Symp. Ser. 2005, 49, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Ohbayashi, T.; Kuwahara, M.; Hasegawa, M.; Kasamatsu, T.; Tamura, T.; Sawai, H. Expansion of repertoire of modified DNAs prepared by PCR using KOD dash DNA polymerase. Org. Biomol. Chem. 2005, 3, 2463–2468. [Google Scholar] [CrossRef] [PubMed]
- Ohmichi, T.; Kuwahara, M.; Sasaki, N.; Hasegawa, M.; Nishikata, T.; Sawai, H.; Sugimoto, N. Nucleic Acid with Guanidinium Modification Exhibits Efficient Cellular Uptake. Angew. Chem. Int. Ed. 2005, 44, 6682–6685. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Hanawa, K.; Ohsawa, K.; Kitagata, R.; Ozaki, H.; Sawai, H. Direct PCR amplification of various modified DNAs having amino acids: Convenient preparation of DNA libraries with high-potential activities for in vitro selection. Bioorg. Med. Chem. 2006, 14, 2518–2526. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Nagashima, J.; Hasegawa, M.; Tamura, T.; Kitagata, R.; Hanawa, K.; Hososhima, S.; Kasamatsu, T.; Ozaki, H.; Sawai, H. Systematic characterization of 2ʹ-deoxynucleoside-5ʹ-triphosphate analogs as substrates for DNA polymerases by polymerase chain reaction and kinetic studies on enzymatic production of modified DNA. Nucleic Acids Res. 2006, 34, 5383–5394. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.; Hipolito, C.; Perrin, D.M. Synthesis and Enzymatic Incorporation of Modified Deoxyadenosine Triphosphates. Eur. J. Org. Chem. 2008, 2008, 4915–4923. [Google Scholar] [CrossRef]
- Lam, C.H.; Hipolito, C.J.; Hollenstein, M.; Perrin, D.M. A divalent metal-dependent self-cleaving DNAzyme with a tyrosine side chain. Org. Biomol. Chem. 2011, 9, 6949–6954. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, K.; Barbas, C.F. Expanding the potential of DNA for binding and catalysis: Highly functionalized dUTP derivatives that are substrates for thermostable DNA polymerases. Angew. Chem. Int. Ed. 1998, 37, 2872–2875. [Google Scholar] [CrossRef]
- Jäger, S.; Rasched, G.; Kornreich-Leshem, H.; Engeser, M.; Thum, O.; Famulok, M. A versatile toolbox for variable DNA functionalization at high density. J. Am. Chem. Soc. 2005, 127, 15071–15082. [Google Scholar] [CrossRef] [PubMed]
- Baccaro, A.; Weisbrod, S.H.; Marx, A. DNA conjugation by the Staudinger ligation: New thymidine analogues. Synthesis-Stuttgart 2007, 13, 1949–1954. [Google Scholar]
- Macickova-Cahova, H.; Hocek, M. Cleavage of adenine-modified functionalized DNA by type II restriction endonucleases. Nucleic Acids Res. 2009, 37, 7612–7622. [Google Scholar] [CrossRef] [PubMed]
- Raindlova, V.; Pohl, R.; Sanda, M.; Hocek, M. Direct Polymerase Synthesis of Reactive Aldehyde-Functionalized DNA and Its Conjugation and Staining with Hydrazines. Angew. Chem. Int. Ed. 2010, 49, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- Raindlova, V.; Pohl, R.; Klepetarova, B.; Havran, L.; Simkova, E.; Horakova, P.; Pivonkova, H.; Fojta, M.; Hocek, M. Synthesis of Hydrazone-Modified Nucleotides and Their Polymerase Incorporation onto DNA for Redox Labeling. Chempluschem 2012, 77, 652–662. [Google Scholar] [CrossRef]
- Hollenstein, M. Nucleoside Triphosphates-Building Blocks for the Modification of Nucleic Acids. Molecules 2012, 17, 13569–13591. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Y.; Zhang, S.; Chaput, J.C. Darwinian evolution of an alternative genetic system provides support for TNA as an RNA progenitor. Nat. Chem. 2012, 4, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Vaught, J.D.; Bock, C.; Carter, J.; Fitzwater, T.; Otis, M.; Schneider, D.; Rolando, J.; Waugh, S.; Wilcox, S.K.; Eaton, B.E. Expanding the Chemistry of DNA for in Vitro Selection. J. Am. Chem. Soc. 2010, 132, 4141–4151. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Sidorov, A.; Gourlain, T.; Mignet, N.; Thorpe, S.J.; Brazier, J.A.; Dickman, M.J.; Hornby, D.P.; Grasby, J.A.; Williams, D.M. Enhancing the catalytic repertoire of nucleic acids: A systematic study of linker length and rigidity. Nucleic Acids Res. 2001, 29, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Gourlain, T.; Sidorov, A.; Mignet, N.; Thorpe, S.J.; Lee, S.E.; Grasby, J.A.; Williams, D.M. Enhancing the catalytic repertoire of nucleic acids. II. Simultaneous incorporation of amino and imidazolyl functionalities by two modified triphosphates during PCR. Nucleic Acids Res. 2001, 29, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M. Synthesis of Deoxynucleoside Triphosphates that Include Proline, Urea, or Sulfonamide Groups and Their Polymerase Incorporation into DNA. Chem. Eur. J. 2012, 18, 13320–13330. [Google Scholar] [CrossRef] [PubMed]
- Perrin, D.M.; Garestier, T.; Hélène, C. Expanding the catalytic repertoire of nucleic acid catalysts: Simultaneous incorporation of two modified deoxyribonucleoside triphosphates bearing ammonium and imidazolyl functionalities. Nucleosides 1999, 18, 377–391. [Google Scholar] [CrossRef]
- Perrin, D.M.; Garestier, T.; Hélène, C. Bridging the gap between proteins and nucleic acids: A metal-independent RNAseA mimic with two protein-like functionalities. J. Am. Chem. Soc. 2001, 123, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Hososhima, S.-i.; Takahata, Y.; Kitagata, R.; Shoji, A.; Hanawa, K.; Ozaki, A.N.; Ozaki, H.; Sawai, H. Simultaneous incorporation of three different modified nucleotides during PCR. Nucleic Acids Res. Suppl. 2003, 3, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M.; Hipolito, C.; Lam, C.; Perrin, D.M. A self-cleaving DNA enzyme modified with amines, guanidines and imidazoles operates independently of divalent metal cations (M2+). Nucleic Acids Res. 2009, 37, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M.; Hipolito, C.J.; Lam, C.H.; Perrin, D.M. A DNAzyme with Three Protein-Like Functional Groups: Enhancing Catalytic Efficiency of M2+-Independent RNA Cleavage. Chem. Biol. Chem. 2009, 10, 1988–1992. [Google Scholar] [CrossRef] [PubMed]
- Hipolito, C.J.; Hollenstein, M.; Lam, C.H.; Perrin, D.M. Protein-inspired modified DNAzymes: Dramatic effects of shortening side-chain length of 8-imidazolyl modified deoxyadenosines in selecting RNaseA mimicking DNAzymes. Org. Biomol. Chem. 2011, 9, 2266–2273. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Ohsawa, K.; Kasamatsu, T.; Shoji, A.; Sawai, H.; Ozaki, H. Screening of a glutamic acid-binding aptamer from arginine-modified DNA library. Nucleic Acids Symp. Ser. (Oxf.) 2005, 49, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Shoji, A.; Kuwahara, M.; Ozaki, H.; Sawai, H. Modified DNA aptamer that binds the (R)-Isomer of a thalidomide derivative with high enantioselectivity. J. Am. Chem. Soc. 2007, 129, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, K.; Kasamatsu, T.; Nagashima, J.I.; Hanawa, K.; Kuwahara, M.; Ozaki, H.; Sawai, H. Arginine-modified DNA aptamers that show enantioselective recognition of the dicarboxylic acid moiety of glutamic acid. Anal. Sci. 2008, 24, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, A.V.; Grasby, J.A.; Williams, D.M. Sequence-specific cleavage of RNA in the absence of divalent metal ions by a DNAzyme incorporating imidazolyl and amino functionalities. Nucleic Acids Res. 2004, 32, 1591–601. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M.; Hipolito, C.J.; Lam, C.H.; Perrin, D.M. Toward the Combinatorial Selection of Chemically Modified DNAzyme RNase A Mimics Active Against all-RNA Substrates. Acs Comb. Sci. 2013, 15, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M.; Hipolito, C.; Lam, C.; Dietrich, D.; Perrin, D.M. A highly selective DNAzyme sensor for mercuric ions. Angew. Chem. Int. Ed. 2008, 47, 4346–4350. [Google Scholar] [CrossRef] [PubMed]
- Ting, R.; Thomas, J.M.; Lermer, L.; Perrin, D.M. Substrate specificity and kinetic framework of a DNAzyme with an expanded chemical repertoire: A putative RNaseA mimic that catalyzes RNA hydrolysis independent of a divalent metal cation. Nucleic Acids Res. 2004, 32, 6660–6672. [Google Scholar] [CrossRef] [PubMed]
- Perrin, D.M. Lifelike but Not Living: Selection of Synthetically Modified Bioinspired Nucleic Acids for Binding and Catalysis. Polym. Sci. 2012, 9, 3–33. [Google Scholar]
- Sample Availability: Samples of the compounds are not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, E.; Lam, C.H.; Perrin, D.M. Synthesis and Enzymatic Incorporation of Modified Deoxyuridine Triphosphates. Molecules 2015, 20, 13591-13602. https://doi.org/10.3390/molecules200813591
Liu E, Lam CH, Perrin DM. Synthesis and Enzymatic Incorporation of Modified Deoxyuridine Triphosphates. Molecules. 2015; 20(8):13591-13602. https://doi.org/10.3390/molecules200813591
Chicago/Turabian StyleLiu, Erkai, Curtis H. Lam, and David M. Perrin. 2015. "Synthesis and Enzymatic Incorporation of Modified Deoxyuridine Triphosphates" Molecules 20, no. 8: 13591-13602. https://doi.org/10.3390/molecules200813591