
DockBench: An Integrated Informatic Platform Bridging the Gap between the Robust Validation of Docking Protocols and Virtual Screening Simulations


 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
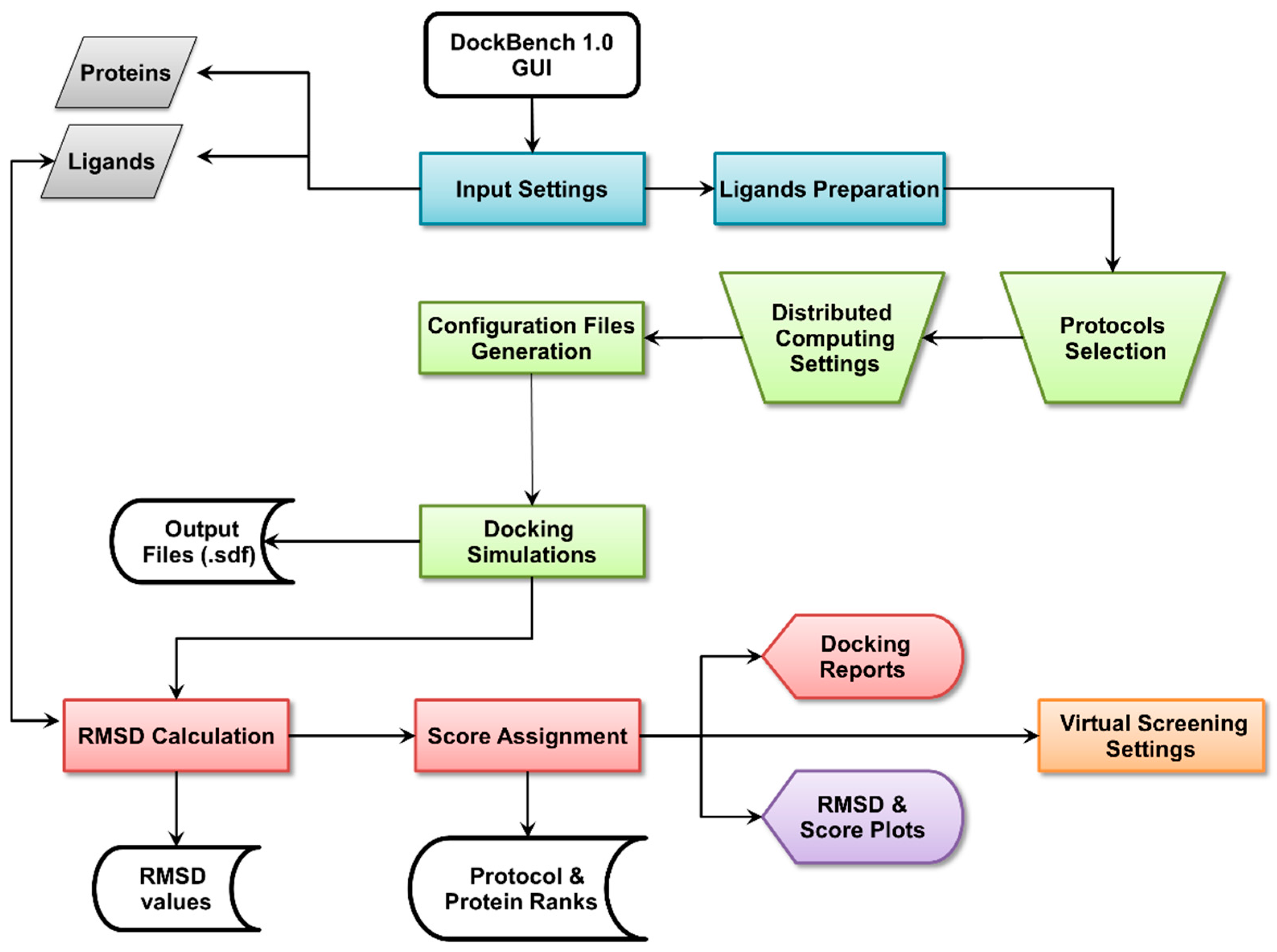
2.1. DockBench General Features
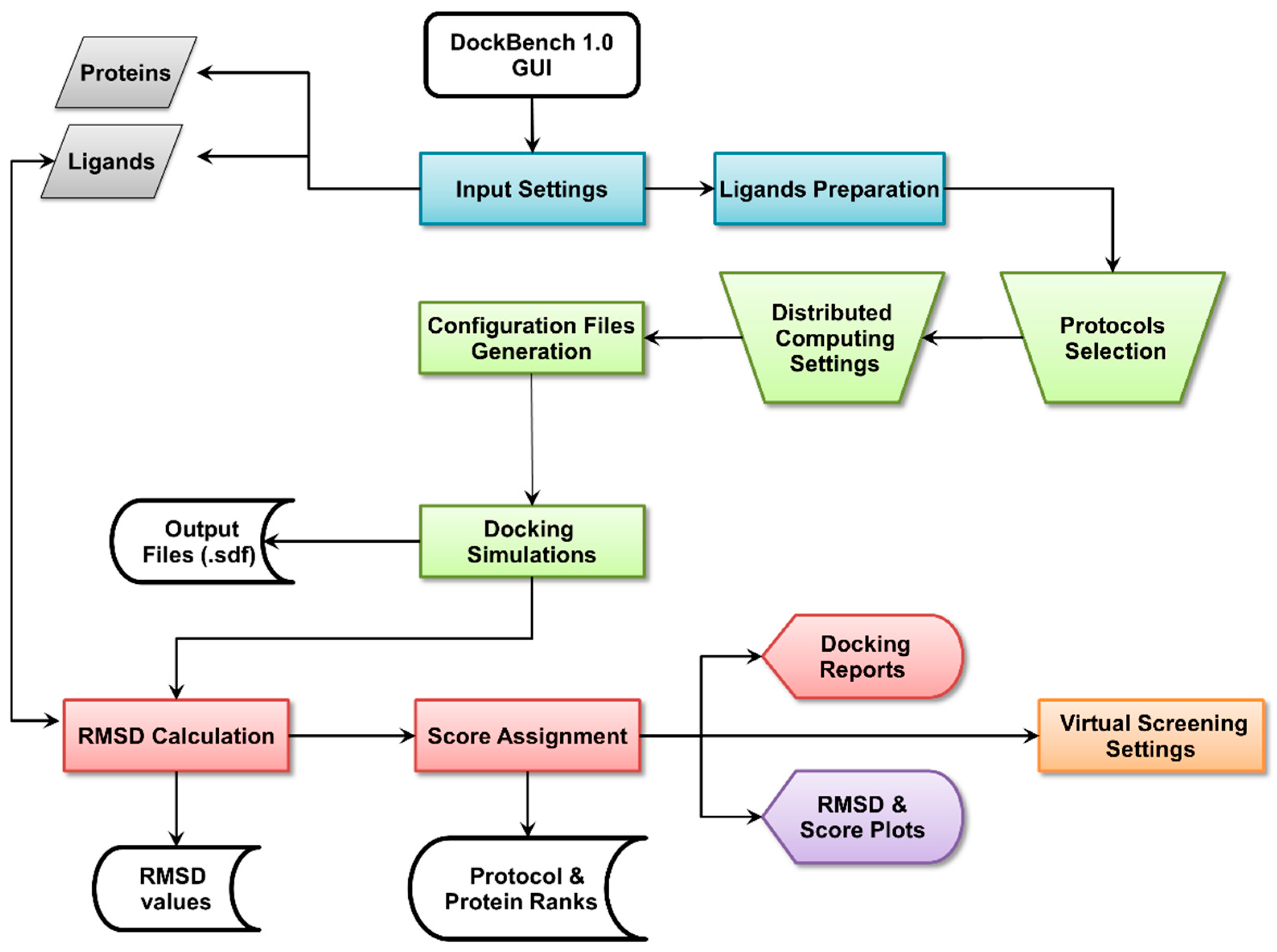
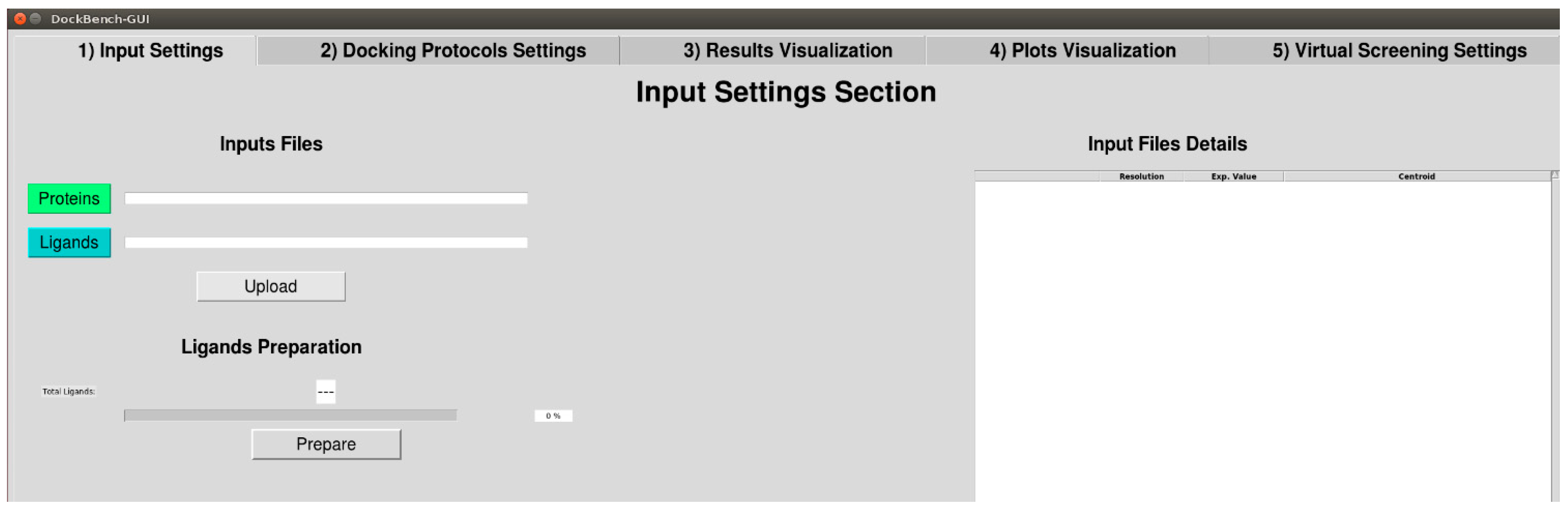
2.1.1. Input Settings
2.1.2. Docking Protocols Settings

{kind=link}
{kind=link}
{kind=link}
| Program | Search Algorithm/Placing Method | Scoring Function | Protocol Abbreviation |
|---|---|---|---|
| Autodock 4.2 | Local Search | AutoDock SF | AUTODOCK-ls |
| Lamarkian GA | AutoDock SF | AUTODOCK-lga | |
| Genetic Algorithm | AutoDock SF | AUTODOCK-ga | |
| AutoDock Vina 1.1.2 | Monte Carlo + BFGS local search | Standard Vina SF | VINA-std |
| Glide 6.5 | Glide Algorithm | Standard Precision | GLIDE-sp |
| GOLD 5.2 | Genetic Algorithm | Goldscore | GOLD-goldscore |
| Genetic Algorithm | Chemscore | GOLD-chemscore | |
| Genetic Algorithm | ASP | GOLD-asp | |
| Genetic Algorithm | PLP | GOLD-plp | |
| MOE 2014.09 | Triangle Matcher | London-dG | MOE-londondg |
| Triangle Matcher | Affinity-dG | MOE-affinitydg | |
| Triangle Matcher | GBIVIWSA | MOE-gbiviwsa | |
| PLANTS 1.2 | ACO Algorithm | PLP | PLANTS-plp |
| ACO Algorithm | PLP95 | PLANTS-plp95 | |
| ACO Algorithm | ChemPLP | PLANTS-chemplp | |
| rDock 2013.1 | Genetic Algorithm + Monte Carlo + Simplex minimization | Standard rDock master SF | RDOCK-std |
| Genetic Algorithm + Monte Carlo + Simplex minimization | Standard rDock master SF + desolvation potential | RDOCK-solv |
2.1.3. Results Visualization
2.1.4. Plots Visualization
2.1.5. Virtual Screening Settings
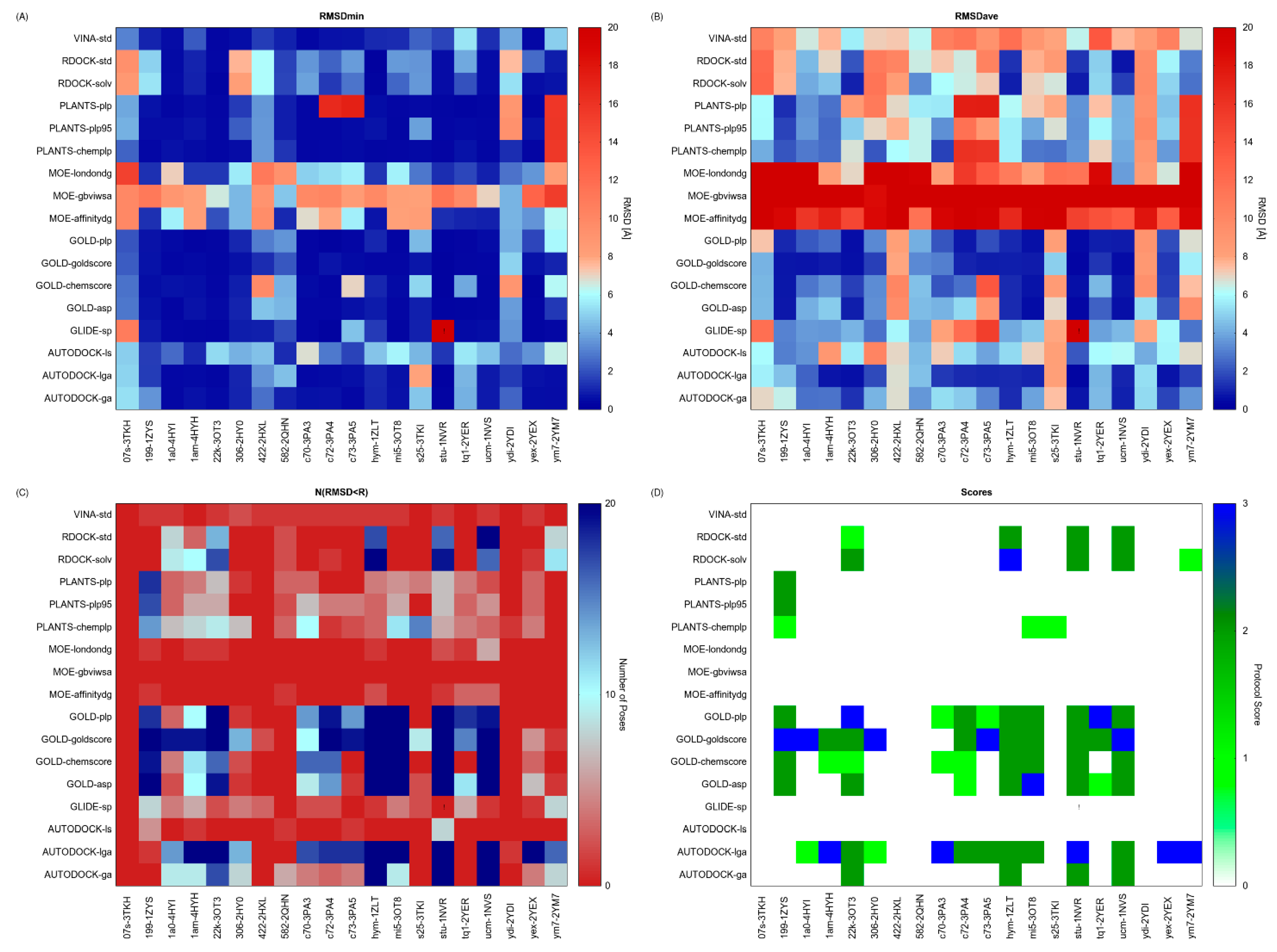
2.2. Case Study
DockBench 1.0 Performances

| Abbreviation | Average Execution Time (s) | Total Time (s) |
|---|---|---|
| AUTODOCK-ga | 973.5 | 20,445.3 |
| AUTODOCK-lga | 633.3 | 13,299.1 |
| AUTODOCK-ls | 7.45 | 156.58 |
| GLIDE-sp | 46.8 | 984.2 |
| GOLD-asp | 133.4 | 2801.8 |
| GOLD-chemscore | 136.2 | 2860.4 |
| GOLD-goldscore | 401.7 | 8436.5 |
| GOLD-plp | 98.6 | 2071.9 |
| PLANTS-chemplp | 61.4 | 1290 |
| PLANTS-plp | 23.3 | 958.1 |
| PLANTS-plp95 | 16.6 | 348.3 |
| MOE-affinitydg | 17.6 | 352.5 |
| MOE-londondg | 18.4 | 368.4 |
| MOE-gbviwsa | 131.9 | 2638.8 |
| RDOCK-std | 20.0 | 426.2 |
| RDOCK-solv | 31.9 | 671.0 |
| VINA-std | 132.7 | 2786.7 |
3. Experimental Section
3.1. Computational Facilities
3.2. DockBench 1.0 Platform
3.2.1. Programming Languages and Software Dependencies
3.2.2. Names Conventions
3.2.3. Implemented Docking Protocols and Standard Settings
| Parameter | Value/Setting |
|---|---|
| Ligand input conformation | Structures generated by minimization |
| Ligand initial partial charges | Provided by the user |
| Water molecules | Excluded |
| Output | 20 conformations (customizable) |
| RMSD threshold | 1.0 Å (customizable) |
| Binding cavity centre (Centroid) | Ligand barycentre in X-ray structure |
| Binding cavity radius (r) | 20 Å (customizable) |
| Grid spacing (for grid-based calculations) | 0.375 Å |
| Refinement and re-scoring | Turned off |
3.3. Case Study Input Files Preparation
3.3.1. Protein Structures
3.2.2. Ligand Structures
| Structure | IUPAC Name | Ligand Abbreviation |
|---|---|---|
 | 1-morpholin-4-yl-2-[4-[2-[(5-pyridin-3-yl-1,3-thiazol-2-yl)amino]pyridin-4-yl]piperazin-1-yl]ethanone | 07s-3TKH |
 | N-{5-[4-(4-methylpiperazin-1-yl)phenyl]-1H-pyrrolo[2,3-b]pyridin-3-yl}pyridine-3-carboxamide | 199-1ZYS |
 | 2-indazol-1-yl-N-(2-piperazin-1-ylphenyl)-1,3-thiazole-4-carboxamide | 1a0-4HYI |
 | 2-(6-methoxy-1-oxoisoindolin-2-yl)-N-(4-(piperazin-1-yl)pyridin-3-yl)thiazole-4-carboxamide | 1am-4HYH |
 | 5-[(1R,3S)-3-azanylcyclohexyl]-6-bromo-3-(1-methylpyrazol-4-yl)pyrazolo[1,5-a]pyrimidin-7-amine | 22k-3OT3 |
 | 3-[5-(piperidin-1-ylmethyl)-1H-indol-2-yl]-6-(1H-pyrazol-4-yl)-1H-quinolin-2-one | 306-2HY0 |
 | 3-[5-[[4-(aminomethyl)piperidin-1-yl]methyl]-1H-indol-2-yl]-1H-indazole-6-carbonitrile | 422-2HXL |
 | 5-ethyl-3-methyl-1H-pyrazolo[4,5-c]quinolin-4-one | 582-2QHN |
 | 2-(4-chlorophenyl)-4-[[(3S)-piperidin-3-yl]amino]thieno[2,3-d]pyridazine-7-carboxamide | c70-3PA3 |
 | 2-(4-chlorophenyl)-4-[[(3S)-piperidin-3-yl]amino]thieno[3,2-c]pyridine-7-carboxamide | c72-3PA4 |
 | 2-(aminocarbonylamino)-5-(4-chlorophenyl)-N-[(3S)-piperidin-3-yl]thiophene-3-carboxamide | c73-3PA5 |
 | (4Z)-4-(2-amino-5-oxo-3H-imidazol-4-ylidene)-2,3-dichloro-1,5,6,7-tetrahydropyrrolo[2,3-c]azepin-8-one | hym-1ZLT |
 | 3-(1-methyl-1H-pyrazol-4-yl)-N-(3-methyl-1,2-thiazol-5-yl)-5-[(3R)-piperidin-3-yl]pyrazolo[1,5-a]pyrimidin-7-amine | mi5-3OT8 |
 | N-(2-azanylethyl)-5-[2-[(4-morpholin-4-ylpyridin-2-yl)amino]-1,3-thiazol-5-yl]pyridine-3-carboxamide | s25-3TKI |
 | (5S,6R,7R,9R)-6-methoxy-5-methyl-7-(methylamino)-6,7,8,9,15,16-hexahydro-17-oxa-4b,9a,15-triaza-5,9 methanodibenzo[b,h]cyclonona[jkl]cyclopenta[e-as-indacen-14(5H)-one | stu-1NVR |
 | 5-(hydroxymethyl)-8-(1H-pyrrol-2-yl)[1,2,4]triazolo[4,3-a]quinolin-1(2H)-one | tq1-2YER |
 | (5R,8S)-5,6,7,8-tetrahydro-13H-5,8-epoxy-4b,8a,14-triazadibenzo[b,h]cycloocta[1,2,3,4-jkl]cyclopenta[e]-as-indacene-13,15(14H)-dione | ucm-1NVS |
 | 2-(carbamoylamino)-5-{4-[2-(dimethylamino)ethoxy]phenyl}thiophene-3-carboxamide | ydi-2YDI |
 | 5-methyl-8-(1H-pyrrol-2-yl)-2H-[1,2,4]triazolo[4,3-a]quinolin-1-one | Yex-2YEX |
 | 5-((6-((piperidin-4-ylmethyl)amino)pyrimidin-4-yl)amino)pyrazine-2-carbonitrile | ym7-2YM7 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Sotriffer, C. Methods and principles in medicinal chemistry. In Virtual Screening: Principles, Challenges, and Practical Guidelines; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Horvath, D. A virtual screening approach applied to the search for trypanothione reductase inhibitors. J. Med. Chem. 1997, 40, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Lill, M. Virtual screening in drug design. In Silico Models for Drug Discovery; Kortagere, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 993, pp. 1–12. [Google Scholar]
- Wilton, D.; Willett, P.; Lawson, K.; Mullier, G. Comparison of Ranking Methods for Virtual Screening in Lead-Discovery Programs. J. Chem. Inf. Model. 2003, 43, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Houston, D.R.; Walkinshaw, M.D. Consensus Docking: Improving the Reliability of Docking in a Virtual Screening Context. J. Chem. Inf. Model. 2013, 53, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.C.; Murray, C.W.; Nissink, J.W.M.; Taylor, R.D.; Taylor, R. Comparing protein-ligand docking programs is difficult. Proteins Struct. Funct. Bioinform. 2005, 60, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Ciancetta, A.; Cuzzolin, A.; Moro, S. Alternative Quality Assessment Strategy to Compare Performances of GPCR-Ligand Docking Protocols: The Human Adenosine A2A Receptor as a Case Study. J. Chem. Inf. Model. 2014, 54, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Converso, A.; Hartingh, T.; Garbaccio, R.M.; Tasber, E.; Rickert, K.; Fraley, M.E.; Yan, Y.; Kreatsoulas, C.; Stirdivant, S.; Drakas, B.; et al. Development of thioquinazolinones, allosteric Chk1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 1240–1244. [Google Scholar] [CrossRef] [PubMed]
- MMs DockBench. Available online: http://mms.dsfarm.unipd.it/mmsdockbench.html (accessed on 25 May 2015).
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Plants: Application of ant colony optimization to structure-based drug design. In Ant Colony Optimization and Swarm Intelligence; Dorigo, M., Gambardella, L.M., Birattari, M., Martinoli, A., Poli, R., Stützle, T., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, Germany, 2006; Volume 4150, pp. 247–258. [Google Scholar]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE), 2014.09. Chemical Computing Group Inc.: 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2015.
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tange, O. GNU Parallel—The Command-Line Power Tool. Login USENIX Mag. 2015, 36, 42–47. [Google Scholar]
- GOLD suite, version 5.2; Cambridge Crystallographic Data Centre: 12 Union Road, Cambridge CB2 1EZ, UK.
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Dudkin, V.Y.; Rickert, K.; Kreatsoulas, C.; Wang, C.; Arrington, K.L.; Fraley, M.E.; Hartman, G.D.; Yan, Y.; Ikuta, M.; Stirdivant, S.M.; et al. Pyridyl aminothiazoles as potent inhibitors of Chk1 with slow dissociation rates. Bioorg. Med. Chem. Lett. 2012, 22, 2609–2612. [Google Scholar] [CrossRef] [PubMed]
- Stavenger, R.A.; Zhao, B.; Zhou, B.-B.S.; Brown, M.J.; Lee, D.; Holt, D.A. Pyrrolo[2,3-b]pyridines Inhibit the Checkpoint Kinase Chk1. Available online: http://www.rcsb.org/pdb/static.do?p=general_information/about_pdb/policies_references.html (accessed on 28 May 2015).
- Huang, X.; Cheng, C.C.; Fischmann, T.O.; Duca, J.S.; Richards, M.; Tadikonda, P.K.; Reddy, P.A.; Zhao, L.; Arshad Siddiqui, M.; Parry, D.; et al. Structure-based design and optimization of 2-aminothiazole-4-carboxamide as a new class of CHK1 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2590–2594. [Google Scholar] [CrossRef] [PubMed]
- Labroli, M.; Paruch, K.; Dwyer, M.P.; Alvarez, C.; Keertikar, K.; Poker, C.; Rossman, R.; Duca, J.S.; Fischmann, T.O.; Madison, V.; et al. Discovery of pyrazolo[1,5-a]pyrimidine-based CHK1 inhibitors: A template-based approach—Part 2. Bioorg. Med. Chem. Lett. 2011, 21, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Garbaccio, R.M.; Fraley, M.E.; Steen, J.; Kreatsoulas, C.; Hartman, G.; Stirdivant, S.; Drakas, B.; Rickert, K.; Walsh, E.; et al. Development of 6-substituted indolylquinolinones as potent Chek1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5907–5912. [Google Scholar] [CrossRef] [PubMed]
- Brnardic, E.J.; Garbaccio, R.M.; Fraley, M.E.; Tasber, E.S.; Steen, J.T.; Arrington, K.L.; Dudkin, V.Y.; Hartman, G.D.; Stirdivant, S.M.; Drakas, B.A.; et al. Optimization of a pyrazoloquinolinone class of Chk1 kinase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5989–5994. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, Y.; Dai, C.; Guzi, T.; Wiswell, D.; Seghezzi, W.; Parry, D.; Fischmann, T.; Siddiqui, M.A. Design, synthesis and SAR of thienopyridines as potent CHK1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 7216–7221. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Ng, K.; Wan, Y.; Gray, N.; Spraggon, G. Crystal Structure of Chk1 Complexed with a Hymenaldisine Analog. Available online: http://www.rcsb.org/pdb/static.do?p=general_information/about_pdb/policies_references.html (accessed on 28 May 2015).
- Dwyer, M.P.; Paruch, K.; Labroli, M.; Alvarez, C.; Keertikar, K.M.; Poker, C.; Rossman, R.; Fischmann, T.O.; Duca, J.S.; Madison, V.; et al. Discovery of pyrazolo[1,5-a]pyrimidine-based CHK1 inhibitors: A template-based approach—Part 1. Bioorg. Med. Chem. Lett. 2011, 21, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B. Structural Basis for Chk1 Inhibition by UCN-01. J. Biol. Chem. 2002, 277, 46609–46615. [Google Scholar] [CrossRef] [PubMed]
- Oza, V.; Ashwell, S.; Brassil, P.; Breed, J.; Ezhuthachan, J.; Deng, C.; Grondine, M.; Horn, C.; Liu, D.; Lyne, P.; et al. Synthesis and evaluation of triazolones as checkpoint kinase 1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 2330–2337. [Google Scholar] [CrossRef] [PubMed]
- Oza, V.; Ashwell, S.; Almeida, L.; Brassil, P.; Breed, J.; Deng, C.; Gero, T.; Grondine, M.; Horn, C.; Ioannidis, S.; et al. Discovery of Checkpoint Kinase Inhibitor (S)-5-(3-Fluorophenyl)-N-(piperidin-3-yl)-3-ureidothiophene-2-carboxamide (AZD7762) by Structure-Based Design and Optimization of Thiophenecarboxamide Ureas. J. Med. Chem. 2012, 55, 5130–5142. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.C.; Matthews, T.P.; Klair, S.; Cheung, K.M.J.; Scanlon, J.; Proisy, N.; Addison, G.; Ellard, J.; Piton, N.; Taylor, S.; et al. Structure-Guided Evolution of Potent and Selective CHK1 Inhibitors through Scaffold Morphing. J. Med. Chem. 2011, 54, 8328–8342. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods II. Applications. J. Comput. Chem. 1989, 10, 221–264. [Google Scholar] [CrossRef]
- Sample Availability: The DockBench 1.0 is available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuzzolin, A.; Sturlese, M.; Malvacio, I.; Ciancetta, A.; Moro, S. DockBench: An Integrated Informatic Platform Bridging the Gap between the Robust Validation of Docking Protocols and Virtual Screening Simulations. Molecules 2015, 20, 9977-9993. https://doi.org/10.3390/molecules20069977
Cuzzolin A, Sturlese M, Malvacio I, Ciancetta A, Moro S. DockBench: An Integrated Informatic Platform Bridging the Gap between the Robust Validation of Docking Protocols and Virtual Screening Simulations. Molecules. 2015; 20(6):9977-9993. https://doi.org/10.3390/molecules20069977
Chicago/Turabian StyleCuzzolin, Alberto, Mattia Sturlese, Ivana Malvacio, Antonella Ciancetta, and Stefano Moro. 2015. "DockBench: An Integrated Informatic Platform Bridging the Gap between the Robust Validation of Docking Protocols and Virtual Screening Simulations" Molecules 20, no. 6: 9977-9993. https://doi.org/10.3390/molecules20069977