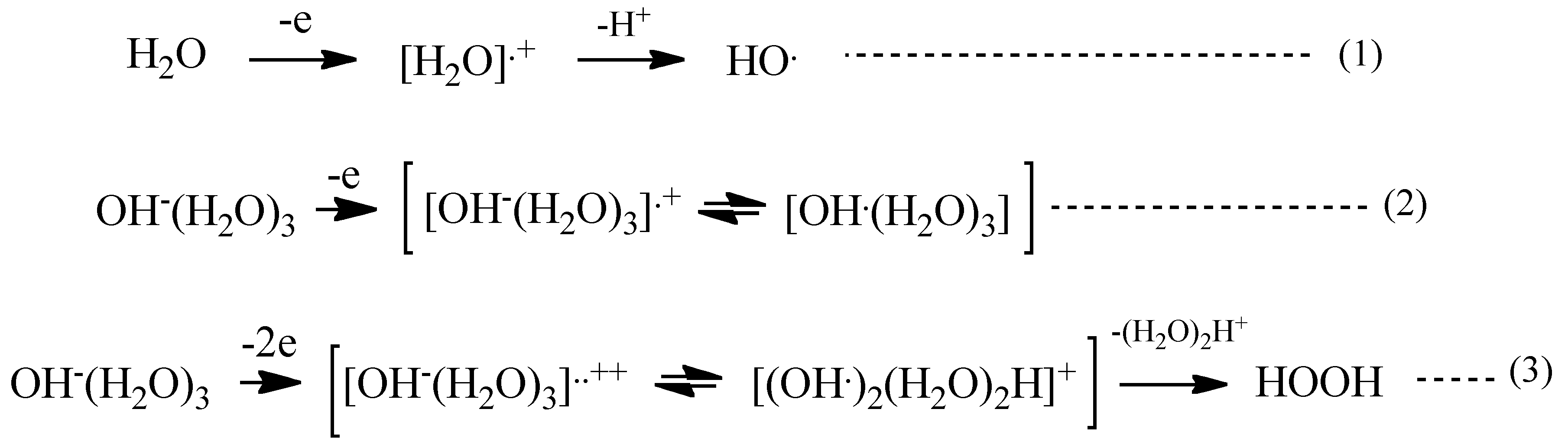2.1. Yamashita/Jono Model for Simulation of PEC-nc-TiO2 Electrodes
Yamashita and Jono’s anatase nc-TiO
2 model (Ti
9O
18H-OH) consists of nine TiO
2 units (TiO
2)
9 derived from the packing unit of the crystalline anatase TiO
2, hydroxide on surface side of the TiO
2 and hydrogen on one side of the TiO
2. To optimize the non-covalent distance between hydroxyl group and nc-TiO
2 unit, all heavy atoms of the nc-TiO
2 unit are frozen and the model was simulated to the energetically optimized geometry (see
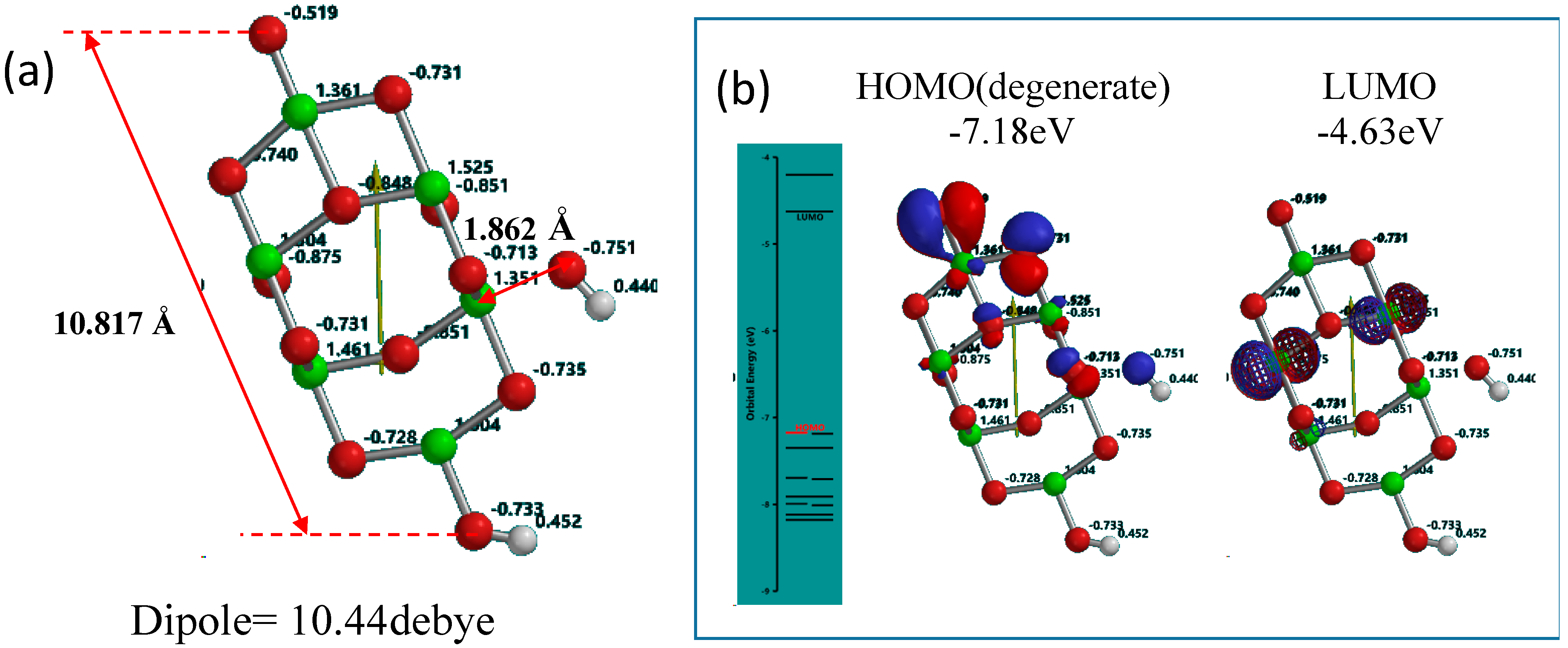
Supplementary Figure S1). The distance (1.862 Å) become shorter, and the refined Yamashita/Jono model is abbreviated as OH(TiO
2)
9H hereafter and size of about 1 nm length, Mulliken charge, the energy structures and configurations of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are shown in
Figure 1.
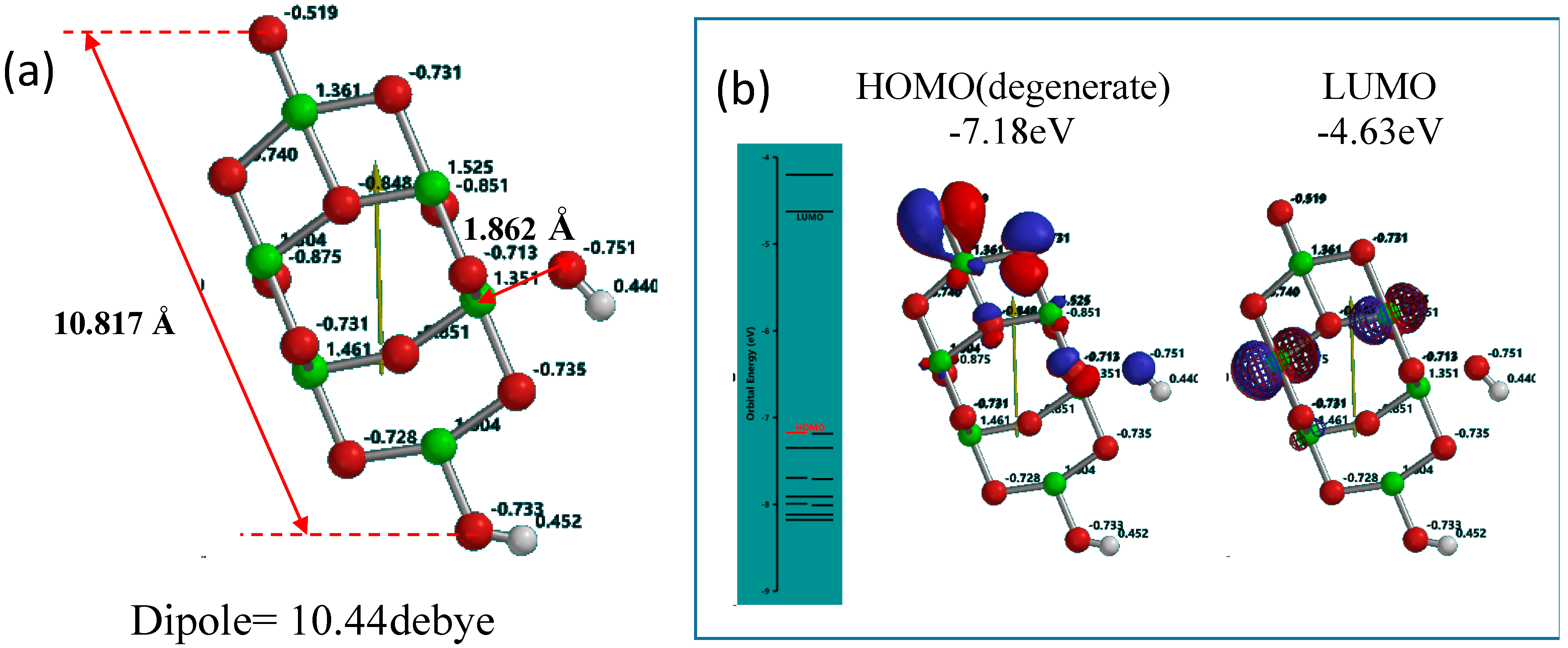
Figure 1.
Yamashita/Jono model of OH(TiO2)9H as a model of PEC-nc-TiO2 electrodes, (a) Size, distance and Mulliken charge; (b) the energy structure and configuration of HOMO and LUMO.
Figure 1.
Yamashita/Jono model of OH(TiO2)9H as a model of PEC-nc-TiO2 electrodes, (a) Size, distance and Mulliken charge; (b) the energy structure and configuration of HOMO and LUMO.
Mulliken charge in OH(TiO
2)
9H indicates that hydroxyl group is charged negative and the protonated nc-TiO
2 unit positive, and then we regard that Yamashita/Jono model is a kind of ion-dipole complex or weak charge transfer complex. The HOMO distributes exclusively on hydroxide ion and the LUMO inside of the nc-TiO
2 unit, giving energy gap 2.55 eV. To know self-association of Yamashita/Jono model, the OH and H-detached (TiO
2)
9 and (TiO
2)
9H units are both DFT-simulated as charge is neutral and cation, respectively (
Supplementary Tables S1 and S2 and Figures S1 and S2).
The DFT simulation data reveals that Mulliken charge on the hydrogen-bearing side is positive and another side negative as well as the OH(TiO
2)
9H model. Further, the HOMO and LUMO are almost degenerate. The association through van der Waals and Coulombic interaction and the comparable dipole to that of the Yamashita/Jono model support that Yamashita/Jono model may self-associate with (TiO
2)
9 and (TiO
2)
9H to yield surface thin-film anatase TiO
2 electrodes as depicted in
Figure 2.
Figure 2.
Association of Yamashita/Jono model via van der Waals and Coulomb interactions, explaining growth of the model to larger size PEC-nc-TiO2 electrodes.
Figure 2.
Association of Yamashita/Jono model via van der Waals and Coulomb interactions, explaining growth of the model to larger size PEC-nc-TiO2 electrodes.
2.3. Photoelectrochemical Oxidation of H2O on PEC-nc-TiO2
As an interface model of nc-TiO
2 electrodes, the Yamashita/Jono model, OH(TiO
2)
9H, is structurally frozen and three H
2O molecules are made manually to interact via hydrogen bonding with the frozen hydroxyl group in Yamashita/Jono model. The (H
2O)
3-hydrogen bonded model structure is optimized by molecular mechanics (MMFF operation in Spartan), and the molecular orbital is verified by DFT-single-point simulation of an interface structure of (H
2O)
3&OH(TiO
2)
9H as a stationary model (
Figure 4). Mulliken charge and electrostatic potential map indicates that negative charge locates on surface oxygen atoms of TiO
2 and of the (H
2O)
3 cluster, and the more negative charge (stronger red color) on the H
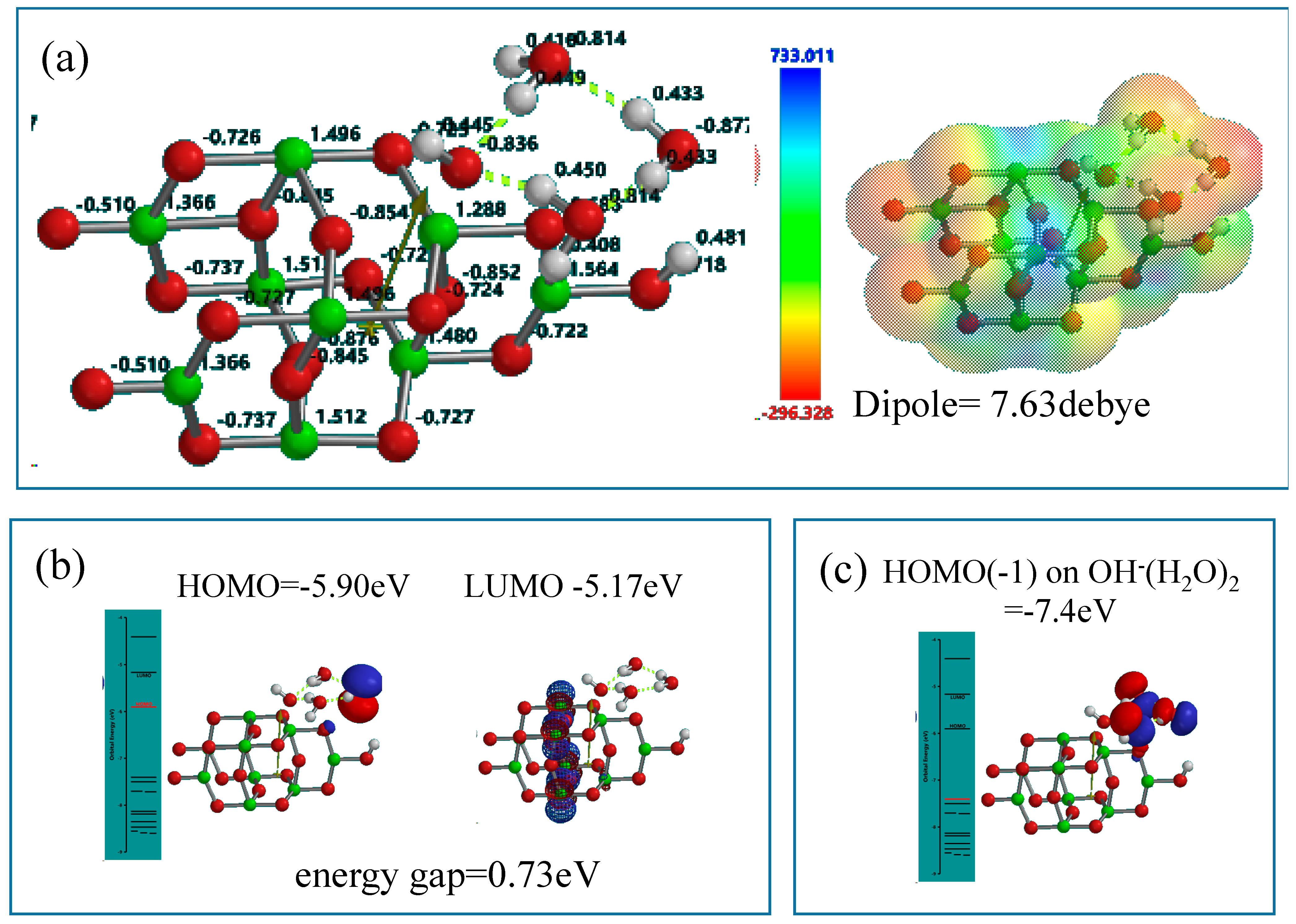
2O molecule in the cluster is worth noting in the stationary state model structure.
In the stationary model, configurations of HOMO distributes on one H
2O of the cluster (H
2O)
3 unit, and LUMO inside the nc-TiO
2 unit. Interestingly, the energy gap (0.73 eV) between HOMO and LUMO is smaller than the energy gap (2.55 eV) in the stationary state of Yamashita/Jono model. This fact suggests that nc-TiO
2 surface binds H
2O molecules via hydrogen bond, forming kinds of charge transfer complexes. In addition, HOMO(−1) (−7.4 eV) distributes on the whole (H
2O)
3 unit, implying more effective H
2O oxidation under negative potential of −7.4 eV (
Figure 4).
For comparison, only one H
2O-associated Yamashita/Jono model, H
2O&OH(TiO
2)
9H, is simulated as the simplest structure of PEC-nc-TiO
2 electrodes (
Supplementary Figure S3). The energy gap (1.75 eV) is given and the hydrogen-bonded H
2O molecule locates HOMO(−1) at −7.8 eV. The simplest orbital energy structure verifies that nc-TiO
2 electrodes intrinsically work as catalytic sites of H
2O oxidation through hydrogen bonding with H
2O molecules.
Figure 4.
DFT-simulation of (H2O)3&OH(TiO2)9H as a stationary state of PEC-nc-TiO2 electrodes, (a) Mulliken charge and electrostatic density map; (b) energy structures of HOMO and LUMO; (c) the configuration of HOMO(−1) on (H2O)3.
Figure 4.
DFT-simulation of (H2O)3&OH(TiO2)9H as a stationary state of PEC-nc-TiO2 electrodes, (a) Mulliken charge and electrostatic density map; (b) energy structures of HOMO and LUMO; (c) the configuration of HOMO(−1) on (H2O)3.
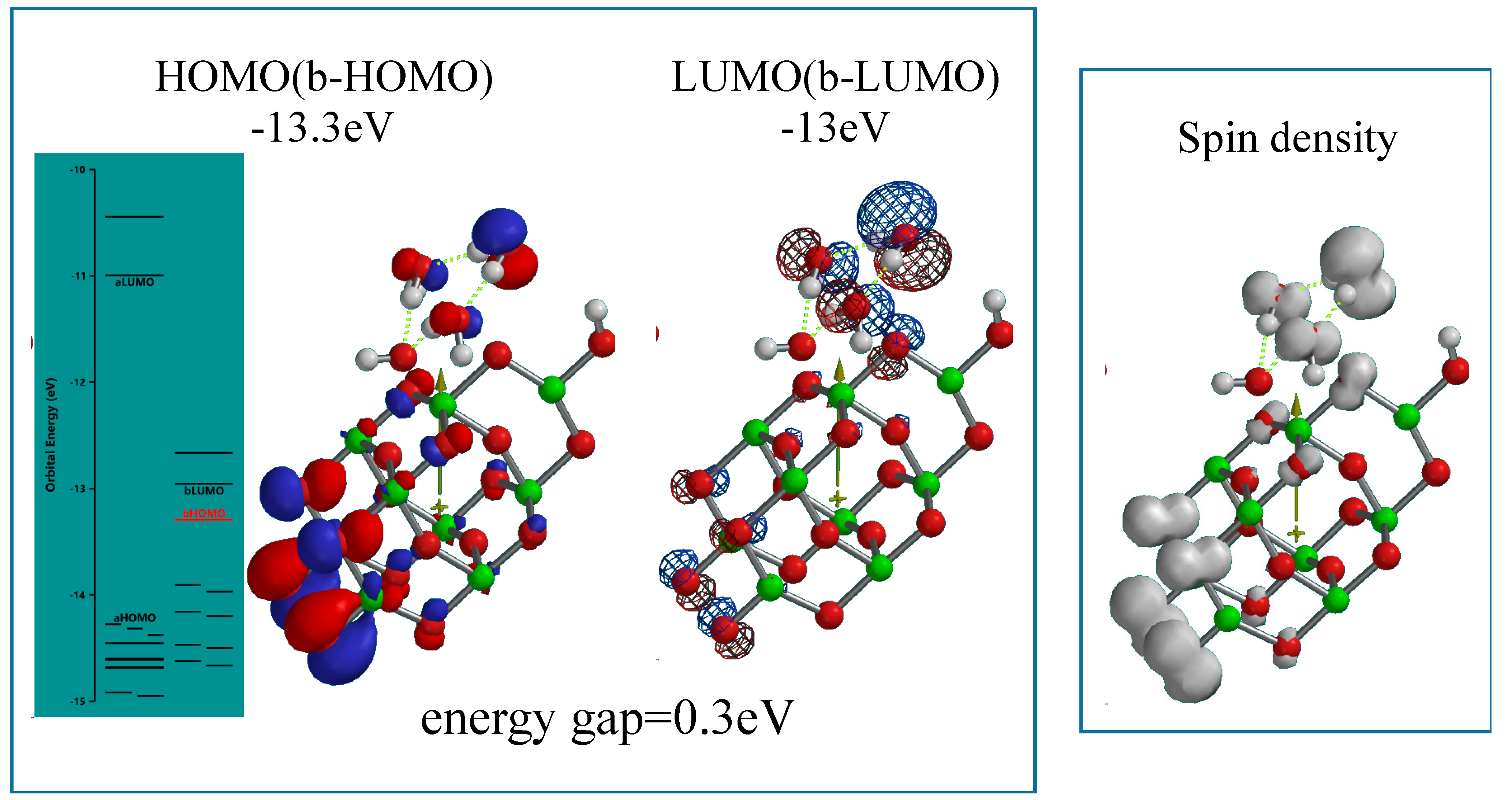
When nc-TiO
2 electrodes are energy-filled by UV-irradiation at short circuit PEC conditions, photoelectron on nc-TiO
2 is ejected to conducting grids instantly, and photocurrent become observable. The (H
2O)
3-associated PEC-nc-TiO
2 model is simulated as radical cation model of [(H
2O)
3 &OH(TiO
2)
9H]
.+ as the energy-filled structure under working conditions (
Figure 5). Interestingly, the radical cation model as the working model is endothermically simulated (ΔE = 192.85 kcal/mol). The orbital energy analysis of the energy-filled model of [(H
2O)
3&OH(TiO
2)
9H]
.+ reveals that the energy gap between HOMO and LUMO potential becomes narrow as small as 0.3 eV with largely negative HOMO (−10.2 eV) and LUMO (−9.9 eV) potential. The HOMO and LUMO distribute in the model with almost the same configurations, and that the spin (unpaired electron) density distributes with the same configuration as the HOMO and LUMO.
The orbital energy analysis of the energy-filled Yamashita/Jono model of [OH(TiO
2)
9H]
.+ reveals that the energy gap is pretty narrow (0.7 eV) (
Supplementary Table S2). These facts suggest that self-organization of Yamashita/Jono model will give photoconductive nc-TiO
2 electrodes when the PEC cell is kept at negative oxidation potential at short circuit conditions and energized by band gap excitation. In fact, the sharp rise in photoconductivity of nc-TiO
2 electrodes was reported and discussed as an insulator-metal (Mott) transition in a donor band of anatase TiO
2 [
11]. It is also worth noting that in studies on dye-sensitized nc-TiO
2 solar cells (DSC), adsorption of cationic species like tetrabutylammonium cation and sensitizing dye molecules enhanced electron transport in nc-TiO
2 electrodes [
12,
13]. Accordingly, the photo-enhanced electron transport is now verified as a key function of nc-TiO
2 electrodes not only in DSC but also in PEC H
2O oxidation.
Figure 5.
Energy structures of (H2O)3&[OH(TiO2)9H]+ as a radical cation model of (H2O)3-interacted PEC-nc-TiO2 electrode under UV-irradiated bias conditions of PEC-nc-TiO2 electrodes, i.e., a photo-energy-driven operational state model.
Figure 5.
Energy structures of (H2O)3&[OH(TiO2)9H]+ as a radical cation model of (H2O)3-interacted PEC-nc-TiO2 electrode under UV-irradiated bias conditions of PEC-nc-TiO2 electrodes, i.e., a photo-energy-driven operational state model.
The HOMO configuration on the H
2O unit at the stationary state of the model, and the spin density distribution on the (H
2O)
3 unit at the energy-filled working state strongly suggest that PEC-nc-TiO
2 electrodes provides catalytic binding sites of H
2O. The same functions of PEC nc-TiO
2 electrodes are confirmed by the molecular orbital simulation of the energy-filled [H
2O&OH(TiO
2)
9H]
+ model (
Tables S1 and S2, Figure S4). The narrowed energy gap (0.3 eV), the comparable configurations of HOMO, LUMO and spin density are quite comparable with those of [(H
2O)
3&OH(TiO
2)
9H]
+.
2.4. DFT Simulation of H2O Oxidation to Hydrogen Peroxide
In PEC H
2O oxidation on nc-TiO
2 electrodes, bias potential is essential to start H
2O oxidation. The HOMO potential of Yamashita/Jono model, −7.18 eV and the average HOMO potential of H
2O clusters, −7.48 eV (
Supplementary Table S3) verify that the bias potential >0.3V is at lease required for PEC H
2O oxidation under neutral working conditions. On the other hand, the HOMO potential (−10.2 eV) of the working model of [(H
2O)
3&OH(TiO
2)
9H]
+ predicts that successive oxidation should occur under more negative bias potential (>2.72 eV) for H
2O oxidation to oxygen through formation of hydrogen peroxide.
In order to verify whether hydroxyl radical may form successively on PEC-nc-TiO
2 electrodes, the two-electron oxidation structure of [H
2O&OH(TiO
2)
9H]
..++ is simulated as dication-diradical model as another working interface model of PEC-nc-TiO
2 electrodes. The more energy-filled model is endothermically (ΔE = 458.43 kcal/mol) simulated as powerful working model (
Figure 6). The energy gap (0.3 eV) and the configuration of HOMO and LUMO are confirmed to verify that such largely energized PEC-nc-TiO
2 electrodes keep photoconductivity with keeping high oxidation potential. The spin density distributes on the (H
2O)
3 unit, suggesting that two-electron H
2O oxidation occurs successively on the catalytic site on nc-TiO
2 electrodes.
Figure 6.
Energy structures of [(H2O)3]&OH(TiO2)9H]..++ as the model of two electron oxidation state of PEC-nc-TiO2 electrodes, i.e., the diradical-dication state of the electrodes.
Figure 6.
Energy structures of [(H2O)3]&OH(TiO2)9H]..++ as the model of two electron oxidation state of PEC-nc-TiO2 electrodes, i.e., the diradical-dication state of the electrodes.
With these simulation analyses, step-wise PEC-H
2O oxidation is shown in
Scheme 1. One-electron oxidation of H
2O molecule yields radical cation of H
2O (H
2O
+) and the removable of proton from the radical cation (deprotonation) leads to hydroxyl radical (HO) (Equations (1) and (2) in
Scheme 1). When H
2O hydroxide ion cluster of OH
−(H
2O)
3 undergoes further oxidation, another hydroxyl radical favorably forms in neighbor on PEC-nc-TiO
2 electrodes, and efficient and effective formation of hydrogen peroxide occurs (Equation (3) in
Scheme 1).
Scheme 1.
PEC-H2O oxidation to hydrogen peroxide as a precursor of oxygen molecule.
Scheme 1.
PEC-H2O oxidation to hydrogen peroxide as a precursor of oxygen molecule.
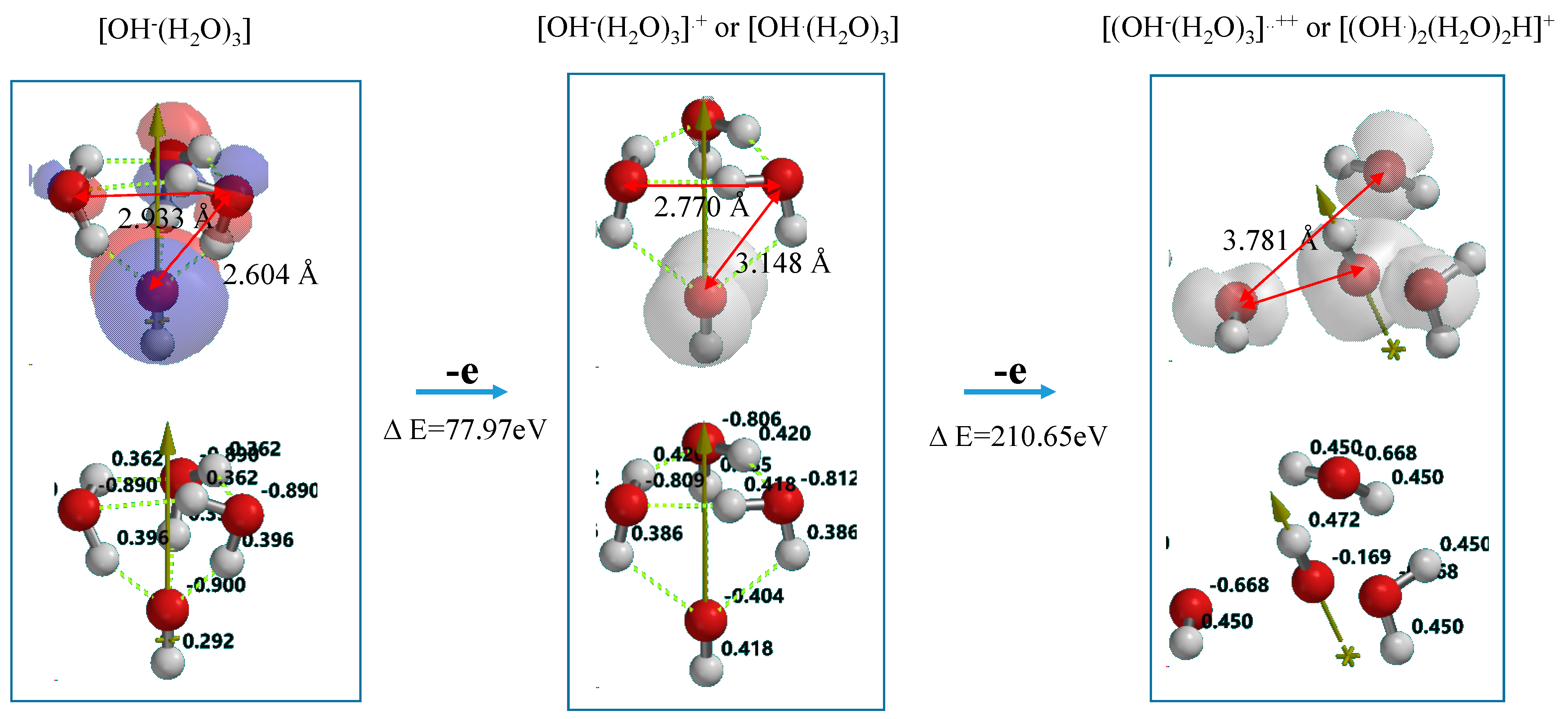
Figure 7 shows DFT-simulation results of oxidation of OH
−(H
2O)
3 to [OH
−(H
2O)
3]
.+ or [OH
.(H
2O)
3] as one-electron oxidation products (Equation (2)), and to [OH
−(H
2O)
3]
..++ or [(OH
.)
2(H
2O)
2H]
+ as two-electron oxidation products. They are simulated endothermically, suggesting that they are in energy filled states (
Supplementary Table S6). In the equilibrium geometry of [OH
.(H
2O)
3]
.+, the spin density distributes only on the hydroxyl group, and the Mulliken charge on hydroxyl group decreases largely from −0.900 to −0.404. Thus the one-electron oxidation product has the structure of [OH
.(H
2O)
3] rather than [OH
−(H
2O)
3]
.+.
Figure 7.
DFT-simulation of step-wise oxidation of the hydroxide ion cluster model of [OH−(H2O)3].
Figure 7.
DFT-simulation of step-wise oxidation of the hydroxide ion cluster model of [OH−(H2O)3].
On the other hand, the two-electron oxidation product shows that the spin density distributes on hydroxyl group and the (H
2O)
3 units. The Mulliken charge on them decreases from −0.806 to −0.668. The distance between hydroxyl group and H
2O is shortened from 3.148 Å to 2.239 Å and the hydrogen bonds observed in [OH
−(H
2O)
3]
.+ disappear. In addition, the hydrogen-oxygen bond distance of H
2O (0.976 Å) in [OH
−(H
2O)
3]
..++ is quite comparable with that (0.988 Å) in [OH
−(H
2O)
3]
.+. The H
2O components in the most energy-filled cluster [OH
−(H
2O)
3]
++ should be tightly aggregated one another. Detachment of (H
2O)
3H
.+ from [(OH
.)
2(H
2O)
2H
+] leaves two hydroxyl radical in neighbor, yielding hydrogen peroxide as a precursor of O
2 molecule (Equation (3) in
Scheme 1). The oxidation of neutral H
2O is verified to occur initially in photocatalytic processes to give effectively hydrogen peroxide on PEC nc-TiO
2 electrodes.
2.5. Verification of the Sakka’s PEC H2O Oxidation under Acidic Conditions
The stationary model of H
3O
+(H
2O)-associated structure of H
3O
+(H
2O)&OH(TiO
2)
9H and two kinds of energy filled models, [H
3O
+(H
2O)&OH(TiO
2)
9H]
.+ and [H
3O
+(H
2O)&OH(TiO
2)
9H]
.++ are simulated as an interface model of PEC-nc-TiO
2 electrodes under Sakka’s acidic conditions (
Supplementary Figures S5 and S6). However, the energy gap are rather wider and the spin density does not localize on H
3O
+(H
2O) in the most energized state of [H
3O
+(H
2O)&OH(TiO
2)
9H]
.++.
The hydronium ion cluster, H
3O
+(H
2O)
2 represent less acidic than H
3O
+(H
2O) (
Supplementary Table S5). The stationary states of H
3O
+(H
2O)
2-associated structure of H
3O
+(H
2O)
2&OH(TiO
2)
9H is simulated as an interface model under Sakka’s less acidic conditions, and analyzed as well in view of molecular orbital energy structure (
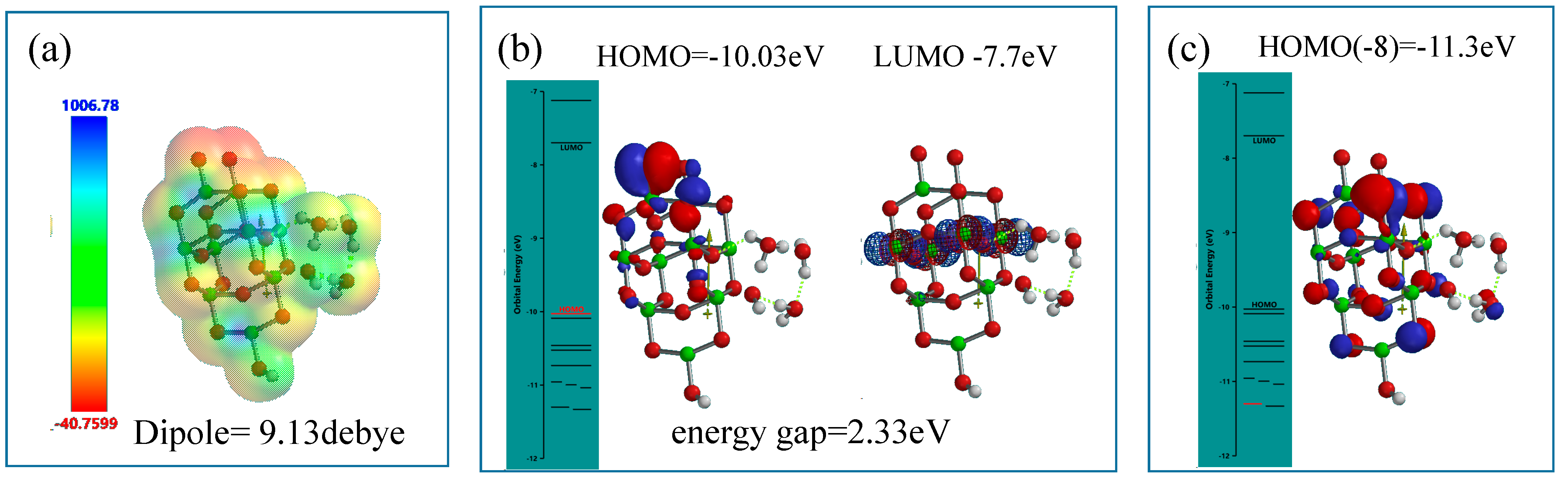
Figure 8).
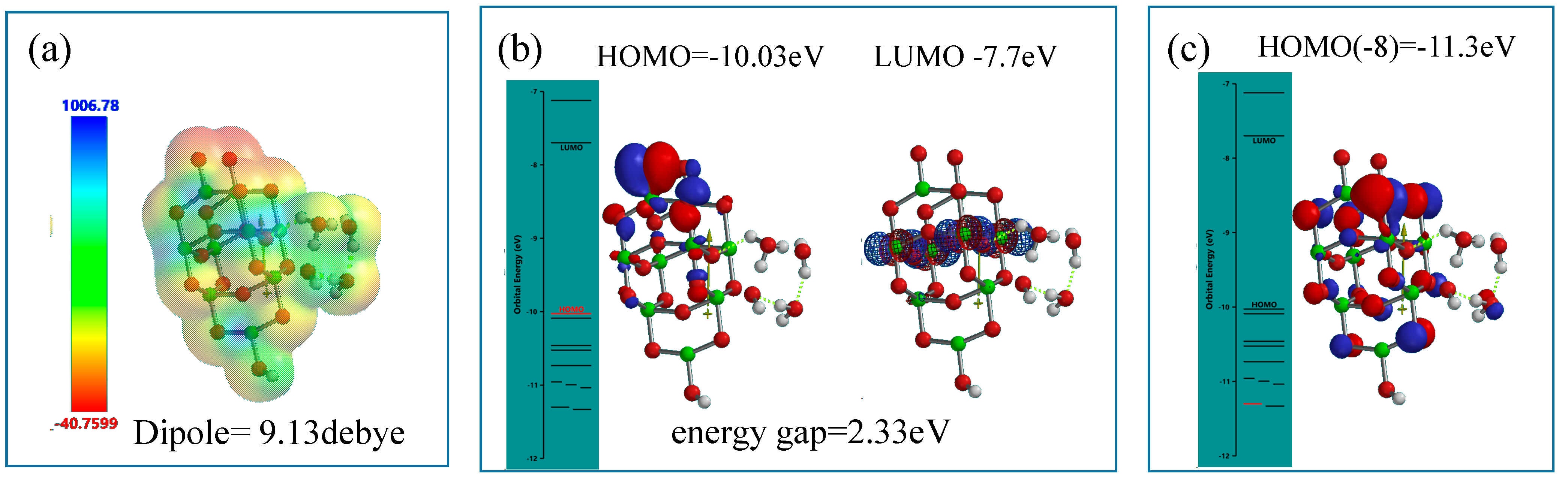
Figure 8.
DFT-simulation of H3O+(H2O)2&OH(TiO2)9H as a model of hydronium ion clusters on PEC-nc-TiO2 electrodes, (a) electrostatic potential map; (b) structures of HOMO and LUMO; (c) Configuration of HOMO(−8).
Figure 8.
DFT-simulation of H3O+(H2O)2&OH(TiO2)9H as a model of hydronium ion clusters on PEC-nc-TiO2 electrodes, (a) electrostatic potential map; (b) structures of HOMO and LUMO; (c) Configuration of HOMO(−8).
Differently from the modeling for the neutral PEC H2O oxidation, electrostatic potential map indicates that negative charge locates much more on the nc-TiO2 unit rather than the H2O unit, and HOMO distributes only on oxygen atoms in the nc-TiO2 unit. The energy gap 2.33 eV implies weak association of acidic H2O on nc-TiO2 electrodes. The orbital energy indicates that HOMO(−8) distributes slightly on the H3O+(H2O)2 unit with very negative potential of −11.3 eV.
The acidic interface model of [H
3O
+(H
2O)
2&OH(TiO
2)
9H] is simulated to [H
3O
+(H
2O)
2& OH(TiO2)
9H]
.+ as the radical cation of the one-electron oxidation state, and to [H
3O
+(H
2O)
2& OH(TiO2)
9H]
..++ as the diradical-dication model of the two electron oxidation sate (
Figure 9). The former radical cation model reveals that configurations of HOMO, LUMO and spin density distribute on the nc-TiO
2 unit and not on the H
3O
+(H
2O)
2, and the energy gap 0.7eV is not favorable in view of photoconductivity compared to that under neutral conditions. However, HOMO(−1) distributes on the (H
2O)
2 unit with orbital potential of −13.9 eV.
Figure 9.
DFT-simulation of oxidation states of H3O+(H2O)2&OH(TiO2)9H, (a) energy structures of the one-electron oxidation state, [H3O+(H2O)2&OH(TiO2)9H].+; (b) energy structures of the two-electron oxidation state, [H3O+(H2O)2&OH(TiO2)9H]..++.
Figure 9.
DFT-simulation of oxidation states of H3O+(H2O)2&OH(TiO2)9H, (a) energy structures of the one-electron oxidation state, [H3O+(H2O)2&OH(TiO2)9H].+; (b) energy structures of the two-electron oxidation state, [H3O+(H2O)2&OH(TiO2)9H]..++.
As for the latter dication-diradical model, the energy gap 0.5 eV and HOMO potential, −16.6 eV are given, and the spin density distributes on H3O+(H2O)2 unit. Accordingly, the DFT-based orbital energy structure verifies that PEC H2O oxidation occurs even under acidic conditions, when nc-TiO2 electrodes are energized by bias potential under energy-gap UV irradiation.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
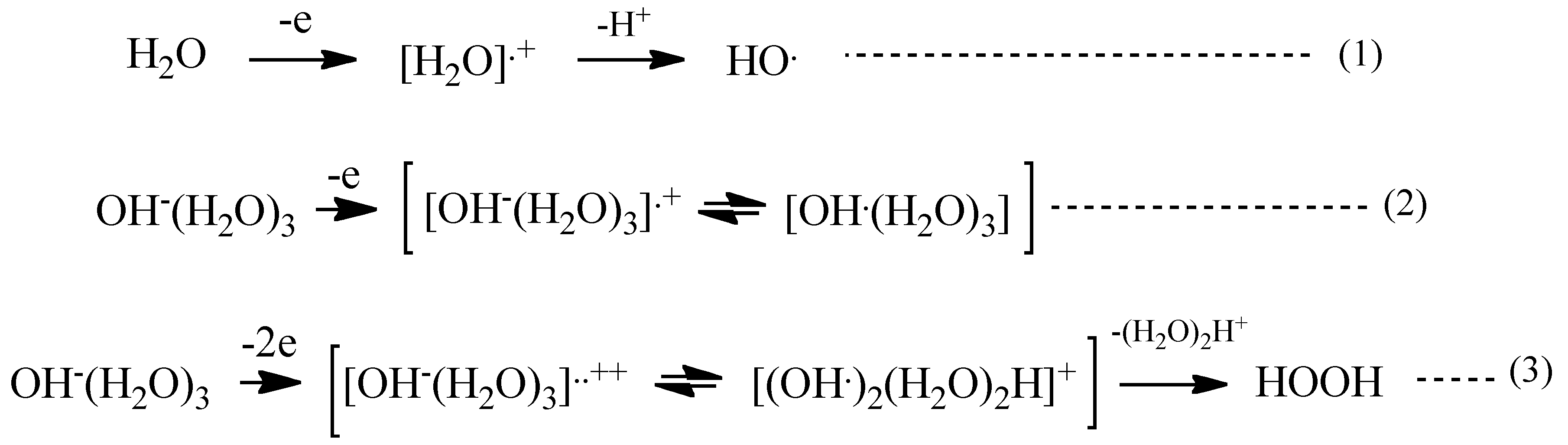{kind=link}