
Microwave-Assisted Condensation Reactions of Acetophenone Derivatives and Activated Methylene Compounds with Aldehydes Catalyzed by Boric Acid under Solvent-Free Conditions

Abstract
:1. Introduction
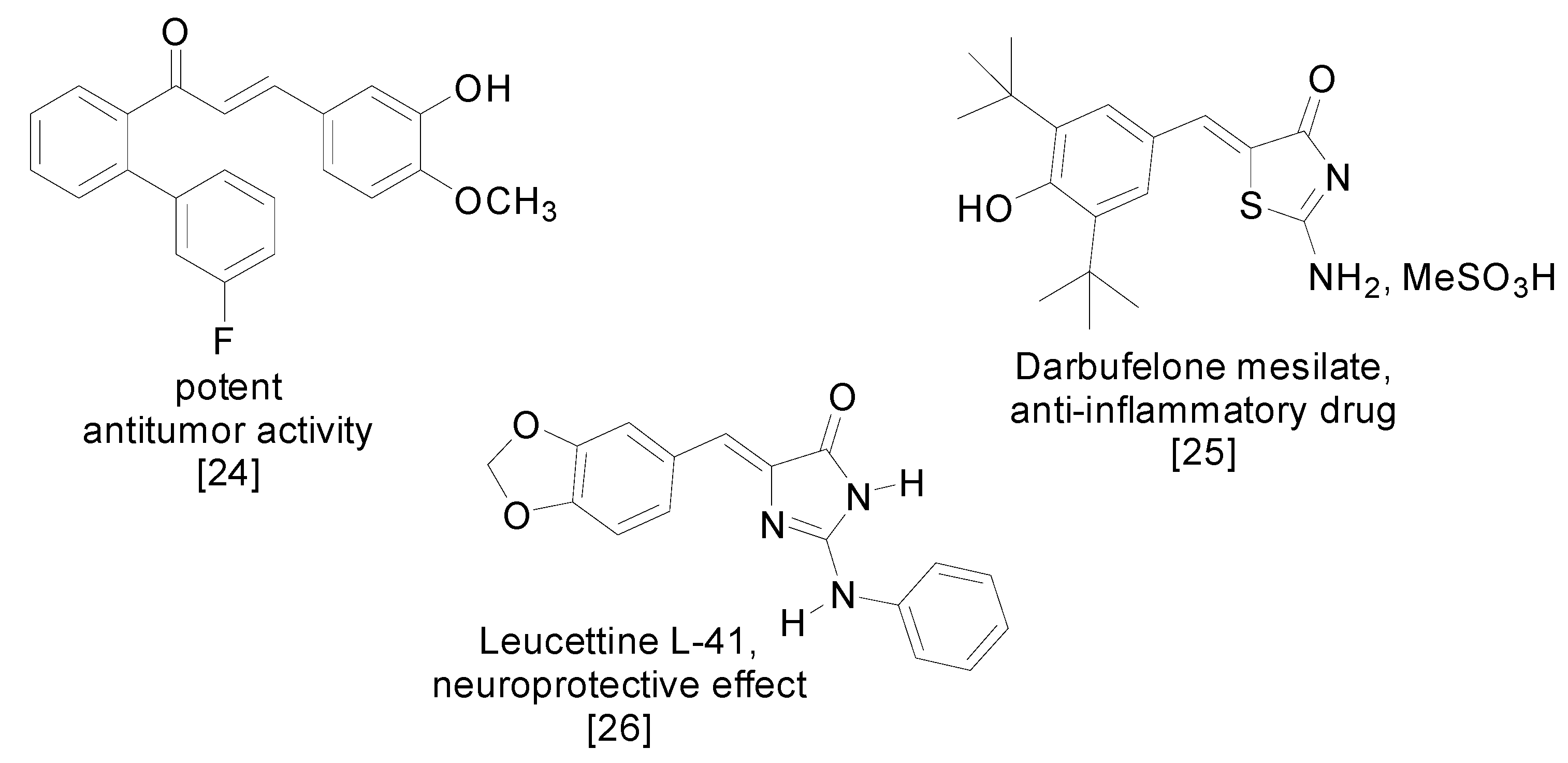
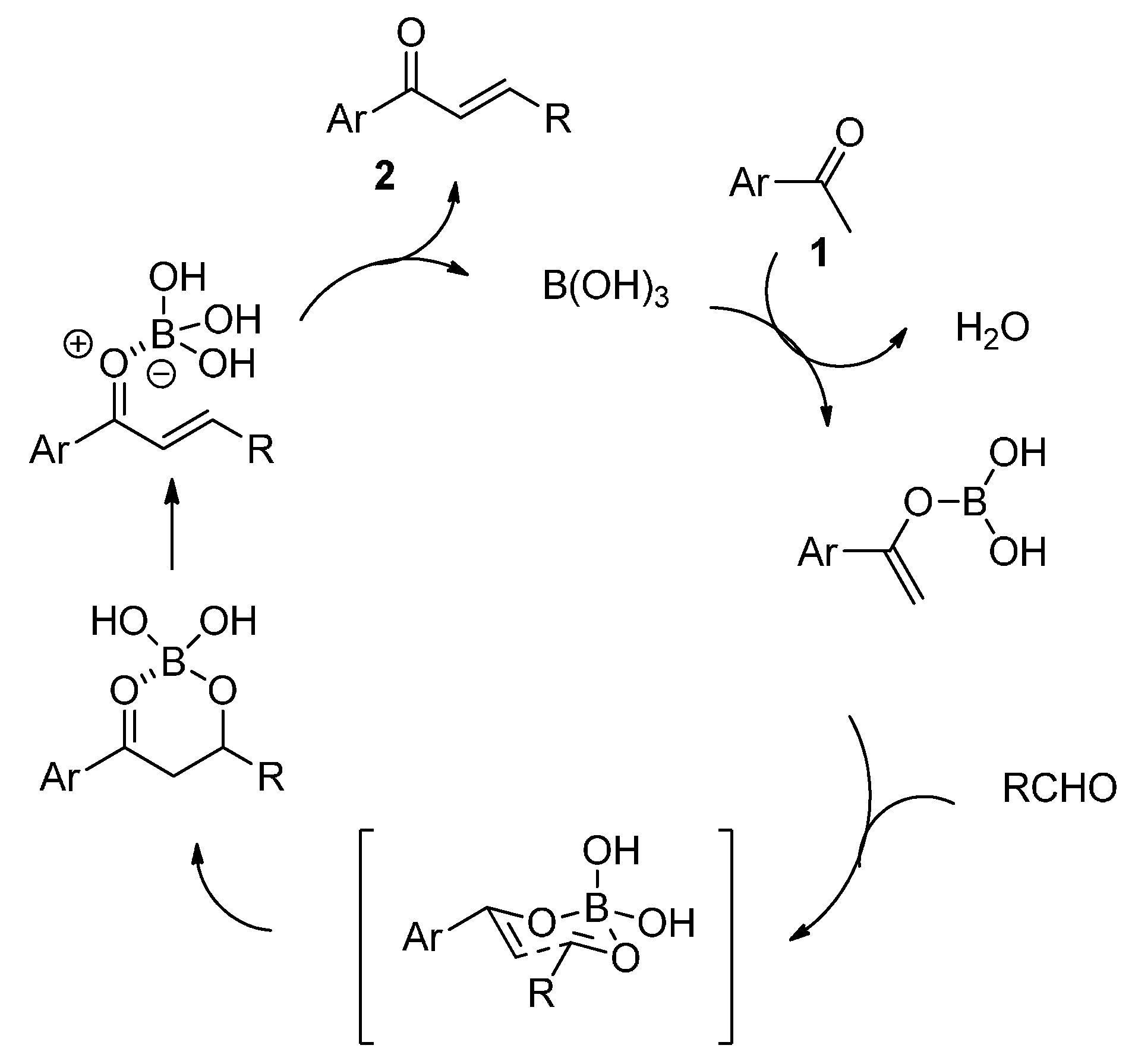
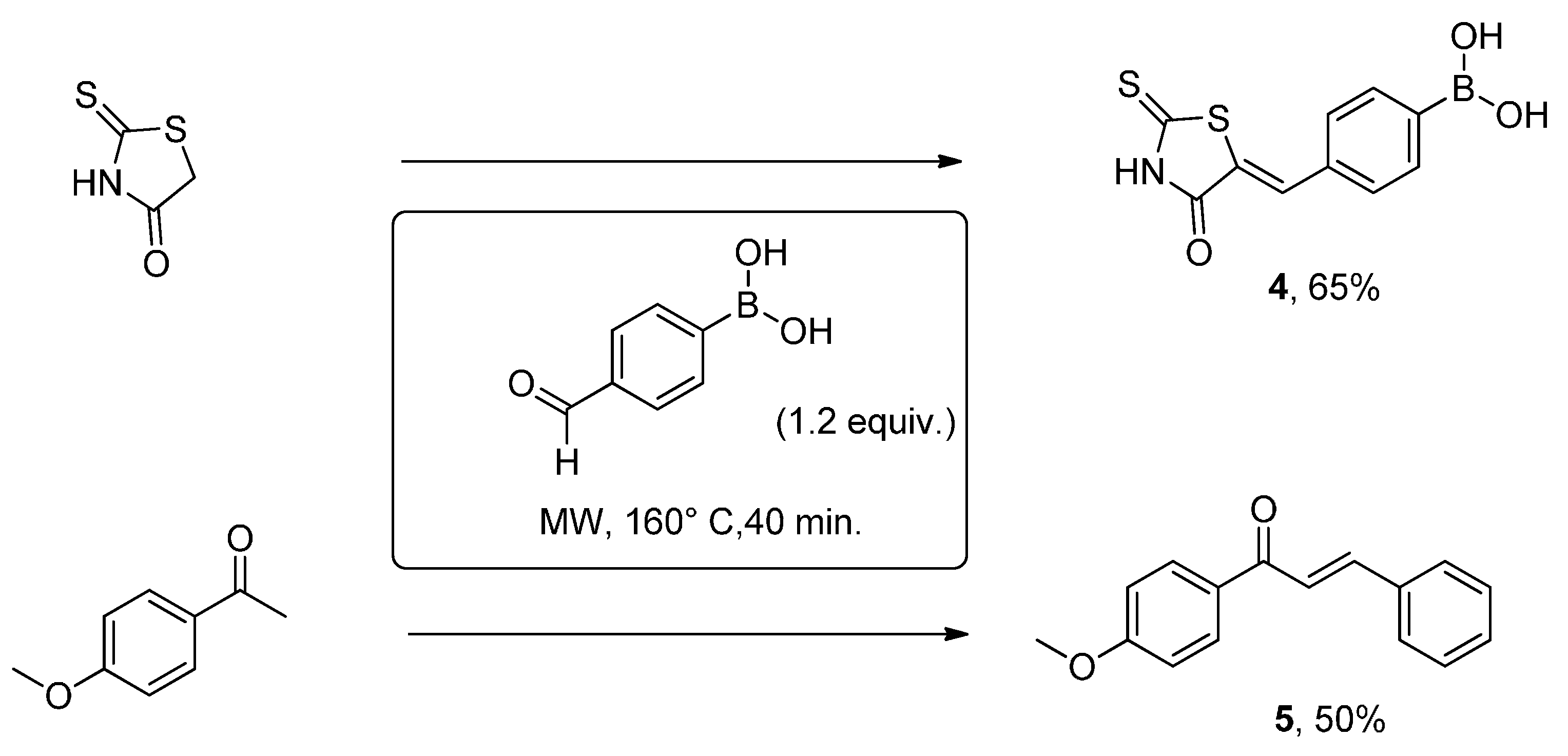
2. Results and Discussion


{kind=link}
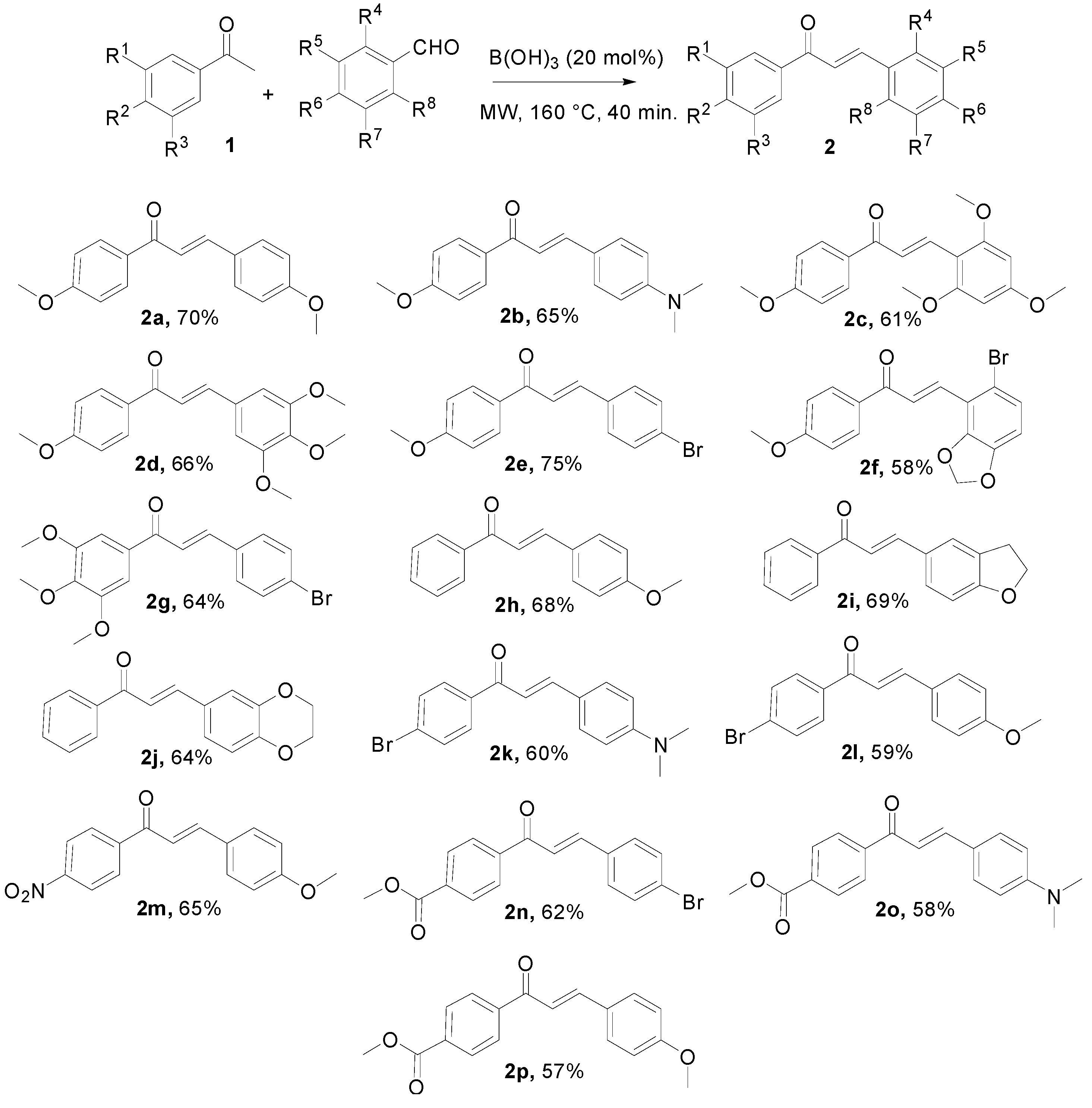{kind=link}
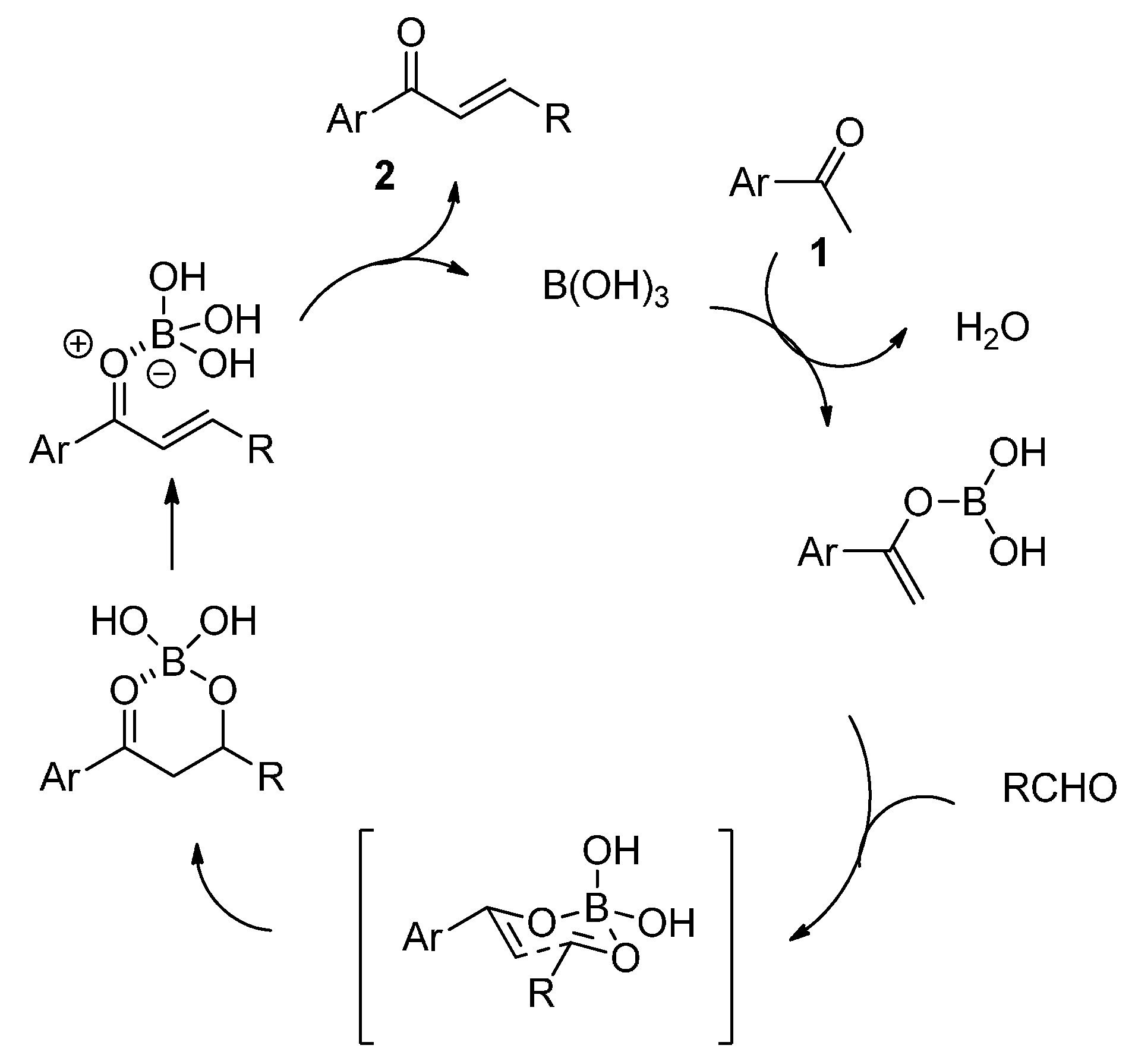{kind=link}
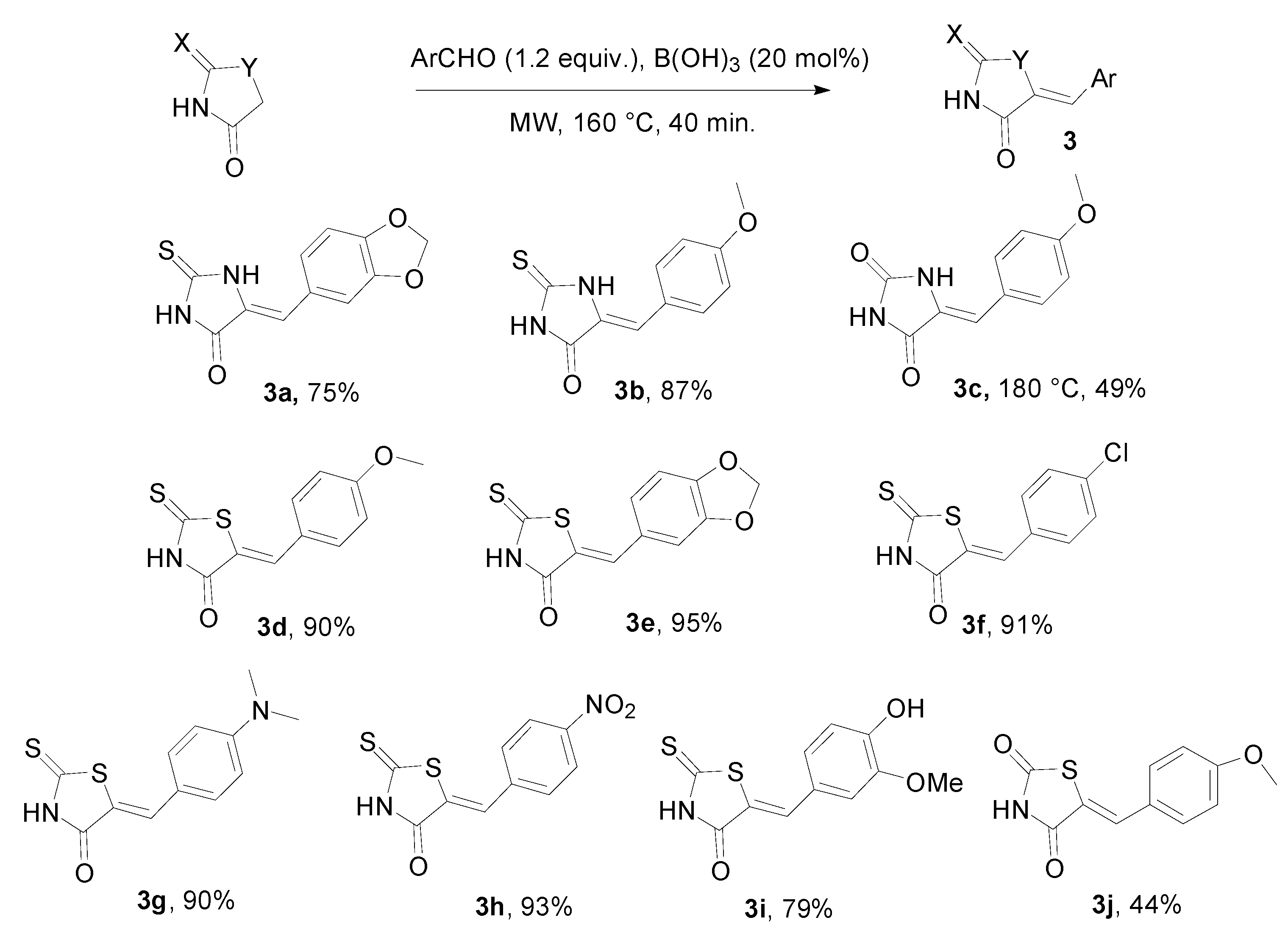{kind=link}
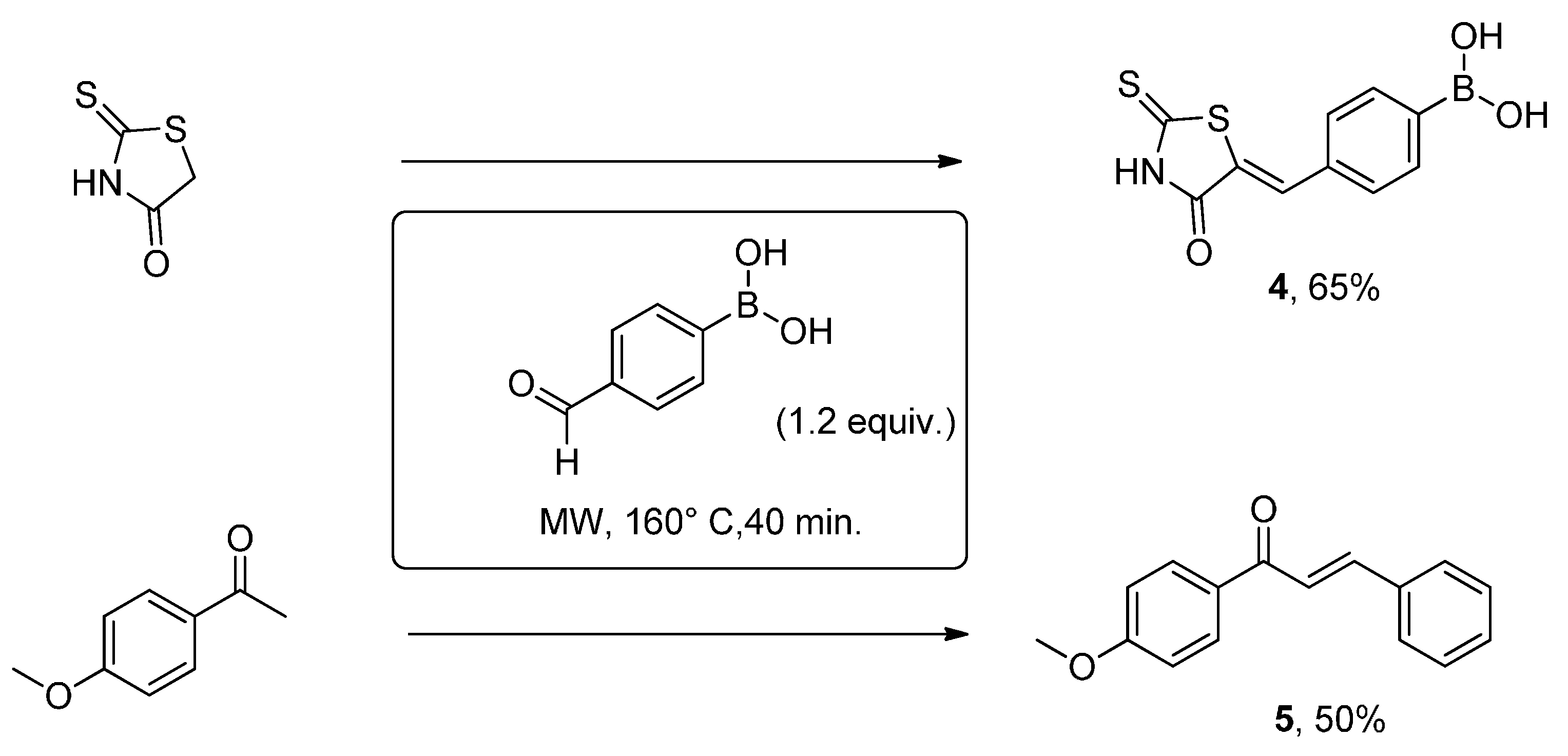{kind=link}
| Entry | Catalyst (mol %) | Conditions | 2a (Conv. %) c | 2a (yield %) d |
|---|---|---|---|---|
| 1 | B(OH)3 (50 mol %) | Toluene, reflux (4 h) | 5% | - |
| 2 | B(OH)3 (50 mol %) | MWI, 160 °C (20 min) b | 95% | - |
| 3 | PhB(OH)2 (50 mol %) | MWI, 160 °C (20 min) b | 100% | - |
| 4 | B(OH)3 (10 mol %) | MWI, 160 °C (40 min) b | 75% | - |
| 5 | B(OH)3 (20 mol %) | MWI, 160 °C (40 min) b | 93% | 70% |

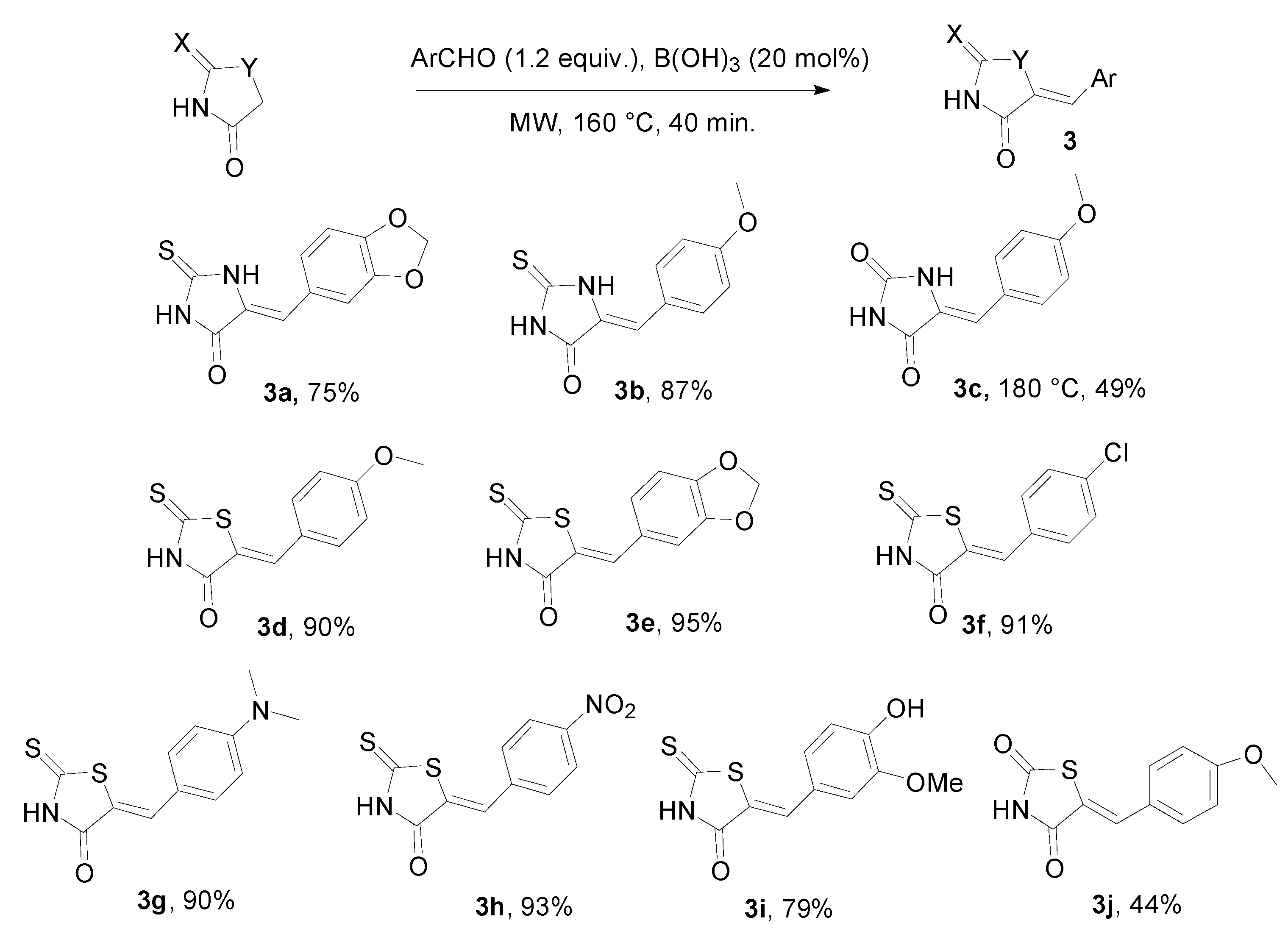
3. Experimental Section
3.1. General Procedure for the Condensation Reaction
3.2. Physical and Spectroscopic Data of Products
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Hall, D.G. Boronic Acids; Wiley VCH: Weinheim, Germany, 2005. [Google Scholar]
- Zheng, H.; Hall, D.G. Boronic acid catalysis: An atom-economical platform for direct activation and functionalization of carboxylic acids and alcohols. Aldrichim. Acta 2014, 47, 41–51. [Google Scholar]
- Houston, T.A.; Wilkinson, B.L.; Blanchfield, J.T. Boric acid catalyzed chemoselective esterification of α-hydroxycarboxylic acids. Org. Lett. 2004, 6, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Ishihara, K.; Yamamoto, H. N-Alkyl-4-boronopyridinium halides versus boric acid as catalysts for the esterification of α-hydroxycarboxylic acids. Org. Lett. 2005, 7, 5047–5050. [Google Scholar] [CrossRef] [PubMed]
- Levonis, S.M.; Bornaghi, L.F.; Houston, T.A. Selective monoesterification of malonic acid catalyzed by boric acid. Aust. J. Chem. 2007, 60, 821–823. [Google Scholar] [CrossRef]
- Tang, P. Boric acid catalyzed amide formation from carboxylic acids and amines: N-benzyl-4-phenylbutyramide. Org. Synth. 2005, 81, 262–272. [Google Scholar]
- Mylavarapu, R.K.; GCM, K.; Kolla, N.; Veeramalla, R.; Koilkonda, P.; Bhattacharya, A.; Bandichhor, R. Boric acid catalyzed amidation in the synthesis of active pharmaceutical ingredients. Org. Process. Res. Dev. 2007, 11, 1065–1068. [Google Scholar] [CrossRef]
- Maras, N.; Kocevar, M. Boric acid catalyzed direct condensation of carboxylic acids with benzene-1,2-diamine into benzimidazoles. Helv. Chim. Acta 2011, 94, 1860–1874. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Sorres, J.; Tran, M.Q.; Ermolenko, L.; Al-Mourabit, A. Boric acid: A highly efficient catalyst for transamidation of carboxamides with amines. Org. Lett. 2012, 14, 3202–3205. [Google Scholar] [CrossRef] [PubMed]
- Rostami, A.; Akradi, J.; Ahmad-Jangi, F. Boric acid as cost-effective and recyclable catalyst for trimethylsilyl protection and deprotection of alcohols and phenols. J. Braz. Chem. Soc. 2010, 21, 1587–1592. [Google Scholar] [CrossRef]
- Delbecq, P.; Celerier, J.P.; Lhommet, G. Decarboxylation of cyclic β-enaminoketoesters with boric acid. Tetrahedron Lett. 1990, 31, 4873–4874. [Google Scholar] [CrossRef]
- Gogoi, K.; Dewan, A.; Gogoi, A.; Borah, G.; Bora, U. Boric acid as highly efficient catalyst for the synthesis of phenols from arylboronic acids. Heteroat. Chem. 2014, 25, 127–130. [Google Scholar] [CrossRef]
- Chaudhuri, M.K.; Hussain, S.; Kantam, M.L.; Neelima, B. Boric acid: A novel and safe catalyst for aza-Michael reactions in water. Tetrahedron Lett. 2005, 46, 8329–8331. [Google Scholar] [CrossRef]
- Chaudhuri, M.K.; Hussain, S. Boric acid catalyzed thia-Michael reactions in water or alcohols. J. Mol. Catal. A Chem. 2007, 269, 214–217. [Google Scholar] [CrossRef]
- Meshram, M.; Rao, N.N.; Kumar, G.S. Boric acid-mediated mild and efficient Friedel-Crafts alkylation of indoles with nitro styrenes. Synth. Commun. 2010, 40, 3496–3500. [Google Scholar] [CrossRef]
- Tu, S.; Fang, F.; Miao, C.; Jiang, H.; Feng, Y.; Shi, D.; Wang, X. One-pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones using boric acid as catalyst. Tetrahedron Lett. 2003, 44, 6153–6155. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, M.Y.; Gao, S.T.; Ma, J.J.; Wang, C.; Liu, C. An efficient synthesis of 1,5-benzodiazepine derivatives catalyzed by boric acid. Chin. Chem. Lett. 2009, 20, 905–908. [Google Scholar] [CrossRef]
- Karimi-Jaberi, Z.; Keshavarzi, M. Efficient one-pot synthesis of 14-substituted-14H-dibenzo[a,j]xanthenes using boric acid under solvent-free conditions. Chin. Chem. Lett. 2010, 21, 547–549. [Google Scholar] [CrossRef]
- Ganguly, N.C.; Roy, S.; Mondal, P. Boric acid-catalyzed one-pot access to 7-aryl-benzopyrano[4,3-b]benzopyran-6,8-diones under aqueous micellar conditions. Synth. Commun. 2014, 44, 433–440. [Google Scholar] [CrossRef]
- Ducki, S. The developments of chalcones as promising anticancer agents. IDrugs 2007, 10, 42–46. [Google Scholar] [PubMed]
- Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: A mini review. Eur. J. Med. Chem. 2014, 85, 756–777. [Google Scholar] [CrossRef] [PubMed]
- Matos, M.J.; Vazquez-Rodriguez, S.; Uriarte, E.; Santana, L. Potential pharmaceutical uses of chalcones: A patent review. Expert Opin. Ther. Pat. 2015, 25, 351–366. [Google Scholar] [CrossRef] [PubMed]
- As example, see: Valdameri, G.; Gauthier, C.; Terreux, R.; Kachadourian, R.; Day, B.J.; Winnischofer, S.M.B.; Rocha, M.E.M.; Frachet, V.; Ronot, X.; Pietro, A.D.; et al. Investigation of chacones as selective inhibitors of the breast cancer resistance protein: Critical role of methoxylation in both inhibition potency and cytotoxicity. J. Med. Chem. 2012, 55, 3193–3200. [Google Scholar]
- Zhu, C.; Zuo, Y.; Wang, R.; Liang, B.; Yue, X.; Wen, G.; Shang, N.; Huang, L.; Chen, Y.; Du, J.; et al. Discovery of potent cytotoxic ortho-aryl chalcones as new scaffold targeting tubulin and mitosis with affinity-based fluorescence. J. Med. Chem. 2014, 57, 6364–6382. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zhou, W.; Li, Y.; Sun, Y.; Zhang, Y.; Ji, H.; Lai, Y. Darbufelone, a novel anti-inflammatory drug, induces growth inhibition of lung cancer cells both in vitro and in vivo. Cancer Chemother. Pharmacol. 2010, 66, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, Cocrystal structures, and neuroprotective properties of leuccettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 56, 9312–9330. [Google Scholar] [CrossRef] [PubMed]
- Mahrwald, R. Modern Aldol Reactions; Wiley VCH: Weinheim, Germany, 2004. [Google Scholar]
- The solubility of boric acid in water is higher than those of arylboronic acids allowing a more simple elimination (1 g in 18 mL according to Sigma-Aldrich information, Saint Louis, MI, USA).
- Offenhauer, R.D.; Nelsen, S.F. Aldehyde and ketone condensation reactions catalyzed by boric acid. J. Org. Chem. 1968, 33, 775–777. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Offenhauer, R.D. Condensation Reactions with Boric Acid. U.S. Patent 3,592,856 A, 13 July 1971. [Google Scholar]
- Deligny, M.; Carreaux, F.; Carboni, B. A concise synthesis of (+)-goniodiol using a catalytic hetero Diels-Alder/allylboration sequence. Synlett 2005, 1462–1464. [Google Scholar] [CrossRef]
- Carreaux, F.; Favre, A.; Carboni, B.; Rouaud, I.; Boustie, J. First synthesis of (+)-8-methoxygoniodiol and its analogue, 8-deoxygoniodiol, using a three component strategy. Tetrahedron Lett. 2006, 47, 4545–4548. [Google Scholar] [CrossRef]
- Favre, A.; Carreaux, F.; Deligny, M.; Carboni, B. Stereoselective synthesis of (+)-goniodiol, (+)-goniotriol, (−)-goniofupyrone and (+)-altholactone using a catalytic asymmetric hetero-Diels-Alder/allylboration approach. Eur. J. Org. Chem. 2008, 29, 4900–4907. [Google Scholar] [CrossRef]
- Touchet, S.; Carreaux, F.; Molander, G.A.; Carboni, B.; Bouillon, A. Iridium-catalyzed allylic amination route to a-aminoboronates: Illustration of the decisive role of boron substituents. Adv. Synth. Catal. 2011, 353, 3391–3396. [Google Scholar] [CrossRef] [PubMed]
- Hemelaere, R.; Carreaux, F.; Carboni, B. A diastereoselective route to trans-2-aryl-2,3-dihydrobenzofurans through sequential cross-metathesis/isomerization/allylboration reactions: Synthesis of bioactive neolignans. Eur. J. Org. Chem. 2015, 2015, 2470–2481. [Google Scholar] [CrossRef]
- As boric acid is coming from the ultimate degradation of phenylboronic acid by oxidation, it can be regarded as the most “green” reagent.
- Wu, X.-F.; Neumann, H.; Beller, M. Palladium-catalyzed oxidative carbonylative coupling reaction of arylboronic acids with styrenes to chalcones under mild aerobic conditions. Chem. Asian. J. 2012, 7, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Mendgen, T.; Steuer, C.; Klein, C.D. Privileged scaffolds or promiscuous binders: A comparative study on rhodanines and related heterocycles in medicinal chemistry. J. Med. Chem. 2012, 55, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Touchet, S.; Carreaux, F.; Carboni, B.; Bouillon, A.; Boucher, J.-L. Aminoboronic acids and esters: From synthetic challenges to the discovery of unique classes of enzyme inhibitors. Chem. Soc. Rev. 2011, 40, 3895–3914. [Google Scholar] [CrossRef] [PubMed]
- Syam, S.; Abdelwahab, S.I.; Al-Mamary, M.A.; Mohan, S. Synthesis of chalcones with anticancer activities. Molecules 2012, 17, 6179–6195. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Mohanakrishmann, D.; Shard, A.; Sharma, A.; Saima; Sinha, A.K.; Sahal, D. Stilbene-chalcone hybrids: Design, synthesis and evaluation as a new class of antimalarial scaffolds that trigger cell death through stage specific apoptosis. J. Med. Chem. 2012, 55, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S.; Rennison, D.; Woo, M.; Kendall, A.; Chabert, J.F.D.; McGown, A.T.; Lawrence, N.J. Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: Synthesis and biological evaluation of antivascular activity. Bioorg. Med. Chem. 2009, 17, 7698–7710. [Google Scholar] [CrossRef] [PubMed]
- Ashtekar, K.D.; Staples, R.J.; Borhan, B. Development of a formal catalytic asymmetric [4 + 2] addition of ethyl-2,3-butadienoate with acyclic enones. Org. Lett. 2011, 13, 5732–5735. [Google Scholar] [CrossRef] [PubMed]
- Salum, L.B.; Altei, W.F.; Chiaradia, L.D.; Cordeiro, M.N.S.; Canevarolo, R.R.; Melo, C.P.S.; Winter, E.; Mattei, B.; Daghestani, H.N.; Santos-Silva, M.C.; et al. Cytotoxic 3,4,5-trimethoxychalcones as mitotic arresters and cell migration inhibitors. Eur. J. Med. Chem. 2013, 63, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Khalilullah, H.; Khan, S.; Ahsan, M.J.; Ahmed, B. Synthesis of antihepatotoxic activity of 5-(2,3-dihydro-1,4-benzodioxane-6-yl)-3-substituted-phenyl-4,5-dihydro-1H-pyrazole derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 7251–7254. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Haratake, M.; Mori, H.; Nakayama, M. Novel chalcones as probes for in vivo imaging of b-amyloid plaques in Alzheimer’s brains. Bioorg. Med. Chem. 2007, 15, 6802–6809. [Google Scholar] [CrossRef] [PubMed]
- Debdab, M.; Carreaux, F.; Renault, S.; Soundarajan, M.; Fedorov, O.; Filippalopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; et al. Leucettines, a class of potent inhibitors of cdc2-like kinases and dual specificity, tyrosine phosphorylation regulated kinases derived from the marine sponge leucettamine B: Modulation of alternative pre-RNA splicing. J. Med. Chem. 2011, 54, 4172–4186. [Google Scholar] [CrossRef] [PubMed]
- Gosling, S.; Rollin, P.; Tatibouët, A. Thiohydantoins: Selective N- and S-functionalization for Liebeskind-Strogl reaction study. Synthesis 2011, 3649–3660. [Google Scholar] [CrossRef]
- Bourahla, K.; Derdour, A.; Rahmouni, M.; Carreaux, F.; Bazureau, J.P. A practical access to novel 2-amino-5-arylidene-1,3-thiazol-4(5H)-ones via sulfur/nitrogen displacement under solvent-free microwave irradiation. Tetrahedron Lett. 2007, 48, 5785–5789. [Google Scholar] [CrossRef]
- Sortino, M.; Delgado, P.; Juarez, S.; Quiroga, J.; Abonia, R.; Insuasty, B.; Nogueras, M.; Rodero, L.; Garibotto, F.M.; Enriz, R.D.; et al. Synthesis and antifungal activity of (Z)-5-arylidenerhodanines. Bioorg. Med. Chem. 2007, 15, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Clay, M.D.; Deyholos, M.K.; Vederas, J.C. Exploration of inhibitors for diaminopimelate aminotransferase. Bioorg. Med. Chem. 2010, 18, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Zidar, N.; Tomasic, T.; Sink, R.; Rupnik, V.; Kovac, A.; Turk, S.; Patin, D.; Blanot, D.; Contreras Martel, C.; Dessen, A.; et al. Discovery of novel 5-benzylidenerhodanine and 5-benzylidenethiazolidine-2,4-dione inhibitors of MurD Ligase. J. Med. Chem. 2010, 53, 6584–6594. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Knaak, C.; Ma, J.; Beharry, Z.M.; McInnes, C.; Wang, W.; Kraft, A.S.; Smith, C.D. Synthesis and evaluation of novel inhibitors of pim-1 and pim-2 protein kinases. J. Med. Chem. 2009, 52, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 2c–f, 2j–l, 2o–p, 3b,d,j are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brun, E.; Safer, A.; Carreaux, F.; Bourahla, K.; L'helgoua'ch, J.-M.; Bazureau, J.-P.; Villalgordo, J.M. Microwave-Assisted Condensation Reactions of Acetophenone Derivatives and Activated Methylene Compounds with Aldehydes Catalyzed by Boric Acid under Solvent-Free Conditions. Molecules 2015, 20, 11617-11631. https://doi.org/10.3390/molecules200611617
Brun E, Safer A, Carreaux F, Bourahla K, L'helgoua'ch J-M, Bazureau J-P, Villalgordo JM. Microwave-Assisted Condensation Reactions of Acetophenone Derivatives and Activated Methylene Compounds with Aldehydes Catalyzed by Boric Acid under Solvent-Free Conditions. Molecules. 2015; 20(6):11617-11631. https://doi.org/10.3390/molecules200611617
Chicago/Turabian StyleBrun, Elodie, Abdelmounaim Safer, François Carreaux, Khadidja Bourahla, Jean-Martial L'helgoua'ch, Jean-Pierre Bazureau, and Jose Manuel Villalgordo. 2015. "Microwave-Assisted Condensation Reactions of Acetophenone Derivatives and Activated Methylene Compounds with Aldehydes Catalyzed by Boric Acid under Solvent-Free Conditions" Molecules 20, no. 6: 11617-11631. https://doi.org/10.3390/molecules200611617