
Transcriptomic Analysis of Flower Blooming in Jasminum sambac through De Novo RNA Sequencing

Abstract
:1. Introduction
2. Results and Discussion
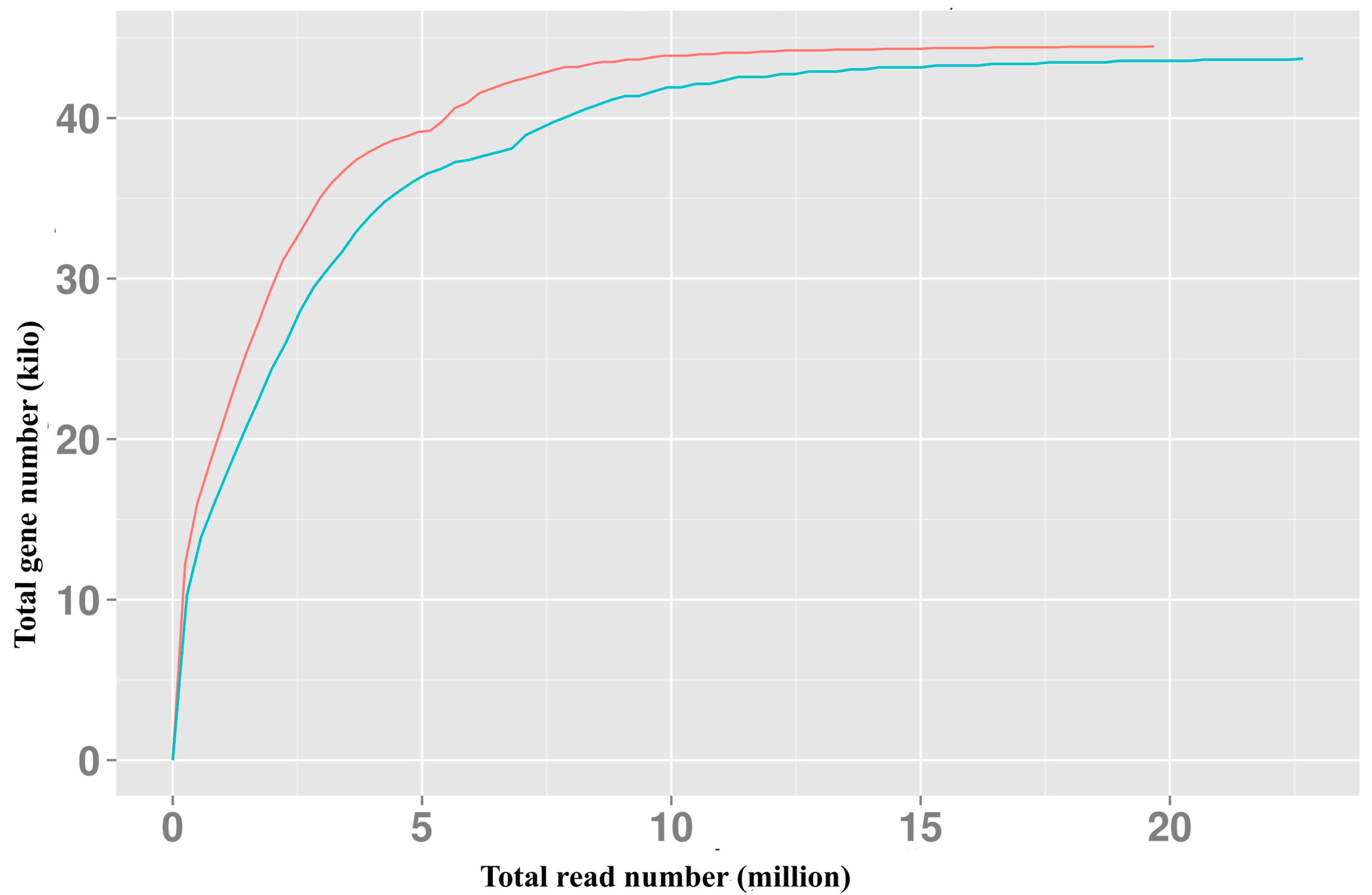
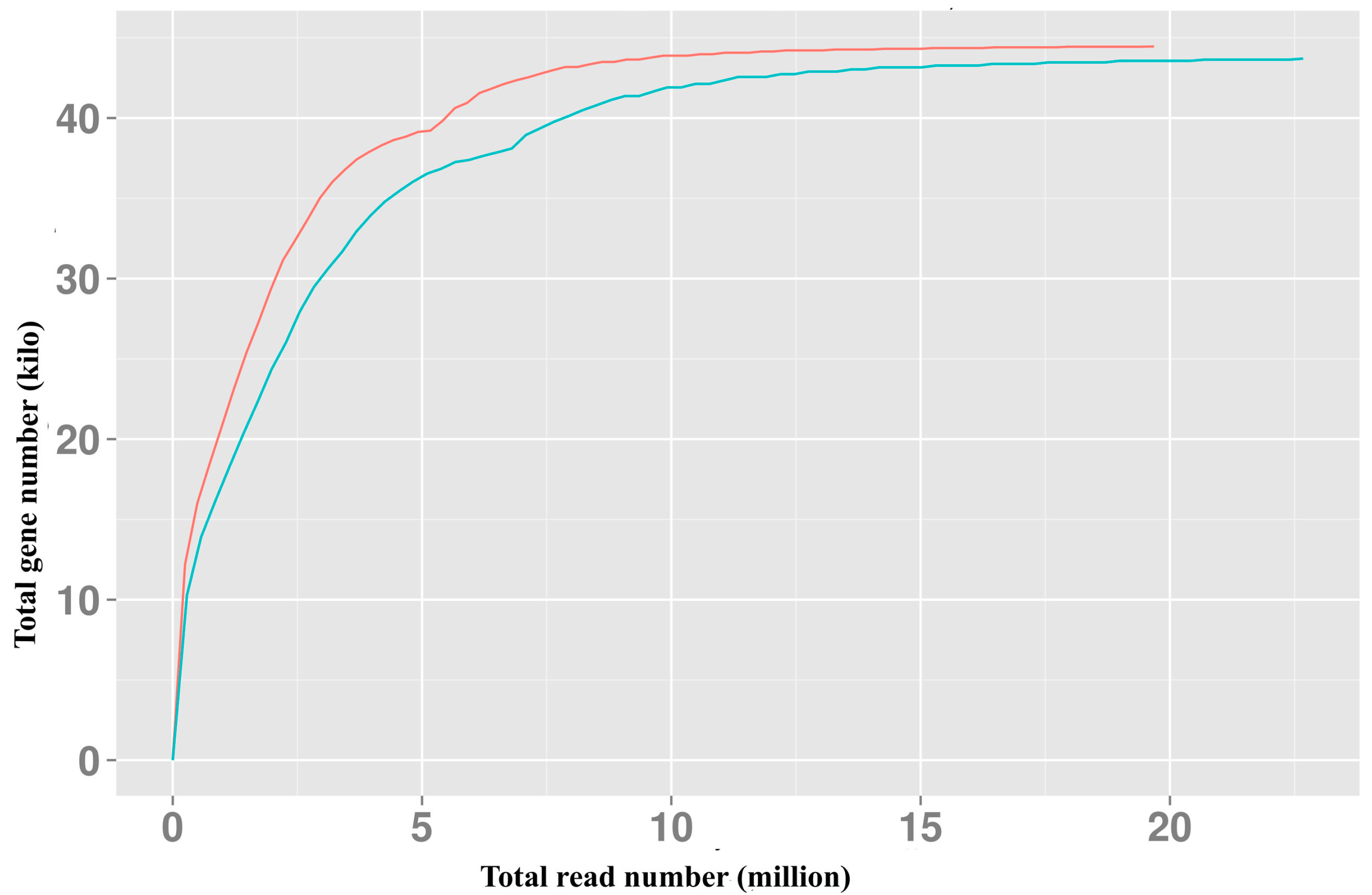
2.1. RNA-Seq and Assembly


{kind=link}
{kind=link}
{kind=link}
| Stage T1 | Stage T2 | |
|---|---|---|
| Read Number | 19,681,589 | 22,671,385 |
| Base Number | 3,975,056,593 | 4,569,637,577 |
| GC Content | 45.27% | 44.64% |
| Q30 | 91.36% | 90.51% |
| Length Range | Contig | Transcript | Unigene |
|---|---|---|---|
| 200–300 | 3,688,028 (98.93%) | 19,287 (17.01%) | 16,596 (33.34%) |
| 300–500 | 14,475 (0.39%) | 15,583 (13.74%) | 11,230 (22.56%) |
| 500–1000 | 11,261 (0.30%) | 19,231 (16.96%) | 8681 (17.44%) |
| 1000–2000 | 9133 (0.24%) | 29,990 (26.45%) | 7938 (15.95%) |
| 2000+ | 4944 (0.13%) | 29,303 (25.84%) | 5327 (10.70%) |
| Total Number | 3727,841 | 113,394 | 49,772 |
| Total Length (bp) | 184,557,344 | 161,608,199 | 42,105,276 |
| Mean Length (bp) | 49.51 | 1425.19 | 845.96 |
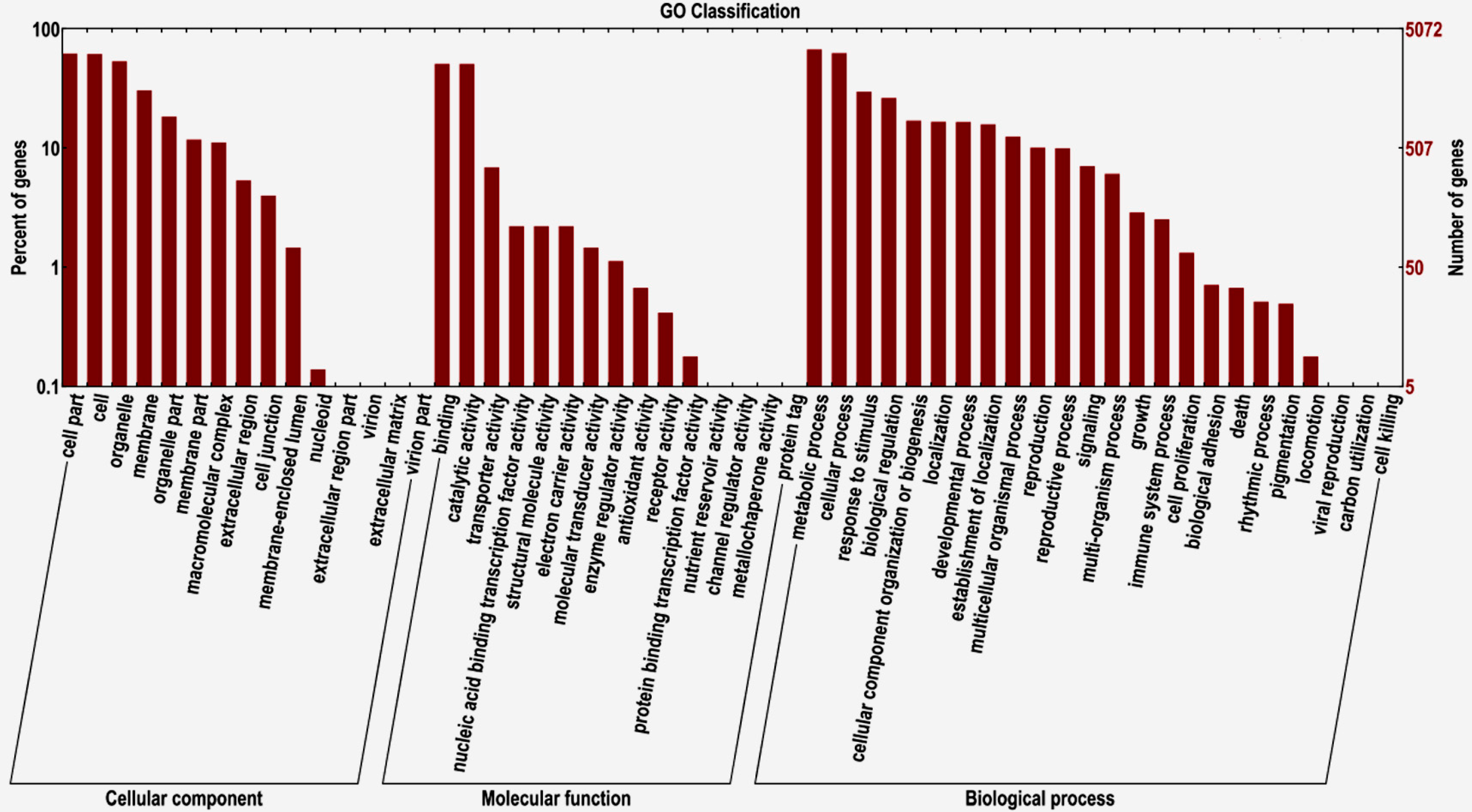
2.2. Transcriptome Functional Annotation
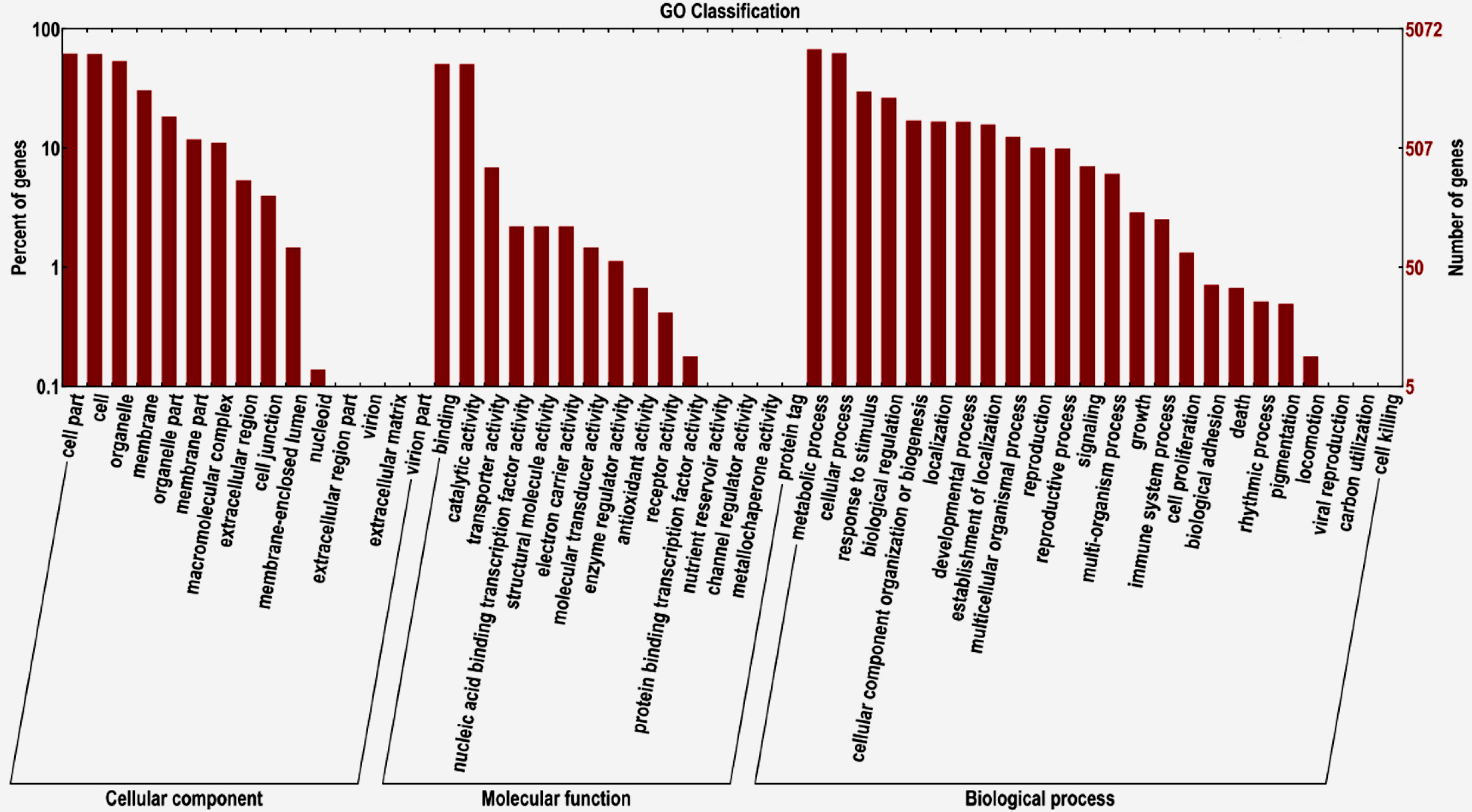
2.3. Differentially-Expressed Genes
2.4. Manual Identification of Flower Development and Floral Scent Genes
2.5. Discussion
| Category of Floral Scent | Aromatic Constituents | Relative Content | No. of Genes | Gene Annotation (Gene ID, Red Letters: Upregulated; Green Letters: Downregulated) |
|---|---|---|---|---|
| Terpenoids | 1. linalool | 20.1002 | 4 | nerolidol/linalool synthase 2 (c19704.graph_c0); |
| nerolidol/linalool synthase 1 (c10900.graph_c0); | ||||
| linalool/myrcene synthase (c47600.graph_c0); | ||||
| (E,E)-geranyllinalool synthase (c22241.graph_c0) | ||||
| 2. alpha-farnesene | 15.3245 | 43 | farnesyltransferase (c29468.graph_c0, c19601.graph_c0, c19799.graph_c0, c18836.graph_c0, c28581.graph_c0, c21749.graph_c1, c44587.graph_c0, c21456.graph_c0, c19099.graph_c0, c26508.graph_c1, c23779.graph_c0, c27539.graph_c0, c19768.graph_c0, c18630.graph_c, c17297.graph_c0, c19238.graph_c0, c13627.graph_c0, c20898.graph_c0, c27921.graph_c0, c26781.graph_c0, c12589.graph_c0, c29535.graph_c0, c8542.graph_c0, c1513.graph_c0, c9983.graph_c0, c28404.graph_c0, c19351.graph_c0, c25991.graph_c0, c27643.graph_c0, c13707.graph_c0, c23982.graph_c0, c29287.graph_c0, c10574.graph_c0, c21749.graph_c0, c40793.graph_c0, c24574.graph_c0); | |
| farnesyl pyrophosphate synthase (c27053.graph_c0); | ||||
| farnesyl-diphosphate farnesyltransferase (c30449.graph_c0, c15924.graph_c0, c36413.graph_c0, c30433.graph_c0, c26200.graph_c0, c30372.graph_c0); | ||||
| farnesyl diphosphate synthase (c22132.graph_c0) | ||||
| 3. Hexanoic acid, 3-hexenyl ester | 2.4303 | 0 | - | |
| 4. alpha-caryophyllene | 2.3001 | 1 | terpene synthase 3 (c29392.graph_c0) | |
| 5. Germacrene D | 1.5928 | 2 | bicyclogermacrene synthase (c35308.graph_c0); germacrene-D synthase (c15826.graph_c1) | |
| Benzenoids/Phenylpropanoids | 6. 3-Hexen-1-ol, benzoate | 30.4966 | 8 | o-succinylbenzoate-CoA ligase (c29960.graph_c0, c11812.graph_c0, c32591.graph_c0, c28597.graph_c0, c35614.graph_c0) |
| hexaprenyldihydroxybenzoate methyltransferase (c24032.graph_c0) | ||||
| (Z)-3-hexen-1-ol acetyltransferase (c30458.graph_c0, c30940.graph_c0) | ||||
| 7. Acetic acid, phenylmenthyl ester | 29.4231 | 8 | phenylalanine ammonia-lyase (c15788.graph_c0, c30394.graph_c0, c16474.graph_c0, c15481.graph_c0, c11285.graph_c0); | |
| phenylacetaldehyde reductase (c30130.graph_c0, c29940.graph_c0, | ||||
| c5943.graph_c0); | ||||
| 8. Methyl salicylate | 8.2246 | 8 | methyl salicylate esterase (c30688.graph_c0, c3125.graph_c0, c29061.graph_c0, c17812.graph_c0, c31454.graph_c0, c11814.graph_c0, c30703.graph_c0); | |
| Salicylate O-methyltransferase (c32548.graph_c0); | ||||
| 9. Benzyl benzoate | 7.8029 | 3 | phenylcoumaran benzylic ether reductase (c22406.graph_c0, c28180.graph_c0, c9894.graph_c0); | |
| 10. Indole | 6.5240 | 3 | Indole-3-acetic acid-amido synthetase (c11944.graph_c0, c25369.graph_c0, c25323.graph_c0) | |
| 11. Benzoic acid, cyclohexyl ester | 5.2911 | 0 | - | |
| 12. Salicylic acid, 3-hexenyl ester | 2.4519 | 1 | - | |
| 13. Benzoic acid, 2-hydroxy-, phenyl | 2.0409 | 0 | - | |
| 14. Benzoic acid, methyl ester | 1.7692 | 0 | - | |
| 15. Benzyl alcohol | 1.5769 | 7 | benzyl alcohol O-benzoyltransferase (c17166.graph_c0, c8299.graph_c0, c26169.graph_c0, c30012.graph_c0); benzyl alcohol benzoyl transferase (c18321.graph_c0, c30940.graph_c0, c24915.graph_c0) |
3. Experimental Section
3.1. Plant Material
3.2. RNA Isolation, Library Construction and Sequencing
3.3. Sequence Cleaning, Assembly and Contig Annotation
3.4. Expression Annotation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Edris, A.E.; Chizzola, R.; Franz, C. Isolation and characterization of the volatile aroma compounds from the concrete headspace and the absolute of Jasminum sambac (L.) Ait. (Oleaceae) flowers grown in Egypt. Eur. Food Res. Technol. 2008, 226, 621–626. [Google Scholar] [CrossRef]
- Liu, J.J.; Zhou, S.Y.; Si, H.Q.; Pang, X.L.; Guo, G.Y. Comparison analysis of aromatic constituents between sunny Jasmine and rainy Jasmine under optimal flowering condition. Southwest China J. Agric. Sci. 2011, 24, 722–727. [Google Scholar]
- Braun, N.A.; Sim, S. Jasminum sambac flower absolutes from India and China-geographic variations. Nat. Prod. Commun. 2012, 7, 645–650. [Google Scholar] [PubMed]
- Chen, Y.P.; Miao, Z.H.; Huang, Y.P.; Zeng, J. Antiarrhythmic effect of aqueous extract of radix jasmini sambac. Chin. Remedies Clin. 2004, 4, 844–846. [Google Scholar]
- Wang, M.; Jiang, Y.L.; Kuang, X.C.; Qin, J.; Ou, B.N.; Li, H.D. Effects of jasmine and jasmine tea extracts on functions of mouse immune cells. Chin. J. Pathophysiol. 2011, 27, 1428–1430. [Google Scholar]
- Sengar, N.; Joshi, A.; Prasad, S.K.; Hemalatha, S. Anti-inflammatory, analgesic and anti-pyretic activities of standardized root extract of Jasminum sambac. J. Ethnopharmacol. 2014, 160, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.T.; Wang, D.L.; Gu, D.Z. Effects of different growth regulators and soil substrates on the rooting of Jasminum sambac ation cuttings. J. Anhui Agric. Sci. 2009, 37, 15817–15818. [Google Scholar]
- Huang, C.M.; Jiang, W.; Wu, J.M.; Yang, L. Floral bud formation and endogenous hormone changes of Jasminum sambac L. With NAA or PP333 treatments. Acta Bot. Boreali-Occidentalia Sin. 2009, 29, 742–748. [Google Scholar]
- He, L.S.; Xia, B.; Meng, X.J.; Wang, C.Y.; Peng, F.; Wang, R. Physiological and biochemical responses of Jasminum sambac L. under natural temperature reduction. J. Nanjing Agric. Univ. 2010, 33, 28–32. [Google Scholar]
- Sun, J.; Chen, G.X.; Ye, N.X.; Lü, S.H.; Liu, Z.Q.; Huang, W.; Lin, Z.D. Cloning and expression analysis of deoxyoxylulose-5-phosphate synthase gene related to aroma from Jasminum sambac and isolation of its promoter. Acta Hortic. Sin. 2014, 41, 1236–1244. [Google Scholar]
- Ou, X.F. Cloning and Analysis on HPL and GDS of Jasminum sambac. Master’s Thesis, Fujian Agriculture and Forestry University, Fuzhou, China, 2012. [Google Scholar]
- Causier, B.; Schwarz-Sommer, Z.; Davies, B. Floral organ identity: 20 years of ABCs. Semin. Cell Dev. Biol. 2010, 21, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.N.; Wei, Y.Z.; Shen, J.Y.; Lai, B.; Huang, X.M.; Ding, F.; Su, Z.X.; Chen, H.B. Transcriptomic analysis of floral initiation in litchi (Litchi chinensis Sonn.) based on de novo RNA sequencing. Plant Cell Rep. 2014, 33, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhu, L.; Xu, L.; Pan, C.; Liu, Y. Comparative transcriptomic analysis of the regulation of flowering in temperate and tropical lotus (Nelumbo nucifera) by RNA-Seq. Ann. Appl. Biol. 2014, 165, 73–95. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, H.; Wan, P.; Kuang, T.; He, Y. Genome-wide transcriptome analysis of gametophyte development in Physcomitrella patens. BMC Plant Biol. 2011, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.; Nayak, S.; May, G.; Jackson, S. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009, 27, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Zhao, L.; Larson-Rabin, Z.; Li, D.Z.; Guo, Z.H. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS ONE 2012, 7, e42082. [Google Scholar] [CrossRef] [PubMed]
- Mizzotti, C.; Galliani, B.M.; Masiero, S. The backstage of the ABC model: The Antirrhinum majus contribution. Plant Biosyst. 2014, 148, 176–186. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, H.; Chen, M.; Jian, H.; Baudino, S.; Caissard, J.C.; Bendahmaned, M.; Li, S.; Zhang, T.; Zhou, N.; et al. Transcriptome and gene expression analysis during flower blooming in Rosa chinensis “Pallida”. Gene 2014, 540, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.D.; Lieber, E.R.; Heftmann, E. Time Course of Steroid Biosynthesis and Metabolism in Haplopappus heterophyllus. Plant Physiol. 1967, 42, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Geuns, J.M.C. Steroid hormones and plant growth and development. Phytochemistry 1978, 17, 1–14. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Z.H. Progress of hormone regulation on flower bud differentiation of fruit trees. Nonwood Forest Res. 2009, 27, 141–146. [Google Scholar]
- Broderick, S.R.; Wijeratne, S.; Wijeratn, A.J.; Chapin, L.J.; Meulia, T.; Jones, M.L. RNA-sequencing reveals early, dynamic transcriptome changes in the corollas of pollinated petunias. BMC Plant Biol. 2014, 14, 307. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Trotochaud, A.E.; Clark, S.E. The Arabidopsis CLAVATA2 gene encodes a receptor-like protein required for the stability of the CLAVATA1 receptor-like kinase. Plant Cell 1999, 11, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.; Mariconti, L.; Rosignol, P.; Perennes, C.; Cella, R.; Bergounioux, C. Two E2F sites in the Arabidopsis MCM3 promoter have different roles in cell cycle activation and meristematic expression. J. Biol. Chem. 2002, 277, 32978–32984. [Google Scholar] [CrossRef] [PubMed]
- Shultz, R.W.; Lee, T.J.; Allen, G.C.; Thompson, W.F.; Hanley-Bowdoin, L. Dynamic localization of the DNA replication proteins MCM5 and MCM7 in plants. Plant Physiol. 2009, 150, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Hauser, B.A.; He, J.Q.; Park, S.O.; Gasser, C.S. TSO1 is a novel protein that modulates cytokinesis and cell expansion in Arabidopsis. Development 2000, 127, 2219–2226. [Google Scholar] [PubMed]
- Fang, S.C.; Fernandez, D.E. Effect of regulated overexpression of the MADS domain factor AGL15 on flower senescence and fruit maturation. Plant Physiol. 2002, 130, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Q.; Habegger, L.; Noisa, P.; Szekely, A.; Qiu, C.; Hutchison, S.; Raha, D.; Egholm, M.; Lin, H.; Weissman, S.; et al. Dynamic transcriptomes during neural differentiation of human embryonic stem cells revealed by short, long, and paired-end sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 5254–5259. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: ftp://ftp.ncbi.nih.gov/blast/db/ (accessed on 5 April 2015).
- Uniprot. Available online: http://www.uniprot.org/ (accessed on 5 April 2015).
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: http://www.genome.jp/kegg/ (accessed on 5 April 2015).
- COGs. Available online: http://www.ncbi.nlm.nih.gov/COG/ (accessed on 5 April 2015).
- Gene Ontology Consortium. Available online: http://geneontology.org/ (accessed on 5 April 2015).
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A. On the interpretation of χ2 from contingency tables, and the calculation of P. J. R. Stat. Soc. 1922, 85, 87–94. [Google Scholar] [CrossRef]
- Sample Availability: Sequence data of J. sambac are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-H.; Zhang, W.; Li, Y. Transcriptomic Analysis of Flower Blooming in Jasminum sambac through De Novo RNA Sequencing. Molecules 2015, 20, 10734-10747. https://doi.org/10.3390/molecules200610734
Li Y-H, Zhang W, Li Y. Transcriptomic Analysis of Flower Blooming in Jasminum sambac through De Novo RNA Sequencing. Molecules. 2015; 20(6):10734-10747. https://doi.org/10.3390/molecules200610734
Chicago/Turabian StyleLi, Yong-Hua, Wei Zhang, and Yong Li. 2015. "Transcriptomic Analysis of Flower Blooming in Jasminum sambac through De Novo RNA Sequencing" Molecules 20, no. 6: 10734-10747. https://doi.org/10.3390/molecules200610734