
Computational Studies of Benzoxazinone Derivatives as Antiviral Agents against Herpes Virus Type 1 Protease

Abstract
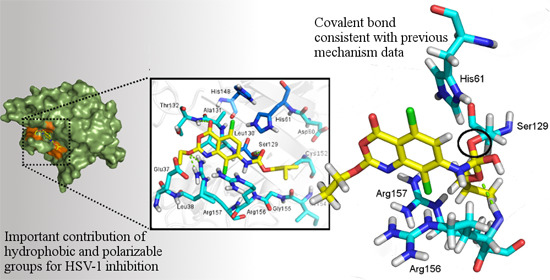
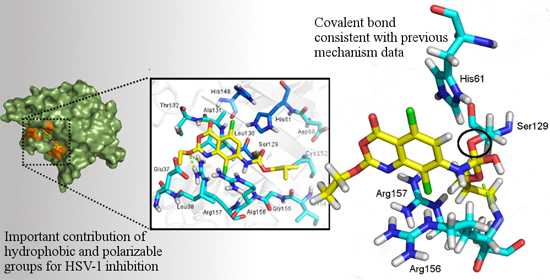
:
1. Introduction
2. Results and Discussion
2.1. Structure-Activity Relationship

{kind=link}
{kind=link}
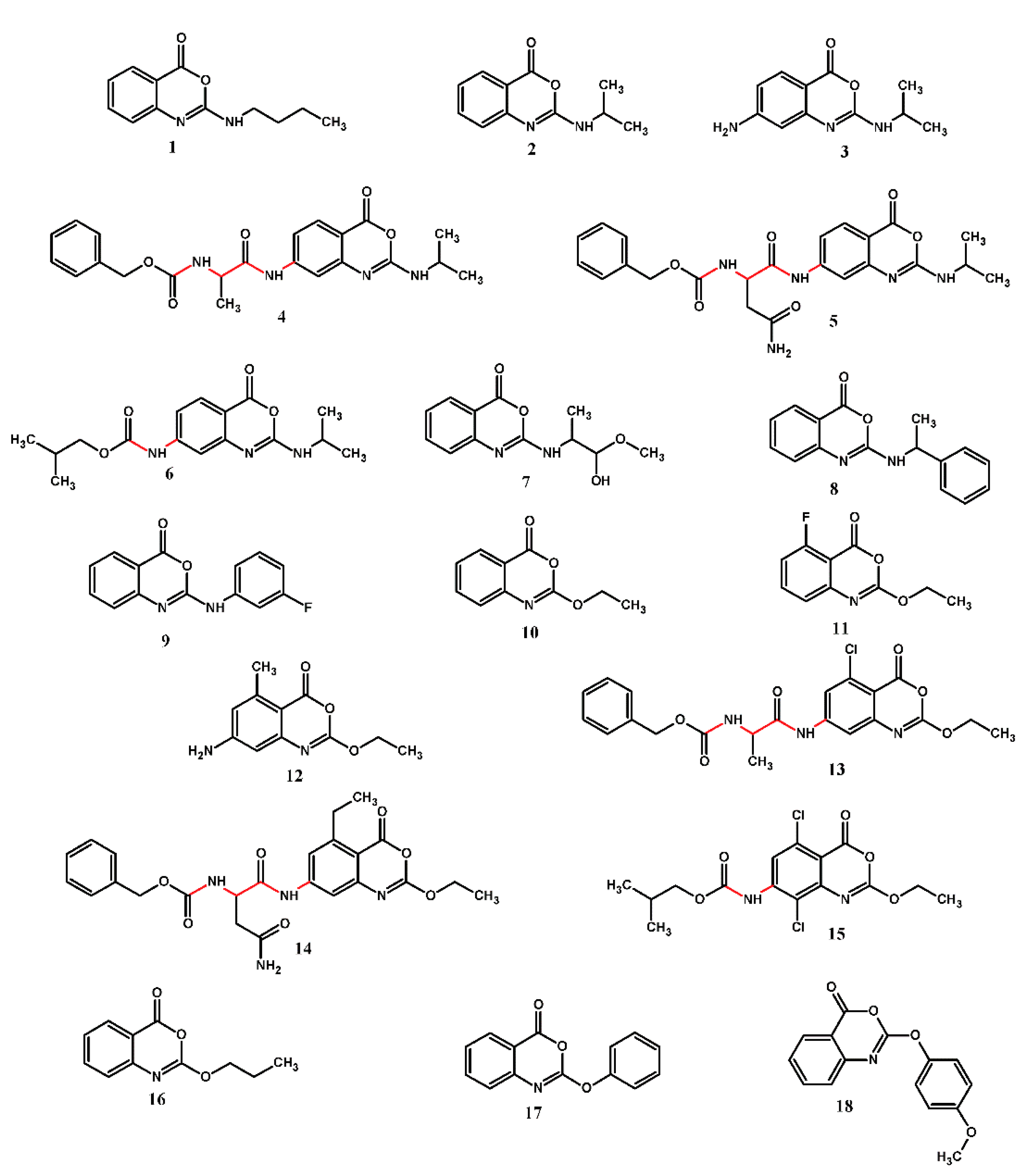{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
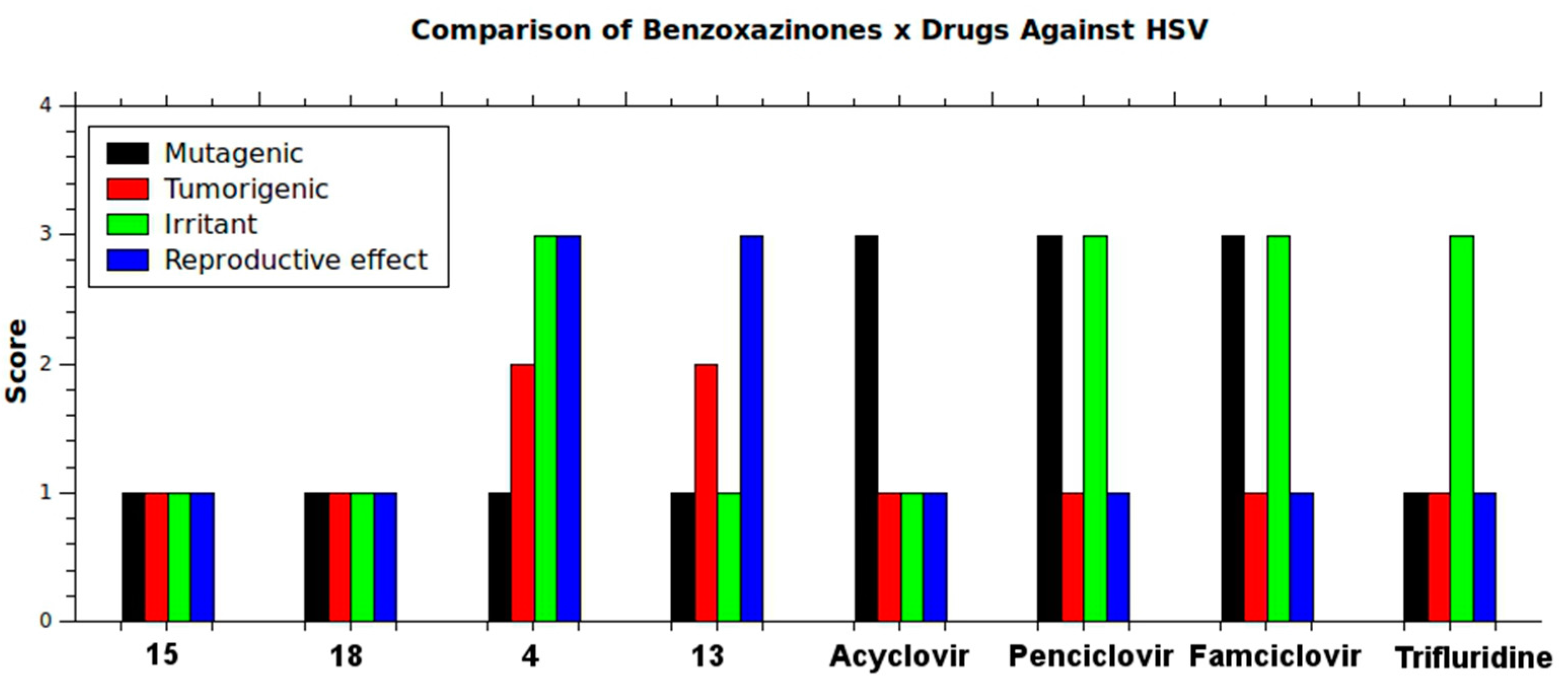{kind=link}
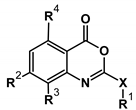
| Compound | X | R1 | R2 | R3 | R4 | IC50 (µM) | MW (amu) | MV (Å3) | cLog P |
|---|---|---|---|---|---|---|---|---|---|
| 1 | N | Bu | H | H | H | 300.0 | 218.26 | 228.46 | 3.10 |
| 2 | N | iPr | H | H | H | 25.0 | 204.23 | 210.10 | 2.52 |
| 3 | N | iPr | NH2 | H | H | 55.0 | 219.24 | 220.39 | 1.71 |
| 4 | N | iPr | Cbz-Ala-NH | H | H | 5.0 | 424.46 | 424.42 | 3.09 |
| 5 | N | iPr | Cbz-Asn-NH | H | H | 15.0 | 467.48 | 453.58 | 1.66 |
| 6 | N | iPr | iBuCOONH | H | H | 50.0 | 319.36 | 325.45 | 3.23 |
| 7 | N | MeO2CCMeH | H | H | H | 25.0 | 248.24 | 240.08 | 1.88 |
| 8 | N | (R)-PhCMeH | H | H | H | 60.0 | 266.30 | 275.00 | 3.91 |
| 9 | N | 3-F-Ph | H | H | H | >300 | 256.24 | 242.65 | 3.68 |
| 10 | O | Et | H | H | H | 25.0 | 191.19 | 188.12 | 2.58 |
| 11 | O | Et | H | H | F | 75.0 | 209.18 | 192.67 | 2.73 |
| 12 | O | Et | NH2 | H | Me | 15.0 | 220.23 | 215.78 | 2.26 |
| 13 | O | Et | Cbz-Ala-NH | H | Cl | 7.0 | 445.86 | 415.55 | 3.71 |
| 14 | O | Et | Cbz-Asn-NH | H | Et | 20.0 | 482.49 | 469.31 | 2.62 |
| 15 | O | Et | iBuCOONH | Cl | Cl | 1.5 | 375.21 | 328.83 | 4.41 |
| 16 | O | Allyl | H | H | H | 15.0 | 203.20 | 202.15 | 2.93 |
| 17 | O | Ph | H | H | H | 10.0 | 239.23 | 234.89 | 3.90 |
| 18 | O | 4-OCH3-Ph | H | H | H | 2.5 | 269.26 | 261.92 | 3.78 |
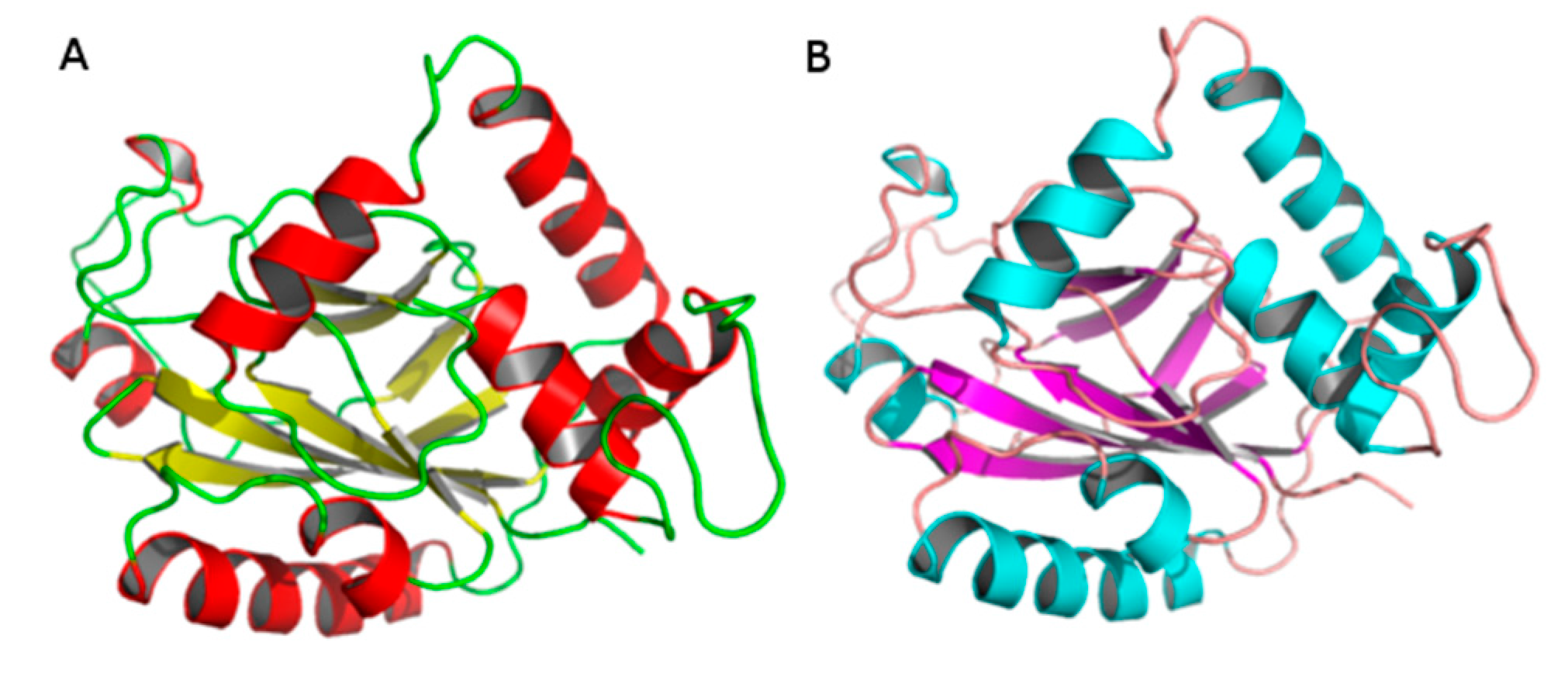
2.2. Comparative Modeling
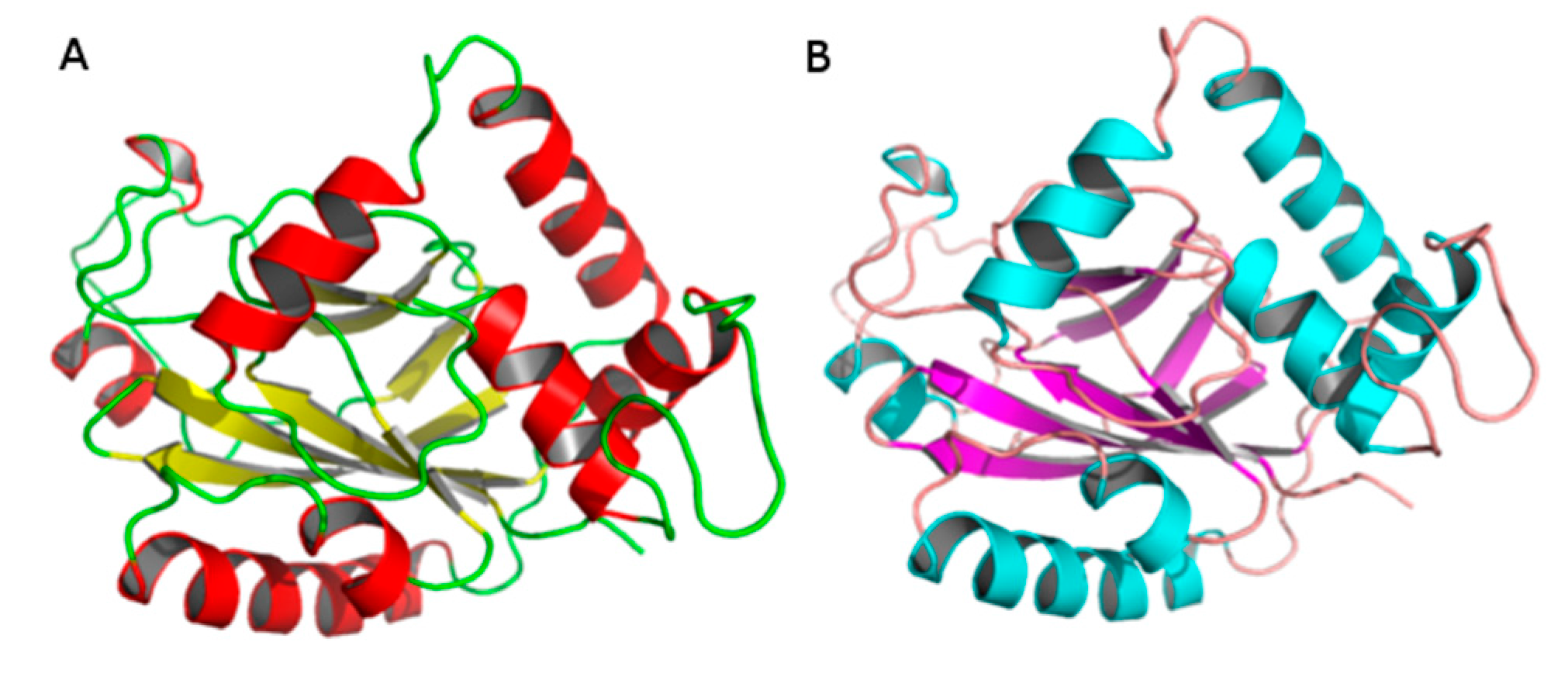
| Crystal Structure | Modeller | SWISS-MODEL |
|---|---|---|
| Ramachandran plot a (%) | 90.80 | 83.7 |
| Ramachandran plot b (%) | 0 | 1.5 |
| Verify 3D c (%) | 87.93 | 81.3 |
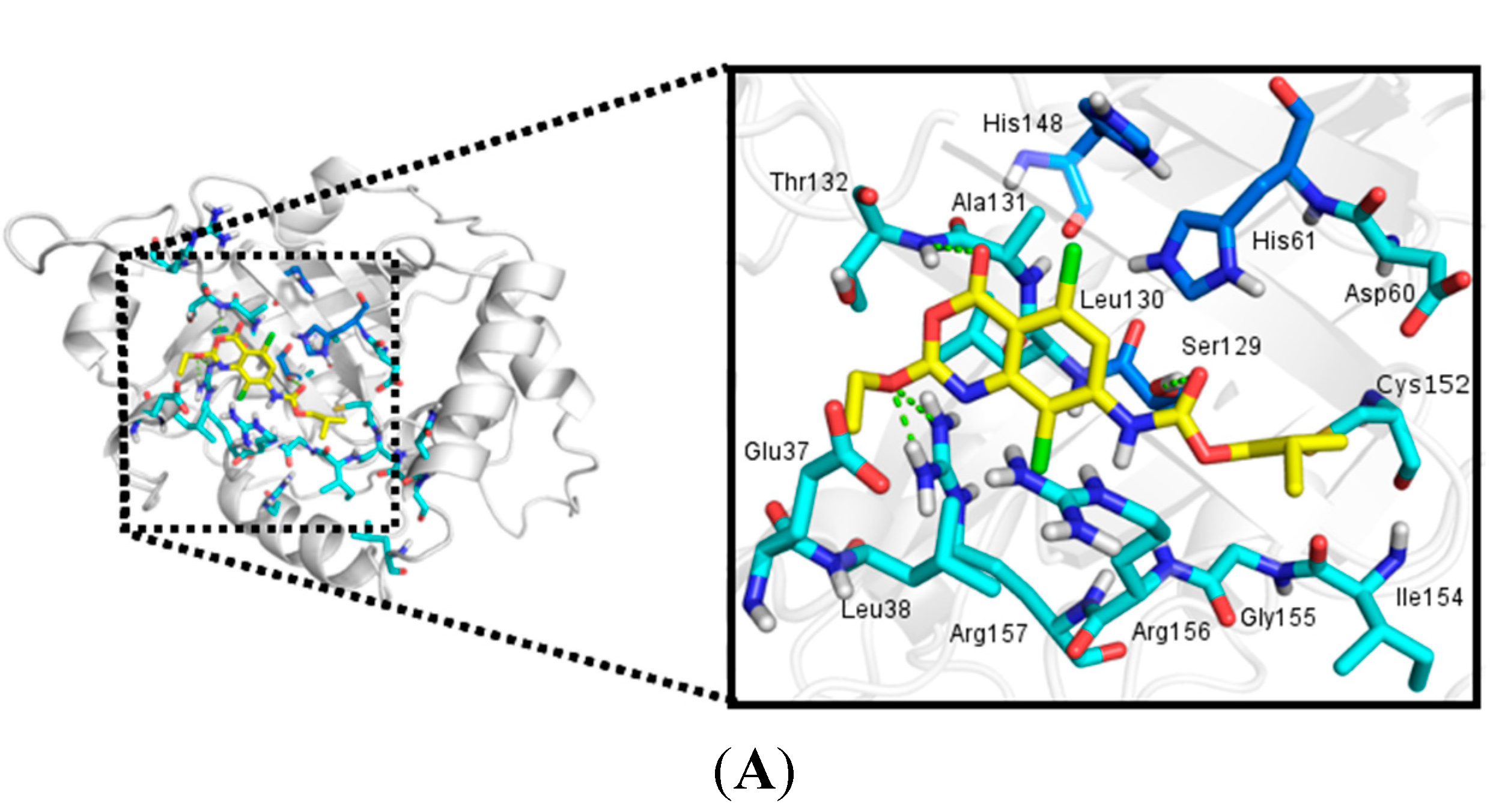
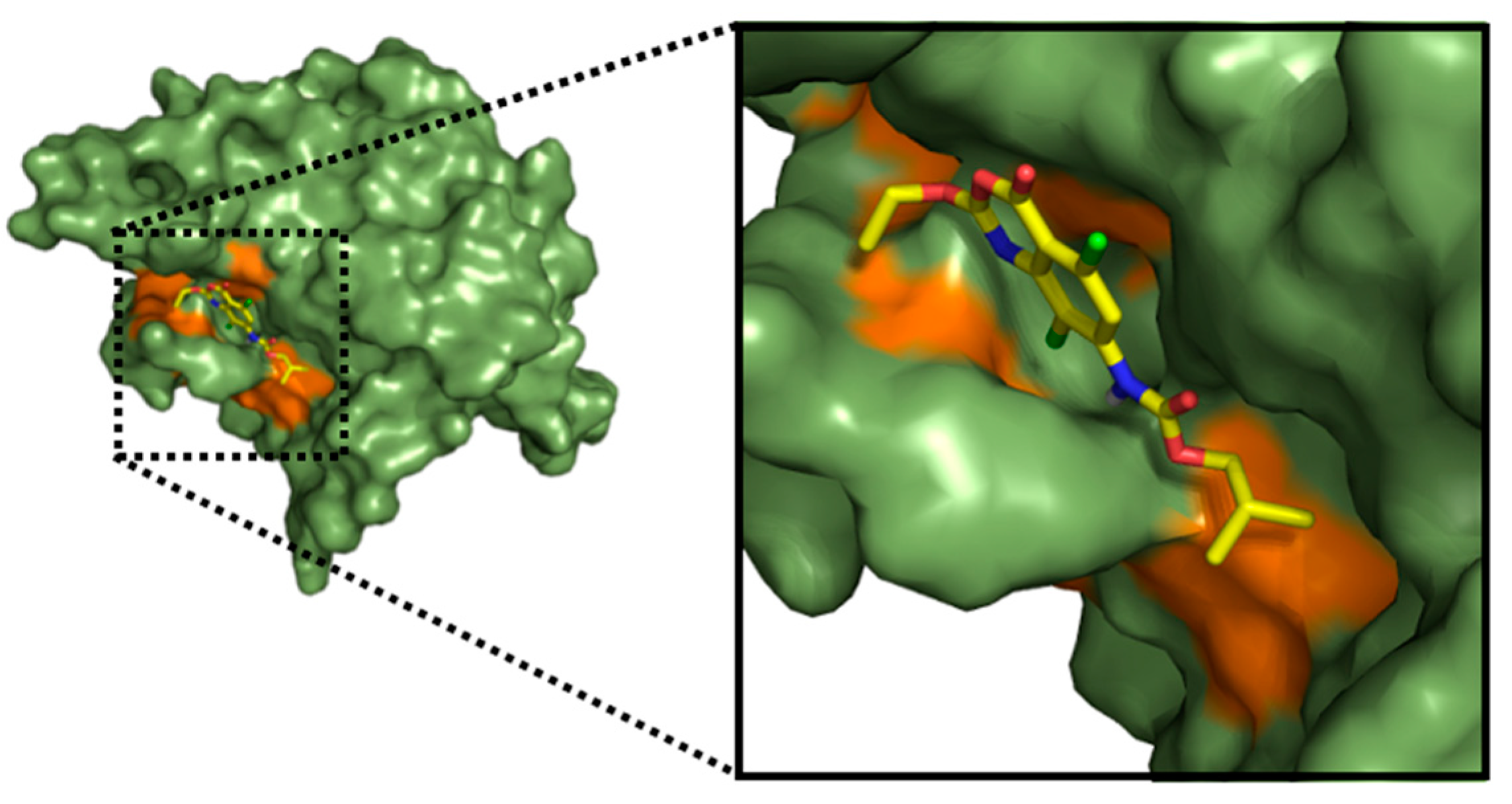
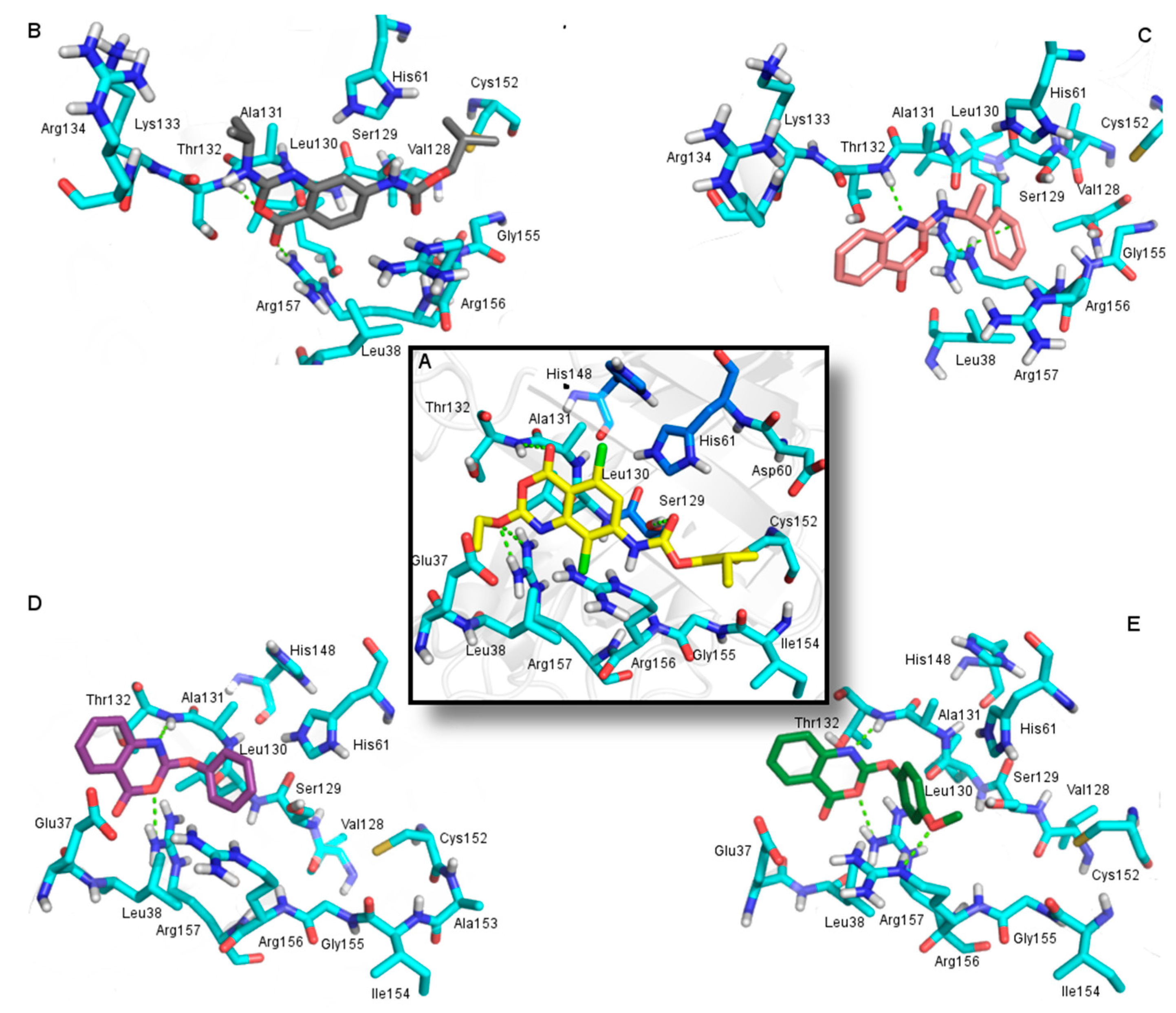
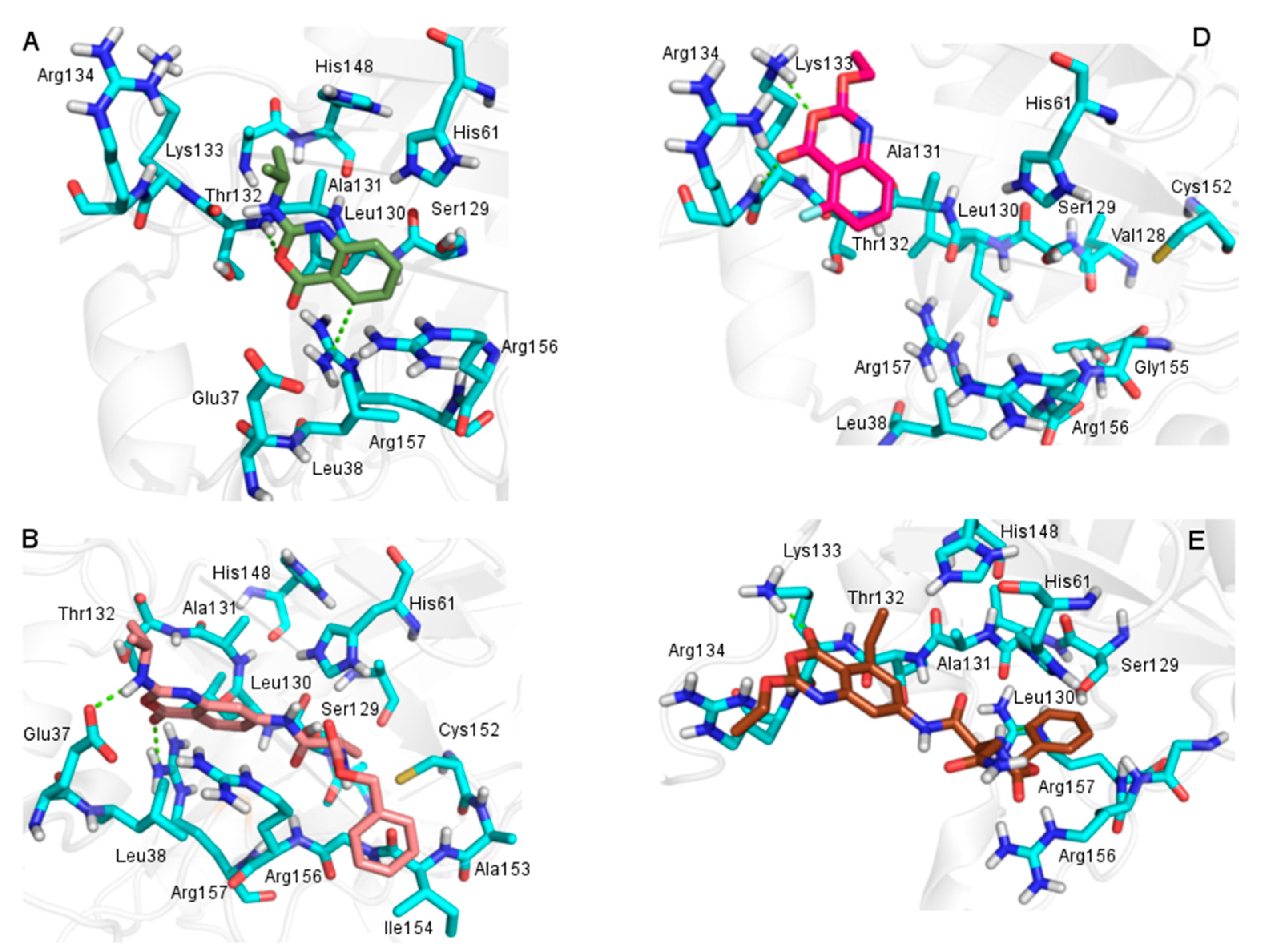
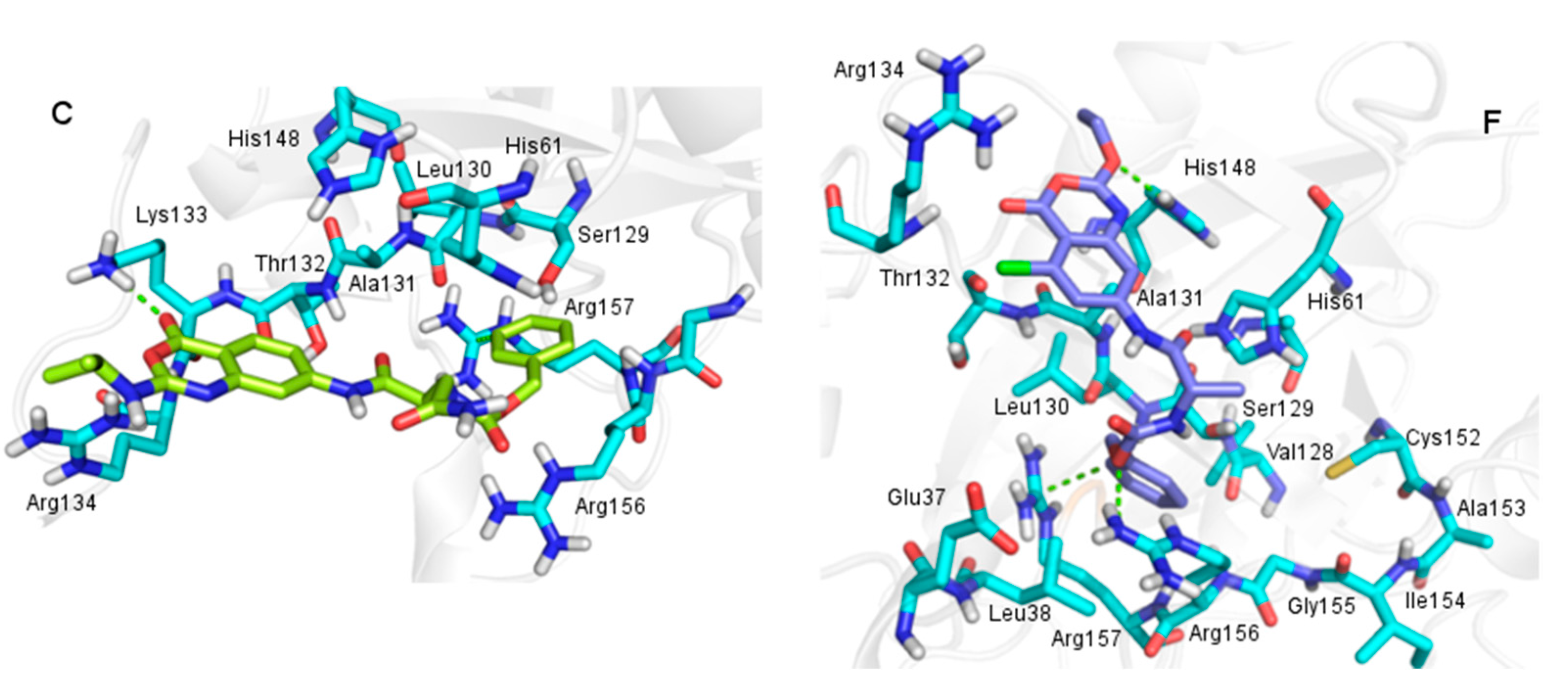
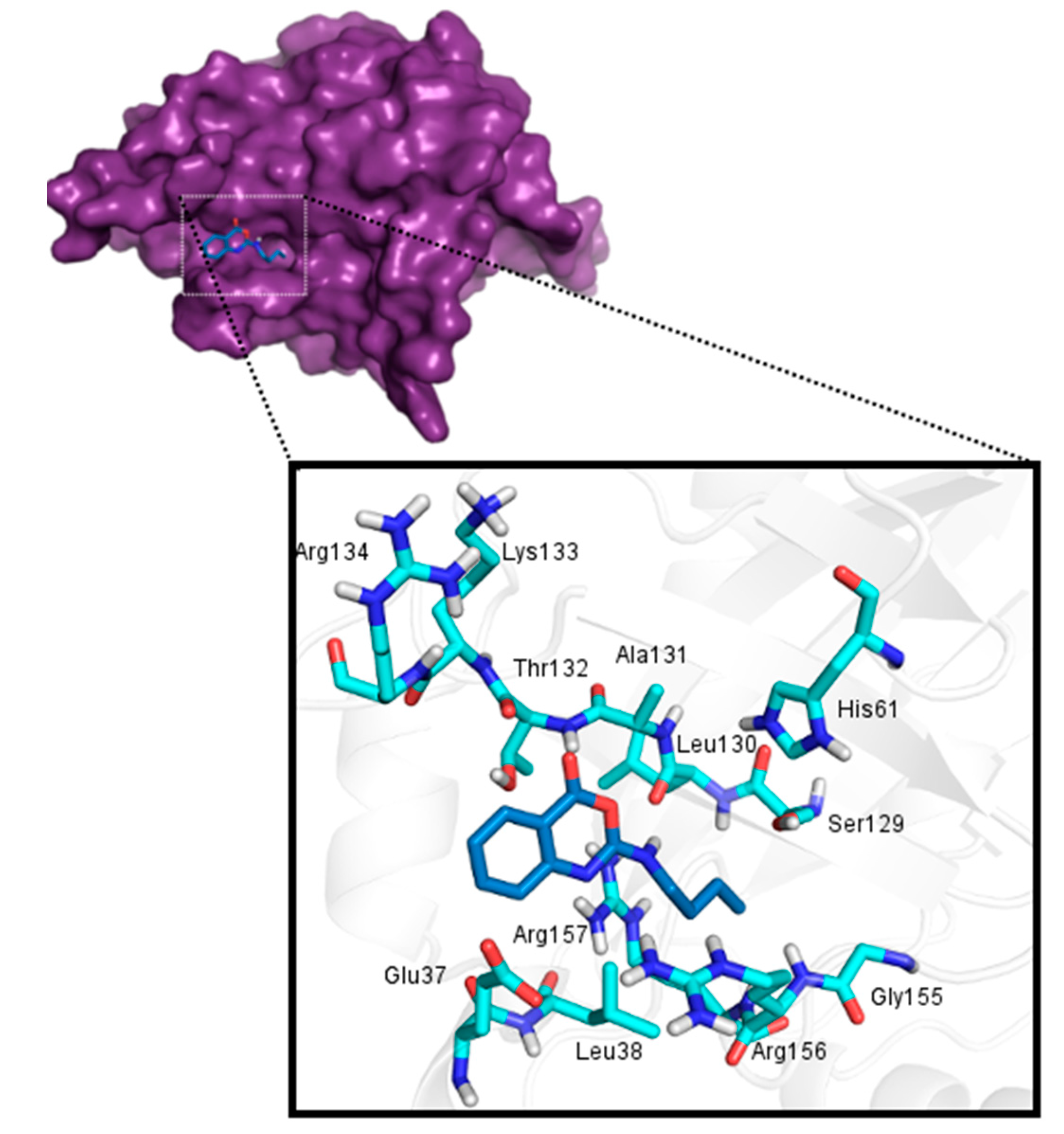
2.3. Molecular Docking
2.3.1. Docking Validation
2.3.2. Docking Analysis

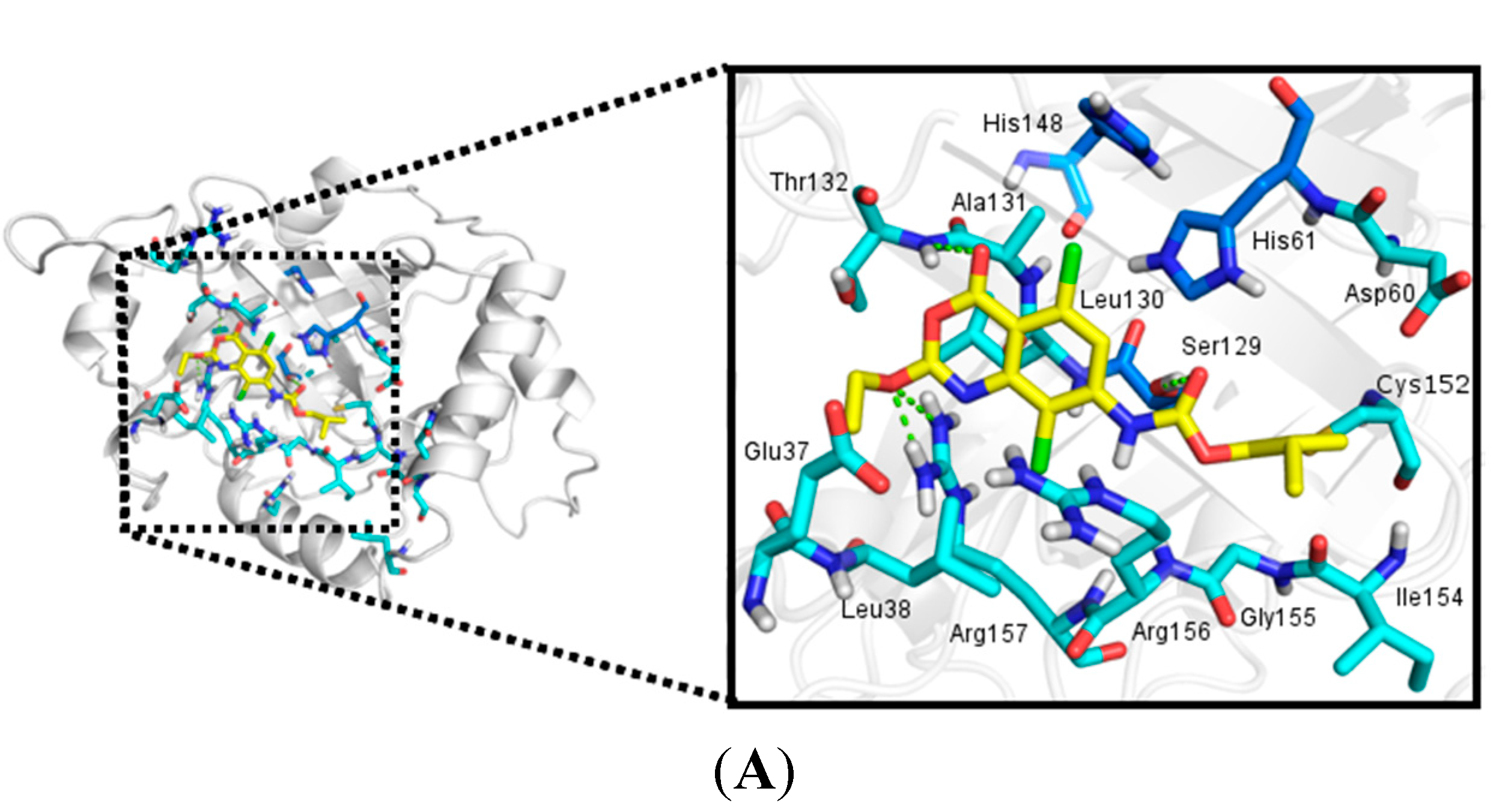

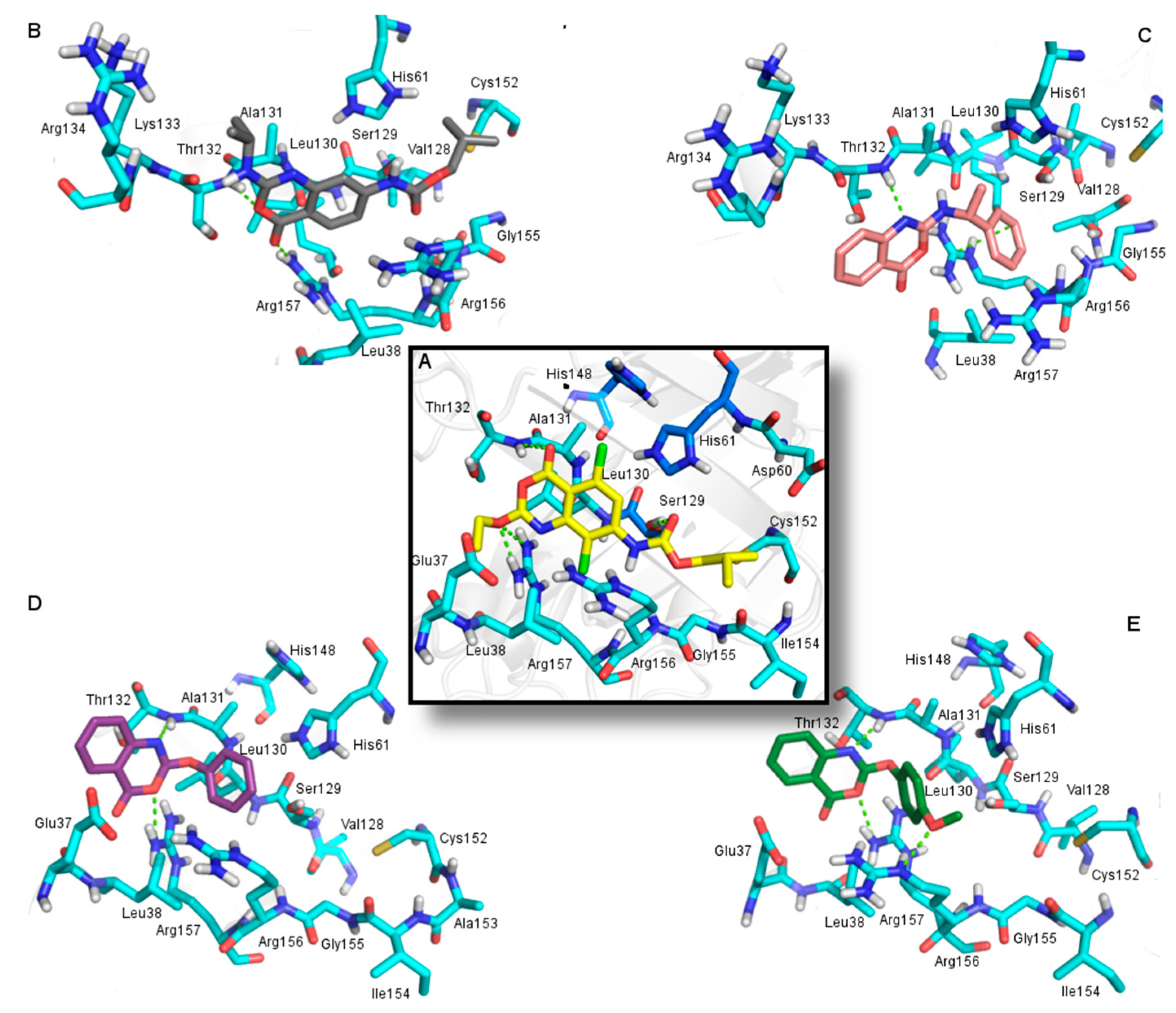
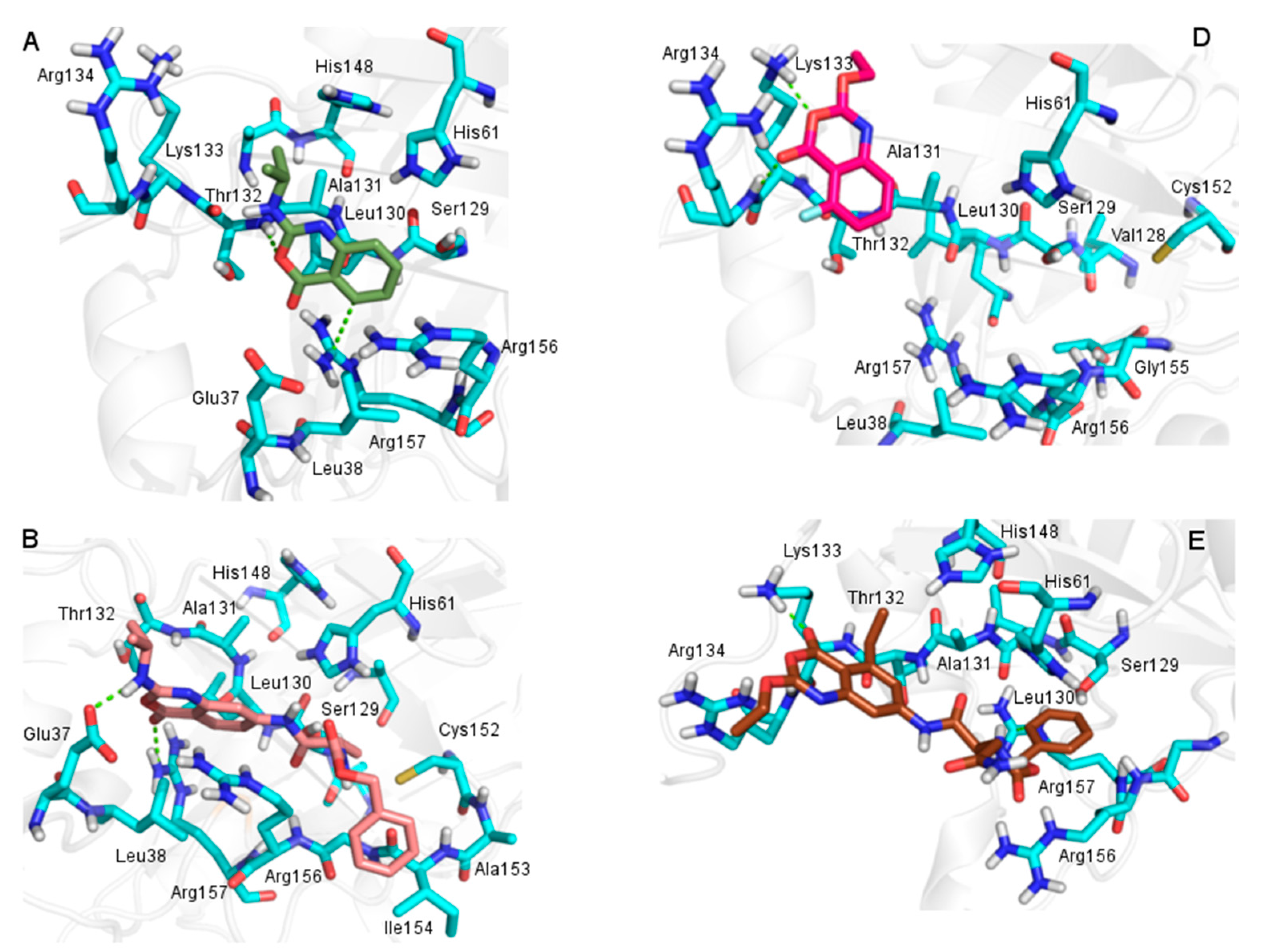


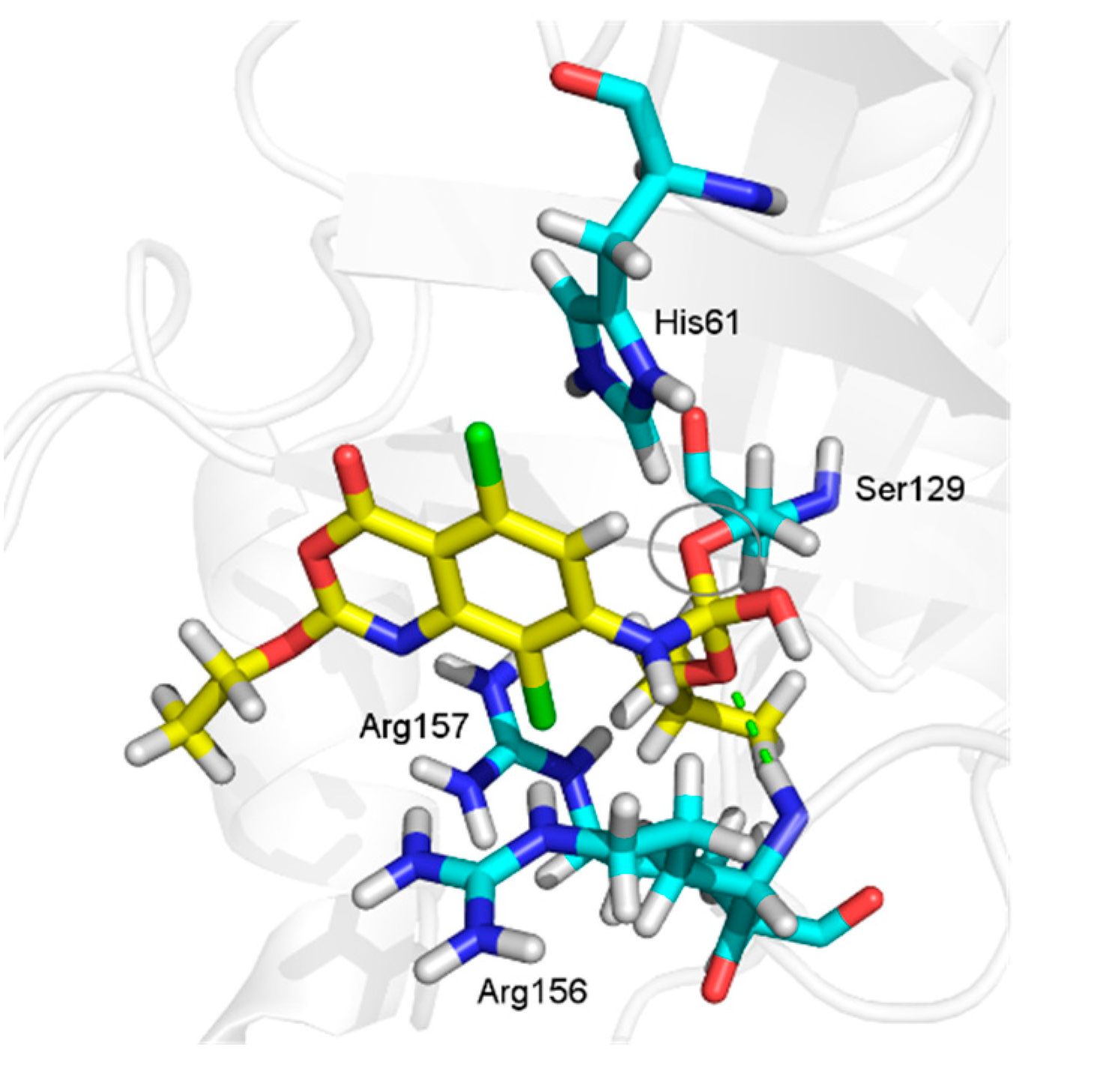
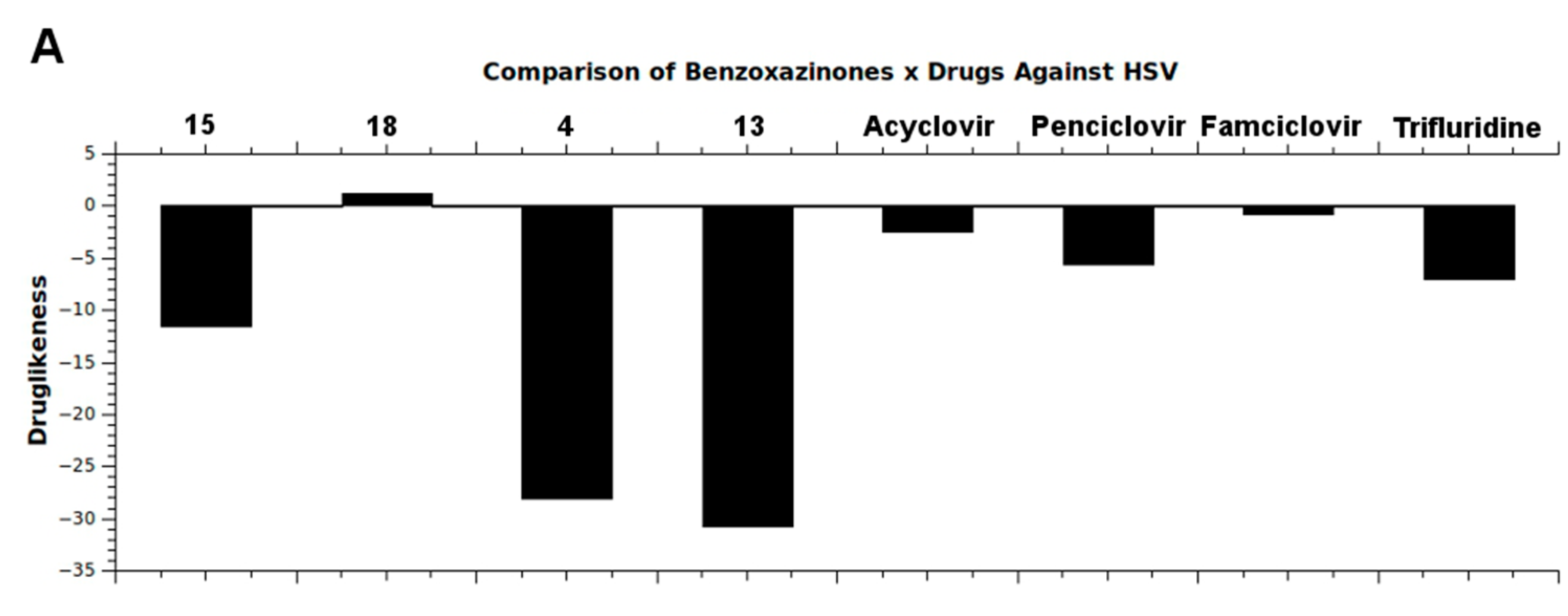
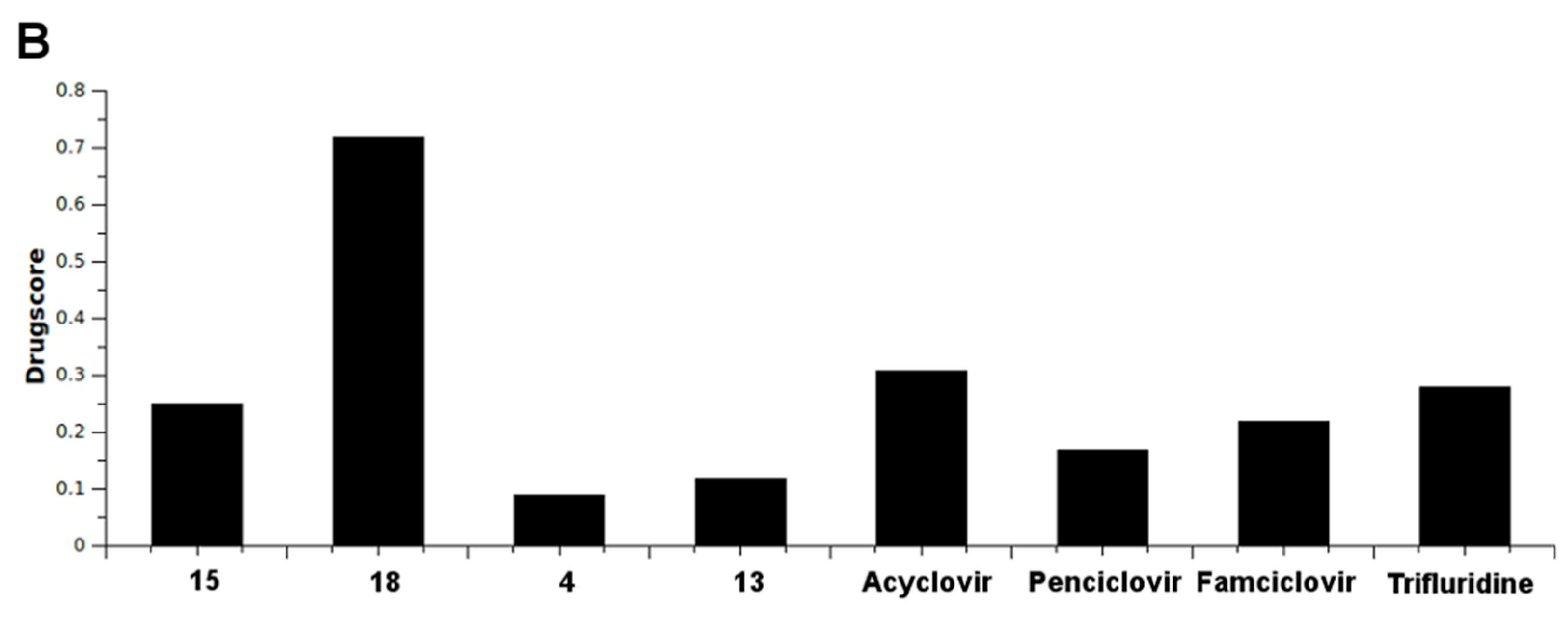
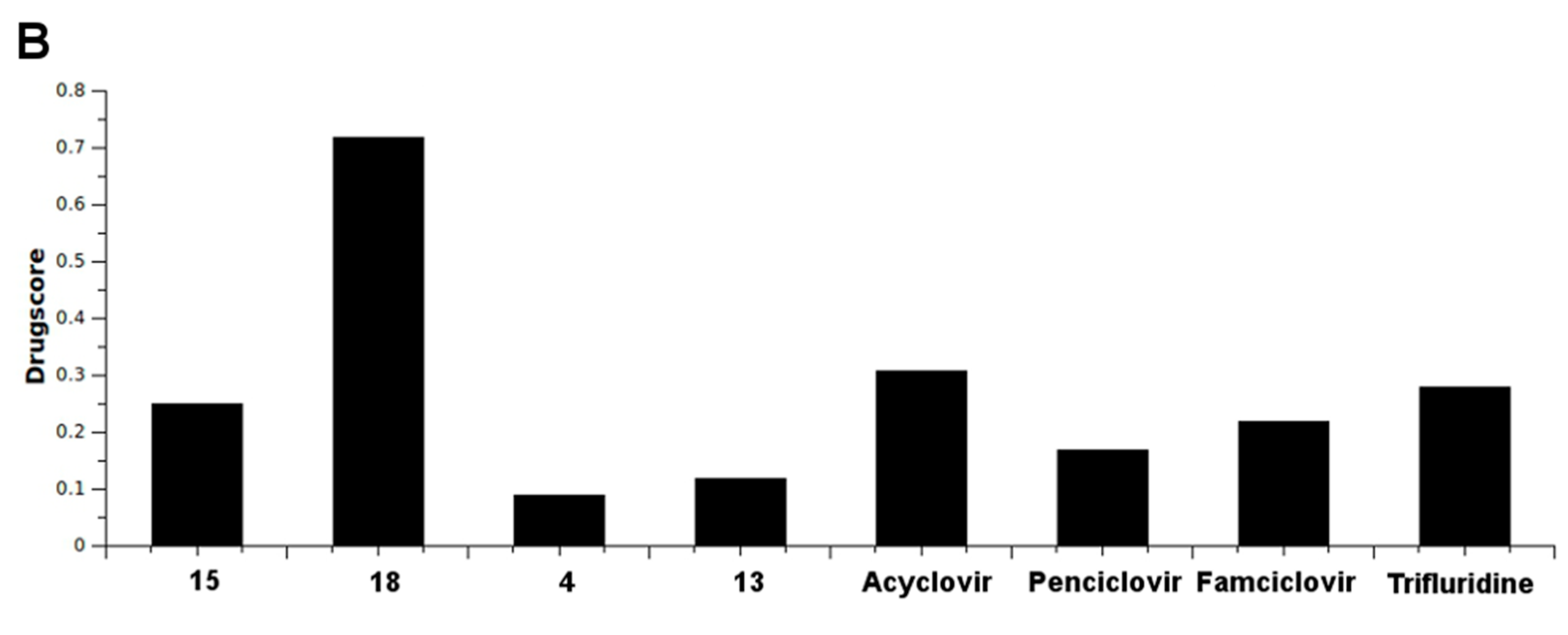
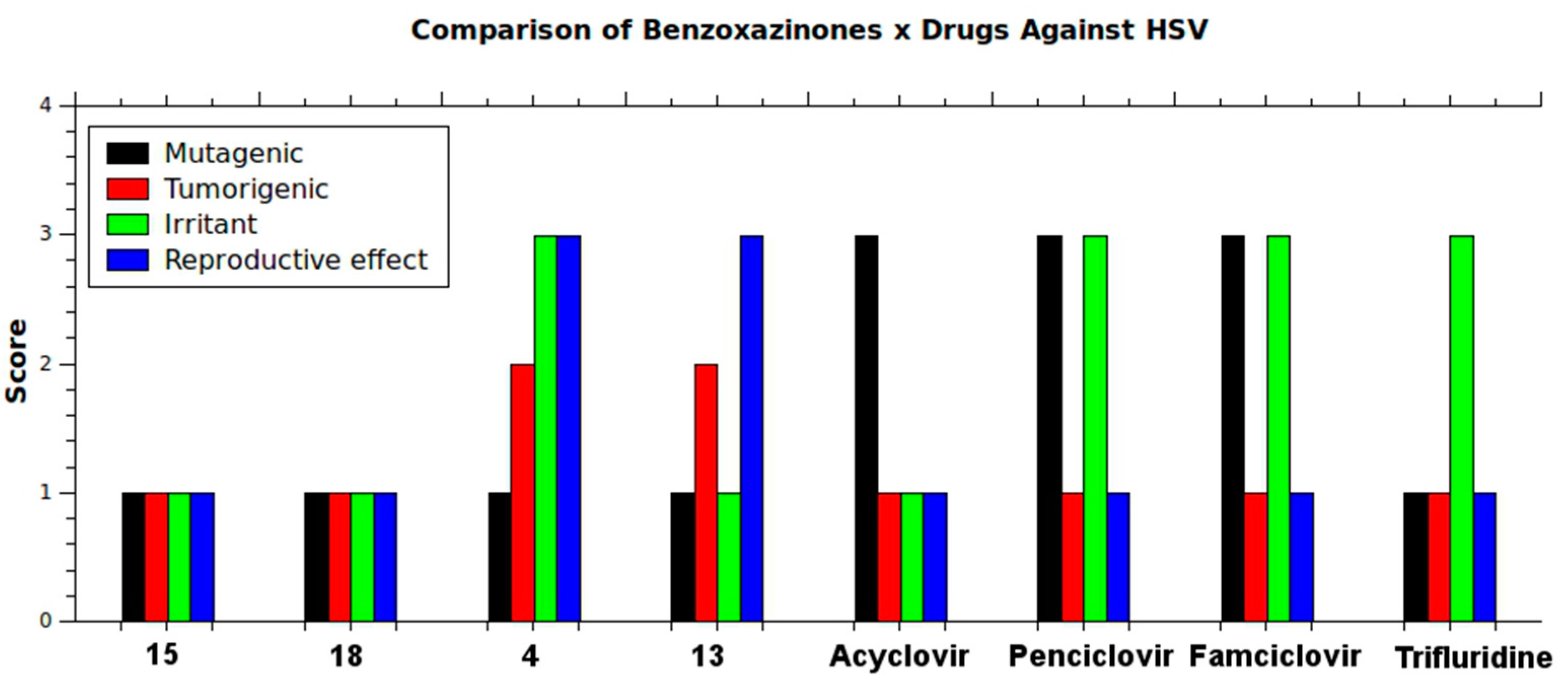
2.4. In Silico Analysis of Pharmacokinetic Properties

3. Experimental Section
3.1. Structure-Activity Relationship
3.2. Comparative Modeling
3.3. Molecular Docking
3.4. In Silico Pharmacokinetic Properties
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marnett, A.B.; Nomura, A.M.; Shimba, N.; Ortiz de Montellano, P.R.; Craik, C.S. Communication between the active sites and dimer interface of a herpesvirus protease revealed by a transition-state inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 6870–6875. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Sanchez, D.J.; Shahangian, A.; Al-Shyoukh, I.; Cheng, G.; Ho, C.M. Cascade search for hsv-1 combinatorial drugs with high antiviral efficacy and low toxicity. Int. J. Nanomed. 2012, 7, 2281–2292. [Google Scholar]
- Hewlett, G.; Hallenberger, S.; Rubsamen-Waigmann, H. Antivirals against DNA viruses (hepatitis b and the herpes viruses). Curr. Opin. Pharmacol. 2004, 4, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Chida, Y.; Mao, X. Does psychosocial stress predict symptomatic herpes simplex virus recurrence? A meta-analytic investigation on prospective studies. Brain Behav. Immun. 2009, 23, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Kinchington, P.R.; Leger, A.J.; Guedon, J.M.; Hendricks, R.L. Herpes simplex virus and varicella zoster virus, the house guests who never leave. Herpesviridae 2012, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. In search of a selective therapy of viral infections. Antiviral Res. 2010, 85, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Eizuru, Y. Development of new antivirals for herpesviruses. Antivir. Chem. Chemother. 2003, 14, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Behrens-Baumann, W. Herpes simplex keratitis. A short overview of the current therapy. Klin. Monbl. Augenheilkd. 2010, 227, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Babe, L.M.; Craik, C.S. Viral proteases: Evolution of diverse structural motifs to optimize function. Cell 1997, 91, 427–430. [Google Scholar] [CrossRef]
- Krausslich, H.G.; Wimmer, E. Viral proteinases. Annu. Rev. Biochem. 1988, 57, 701–754. [Google Scholar] [CrossRef] [PubMed]
- Hoog, S.S.; Smith, W.W.; Qiu, X.; Janson, C.A.; Hellmig, B.; McQueney, M.S.; O’Donnell, K.; O’Shannessy, D.; DiLella, A.G.; Debouck, C.; et al. Active site cavity of herpesvirus proteases revealed by the crystal structure of herpes simplex virus protease/inhibitor complex. Biochemistry 1997, 36, 14023–14029. [Google Scholar] [CrossRef] [PubMed]
- Reiling, K.K.; Pray, T.R.; Craik, C.S.; Stroud, R.M. Functional consequences of the kaposi’s sarcoma-associated herpesvirus protease structure: Regulation of activity and dimerization by conserved structural elements. Biochemistry 2000, 39, 12796–12803. [Google Scholar] [CrossRef] [PubMed]
- Shahiduzzaman, M.; Coombs, K.M. Activity based protein profiling to detect serine hydrolase alterations in virus infected cells. Front. Microbiol. 2012, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tong, L. Viral proteases. Chem. Rev. 2002, 102, 4609–4626. [Google Scholar] [CrossRef] [PubMed]
- Waxman, L.; Darke, P.L. The herpesvirus proteases as targets for antiviral chemotherapy. Antivir. Chem. Chemother. 2000, 11, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, E.; Chinchilla, N.; Molinillo, J.M.; Macias, F.A.; Astola, A.; Ortiz, M.; Valdivia, M.M. Aneugenic effects of benzoxazinones in cultured human cells. Mutat. Res. 2010, 695, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L.; Moorman, A.R.; Dobbs, J.; Abeles, R.H. Suicide inactivation of chymotrypsin by benzoxazinones. Biochemistry 1984, 23, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Jarvest, R.L.; Parratt, M.J.; Debouck, C.M.; Gorniak, J.G.; Jennings, L.J.; Serafinowska, H.T.; Strickler, J.E. Inhibition of hsv-1 protease by benzoxazinones. Bioorg. Med. Chem. Lett. 1996, 6, 2463–2466. [Google Scholar] [CrossRef]
- Hemmateenejad, B.; Elyasi, M. A segmented principal component analysis-regression approach to quantitative structure-activity relationship modeling. Anal. Chim. Acta 2009, 646, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Sant’Anna, C.M.R. Molecular modeling methods in the study and design of bioactive compound: An introduction. Rev. Virtual Quim. 2009, 1, 49–57. [Google Scholar] [CrossRef]
- Zuegg, J.; Cooper, M.A. Drug-likeness and increased hydrophobicity of commercially available compound libraries for drug screening. Curr. Top. Med. Chem. 2012, 12, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist's guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Consortium, T.U. Reorganizing the protein space at the universal protein resource (uniprot). Nucleic Acids Res. 2012, 40, D71–D75. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Peitsch, M.C. Protein modeling by e-mail. Nat. Biotech. 1995, 13, 658–660. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; John, B.; Mirkovic, N.; Fiser, A.; Ilyin, V.A.; Pieper, U.; Stuart, A.C.; Marti-Renom, M.A.; Madhusudhan, M.S.; Yerkovich, B.; et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003, 31, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Marti-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sanchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Marti-Renom, M.A.; Yerkovich, B.; Sali, A. Comparative Protein Structure Prediction. Curr. Protoc. Protein Sci. 2:2.9; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Melo, F.; Sanchez, R.; Sali, A. Statistical potentials for fold assessment. Protein Sci. 2002, 11, 430–448. [Google Scholar] [CrossRef] [PubMed]
- Pieper, U.; Eswar, N.; Braberg, H.; Madhusudhan, M.S.; Davis, F.P.; Stuart, A.C.; Mirkovic, N.; Rossi, A.; Marti-Renom, M.A.; Fiser, A.; et al. Modbase, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2004, 32, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Overington, J.P. Derivation of rules for comparative protein modeling from a database of protein structure alignments. Protein Sci. 1994, 3, 1582–1596. [Google Scholar] [CrossRef] [PubMed]
- Doreleijers, J.F.; Raves, M.L.; Rullmann, T.; Kaptein, R. Completeness of noes in protein structure: A statistical analysis of NMR. J. Biomol. NMR 1999, 14, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Doreleijers, J.F.; Rullmann, J.A.; Kaptein, R. Quality assessment of nmr structures: A statistical survey. J. Mol. Biol. 1998, 281, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. Aqua and procheck-nmr: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Bowie, J.U.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Luthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor-sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Nissink, J.W.M.; Murray, C.; Hartshorn, M.; Verdonk, M.L.; Cole, J.C.; Taylor, R. A new test set for validating predictions of protein-ligand interaction. Proteins Struct. Funct. Genet. 2002, 49, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using gold. Proteins Struct. Funct. Genet. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Organic Chemistry Portal. Available online: http://www.organic-chemistry.org/prog/peo (accessed on 20 May 2014).
- Samples Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mello, J.F.R.; Botelho, N.C.; Souza, A.M.T.; Oliveira, R.; Brito, M.A.; Abrahim-Vieira, B.D.A.; Sodero, A.C.R.; Castro, H.C.; Cabral, L.M.; Miceli, L.A.; et al. Computational Studies of Benzoxazinone Derivatives as Antiviral Agents against Herpes Virus Type 1 Protease. Molecules 2015, 20, 10689-10704. https://doi.org/10.3390/molecules200610689
Mello JFR, Botelho NC, Souza AMT, Oliveira R, Brito MA, Abrahim-Vieira BDA, Sodero ACR, Castro HC, Cabral LM, Miceli LA, et al. Computational Studies of Benzoxazinone Derivatives as Antiviral Agents against Herpes Virus Type 1 Protease. Molecules. 2015; 20(6):10689-10704. https://doi.org/10.3390/molecules200610689
Chicago/Turabian StyleMello, Juliana F. R., Nathália C. Botelho, Alessandra M. T. Souza, Riethe Oliveira, Monique A. Brito, Bárbara De A. Abrahim-Vieira, Ana Carolina R. Sodero, Helena C. Castro, Lucio M. Cabral, Leonardo A. Miceli, and et al. 2015. "Computational Studies of Benzoxazinone Derivatives as Antiviral Agents against Herpes Virus Type 1 Protease" Molecules 20, no. 6: 10689-10704. https://doi.org/10.3390/molecules200610689