
Photorelease of Pyridyl Esters in Organometallic Ru(II) Arene Complexes

Abstract
:
1. Introduction
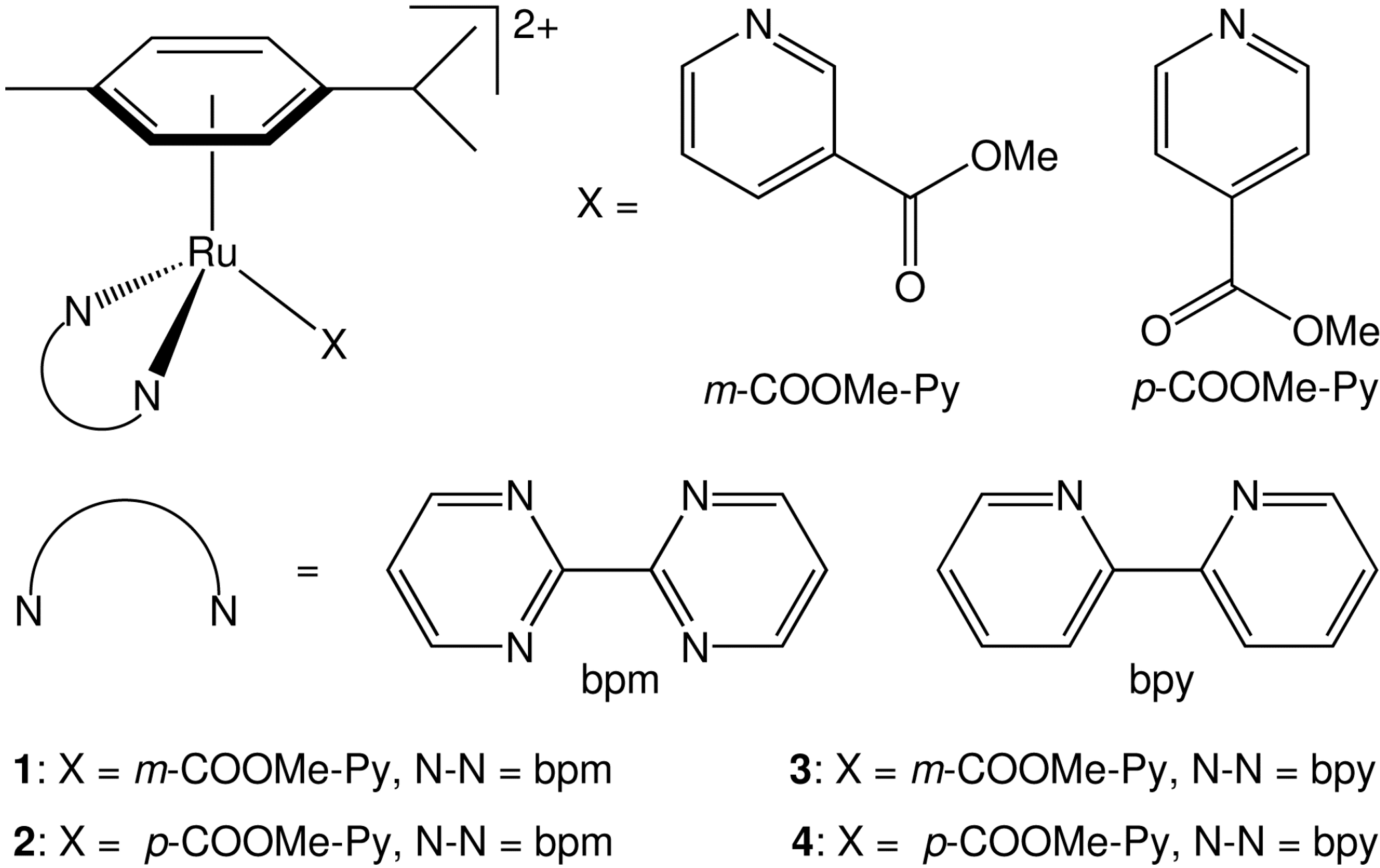
2. Results and Discussion
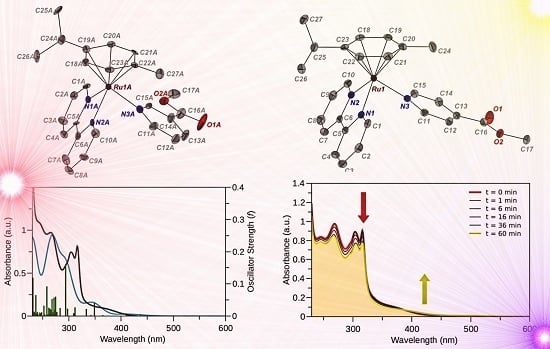
2.1. Synthesis and X-ray Crystal Structures
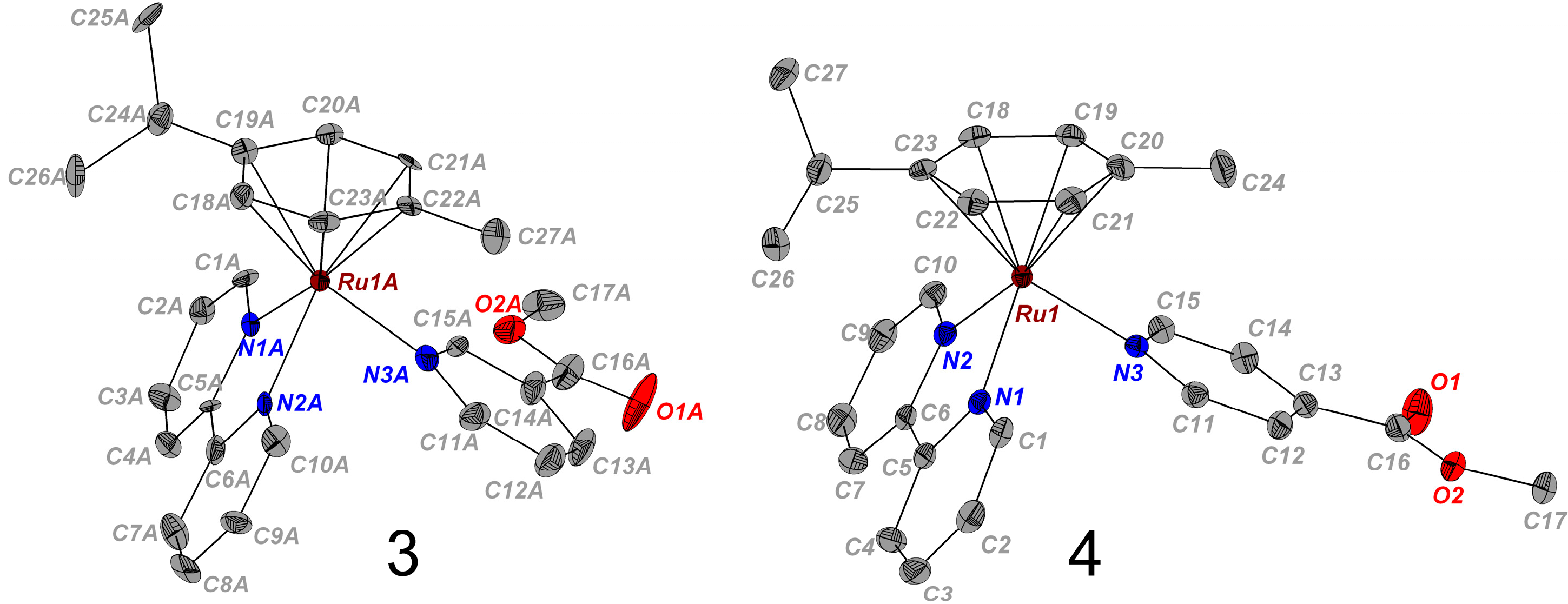

{kind=link}
{kind=link}
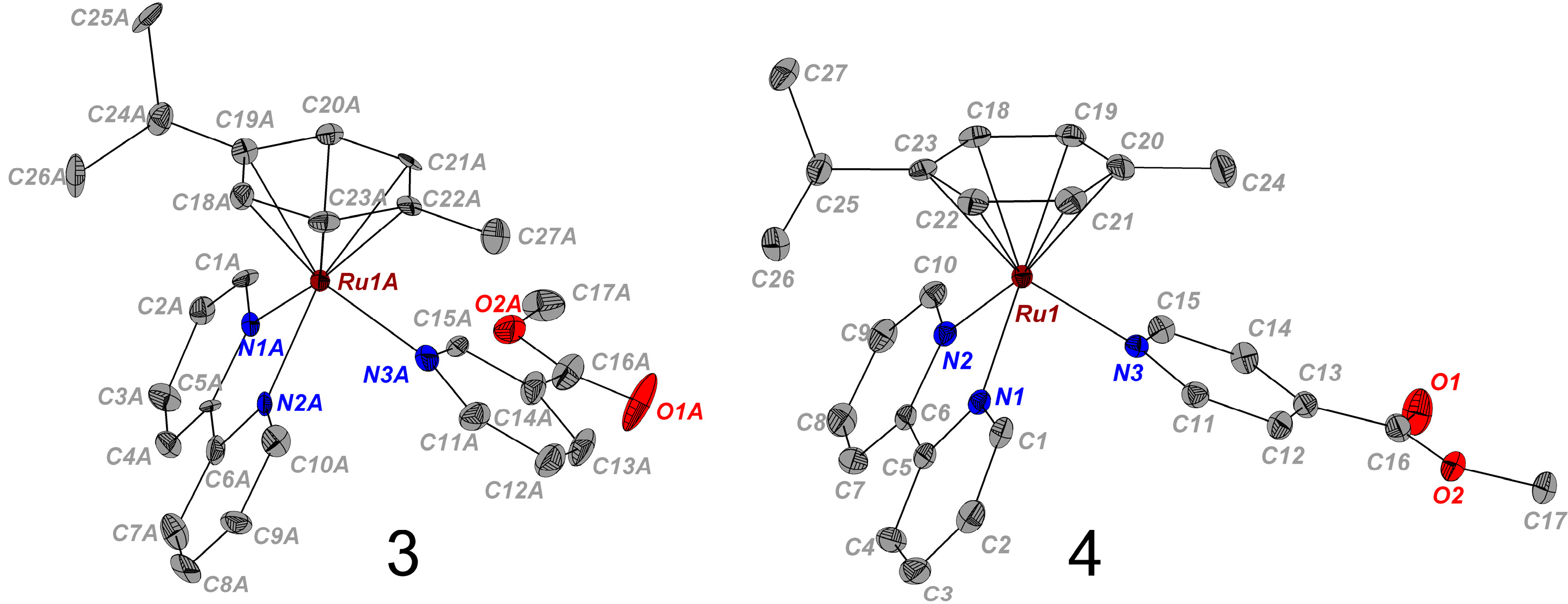{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3(PF6)2 | 4(PF6)2 | |
|---|---|---|
| Empirical Formula | C27H29F12N3O2P2Ru | C27H29F12N3O2P2Ru |
| Formula weight (g·mol−1) | 818.54 | 818.54 |
| Crystal system | Monoclinic | Monoclinic |
| Crystal size/mm | 0.52 × 0.20 × 0.10 | 0.50 × 0.21 × 0.16 |
| Space group | P2(1) | P2(1)/c |
| Crystal | Red block | Dark-red block |
| a/Å | 18.2866(3) | 14.14530(10) |
| b/Å | 9.55390(12) | 18.6107(2) |
| c/Å | 36.8439(5) | 11.70990(10) |
| α/deg | 90.00 | 90.00 |
| β/deg | 102.5101(10) | 101.8020(10) |
| γ/deg | 90.00 | 90.00 |
| Volume/Å3 | 6284.10(15) | 3017.51(5) |
| Temperature/K | 100(1) | 100(1) |
| Z | 8 | 4 |
| μ (CuKα) [mm−1] | 0.704 | 0.733 |
| Reflections collected | 47851 | 20749 |
| Independent reflections [Rint] | 18934[0.032] | 5932[0.028] |
| Parameters/restraints | 2176/434 | 491/96 |
| R1 [a], wR2 [b] [I > 2σ (I)] | 0.0487, 0.1027 | 0.0255, 0.0552 |
| R1 [a], wR2 [b] (all data) | 0.0516, 0.1043 | 0.0295, 0.0577 |
| GoF [c] | 1.108 | 1.108 |
| Δρ max and min/eÅ−3 | 1.85 and −0.713 | 0.573 and −0.433 |
| Bond Length (Å)/Angle (°) | 3 | 4 |
|---|---|---|
| Ru–arene(centroid) | 1.708 | 1.702 |
| Ru(1)–N(1) | 2.084(6) | 2.0851(17) |
| Ru(1)–N(2) | 2.083(6) | 2.0917(17) |
| Ru(1)–N(3) | 2.137(7) | 2.1242(17) |
| C(5)–C(6) | 1.457(10) | 1.471(3) |
| N(1)–Ru(1)–N(2) | 77.1(3) | 77.30(7) |
| N(1)–Ru(1)–N(3) | 84.8(2) | 88.16(7) |
| N(2)–Ru(1)–N(3) | 86.6(2) | 86.80(7) |
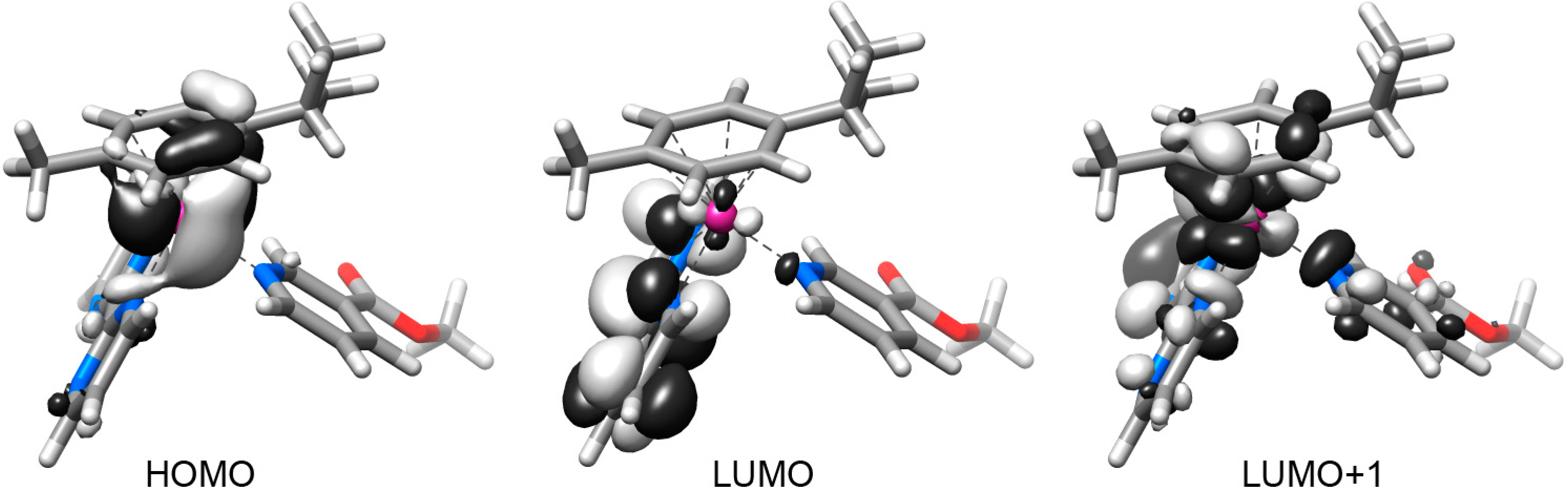
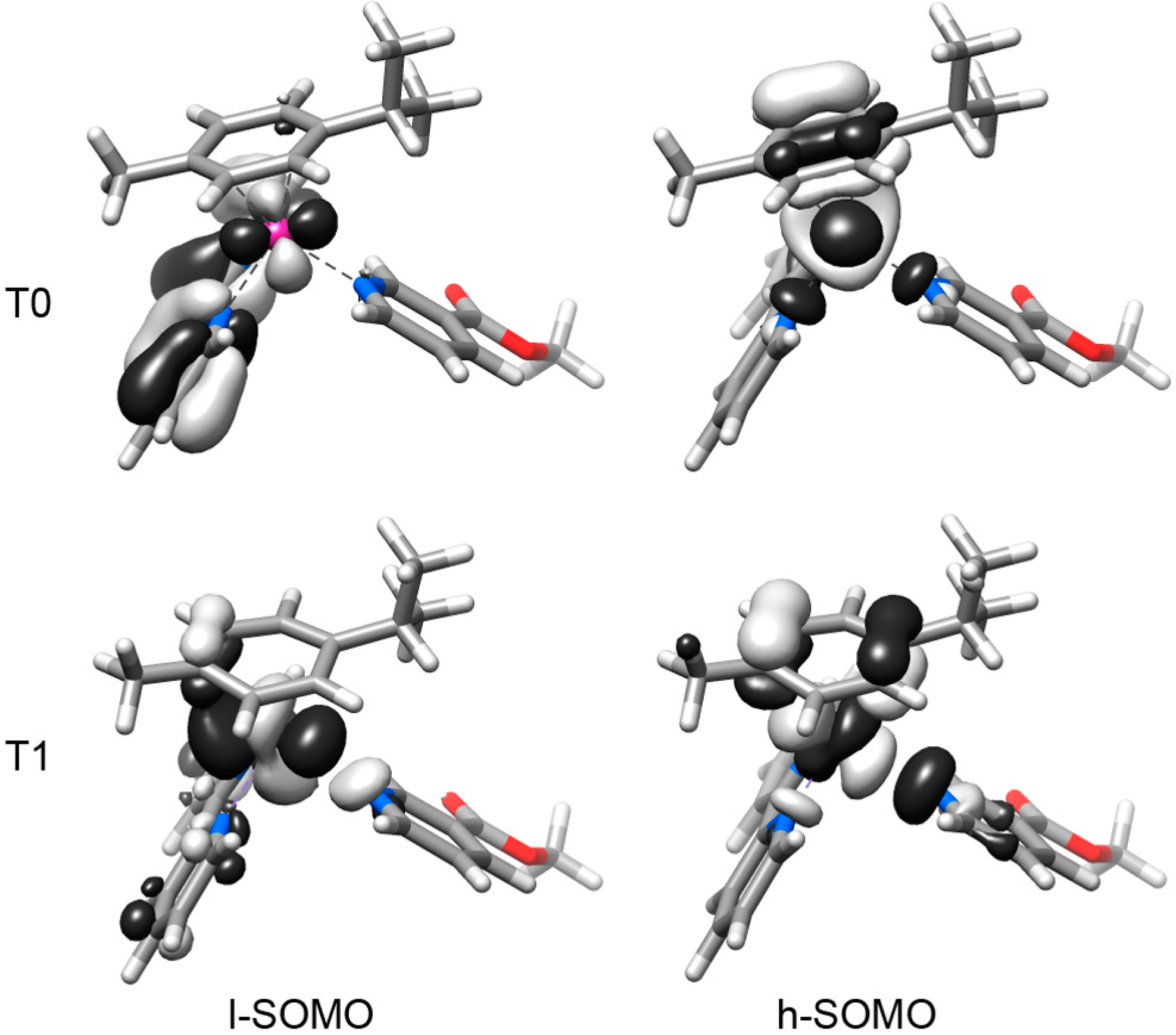
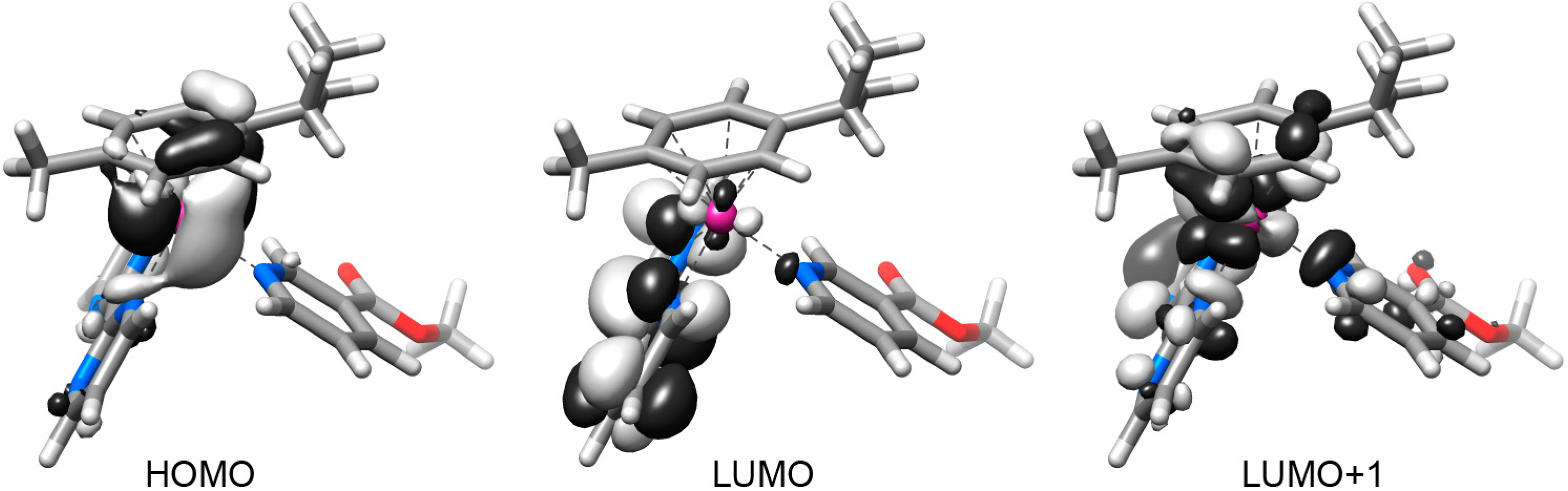
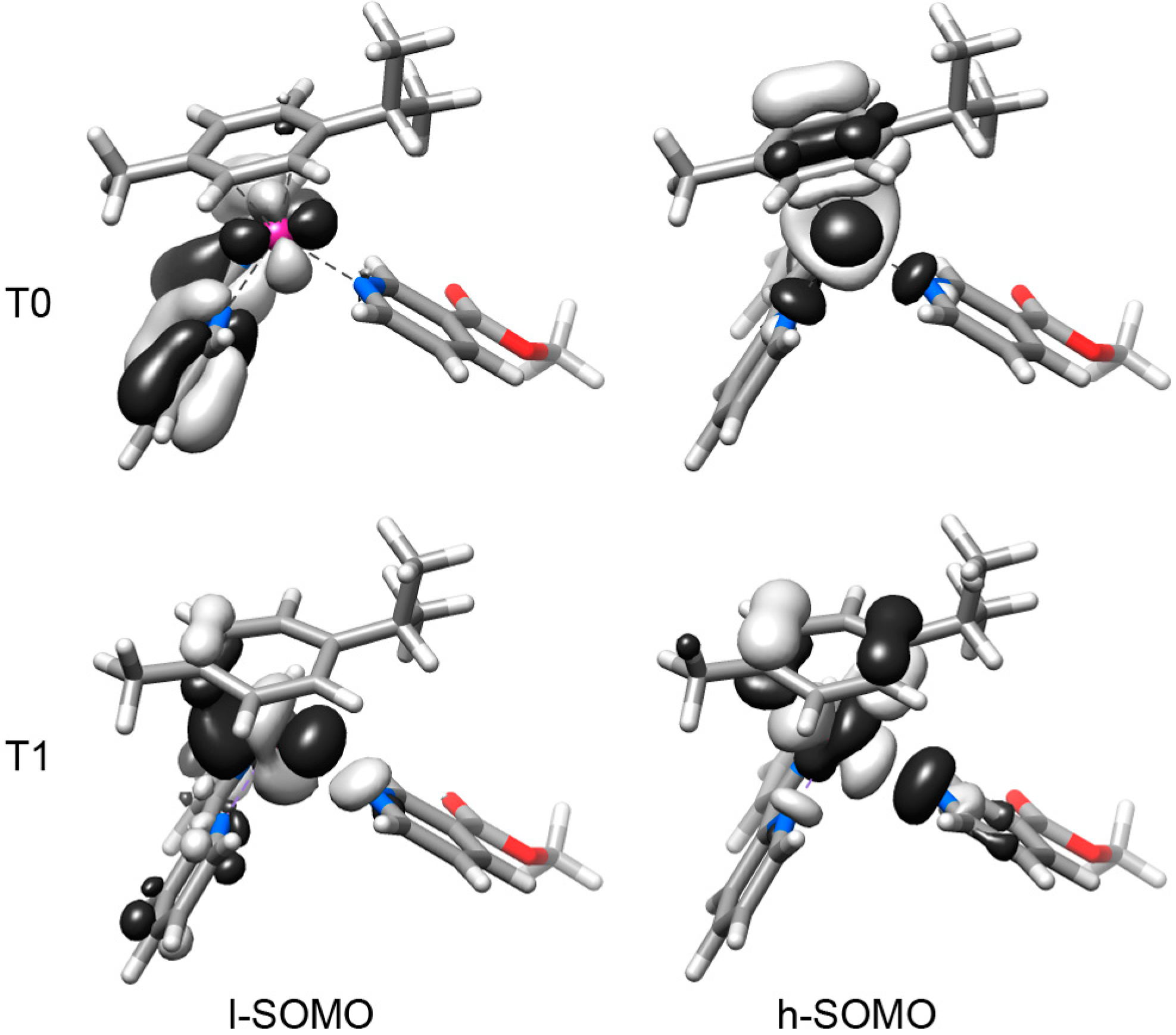
2.2. DFT Geometries and Electronic Structures of 1–4
| Compound | Ru–N(L) | Ru–N(N-N) | Ru–N(N-N) | Ru–arene(centroid) |
|---|---|---|---|---|
| S0 | ||||
| 1 | 2.157 | 2.111 | 2.109 | 1.845 |
| 2 | 2.147 | 2.113 | 2.111 | 1.850 |
| 3 | 2.160 | 2.102 | 2.098 | 1.848 |
| 4 | 2.148 | 2.104 | 2.102 | 1.853 |
| T0 | ||||
| 1 | 2.140 | 2.439 | 2.130 | 2.083 |
| 2 | 2.136 | 2.454 | 2.137 | 2.094 |
| 3 | 2.152 | 2.386 | 2.112 | 2.092 |
| 4 | 2.153 | 2.391 | 2.110 | 2.096 |
| T1 | ||||
| 1 | 2.556 | 2.103 | 2.099 | 2.123 |
| 2 | 2.522 | 2.105 | 2.093 | 2.137 |
| 3 | 2.565 | 2.084 | 2.087 | 2.142 |
| 4 | 2.532 | 2.088 | 2.082 | 2.153 |
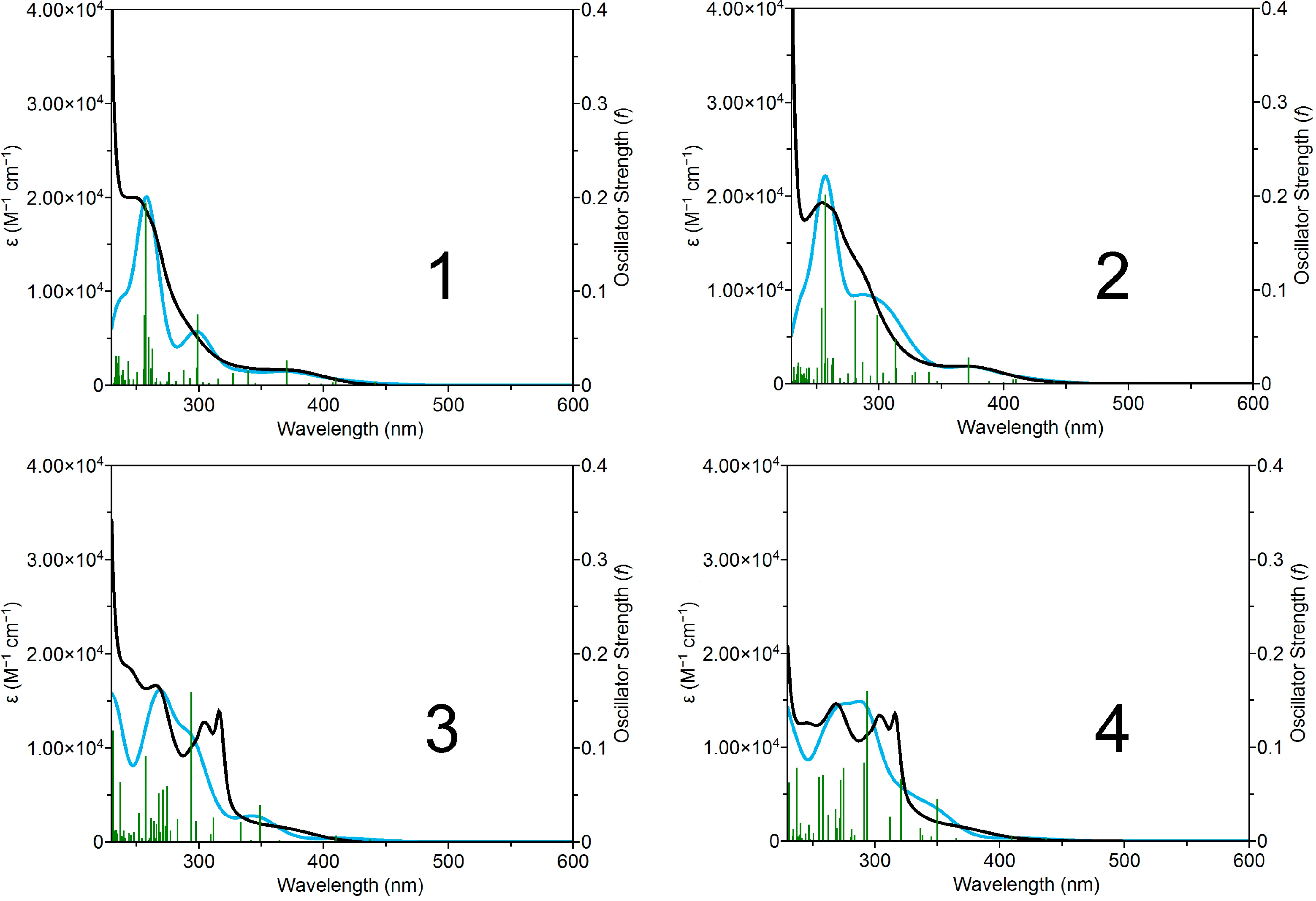
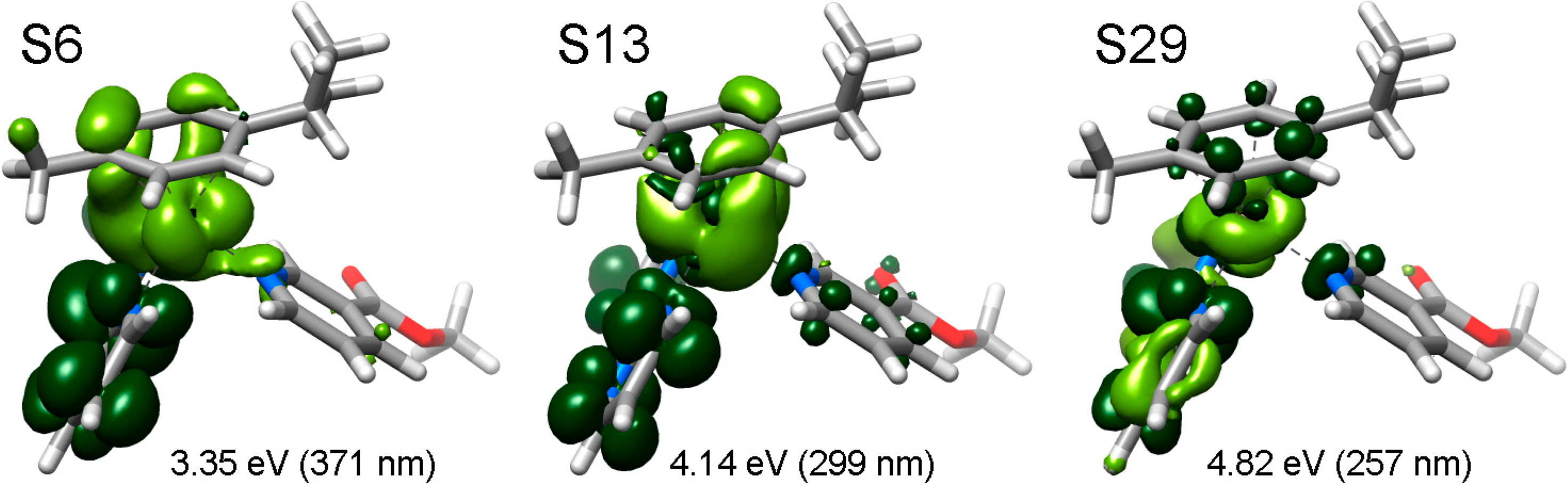
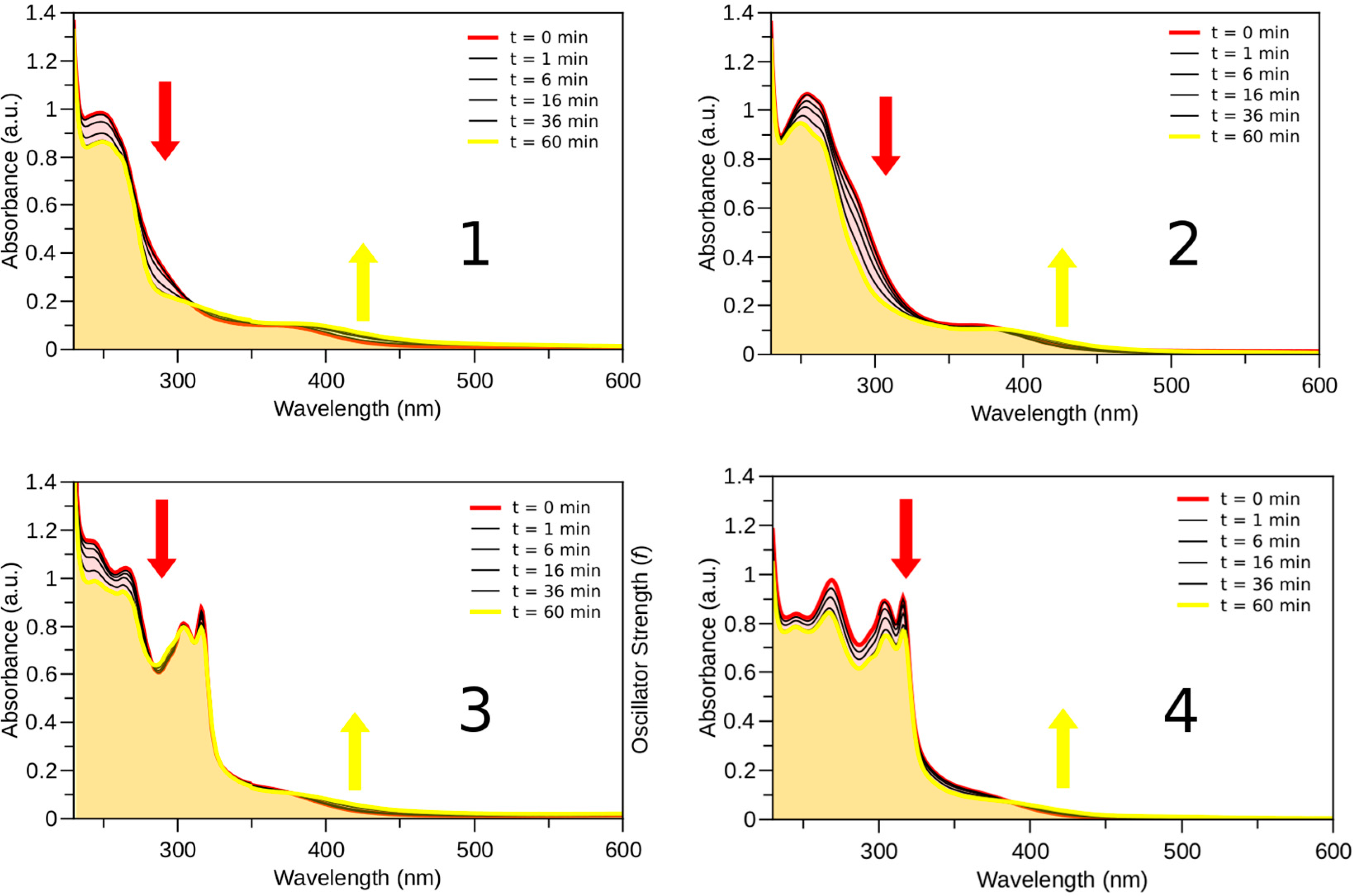
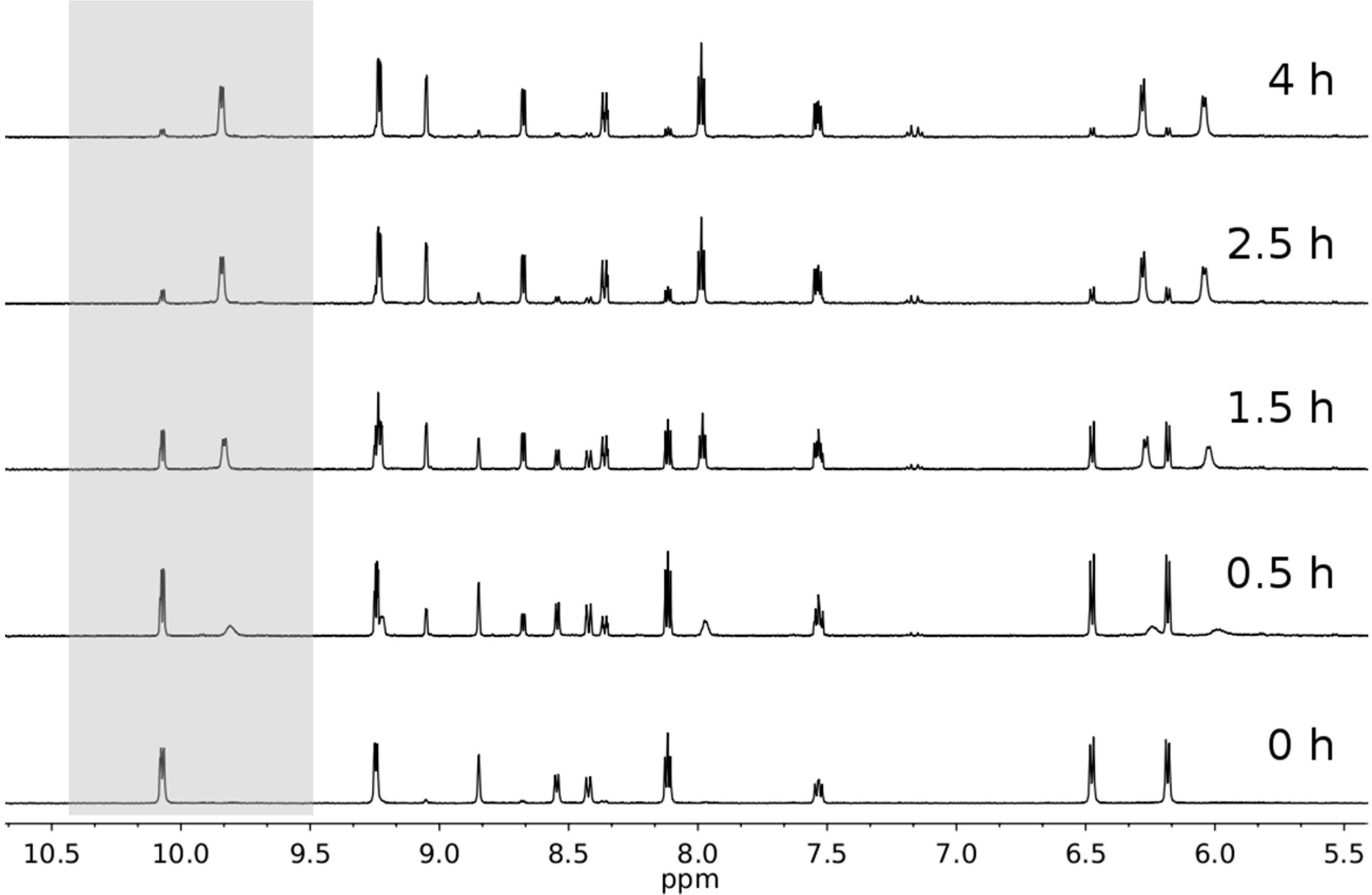
2.3. Photophysical and Photochemical Properties of 1–4
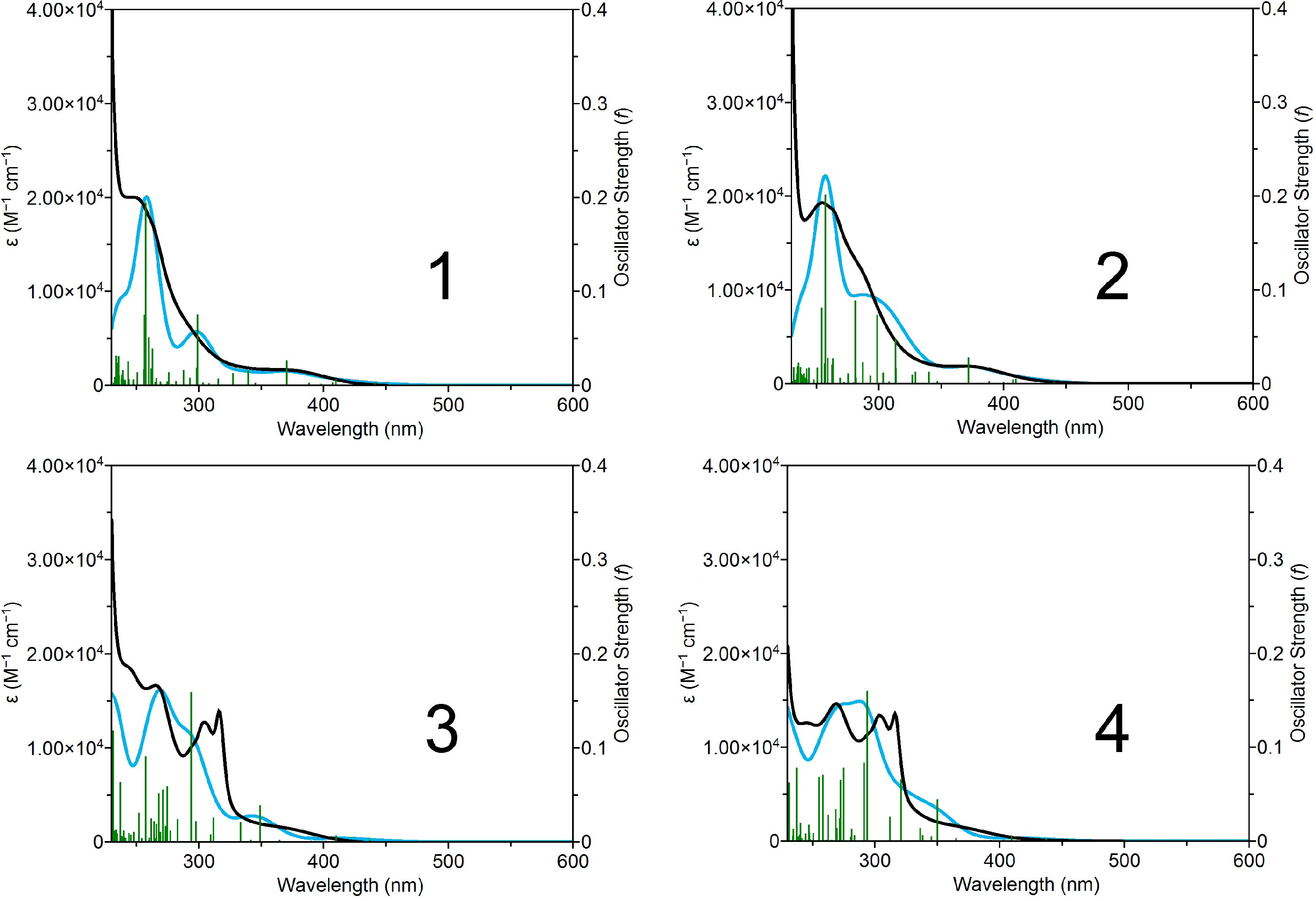
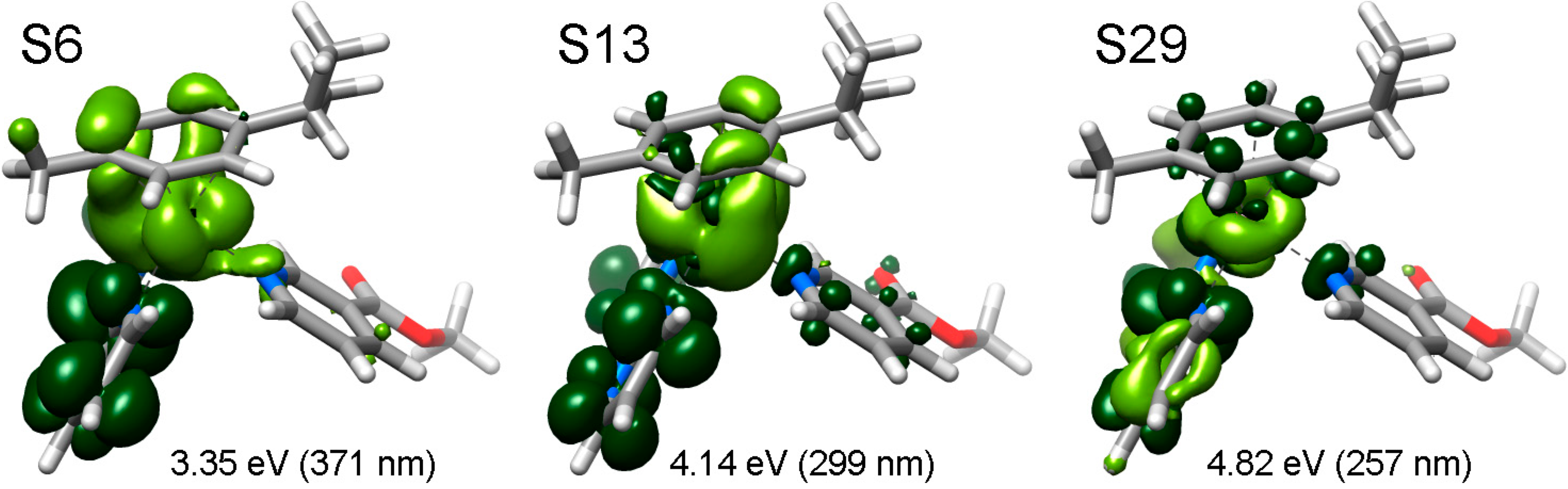
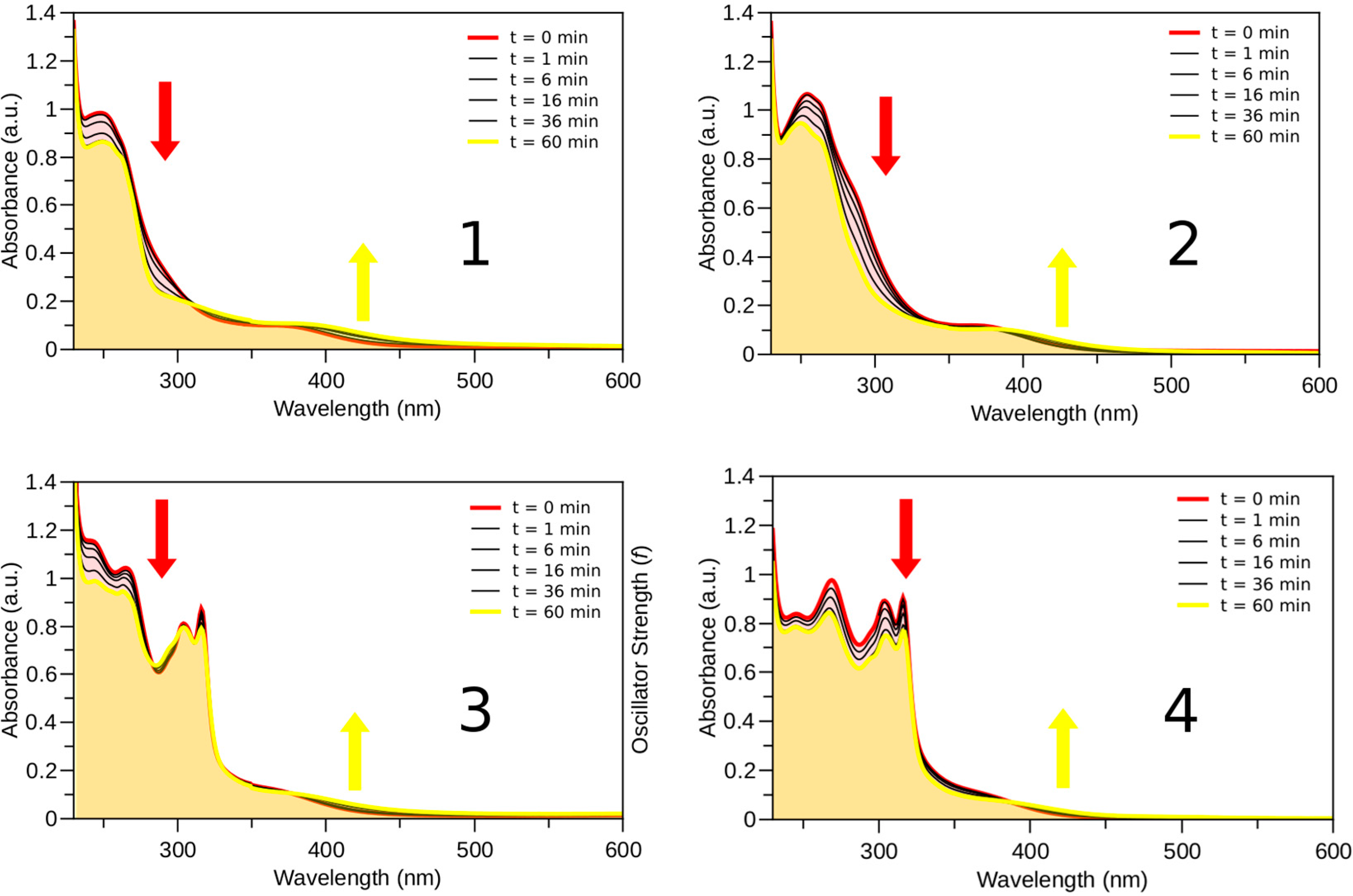
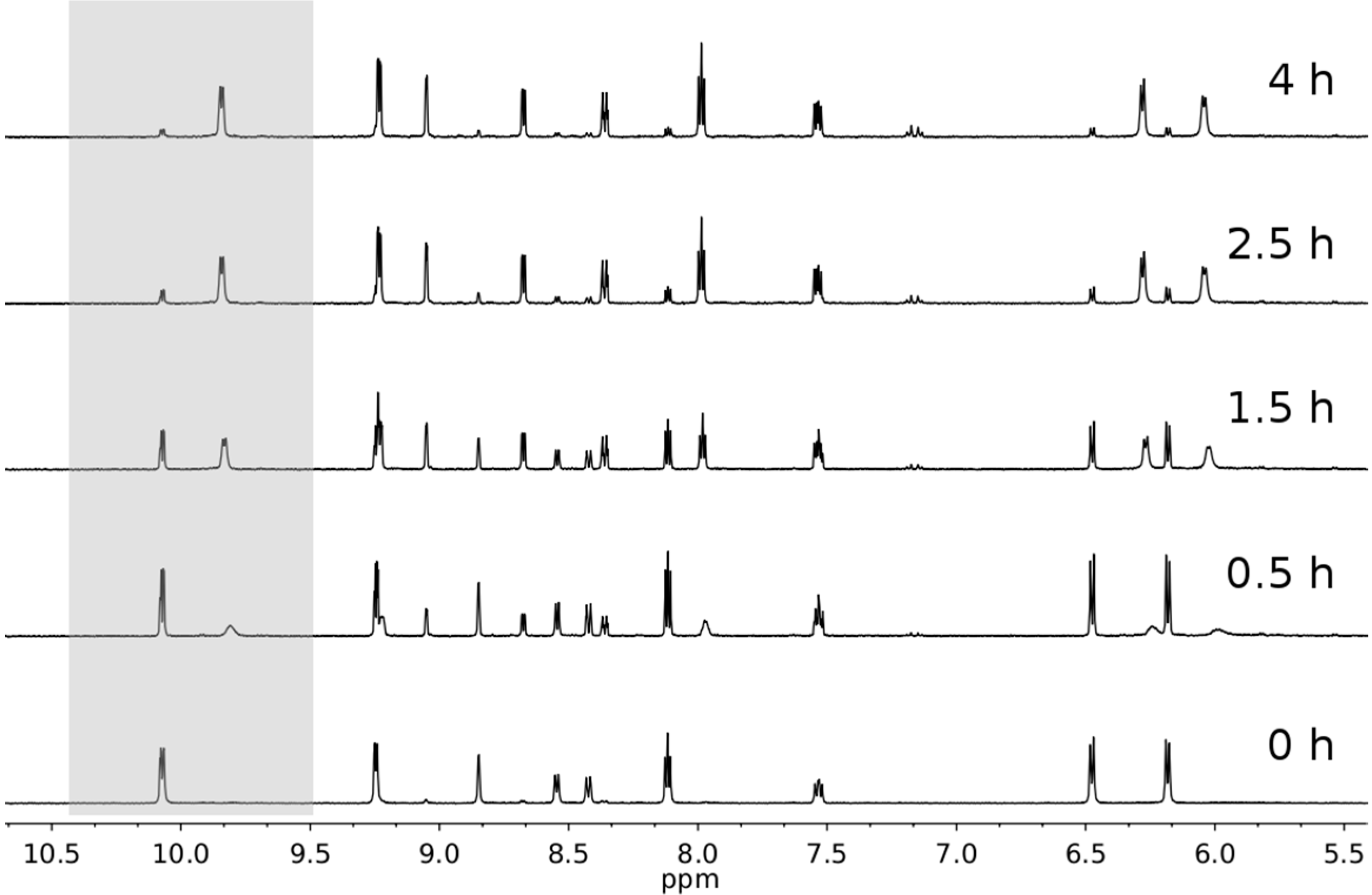
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.2.1. [(η6-p-cym)Ru(bpm)(m-COOMe-Py)]2+ (1)
3.2.2. [(η6-p-cym)Ru(bpm)(p-COOMe-Py)]2+ (2)
3.2.3. [(η6-p-cym)Ru(bpy)(m-COOMe-Py)]2+ (3)
3.2.4. [(η6-p-cym)Ru(bpy)(p-COOMe-Py)]2+ (4)
3.3. X-ray Crystallography
3.4. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Bown, S.G. Photodynamic therapy for photochemists. Philos. Trans. A-Math. Phys. Eng. Sci. 2013, 371. [Google Scholar] [CrossRef]
- Schatzschneider, U. Photoactivated biological activity of transition-metal complexes. Eur. J. Inorg. Chem. 2010, 1451–1467. [Google Scholar] [CrossRef]
- Farrer, N.J.; Salassa, L.; Sadler, P.J. Photoactivated chemotherapy (PACT): The potential of excited-state d-block metals in medicine. Dalton Trans. 2009, 10690–10701. [Google Scholar] [CrossRef]
- Westendorf, A.F.; Woods, J.A.; Korpis, K.; Farrer, N.J.; Salassa, L.; Robinson, K.; Appleyard, V.; Murray, K.; Grunert, R.; Thompson, A.M.; et al. Trans,trans,trans-[Pt-IV(N3)2(OH)2(py)(NH3)]: A light-activated antitumor platinum complex that kills human cancer cells by an apoptosis-independent mechanism. Mol. Cancer Ther. 2012, 11, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Howerton, B.S.; Heidary, D.K.; Glazer, E.C. Strained ruthenium complexes are potent light-activated anticancer agents. J. Am. Chem. Soc. 2012, 134, 8324–8327. [Google Scholar] [CrossRef] [PubMed]
- Dolmans, D.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Zayat, L.; Filevich, O.; Baraldo, L.M.; Etchenique, R. Ruthenium polypyridyl phototriggers: From beginnings to perspectives. Philos. Trans. A-Math. Phys. Eng. Sci. 2013, 371. [Google Scholar] [CrossRef]
- Filevich, O.; Etchenique, R. RuBiGABA-2: A hydrophilic caged GABA with long wavelength sensitivity. Photochem. Photobiol. Sci. 2013, 12, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Ford, P.C. Polychromophoric metal complexes for generating the bioregulatory agent nitric oxide by single- and two-photon excitation. Acc. Chem. Res. 2008, 41, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Fry, N.L.; Mascharak, P.K. Photoactive ruthenium nitrosyls as NO donors: How to sensitize them toward visible light. Acc. Chem. Res. 2011, 44, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Schatzschneider, U. PhotoCORMs: Light-triggered release of carbon monoxide from the coordination sphere of transition metal complexes for biological applications. Inorg. Chim. Acta 2011, 374, 19–23. [Google Scholar] [CrossRef]
- Pizarro, A.M.; Habtemariam, A.; Sadler, P.J. Activation mechanisms for organometallic anticancer complexes. In Medicinal Organometallic Chemistry; Springer-Verlag Berlin: Berlin, Germany, 2010; Volume 32, pp. 21–56. [Google Scholar]
- Betanzos-Lara, S.; Salassa, L.; Habtemariam, A.; Novakova, O.; Pizarro, A.M.; Clarkson, G.J.; Liskova, B.; Brabec, V.; Sadler, P.J. Photoactivatable organometallic pyridyl ruthenium(II) arene complexes. Organometallics 2012, 31, 3466–3479. [Google Scholar] [CrossRef]
- Betanzos-Lara, S.; Salassa, L.; Habtemariam, A.; Sadler, P.J. Photocontrolled nucleobase binding to an organometallic Ru-II arene complex. Chem. Commun. 2009, 6622–6624. [Google Scholar] [CrossRef]
- Habtemariam, A.; Melchart, M.; Fernandez, R.; Parsons, S.; Oswald, I.D.H.; Parkin, A.; Fabbiani, F.P.A.; Davidson, J.E.; Dawson, A.; Aird, R.E.; et al. Structure-activity relationships for cytotoxic ruthenium(II) arene complexes containing N,N-, N,O-, and O,O-chelating ligands. J. Med. Chem. 2006, 49, 6858–6868. [Google Scholar] [CrossRef] [PubMed]
- Salassa, L.; Garino, C.; Salassa, G.; Gobetto, R.; Nervi, C. Mechanism of ligand photodissociation in photoactivable [Ru(bpy)2L2]2+ complexes: A density functional theory study. J. Am. Chem. Soc. 2008, 130, 9590–9597. [Google Scholar] [CrossRef] [PubMed]
- Camilo, M.R.; Cardoso, C.R.; Carlos, R.M.; Lever, A.B.P. Photosolvolysis of cis-[Ru(α-diimine)2(4-aminopyridine)2]2+ complexes: Photophysical, spectroscopic, and density functional theory analysis. Inorg. Chem. 2014, 53, 3694–3708. [Google Scholar] [CrossRef] [PubMed]
- Zelonka, R.A.; Baird, M.C. Benzene complexes of ruthenium(II). Can. J. Chem. 1972, 50, 3063–3072. [Google Scholar] [CrossRef]
- Bennett, M.A.; Smith, A.K. Arene ruthenium(II) complexes formed by dehydrogenation of cyclohexadienes with ruthenium(III) trichloride. J. Chem. Soc. Dalton Trans. 1974, 233–241. [Google Scholar] [CrossRef]
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2006; pp. 601–616. [Google Scholar]
- Wang, F.Y.; Habtemariam, A.; van der Geer, E.P.L.; Fernandez, R.; Melchart, M.; Deeth, R.J.; Aird, R.; Guichard, S.; Fabbiani, F.P.A.; Lozano-Casal, P.; et al. Controlling ligand substitution reactions of organometallic complexes: Tuning cancer cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2005, 102, 18269–18274. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXL97, University of Göttingen: Göttingen, Germany, 1997.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Cossi, M.; Scalmani, G.; Rega, N.; Barone, V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002, 117, 43–54. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations—Potentials for the transition-metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- O'Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Browne, W.R.; O’Boyle, N.M.; McGarvey, J.J.; Vos, J.G. Elucidating excited state electronic structure and intercomponent interactions in multicomponent and supramolecular systems. Chem. Soc. Rev. 2005, 34, 641–663. [Google Scholar] [CrossRef] [PubMed]
- Headgordon, M.; Grana, A.M.; Maurice, D.; White, C.A. Analysis of electronic-transitions as the difference of electron-attachment and detachment densities. J. Phys. Chem. 1995, 99, 14261–14270. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Adhireksan, Z.; Davey, G.E.; Campomanes, P.; Groessl, M.; Clavel, C.M.; Yu, H.J.; Nazarov, A.A.; Yeo, C.H.F.; Ang, W.H.; Droge, P.; et al. Ligand substitutions between ruthenium-cymene compounds can control protein versus DNA targeting and anticancer activity. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Carrone, G.; Gantov, F.; Slep, L.D.; Etchenique, R. Fluorescent ligands and energy transfer in photoactive ruthenium–bipyridine complexes. J. Phys. Chem. A 2014, 118, 10416–10424. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habtemariam, A.; Garino, C.; Ruggiero, E.; Alonso-de Castro, S.; Mareque-Rivas, J.C.; Salassa, L. Photorelease of Pyridyl Esters in Organometallic Ru(II) Arene Complexes. Molecules 2015, 20, 7276-7291. https://doi.org/10.3390/molecules20047276
Habtemariam A, Garino C, Ruggiero E, Alonso-de Castro S, Mareque-Rivas JC, Salassa L. Photorelease of Pyridyl Esters in Organometallic Ru(II) Arene Complexes. Molecules. 2015; 20(4):7276-7291. https://doi.org/10.3390/molecules20047276
Chicago/Turabian StyleHabtemariam, Abraha, Claudio Garino, Emmanuel Ruggiero, Silvia Alonso-de Castro, Juan C. Mareque-Rivas, and Luca Salassa. 2015. "Photorelease of Pyridyl Esters in Organometallic Ru(II) Arene Complexes" Molecules 20, no. 4: 7276-7291. https://doi.org/10.3390/molecules20047276