
Efficient Synthesis of Readily Water-Soluble Sulfonic Acid Carbamates

Abstract
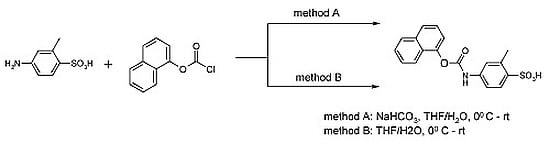
:
1. Introduction
2. Results and Discussion
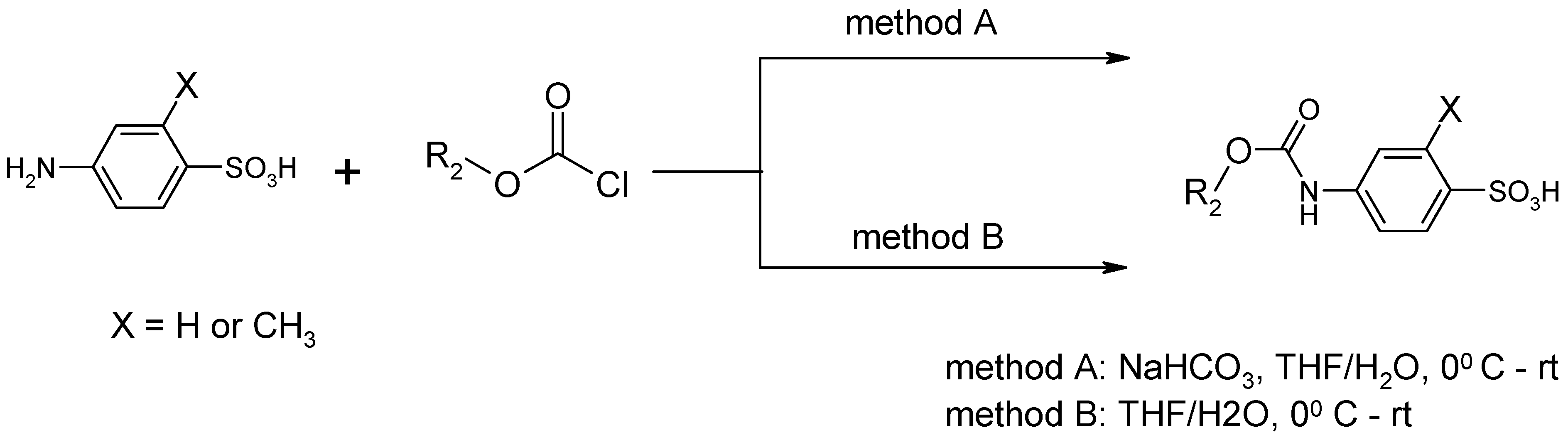

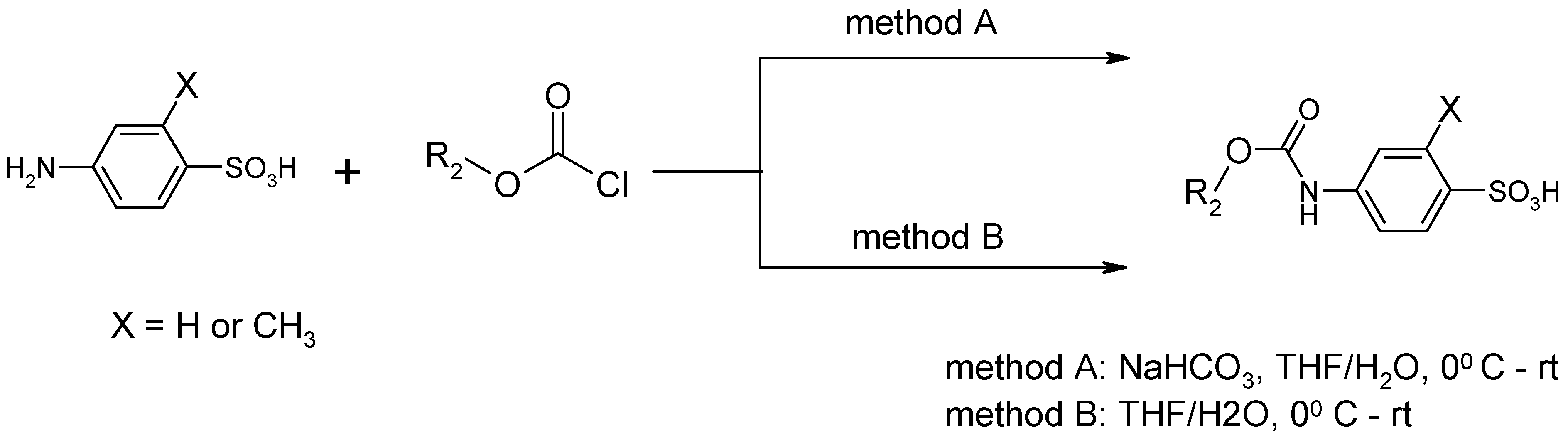
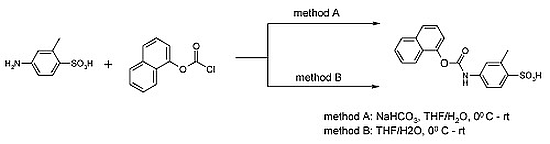{kind=link}
{kind=link}
| Entry | Substrate 1 (R1-NH2) | Substrate 2 (R2-OCOCl) | Product (R1-NHOCO-R2) | Yield (%) | ||
|---|---|---|---|---|---|---|
| A | B | |||||
| 1 |  |  |  | 99 | 99 | |
| 2 |  |  |  | 98 | 98 | |
| 3 |  |  |  | 98 | 97 | |
| 4 |  |  |  | 99 | 98 | |
| 5 |  |  |  | 98 | 96 | |
| 6 |  |  |  | 98 | 98 | |
| 7 |  |  |  | 97 | 94 | |
| 8 |  |  |  | 99 | 95 | |
| 9 |  |  |  | 97 | 97 | |
| 10 |  |  |  | 97 | 96 | |
3. Experimental Section
3.1. General
3.2. Synthesis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kim, M.K.; Park, K.S.; Yeo, W.S.; Choo, H.; Chong, Y. In vitro solubility, stability and permeability of novel quercetin-amino acid conjugates. Bioorg. Med. Chem. 2009, 17, 1164–1171. [Google Scholar]
- Mulholland, P.J.; Ferry, D.R.; Anderson, D.; Hussain, S.A.; Young, A.M.; Cook, J.E.; Hodgkin, E.; Seymour, L.W.; Kerr, D.J. Pre-clinical and clinical study of QC12, a water-soluble, pro-drug of quercetin. Ann. Oncol. 2001, 12, 245–248. [Google Scholar]
- Ninomiya, M.; Tanaka, K.; Tsuchida, Y.; Muto, Y.; Koketsu, M.; Watanabe, K. Increased bioavailability of tricin-amino acid conjugates via a prodrug approach. J. Med. Chem. 2011, 54, 1529–1536. [Google Scholar]
- Rubio-Aliaga, I.; Daniel, H. Mammalian peptide transporters as targets for drug delivery. Trends Pharmacol. Sci. 2002, 23, 434–440. [Google Scholar]
- Cao, F.; Guo, J.X.; Ping, Q.N.; Liao, Z.G. Prodrugs of scutellarin: ethyl, benzyl and N,N-diethylglycolamide ester synthesis, physicochemical properties, intestinal metabolism and oral bioavailability in the rats. Eur. J. Pharm. Sci. 2006, 29, 385–393. [Google Scholar]
- Labanca, R.A.; Gloria, M.B.A. Spectrophotometric determination of urea in sugar cane distilled spirits. J. Agric. Food Chem. 2008, 56, 5211–5215. [Google Scholar]
- Esti, M.; Fidaleo, M.; Moresi, M.; Tamborra, P. Modeling of urea degradation in white and rosé wines by acid urease. J. Agric. Food Chem. 2007, 55, 2590–2596. [Google Scholar]
- Lachenmeier, D.W.; Lima, M.C.P.; Nobrega, I.C.C.; Pereira, J.A.P.; Kerr-Correa, F.; Kanteres, F.; Rehm, J. Cancer risk assessment of ethyl carbamate in alcoholic beverages from Brazil with special consideration to the spirits cachaça and tiquira. BMC Cancer 2010, 10, 266–280. [Google Scholar]
- Araque, I.; Gil, J.; Carrete, R.; Bordons, A.; Reguant, C. Detection of arc genes related with the ethyl carbamate precursors in wine lactic acid bacteria. J. Agric. Food Chem. 2009, 57, 1841–1847. [Google Scholar]
- Stokes, K.; McVenes, R.; Anderson, J.M. Polyurethane elastomer biostability. J. Biomater. Appl. 1995, 9, 321–354. [Google Scholar]
- Choi, T.; Weksler, J.; Padsalgikar, A.; Runt, J. Novel hard-block polyurethanes with high strength and transparency for biomedical applications. J. Biomater. Sci. Polym. Ed. 2011, 22, 973–980. [Google Scholar]
- Szycher, M. Szycher’s Handbook of Polyurethanes; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Hirsch, D.J.; Bergen, P.; Jindal, K.K. Polyurethane catheters for long-term hemodialysis access. Artif. Organs 1997, 21, 349–354. [Google Scholar]
- Browning, M.B.; Dempsey, D.; Guiza, V.; Becerra, S.; Rivera, J.; Russell, B.; Clubb, F.; Miller, M.; Fossum, T.; Dong, J.F.; et al. Multilayer vascular grafts based on collagen-mimetic proteins. Acta Biomater. 2012, 8, 1010–1021. [Google Scholar]
- Smith, S.L.; Ash, H.E.; Unsworth, A. A tribological study of UHMWPE acetabular cups and polyurethane compliant layer acetabular cups. J. Biomed. Mater. Res. Part B 2000, 53, 710–716. [Google Scholar]
- McCoy, T.J.; Wabers, H.D.; Cooper, S.L. Series shunt evaluation of polyurethane vascular graft materials in chronically AV-shunted canines. J. Biomed. Mater. Res. 1990, 24, 107–129. [Google Scholar]
- Fernandez, M.; Pico, Y.; Manes, J. Determination of carbamate residues in fruit and vegetables by matrix solid-phase dispersion and liquid chromatography-mass spectrometry. J. Chromatogr. A 2000, 871, 43–56. [Google Scholar]
- US Environmental Protection Agency. National Survey of Pesticides in Drinking Water Wells, Phase II Report; EPA 570/9-91-020; National Technical Information Service; US Environmental Protection Agency: Springfield, VA, USA, 1992.
- Directive 2006/118/EC of the European Parliament and of the Council of 12 December 2006 on the protection of groundwater against pollution and deterioration. Off. J. Eur. Union 2006, L372, 19–31.
- Nottebohm, M.; Licha, T.; Sauter, M. Tracer design for tracking thermal fronts in geothermal reservoirs. Geothermics 2012, 43, 37–44. [Google Scholar]
- Tester, J.W.; Robinson, B.A.; Ferguson, J.H. The theory and selection of chemically reactive tracers for reservoir thermal capacity production. In Proceedings of the Twelfth Workshop on Geothermal Reservoir Engineering, Stanford University, Stanford, CA, USA, 20–22 January 1987. SGP-TR-109.
- Houben-Weyl. In Methods of Organic Chemistry; Georg Thieme Publishers: Stuttgart, Germany, 1952; Volume 8, pp. 137, 120 and 101.
- Spegazzini, N.; Siesler, H.W.; Ozak, Y. Modeling of isomeric structure of diphenyl urethane by FT-IR spectroscopy during synthesis from phenylisocyanate and phenol as an inverse kinetic problem. J. Phys. Chem. A 2011, 115, 8832–8844. [Google Scholar]
- Spegazzini, N.; Siesler, H.W.; Ozak, Y. Sequential identification of model parameters by derivative double two-dimensional correlation spectroscopy and calibration-free approach for chemical reaction systems. Anal. Chem. 2012, 84, 8330–8339. [Google Scholar]
- Hutchby, M.; Houlden, C.E.; Ford, J.G.; Tyler, S.N.G.; Gagn, M.R.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Hindered ureas as masked isocyanates: Facile carbamoylation of nucleophiles under neutral conditions. Angew. Chem. Int. Ed. 2009, 48, 8721–8724. [Google Scholar]
- Gurgiolo, A.E.; Jackson, L. Preparation of Carbamates from Aromatic Amines and Organic Carbonates. U.S. Patent 4,268,683, 19 May 1981. [Google Scholar]
- Franz, R.A.; Applegath, F.; Morriss, F.V.; Breed, L.W. A new synthesis of ureas. The preparation of methyl-N-phenylurethane from carbon monoxide, sulfur, aniline, and methanol. J. Org. Chem. 1963, 28, 585–586. [Google Scholar]
- Putnam Bennett, R.; Hardy, W.B. Process of Producing Urethanes. U.S. Patent 3,467,694, 16 September 1969. [Google Scholar]
- Idzik, K.R.; Noedler, K.; Licha, T. Efficient synthesis of readily water soluble amides containing sulfonic groups. Synth. Commun. 2014, 44, 133–140. [Google Scholar]
- Idzik, K.R.; Noedler, K.; Friedrich, M.; Licha, T. Efficient synthesis and reaction kinetics of readily water soluble esters containing sulfonic groups. Molecules 2014, 19, 21022–21033. [Google Scholar]
- Nödler, K.; Licha, T.; Bester, K.; Sauter, M. Development of a multi-residue analytical method, based on liquid chromatography-tandem mass spectrometry, for the simultaneous determination of 46 micro-contaminants in aqueous samples. J. Chromatogr. A 2010, 1217, 6511–6521. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idzik, K.R.; Nödler, K.; Licha, T. Efficient Synthesis of Readily Water-Soluble Sulfonic Acid Carbamates. Molecules 2015, 20, 6856-6865. https://doi.org/10.3390/molecules20046856
Idzik KR, Nödler K, Licha T. Efficient Synthesis of Readily Water-Soluble Sulfonic Acid Carbamates. Molecules. 2015; 20(4):6856-6865. https://doi.org/10.3390/molecules20046856
Chicago/Turabian StyleIdzik, Krzysztof R., Karsten Nödler, and Tobias Licha. 2015. "Efficient Synthesis of Readily Water-Soluble Sulfonic Acid Carbamates" Molecules 20, no. 4: 6856-6865. https://doi.org/10.3390/molecules20046856
APA StyleIdzik, K. R., Nödler, K., & Licha, T. (2015). Efficient Synthesis of Readily Water-Soluble Sulfonic Acid Carbamates. Molecules, 20(4), 6856-6865. https://doi.org/10.3390/molecules20046856