
β–Cyclodextrin–Propyl Sulfonic Acid Catalysed One-Pot Synthesis of 1,2,4,5-Tetrasubstituted Imidazoles as Local Anesthetic Agents

Abstract
:
1. Introduction
2. Results and Discussion
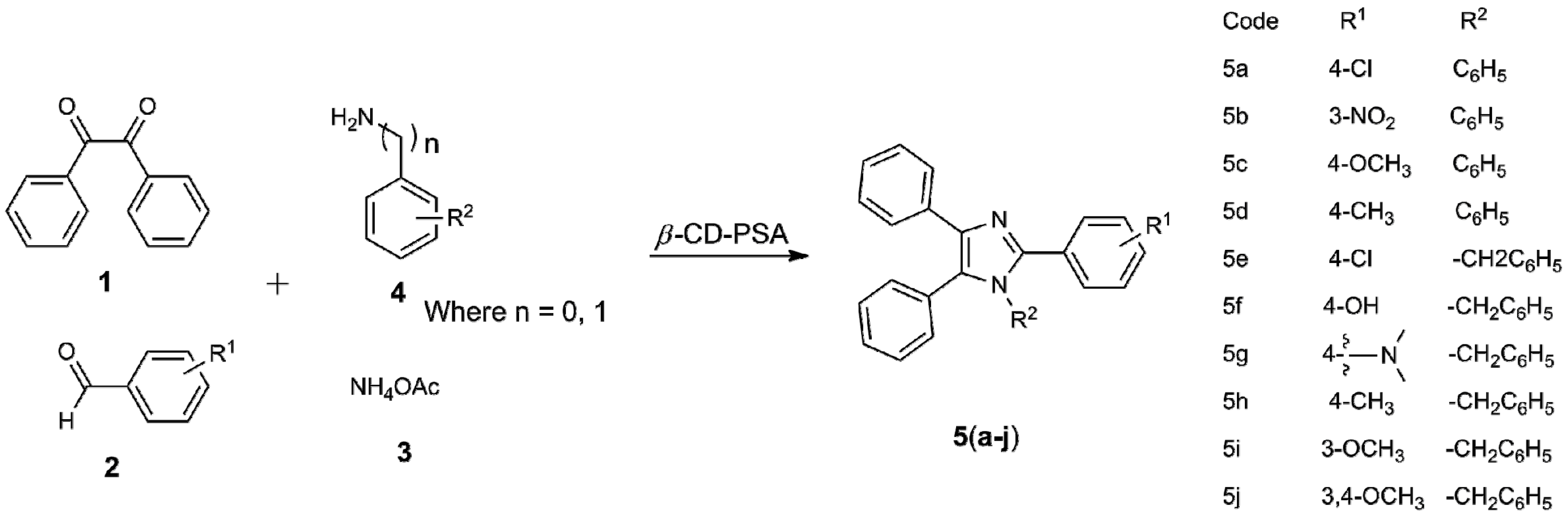
2.1. Chemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvents | Temp (°C) | Amount (mol %) | Time (min) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | No Solvent | rt. | -- | 150 | No reaction |
| 2 | No Solvent | 80 | -- | 150 | No reaction |
| 3 | No Solvent | rt. | 2 | 150 | 28 |
| 4 | No Solvent | 50 | 2 | 40 | 55 |
| 5 | No Solvent | 80 | 2 | 30 | 85 |
| 6 | No Solvent | 100 | 2 | 20 | 96 |
| 7 | No Solvent | 80 | 1 | 70 | 74 |
| 8 | No Solvent | 80 | 3 | 50 | 92 |
| 9 | Water | 80 | 2 | 30 | 63 |
| 10 | CH3CH2OH | reflux | 2 | 40 | 78 |
| 11 | ClCH2CH2Cl | reflux | 2 | 40 | 45 |
| 12 | EtOAc | reflux | 2 | 40 | 70 |
| 13 | THF | reflux | 2 | 40 | 56 |
| 14 | CH3CN | 100 | 2 | 40 | 64 |
| 15 | DMF | 100 | 2 | 40 | 72 |
| Entry | Catalyst | Solvent | Condition | Yield (in %) b | Time | Ref. |
|---|---|---|---|---|---|---|
| 1 | Zeolite HY | - | MW | 42–85 | 6 min | [12] |
| 2 | Silica gel | - | MW | 60–90 | 6 min | [12] |
| 3 | Silica gel/NaHSO4 | DCM | reflux | 85–90 | 2.5 h | [13] |
| 4 | HClO4–SiO2 | - | 140 °C | 60–94 | 15–20 min | [14] |
| 5 | I2 | Ethanol | 75°C | 97–99 | 15–20 min | [15] |
| 6 | K5CoW12O40·3H2O | DCM | 140 °C | 15–95 | 2.5 h | [8] |
| 7 | BF3·SiO2 | - | 140 °C | 80–96 | 2 h | [16] |
| 8 | InCl3·3H2O | MeOH | r.t. | 47–84 | 6–9 h | [17] |
| 10 | [(CH2)4SO3HMIM] | - | 140 °C | 85–95 | 2–2.5 h | [20] |
| 11 | MCM-41 | - | 140 °C | 74–82 | 1.92–2.25 h | [21] |
| 12 | MCM-41 | AcOH | reflux | 75–85 | 25–35 min | [21] |
| 13 | p-TsOH | - | 140 °C | 75–82 | 1.92–2.17 h | [21] |
| 14 | p-TsOH | EtOH | reflux | 73–83 | 13–23 min | [21] |
| 15 | - | 1-Butyl-3-methylimidazolium bromide | 140 °C | 82–93 | 1.5–5 h | [22] |
| 16 | - | 1-Butyl-3-methylimidazolium bromide | MW | 82–93 | 3–8 min | [22] |
| 17 | P2O5/SiO2 | - | 100 °C | 87–98 | 15–55 min | [23] |
| 18 | - | - | 140 °C | 0 | 3 h | [20] |
| 19 | SiO2-Pr-SO3H | - | 140 °C | 85–98 | 10 min–3 h | [19] |
| 20 | β-CD-Pr-SO3H | - | 100 °C | 68–96 | 15 min–4 h | (This work) |
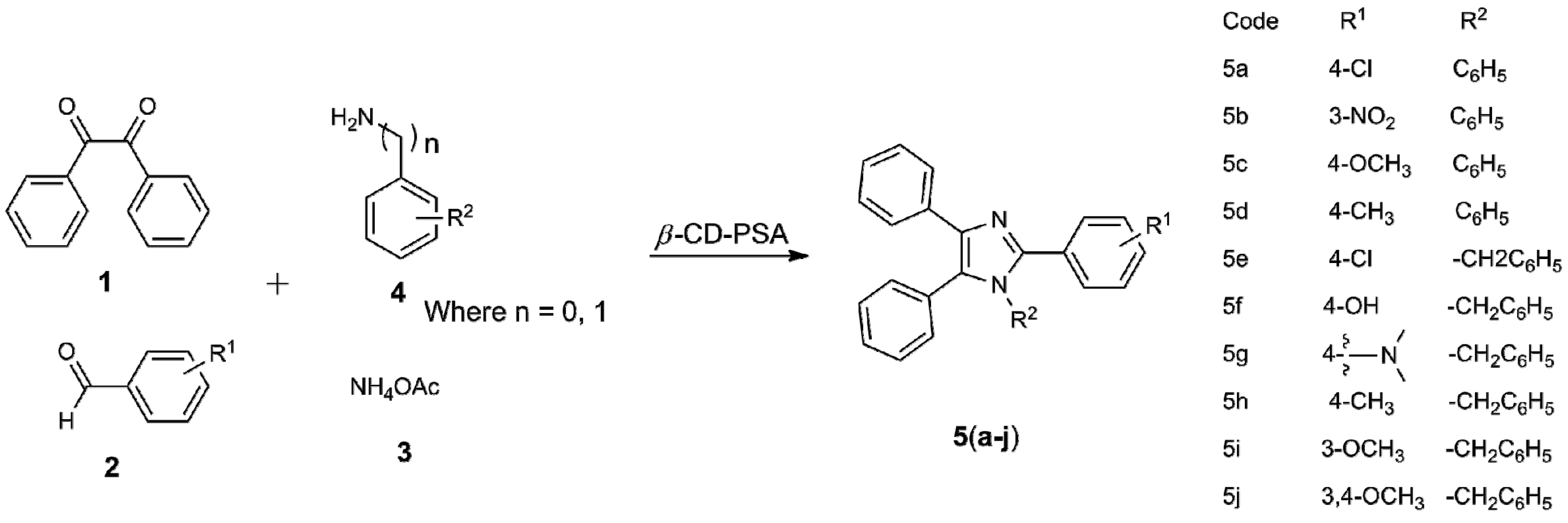
| Entry | R1 | R2 | Product | Time | Yield (in %) b | M.P. in °C (Observed) | M.P. in °C (in Literature) |
|---|---|---|---|---|---|---|---|
| 1 | 4-Cl | C6H5 | 5a | 30 min | 96 | 158–160 | 157–159 |
| 2 | 3-NO2 | C6H5 | 5b | 15 min | 93 | 247–249 | 249–250 |
| 3 | 4-OCH3 | C6H5 | 5c | 3 h | 90 | 180–182 | 181–183 |
| 4 | 4-CH3 | C6H5 | 5d | 2.5 h | 90 | 188–190 | 189–190 |
| 5 | 4-Cl | -CH2C6H5 | 5e | 3 h | 89 | 166–168 | 163–165 |
| 6 | 4-OH | -CH2C6H5 | 5f | 3 h | 80 | 132–134 | 131–132 |
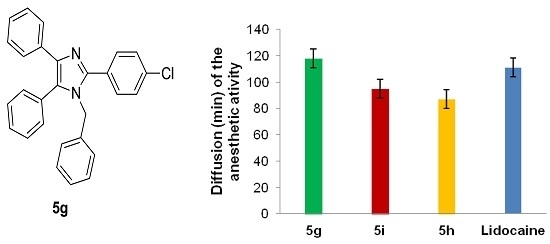
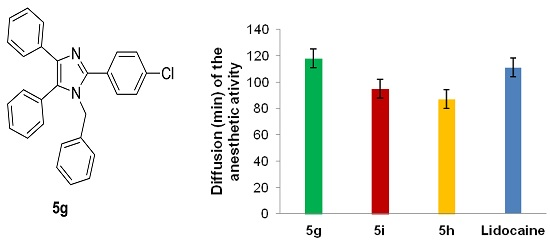
| 7 | 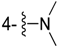 | -CH2C6H5 | 5g | 3.5 h | 73 | 154–155 | 155–157 |
| 8 | 4-CH3 | -CH2C6H5 | 5h | 3.5 h | 75 | 160–162 | 163–165 |
| 9 | 3-OCH3 | -CH2C6H5 | 5i | 4 h | 70 | 115–117 | 128–129 |
| 10 | 3,4-OCH3 | -CH2C6H5 | 5j | 4 h | 68 | 189–191 | 188–190 |
2.2. Reusability of the Catalyst
| No. of Runs | Percentage Yield |
|---|---|
| 1 | 96 |
| 2 | 94 |
| 3 | 90 |
| 4 | 90 |
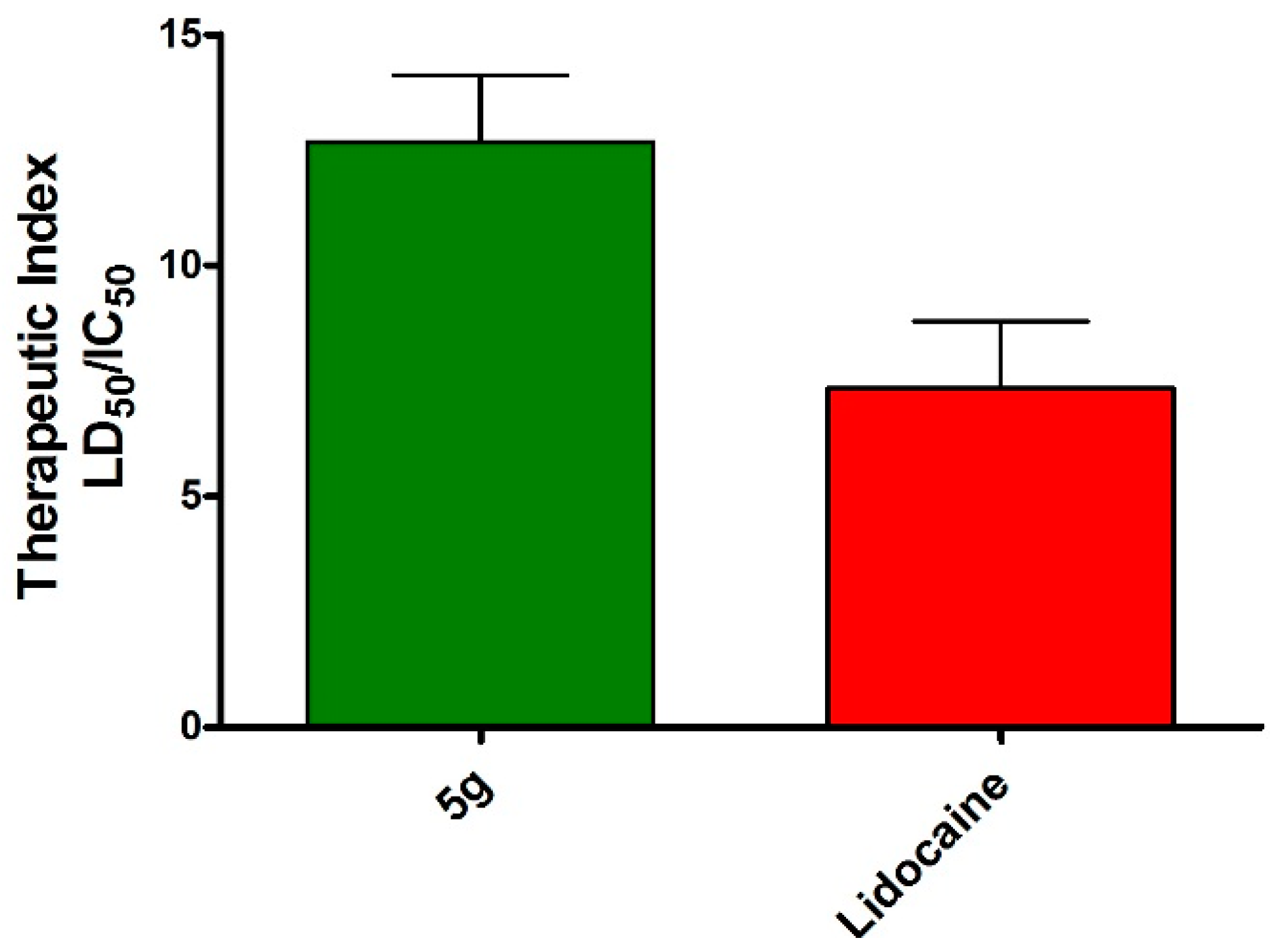
2.3. Pharmacology
Local Anesthetic Activity
| Compound | Corneal Anesthesia a | Mouse Tail Anesthesia b |
|---|---|---|
| 5a | 20.3 ± 8.6 | 11.3 (±0.11) × 10−2 |
| 5b | 12.4 ± 9.3 | 5.7 (±0.42) × 10−2 |
| 5c | 15.5 ± 12.3 | 6.5 (±0.23) × 10−2 |
| 5d | 10.32 ± 0.12 | 4.3 (±0.54) × 10−2 |
| 5e | 21.65 ± 4.7 | 3.4 (±0.32) × 10−2 |
| 5f | 5.5 ± 3.5 | 5.4 (±0.14) × 10−2 |
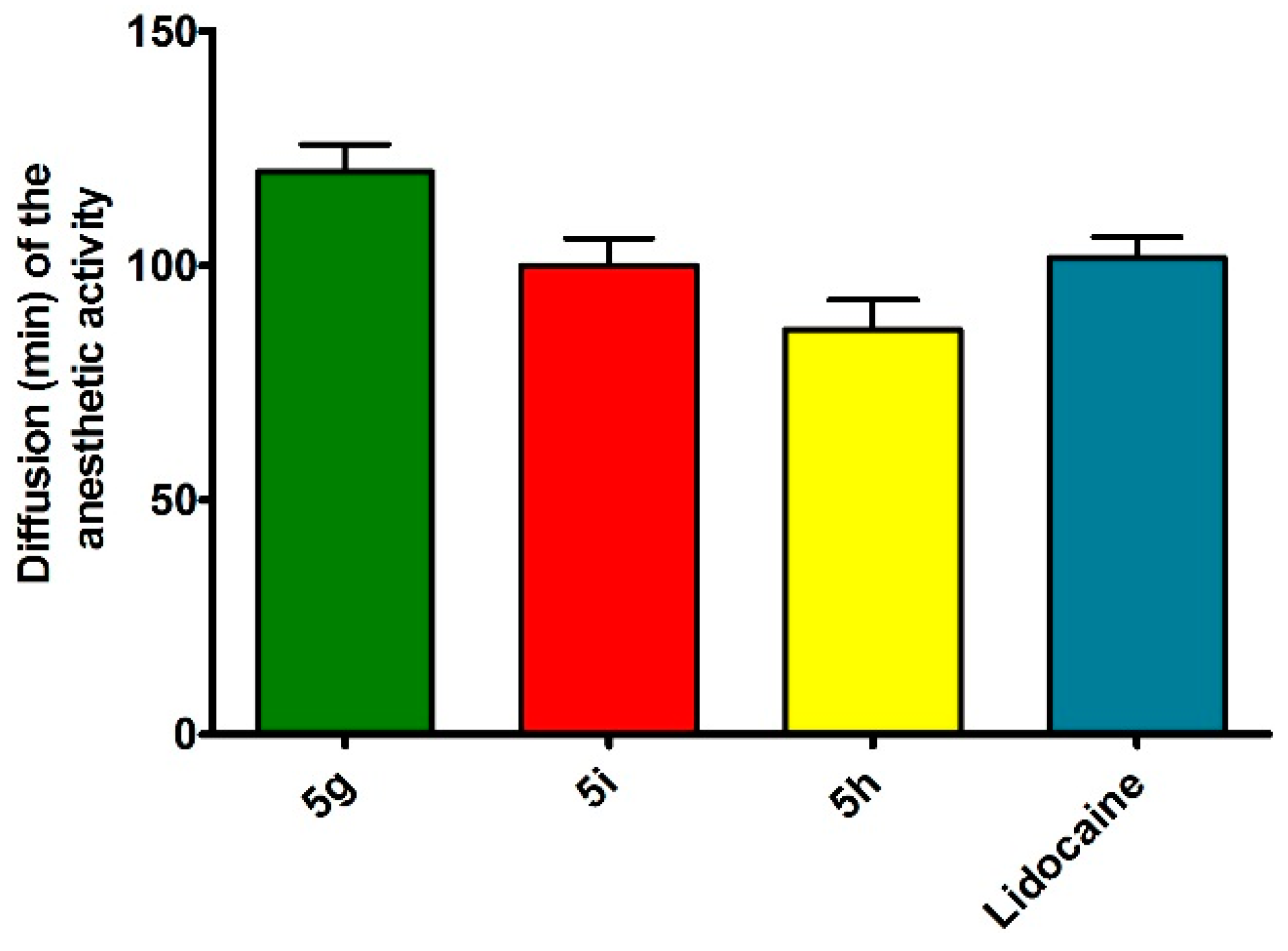
| 5g | 121.23 ± 12.3 | 1.6 (±0.26) × 10−2 |
| 5h | 65.65 ± 5.8 | 9.2 (±0.16) × 10−2 |
| 5i | 95.4 ± 13.2 | 7.4 (±0.18) × 10−2 |
| 5j | 20.3 ± 10.3 | 4.1 (±0.45) × 10−2 |
| Lidocaine HCl c | 100 | 2.1 (±0.25) × 10−2 |
3. Experimental Section
3.1. General Information
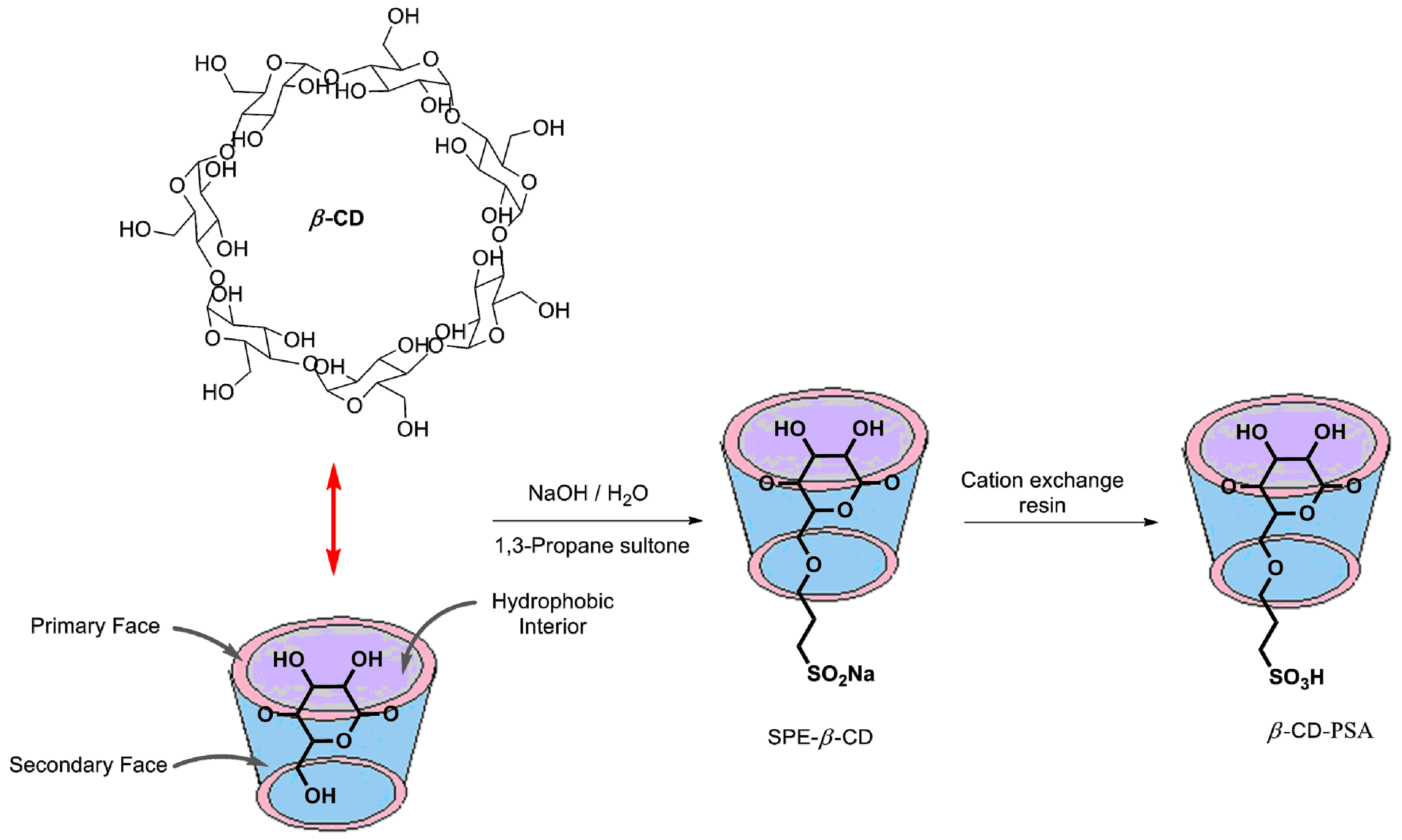
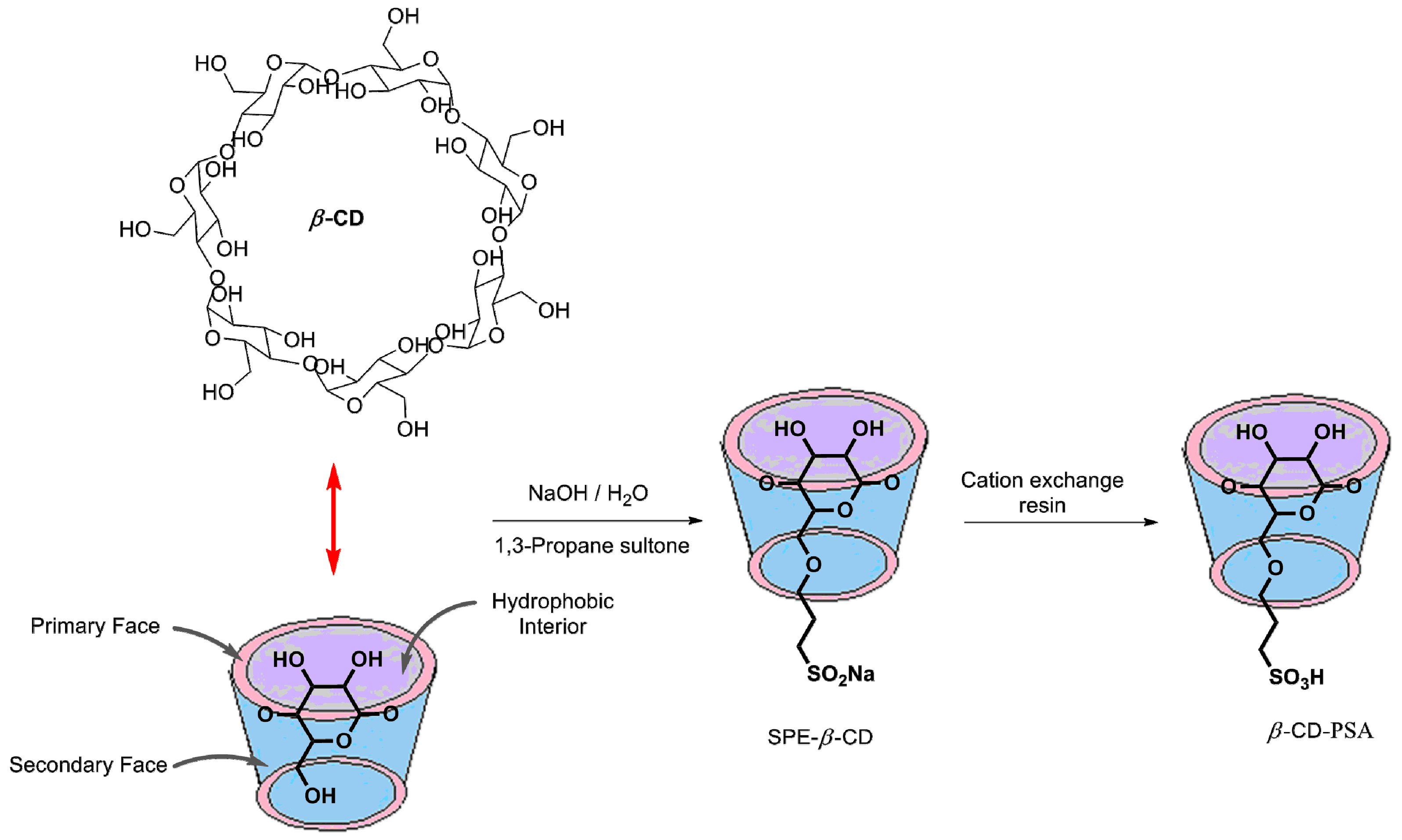
3.1.1. Synthesis of β-CD-PSA
3.1.2. General Procedure for the Synthesis of 1,2,4,5-Tetrasubstituted Imidazoles 5(a–j)
3.2. Pharmacology
3.2.1. Corneal Anesthesia
3.2.2. Mouse Tail Anesthesia
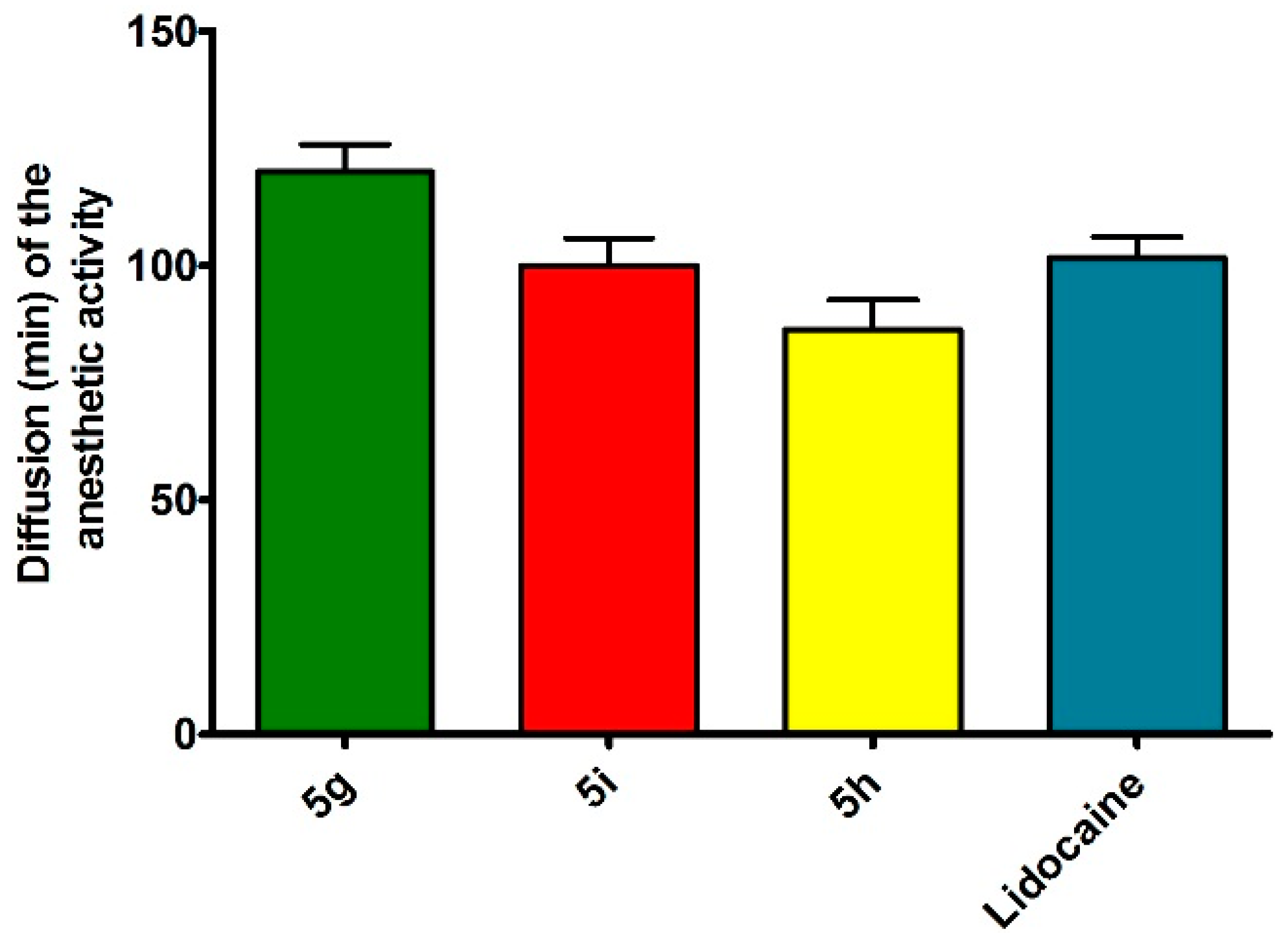
3.2.3. Rat Sciatic Nerve Block
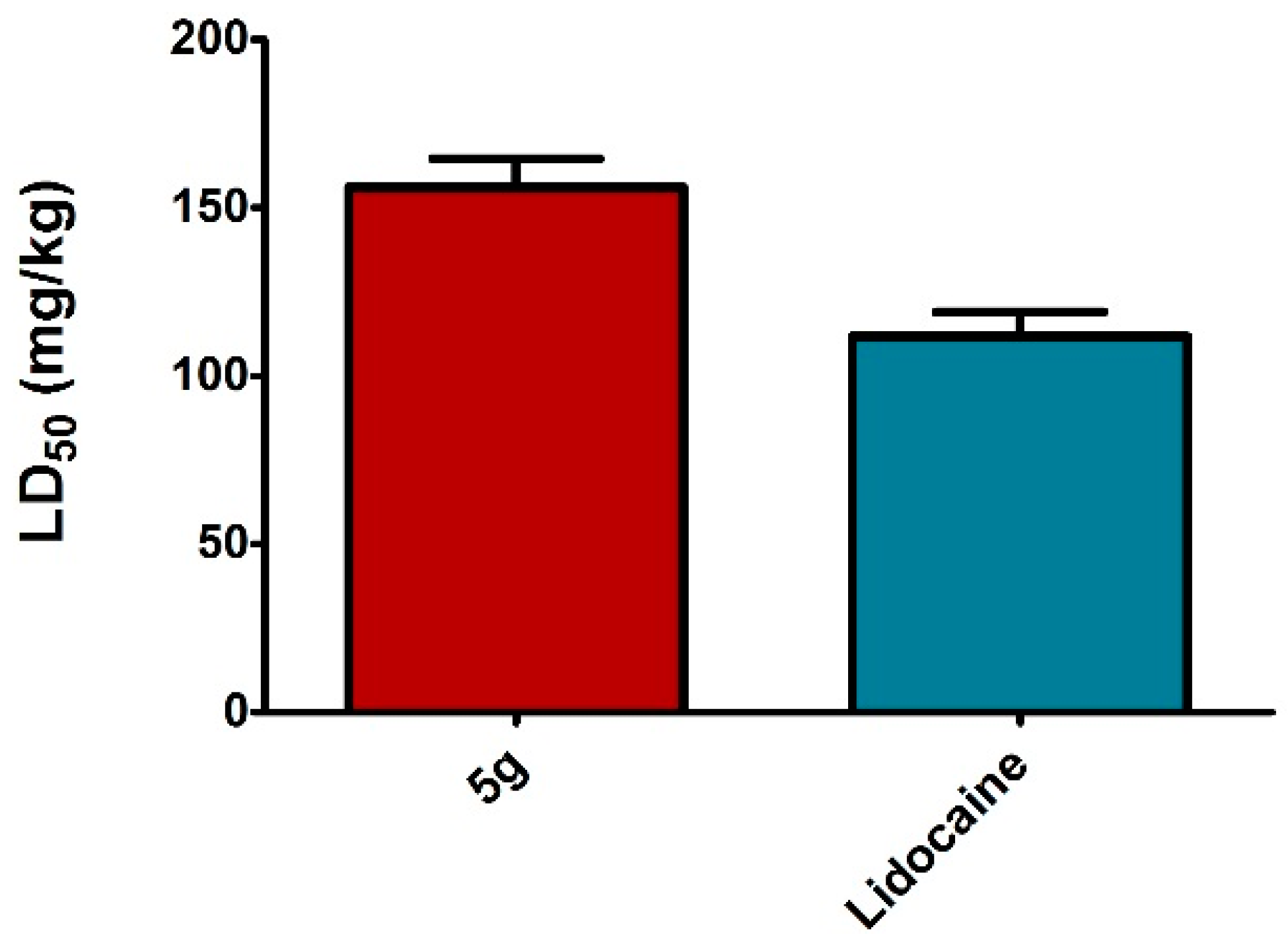
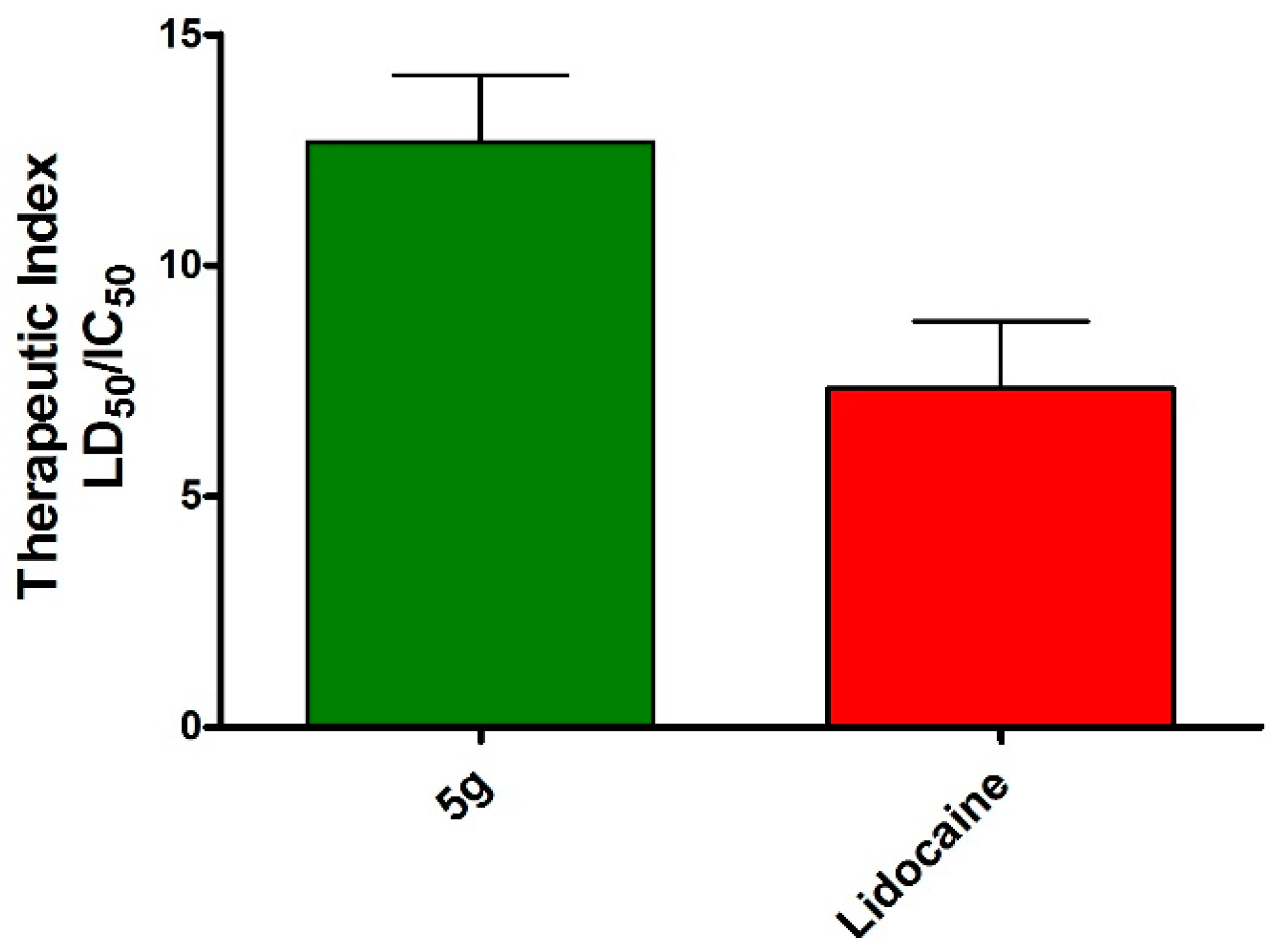
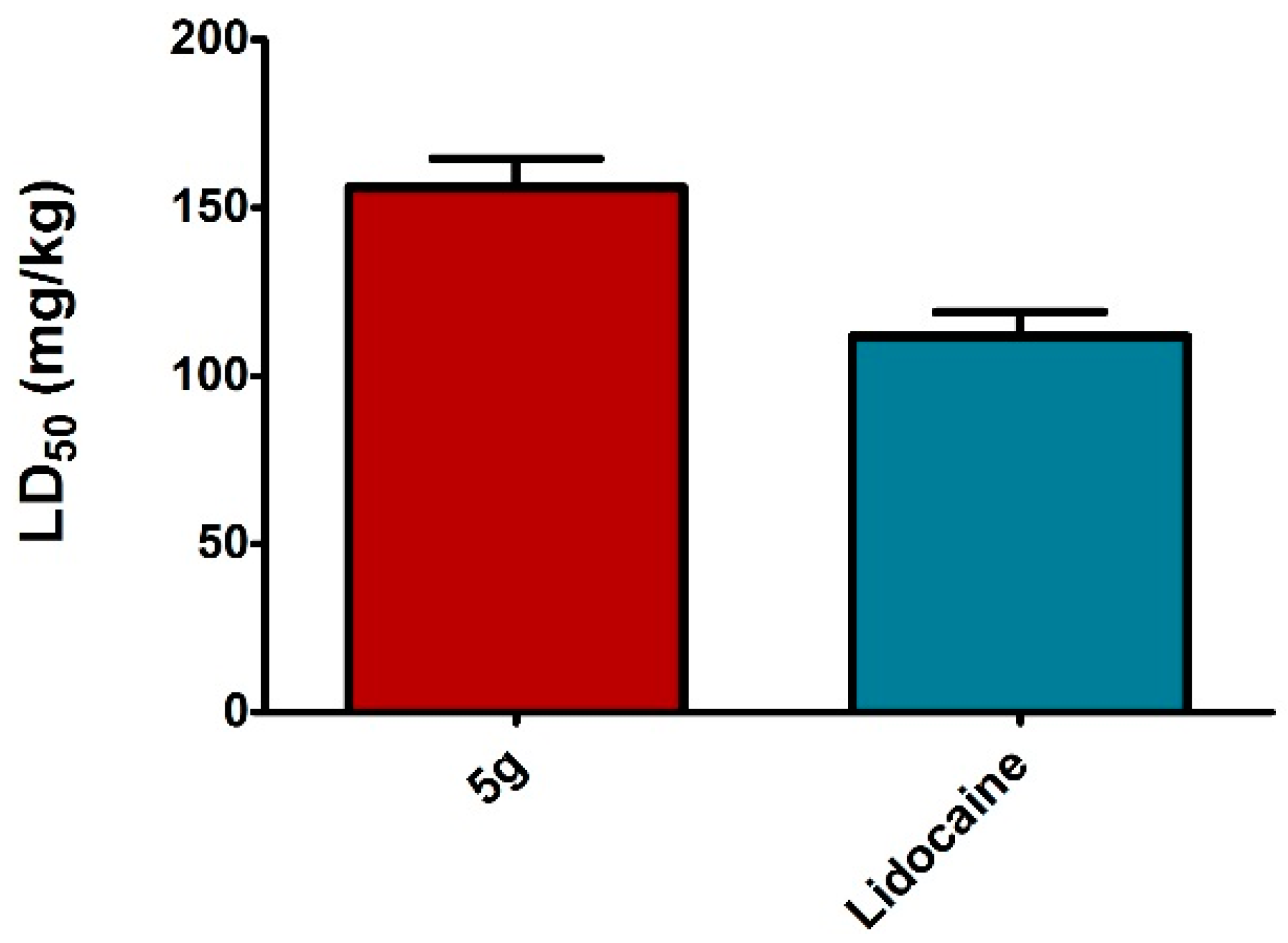
3.2.4. Acute Toxicity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dua, R.; Shrivastava, S.; Sonwane, S.K.; Srivastava, S.K. Pharmacological significance of synthetic heterocycles scaffold: A Review. Adv. Biol. Res. 2011, 5, 120–144. [Google Scholar]
- Zhang, L.; Peng, X.M.; Damu, G.L.; Geng, R.X.; Zhou, C.H. Comprehensive review in current developments of imidazole-based medicinal chemistry. Med. Res. Rev. 2014, 34, 340–437. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Nagle, A.; Kuhen, K.; Gagaring, K.; Borboa, R.; Francek, C.; Chen, Z.; Plouffe, D.; Goh, A.; Lakshminarayana, S.B.; et al. Imidazolopiperazines: Hit to lead optimization of new antimalarial agents. J. Med. Chem. 2011, 54, 5116–5130. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, S.; Selvan, P.S.; Gopal, N.; Gupta, J.K.; De, B. Synthesis and antibacterial activity of some Imidazole-5-(4H)one derivatives. Arch. Pharm. 2005, 338, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.C.; Adamus, W.S.; Kuthan, H. Antihistaminic activity and side effect profile of epinastine and terfenadine in healthy volunteers. Int. J. Clin. Pharmacol. Ther. Toxicol. 1990, 28, 493–497. [Google Scholar] [PubMed]
- Silva, V.G.; Silva, R.O.; Damasceno, S.R.; Carvalho, N.S.; Prudêncio, R.S.; Aragão, K.S.; Guimarães, M.A.; Campos, S.A.; Véras, L.M.; Godejohann, M.; et al. Anti-inflammatory and antinociceptive activity of epiisopiloturine, an imidazole alkaloid isolated from Pilocarpus microphyllus. J. Nat. Prod. 2013, 76, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Sondhi, S.M.; Jain, S.; Dinodia, M.; Kumar, A. Synthesis of some thiophene, imidazole and pyridine derivatives exhibiting good anti-inflammatory and analgesic activities. Med. Chem. 2008, 4, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Pandey, J.; Tiwari, V.K.; Verma, S.S.; Chaturvedi, V.; Bhatnagar, S.; Sinha, S.; Gaikwad, A.N.; Tripathi, R.P. Synthesis and antitubercular screening of imidazole derivatives. Eur. J. Med. Chem. 2009, 44, 3350–3355. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, G.; Boiani, M.; Cerecetto, H.; Gerpe, A.; González, M.; Sainz, Y.F.; Denicola, A.; de Ocáriz, C.O.; Nogal, J.J.; Montero, D.; et al. Novel antiprotozoal products: Imidazole and benzimidazole N-oxide derivatives and related compounds. Arch. Pharm. 2004, 337, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S. Synthesis and anthelmintic activity of some novel 2-substituted-4,5-diphenyl imidazoles. Acta Pharm. 2010, 60, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Tamas, G.; Xue, L.; Simon, S.L.; Quitevis, E.L. Thermophysical properties of Imidazolium-based ionic liquids: The effect of aliphatic versus aromatic functionality. J. Chem. Eng. Data 2014, 59, 2717–2724. [Google Scholar] [CrossRef]
- Balalaei, S.; Arabanian, A. One-pot synthesis of tetrasubstituted imidazoles catalyzed by zeolite HY and silica gel under microwave irradiation. Green Chem. 2000, 2, 274–276. [Google Scholar] [CrossRef]
- Karimi, A.R.; Alimohammadi, Z.; Azizian, J.; Mohammadi, A.A.; Mohammadizadeh, M.R. Solvent-free synthesis of tetrasubstituted imidazoles on silica gel/NaHSO4 support. Catal. Commun. 2006, 7, 728–732. [Google Scholar] [CrossRef]
- Kantevari, S.; Vuppalapati, S.V.N.; Biradar, D.O.; Nagarapu, L. Highly efficient, one-pot, solvent-free synthesis of tetrasubstituted imidazoles using HClO4–SiO2 as novel heterogeneous catalyst. J. Mol. Catal. A Chem. 2007, 266, 109–113. [Google Scholar] [CrossRef]
- Kidwai, M.; Mothsra, P.; Bansal, V.; Somvanshi, R.K.; Ethayathulla, A.S.; Dey, S.; Singh, T.P. One-pot synthesis of highly substituted imidazoles using molecular iodine: A versatile catalyst. J. Mol. Catal. A Chem. 2007, 265, 177–182. [Google Scholar] [CrossRef]
- Sadeghi, B.; Mirjalili, B.B.F.; Hashemi, M.M. BF3·SiO2: An efficient reagent system for the one-pot synthesis of 1,2,4,5-tetrasubstituted imidazoles. Tetrahedron Lett. 2008, 49, 2575–2577. [Google Scholar] [CrossRef]
- Sharma, D.; Hazarika, P.; Konwar, D. An efficient and onepot synthesis of 2,4,5-trisubstituted and 1,2,4,5-tetrasubstituted imidazoles catalyzed by InCl3·3H2O. Tetrahedron Lett. 2008, 49, 2216–2220. [Google Scholar] [CrossRef]
- Nagarapu, L.; Apuri, S.; Kantevari, S. Potassium dodecatugstocobaltate trihydrate (K5CoW12O40·3H2O): A mild and efficient reusable catalyst for the one-pot synthesis of 1,2,4,5-tetrasubstituted imidazoles under conventional heating and microwave irradiation. J. Mol. Catal. A Chem. 2007, 266, 104–108. [Google Scholar] [CrossRef]
- Ziarani, M.G.; Dashtianeh, Z.; Nahad, M.S.; Badiei, A. One-pot synthesis of 1,2,4,5-tetra substituted imidazoles using sulfonic acid functionalized silica (SiO2-Pr-SO3H). Arabian J. Chem. 2015, 8, 692–697. [Google Scholar] [CrossRef]
- Davoodnia, A.; Heravi, M.M.; Safavi-Rad, Z.; Tavakoli-Hoseini, N. Green, one-pot, solvent-free synthesis of 1,2,4,5-tetrasubstituted imidazoles using a Bronsted acidic ionic liquid as novel and reusable catalyst. Synth. Commun. 2010, 40, 2588–2597. [Google Scholar] [CrossRef]
- Shoar, R.H.; Rahimzadeh, G.; Derikvand, F.; Farzaneh, M. Four-component, one-pot synthesis of tetra-substituted imidazoles using a catalytic amount of MCM-41 or p-TsOH. Synth. Commun. 2010, 40, 1270–1275. [Google Scholar] [CrossRef]
- Hasaninejad, A.; Zare, A.; Shekouhy, M.; Ameri Rad, J. Catalyst-free one-pot four component synthesis of polysubstituted imidazoles in neutral ionic liquid 1-butyl-3-methylimidazolium bromide. J. Comb. Chem. 2010, 12, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Shaterian, H.R.; Ranjbar, M.; Azizi, K. Efficient multicomponent synthesis of highly substituted imidazoles utilizing P2O5/SiO2 as a reusable catalyst. Chin. J. Chem. 2011, 29, 1635–1645. [Google Scholar] [CrossRef]
- Uçucu, Ŭ.; Karaburun, N.G.; Işikdağ, Ĭ. Synthesis and analgesic activity of some 1-benzyl-2-substituted-4,5-diphenyl-1H-imidazole derivatives. IL Farmaco 2001, 56, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Regnier, J. Essai de mesure de l’anesthésie produite sur les terminaisons nerveuses (cornée, muqueuse linguale) par les anesthésiques locaux. Comparaison des pouvoirs anesthésiques. Bull. Sci. Pharm. 1923, 30, 580–586. [Google Scholar]
- Bianchi, C. A simple new quantitative method for testing local anaesthetics. Br. J. Pharmacol. 1956, 11, 104–106. [Google Scholar] [CrossRef]
- Tallarida, R.J.; Murray, R.B. Manual of Pharmacologic Calculations with Computer Programs, 2nd ed.; Springer: New York, NY, USA, 1981. [Google Scholar]
- Al Saadi, D.; Sneader, W.E. Pharmacological evaluation of certain novel prolonged-acting local anaesthetics. In vivo rat sciatic nerve block. Arzneim Forsch. 1991, 41, 195–198. [Google Scholar]
- Sample Availability: Samples of the compounds 5a–5j are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ran, Y.; Li, M.; Zhang, Z.-Z. β–Cyclodextrin–Propyl Sulfonic Acid Catalysed One-Pot Synthesis of 1,2,4,5-Tetrasubstituted Imidazoles as Local Anesthetic Agents. Molecules 2015, 20, 20286-20296. https://doi.org/10.3390/molecules201119696
Ran Y, Li M, Zhang Z-Z. β–Cyclodextrin–Propyl Sulfonic Acid Catalysed One-Pot Synthesis of 1,2,4,5-Tetrasubstituted Imidazoles as Local Anesthetic Agents. Molecules. 2015; 20(11):20286-20296. https://doi.org/10.3390/molecules201119696
Chicago/Turabian StyleRan, Yan, Ming Li, and Zong-Ze Zhang. 2015. "β–Cyclodextrin–Propyl Sulfonic Acid Catalysed One-Pot Synthesis of 1,2,4,5-Tetrasubstituted Imidazoles as Local Anesthetic Agents" Molecules 20, no. 11: 20286-20296. https://doi.org/10.3390/molecules201119696
APA StyleRan, Y., Li, M., & Zhang, Z.-Z. (2015). β–Cyclodextrin–Propyl Sulfonic Acid Catalysed One-Pot Synthesis of 1,2,4,5-Tetrasubstituted Imidazoles as Local Anesthetic Agents. Molecules, 20(11), 20286-20296. https://doi.org/10.3390/molecules201119696