
Plasmin Regulation through Allosteric, Sulfated, Small Molecules


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Rationale for Screening a Focused Library of Sulfated Small Molecules against Human Plasmin
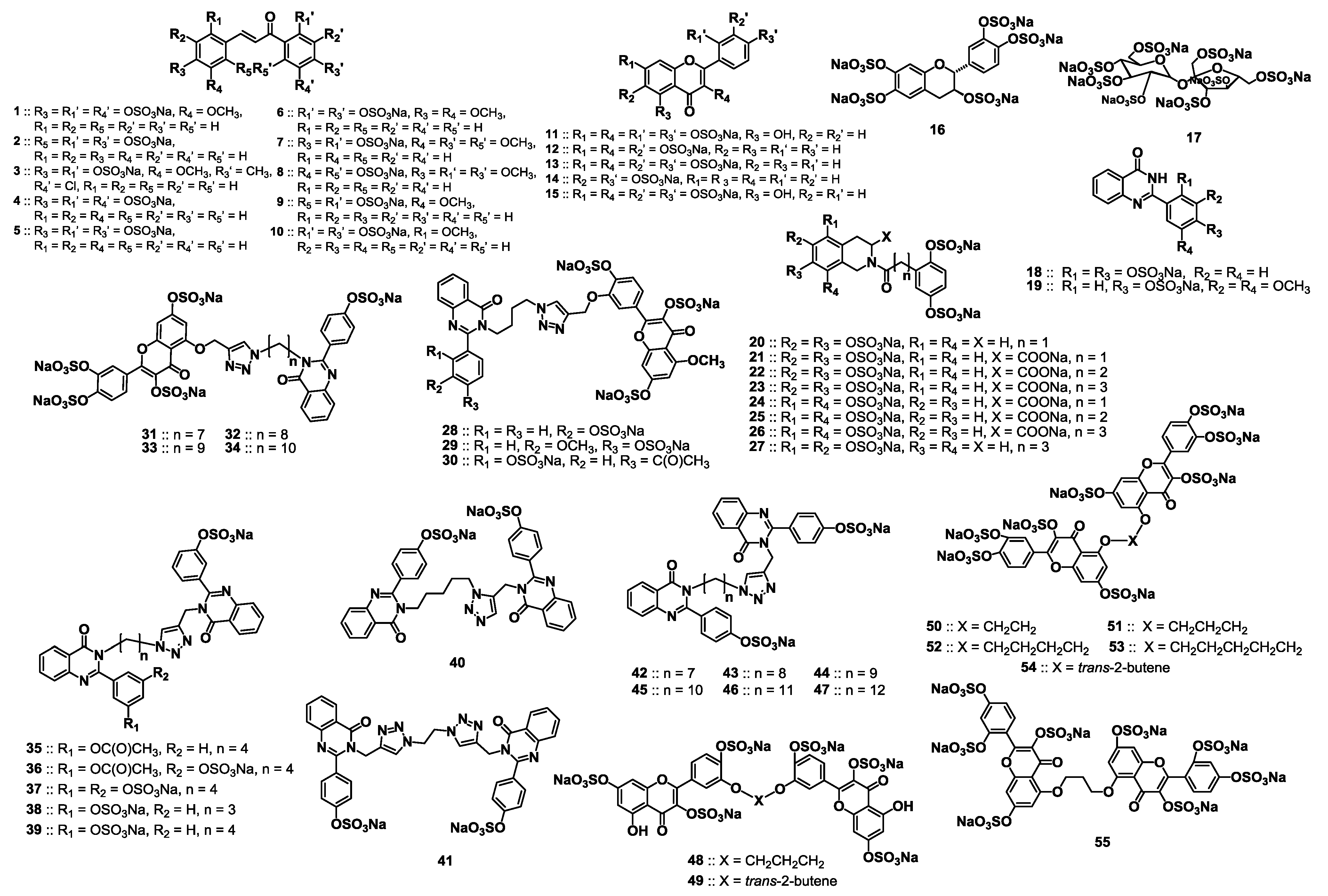
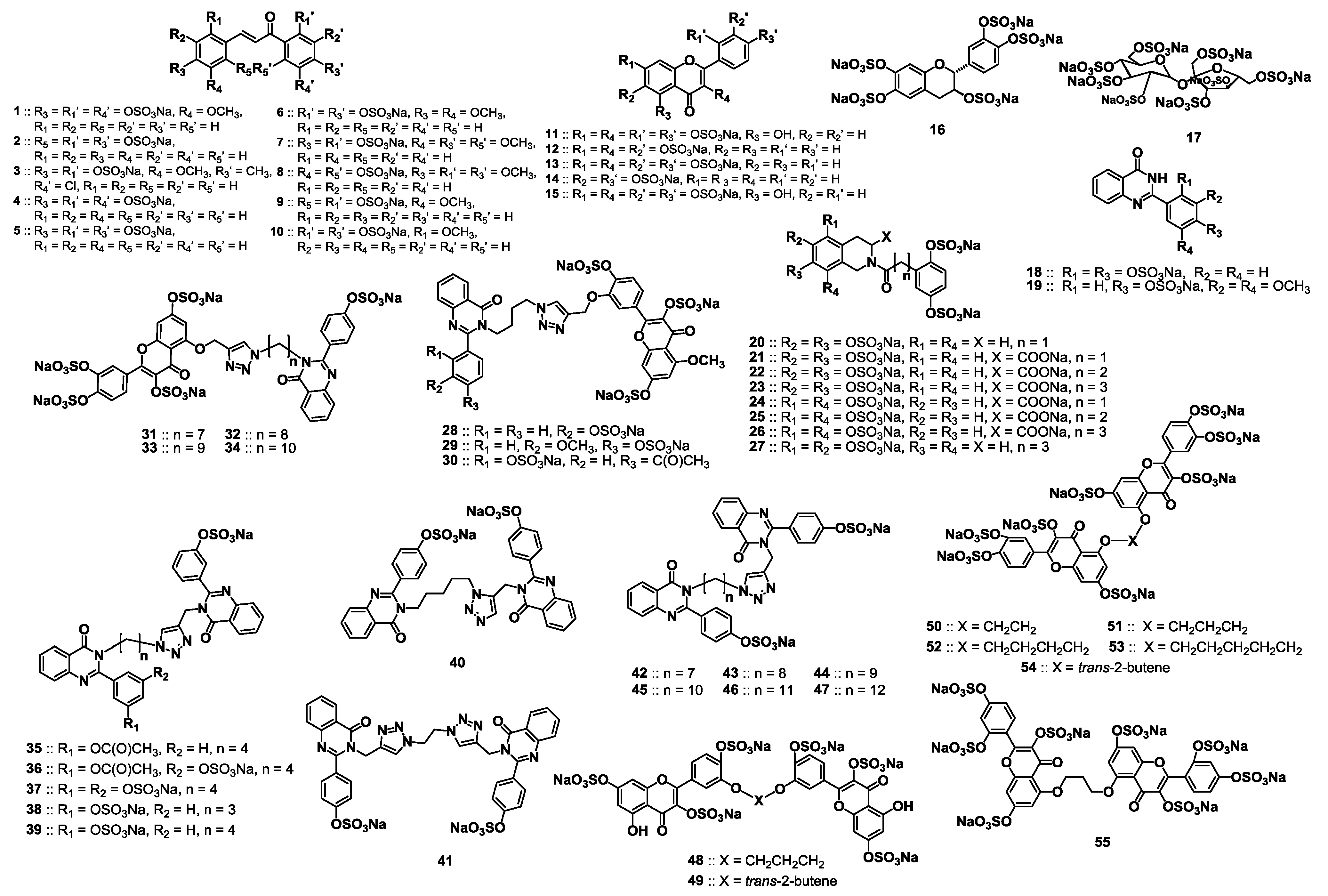
2.2. Chemical Synthesis of the Library of NSGMs

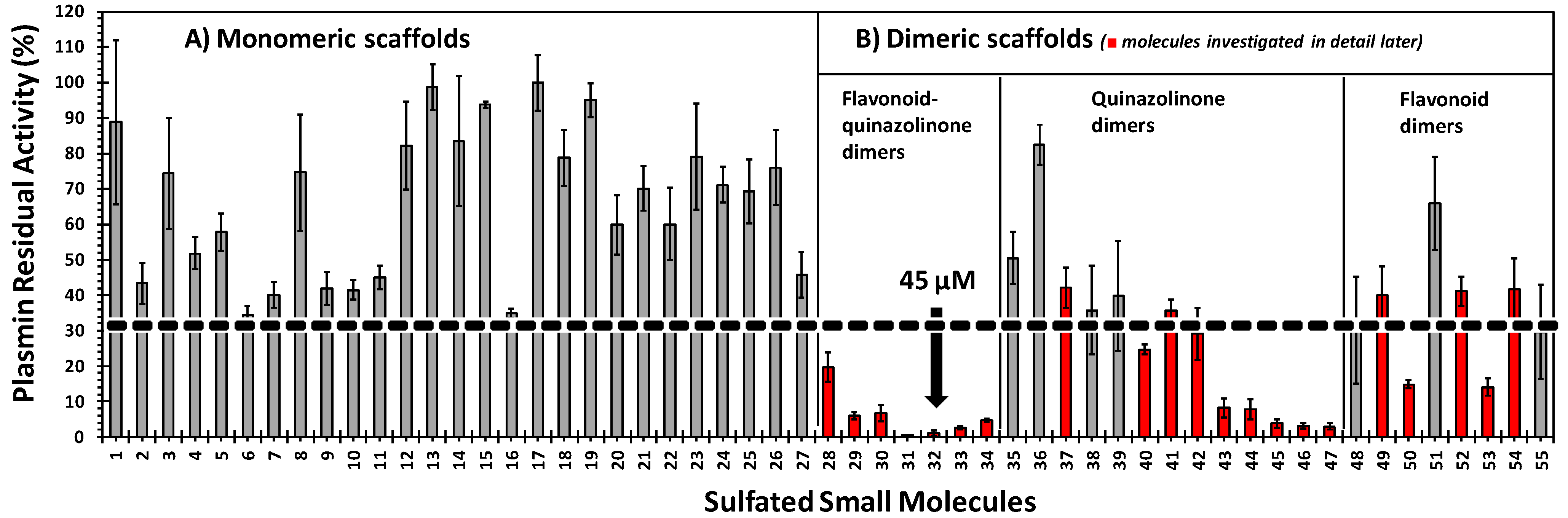
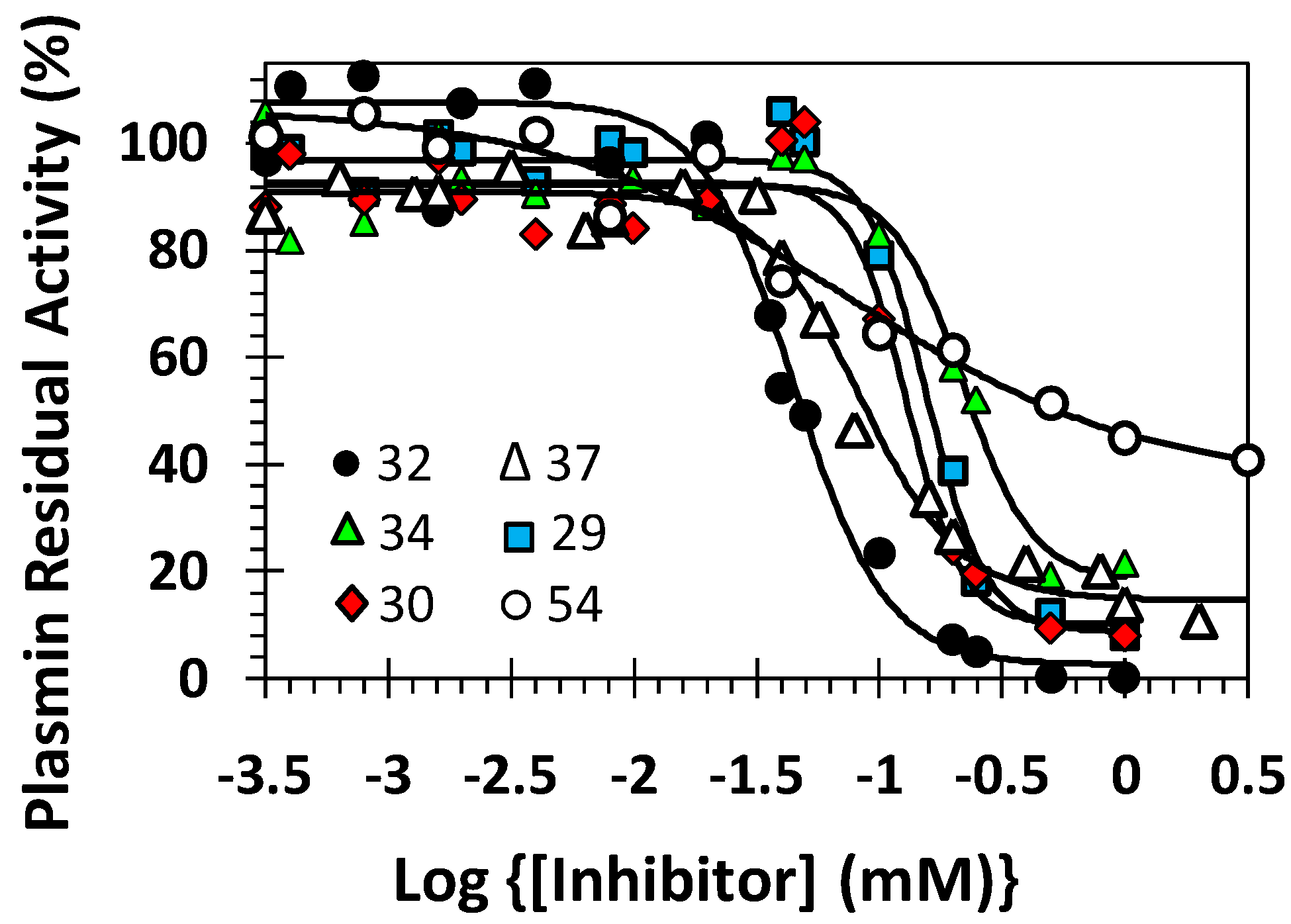
2.3. Inhibition of Human Plasmin by the Library of NSGMs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmin Inhibitor | IC50 (μM) | HS | ΔY% |
|---|---|---|---|
| 28 | 149 ± 5.6 b | 3.3 ± 0.7 | 82 ± 4 |
| 29 | 157 ± 5.0 | 3.6 ± 0.7 | 88 ± 4 |
| 30 | 220 ± 11 | 3.0 ± 1.3 | 74 ± 7 |
| 31 | 56 ± 2 | 3.2 ± 0.8 | 87 ± 4 |
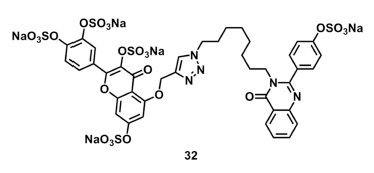
| 32 | 45 ± 2 | 2.3 ± 0.7 | 105 ± 6 |
| 33 | 128 ± 8 | 1.8 ± 0.4 | 89 ± 7 |
| 34 | 130 ± 6 | 3.9 ± 1.0 | 83 ± 5 |
| 35 | 642 ± 78 | 1.4 ± 0.4 | 88 ± 11 |
| 36 | >1000 | ND c | ND |
| 37 | 84 ± 4 | 2.1 ± 0.4 | 76 ± 3 |
| 38 | >400 | ND | ND |
| 39 | >400 | ND | ND |
| 40 | 239 ± 73 | 1.1 ± 0.6 | 87 ±29 |
| 41 | 125 ± 9 | 4.0 ± 1.8 | 54 ± 5 |
| 42 | 161 ± 24 | 1.0 ± 0.2 | 92 ± 4 |
| 43 | 183 ± 43 | 1.0 ± 0.3 | 94 ± 21 |
| 44 | 137 ± 11 | 1.0 ± 0.2 | 102 ± 9 |
| 45 | 98 ± 9 | 1.7 ± 0.6 | 94 ± 6 |
| 46 | 111 ± 7 | 2.4 ± 0.4 | 95 ± 2 |
| 47 | 89 ± 7 | 1.9 ± 0.6 | 98 ± 7 |
| 48 | 621 ± 185 | 1.3 ± 0.4 | 89 ± 36 |
| 49 | 277 ± 61 | 1.8 ± 1.0 | 75 ± 22 |
| 50 | 185 ± 68 | 1.4 ± 0.2 | 38 ± 15 |
| 51 | ~2830 | ND | ND |
| 52 | 76 ± 12 | 1.0 ± 0.2 | 72 ± 9 |
| 53 | 209 ± 25 | 0.6 ± 0.1 | 105 ± 8 |
| 54 | 75 ± 25 | 0.7 ± 0.3 | 71 ± 18 |
| 55 | >400 | ND | ND |
2.4. Structure-Activity Relationship of Plasmin Inhibition
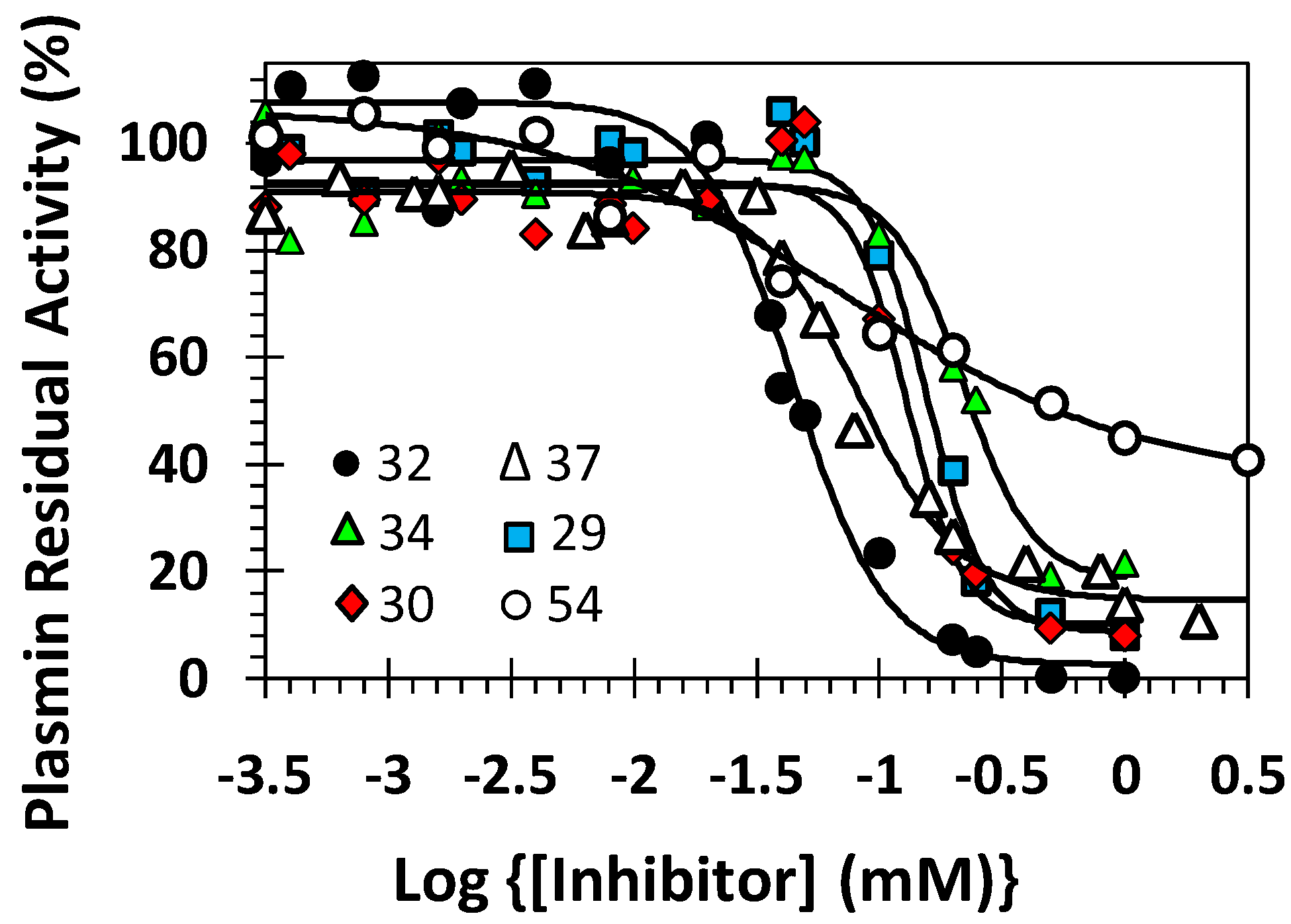
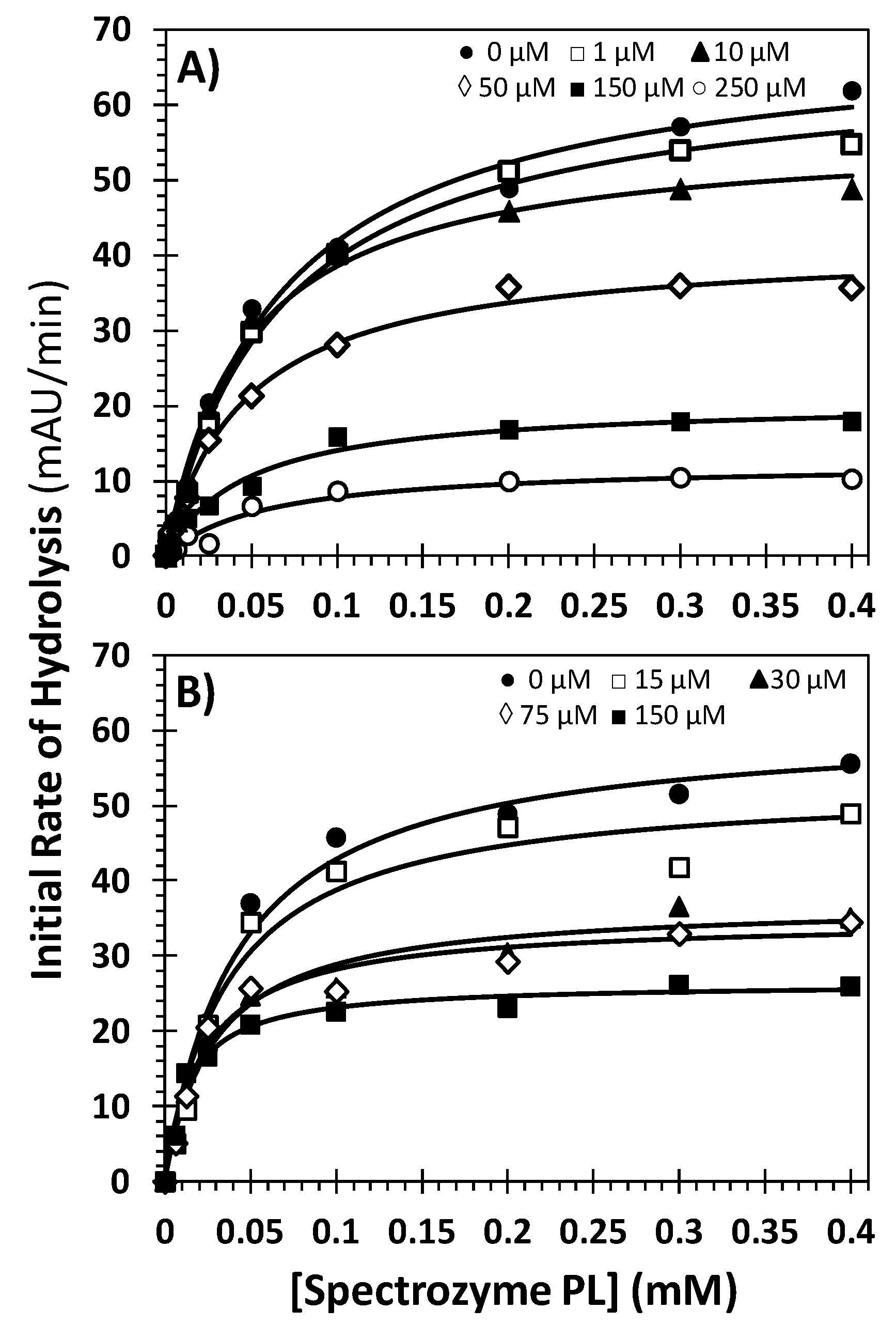
2.5. Mechanism of Plasmin Inhibition by NSGMs 32 and 52
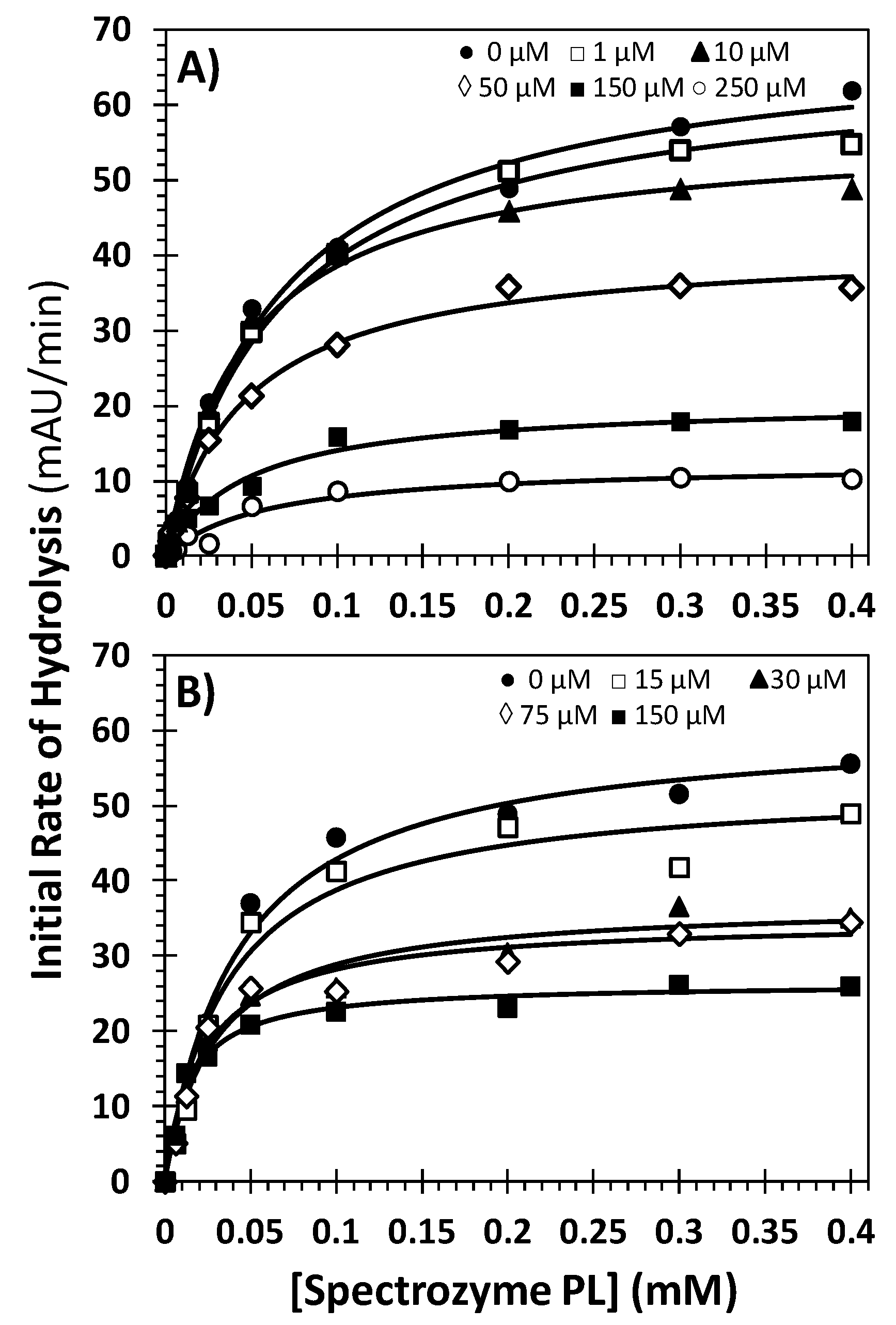
| Inhibitor | Conc. (μM) | KM (mM) | VMAX (mAU/min) |
|---|---|---|---|
| 32 | 0 | 0.07 ± 0.01 b | 69.6 ± 3.3 |
| 1 | 0.07 ± 0.01 | 65.8 ± 2.1 | |
| 10 | 0.05 ± 0.01 | 56.4 ± 2.3 | |
| 50 | 0.05 ± 0.01 | 41.4 ± 1.6 | |
| 150 | 0.05 ± 0.01 | 20.7 ± 1.1 | |
| 250 | 0.06 ± 0.01 | 12.5 ± 1.3 | |
| 52 | 0 | 0.04 ± 0.01 | 61.0 ± 2.4 |
| 15 | 0.04 ± 0.01 | 52.9 ± 2.9 | |
| 30 | 0.03 ± 0.01 | 37.0 ± 1.5 | |
| 75 | 0.023 ± 0.004 | 34.8 ± 1.5 | |
| 150 | 0.014 ± 0.002 | 26.4 ± 0.8 |
2.6. Selectivity Studies: Direct Inhibition of Thrombin and Factor Xa of the Coagulation Cascade
3. Experimental Section
3.1. Chemicals, Reagents, Analytical Chemistry, Enzymes, Peptides
3.2. Chemical Characterization of Compounds
3.3. General Procedure of Chemical Sulfation of Small Molecules
3.4. Direct Inhibition of Human Plasmin by Sulfated Small Molecules
3.5. Michaelis-Menten Kinetics of Spectrozyme PL Hydrolysis by Plasmin in the Presence of Molecules 32 and 52
3.6. Selectivity Studies: Direct Inhibition of Thrombin and Factor Xa of the Coagulation Cascade
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Al-Horani, R.A.; Desai, U.R. Recent Advances on plasmin inhibitors for the treatment of fibrinolysis-related disorders. Med. Res. Rev. 2014, 34, 1168–1216. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, J.E.; Harris, J.M. Natural and engineered plasmin inhibitors: Applications and design strategies. Chembiochem 2012, 13, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Royston, D. Blood-sparing drugs: Aprotinin, tranexamic acid, and epsilon-aminocaproic acid. Int. Anesthesiol. Clin. 1995, 33, 155–179. [Google Scholar] [CrossRef] [PubMed]
- Makhija, N.; Sarupria, A.; Kumar Choudhary, S.; Das, S.; Lakshmy, R.; Kiran, U. Comparison of epsilon aminocaproic acid and tranexamic acid in thoracic aortic surgery: Clinical efficacy and safety. J. Cardiothorac. Vasc. Anesth. 2013, 27, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Mangano, D.T.; Tudor, I.C.; Dietzel, C. The risk associated with aprotinin in cardiac surgery. N. Engl. J. Med. 2006, 354, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Schneeweiss, S.; Seeger, J.D.; Landon, J.; Walker, A.M. Aprotinin during coronary-artery bypass grafting and risk of death. N. Engl. J. Med. 2008, 358, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Lecker, I.; Wang, D.S.; Romaschin, A.D.; Peterson, M.; Mazer, C.D.; Orser, B.A. Tranexamic acid concentrations associated with human seizures inhibit glycine receptors. J. Clin. Investig. 2012, 122, 4654–4666. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A. Antifibrinolytics in cardiac surgery. Ann. Card. Anaesth. 2013, 16, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Perona, J.J.; Craik, C.S. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995, 4, 337–360. [Google Scholar] [CrossRef] [PubMed]
- Sanders, T.C.; Seto, C.T. 4-Heterocyclohexanone-based inhibitors of the serine protease plasmin. J. Med. Chem. 1999, 42, 2969–2976. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Seto, C.T. Selective inhibitors of the serine protease plasmin: Probing the S3 and S3' subsites using a combinatorial library. J. Med. Chem. 2005, 48, 6908–6917. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Seto, C.T. Structure-activity studies of cyclic ketone inhibitors of the serine protease plasmin: Design, synthesis, and biological activity. Bioorg. Med. Chem. 2006, 14, 8467–8487. [Google Scholar] [CrossRef] [PubMed]
- Saupe, S.M.; Leubner, S.; Betz, M.; Klebe, G.; Steinmetzer, T. Development of new cyclic plasmin inhibitors with excellent potency and selectivity. J. Med. Chem. 2013, 56, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Saupe, S.M.; Steinmetzer, T. A new strategy for the development of highly potent and selective plasmin inhibitors. J. Med. Chem. 2012, 55, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, M.S.; Ogueli, G.I.; Kumar, Y.; Vadivel, K.; Lawson, G.; Shanker, S.; Schmidt, A.E.; Bajaj, S.P. Engineering kunitz domain 1 (KD1) of human tissue factor pathway inhibitor-2 to selectively inhibit fibrinolysis: properties of KD1-L17R variant. J. Biol. Chem. 2011, 286, 4329–4340. [Google Scholar] [CrossRef] [PubMed]
- Flight, S.M.; Johnson, L.A.; Du, Q.S.; Warner, R.L.; Trabi, M.; Gaffney, P.J.; Lavin, M.F.; de Jersey, J.; Masci, P.P. Textilinin-1, an alternative anti-bleeding agent to aprotinin: Importance of plasmin inhibition in controlling blood loss. Br. J. Haematol. 2009, 145, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Devy, L.; Rabbani, S.A.; Stochl, M.; Ruskowski, M.; Mackie, I.; Naa, L.; Toews, M.; van Gool, R.; Chen, J.; Ley, A.; et al. PEGylated DX-1000: Pharmaco-kinetics and antineoplastic activity of a specific plasmin inhibitor. Neoplasia 2007, 9, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Swedberg, J.E.; Harris, J.M. Plasmin substrate binding site cooperativity guides the design of potent peptide aldehyde inhi-bitors. Biochemistry 2011, 50, 8454–8462. [Google Scholar] [CrossRef] [PubMed]
- Teno, N.; Gohda, K.; Wanaka, K.; Sueda, T.; Tsuda, Y. Identi-fication of novel plasmin inhibitors possessing nitrile moiety as warhead. Bioorg. Med. Chem. Lett. 2011, 21, 6305–6309. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Pettersen, D.; Ohlsson, B.; Schell, P.; Karle, M.; Evertsson, E.; Pahlén, S.; Jonforsen, M.; Plowright, A.T.; Boström, J.; et al. Discovery of the fibrinolysis inhibitor AZD6564, acting via interference of a protein-protein interaction. ACS Med. Chem. Lett. 2014, 5, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Boström, J.; Grant, J.A.; Fjellström, O.; Thelin, A.; Gustafsson, D. Potent fibrinolysis inhibitor discovered by shape and electrostatic complementarity to the drug tranexamic acid. J. Med. Chem. 2013, 56, 3273–3280. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.F.; Sundboom, J.L. Heparin and protease inhibition. II. The role of heparin in the ATIII inactivation of thrombin, plasmin, and trypsin. Thromb. Res. 1981, 22, 115–133. [Google Scholar] [CrossRef] [PubMed]
- Jordan, R.E.; Oosta, G.M.; Gardner, W.T.; Rosenberg, R.D. The binding of low molecular weight heparin to hemostatic enzymes. J. Biol. Chem. 1980, 255, 10073–10080. [Google Scholar] [PubMed]
- Yomtova, V.M.; Stambolieva, N.A.; Blagoev, B.M. Kinetic study of the effect of heparin on the amidase activity of trypsin, plasmin and urokinase. Thromb. Haemost. 1983, 49, 199–203. [Google Scholar] [PubMed]
- Bauer, P.I.; Pozsgay, M.; Machovich, R.; Elödi, P.; Horváth, I. The interaction of heparin with human plasmin. Int. J. Biochem. 1983, 15, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Legras, S.; Diczhazi, C.; Moczar, M. N-oleoyl heparin inhibits the amidolytic activity of plasmin and urokinase. Int. J. Biol. Macromol. 1992, 14, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.L.; Abdel Aziz, M.H.; Zhou, Q.; Desai, U.R. Sulfated, low-molecular-weight lignins are potent inhibitorsof plasmin, in addition to thrombin and factor Xa: Novel opportunity for con-trolling complex pathologies. Thromb. Haemost. 2010, 103, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, D.; Papy-Garcia, D.; Escartin, Q.; Sagot, M.A.; Cao, Y.; Barritault, D.; Courtois, J.; Hornebeck, W.; Caruelle, J.P. Human plasmin enzymatic activity is inhibited by chemically modified dextrans. J. Biol. Chem. 2000, 275, 29383–29390. [Google Scholar] [CrossRef] [PubMed]
- Vörös, G.; Kolev, K.; Csomor, K.; Machovich, R. Inhibition of plasmin activity by sulfated polyvinylalcohol-acrylate copolymers. Thromb. Res. 2000, 100, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Raman, K.; Karuturi, R.; Swarup, V.P.; Desai, U.R.; Kuberan, B. Discovery of novel sulfonated small molecules that inhibit vascular tube formation. Bioorg. Med. Chem. Lett. 2012, 22, 4467–4470. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, G.T.; Desai, U.R. Designing small, nonsugar activators of antithrombin using hydropathic interaction analyses. J. Med. Chem. 2002, 45, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, G.T.; Desai, U.R. Exploring new non-sugar sulfated molecules as activators of antithrombin. Bioorg. Med. Chem. Lett. 2003, 13, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.J.; Karuturi, R.; Al-Horani, R.A.; Baranwal, S.; Patel, J.; Desai, U.R.; Patel, B.B. Synthetic, non-saccharide, glycosaminoglycan mimetics selectively target colon cancer stem cells. ACS Chem. Biol. 2014, 9, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, P.S.; Liang, A.; Mehta, A.Y.; Abdel Aziz, M.H.; Zhou, Q.; Desai, U.R. Rational design of potent, small, synthetic allosteric inhibitors of thrombin. J. Med. Chem. 2011, 54, 5522–5531. [Google Scholar] [CrossRef] [PubMed]
- Abdel Aziz, M.H.; Sidhu, P.S.; Liang, A.; Kim, J.Y.; Mosier, P.D.; Zhou, Q.; Farrell, D.H.; Desai, U.R. Designing allosteric regulators of thrombin. Monosulfated benzofuran dimers selectively interact with Arg173 of exosite 2 to induce inhibition. J. Med. Chem. 2012, 55, 6888–6897. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Liang, A.; Desai, U.R. Designing nonsaccharide, allosteric activators of antithrombin for accelerated inhibition of factor Xa. J. Med. Chem. 2011, 54, 6125–6138. [Google Scholar] [CrossRef] [PubMed]
- Karuturi, R.; Al-Horani, R.A.; Mehta, S.C.; Gailani, D.; Desai, U.R. Discovery of allosteric modulators of factor XIa by targeting hydrophobic domains adjacent to its heparin-binding site. J. Med. Chem. 2013, 56, 2415–2428. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Ponnusamy, P.; Mehta, A.Y.; Gailani, D.; Desai, U.R. Sulfated pentagalloylglucoside is a potent, allosteric, and selective inhibitor of factor XIa. J. Med. Chem. 2013, 56, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Desai, U.R. Designing allosteric inhibitors of factor XIa. Lessons from the interactions of sulfated pentagalloylglucopyranosides. J. Med. Chem. 2014, 57, 4805–4818. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.J.; Boothello, R.S.; Mehta, A.Y.; Scarsdale, J.N.; Wright, H.T.; Desai, U.R. Interaction of thrombin with sucrose octasulfate. Biochemistry 2011, 50, 6973–6982. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Karuturi, R.; Verespy Ш, S.; Desai, U.R. Synthesis of glycosaminoglycan mimetics through sulfation of polyphenols. Methods Mol. Biol. 2015, 1229, 49–67. [Google Scholar] [PubMed]
- Al-Horani, R.A.; Desai, U.R. Chemical sulfation of small molecules—Advances and challenges. Tetrahedron 2010, 66, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Al-Horani, R.A.; Mehta, A.Y.; Desai, U.R. Potent direct inhibitors of factor Xa based on the tetrahydroisoquinoline scaffold. Eur. J. Med. Chem. 2012, 54, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, P.S.; Abdel Aziz, M.H.; Sarkar, A.; Mehta, A.Y.; Zhou, Q.; Desai, U.R. Designing allosteric regulators of thrombin. Exosite 2 features multiple subsites that can be targeted by sulfated small molecules for inducing inhibition. J. Med. Chem. 2013, 56, 5059–5070. [Google Scholar] [CrossRef] [PubMed]
- Liang, A.; Thakkar, J.N.; Desai, U.R. Study of physico-chemical properties of novel highly sulfated, aromatic, mimetics of heparin and heparan sulfate. J. Pharm Sci. 2010, 99, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Desai, U.R. The promise of sulfated synthetic small molecules as modulators of glycosaminoglycan function. Future Med. Chem. 2013, 5, 1363–1366. [Google Scholar] [CrossRef] [PubMed]
- Correia-da-Silva, M.; Sousa, E.; Pinto, M.M. Emerging sulfated flavonoids and other polyphenols as drugs: Nature as an inspiration. Med. Res. Rev. 2014, 34, 223–279. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Horani, R.A.; Karuturi, R.; White, D.T.; Desai, U.R. Plasmin Regulation through Allosteric, Sulfated, Small Molecules. Molecules 2015, 20, 608-624. https://doi.org/10.3390/molecules20010608
Al-Horani RA, Karuturi R, White DT, Desai UR. Plasmin Regulation through Allosteric, Sulfated, Small Molecules. Molecules. 2015; 20(1):608-624. https://doi.org/10.3390/molecules20010608
Chicago/Turabian StyleAl-Horani, Rami A., Rajesh Karuturi, Domonique T. White, and Umesh R. Desai. 2015. "Plasmin Regulation through Allosteric, Sulfated, Small Molecules" Molecules 20, no. 1: 608-624. https://doi.org/10.3390/molecules20010608