Conformational Characterization of Ipomotaosides and Their Recognition by COX-1 and 2

Abstract
:1. Introduction
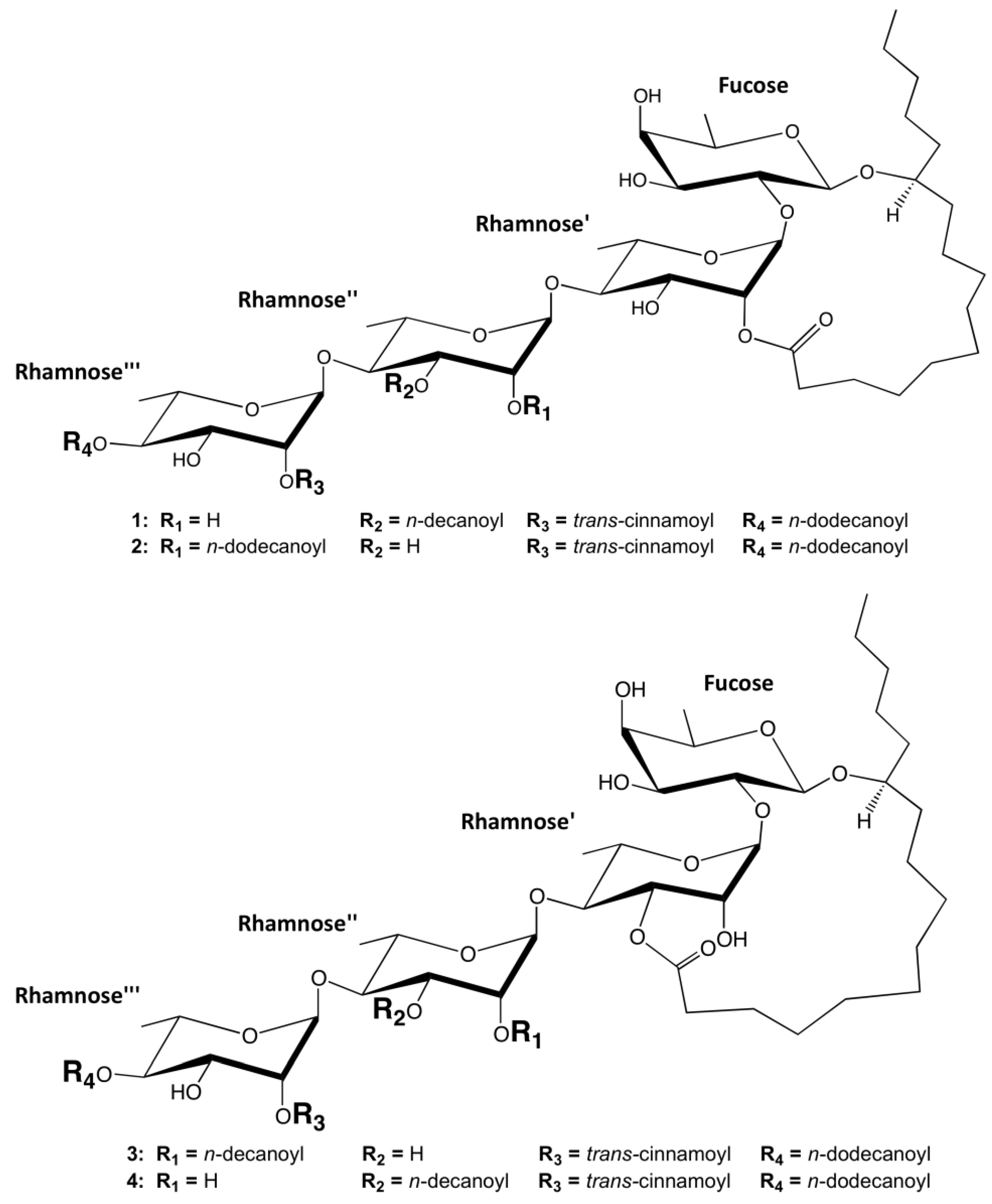
2. Results and Discussion
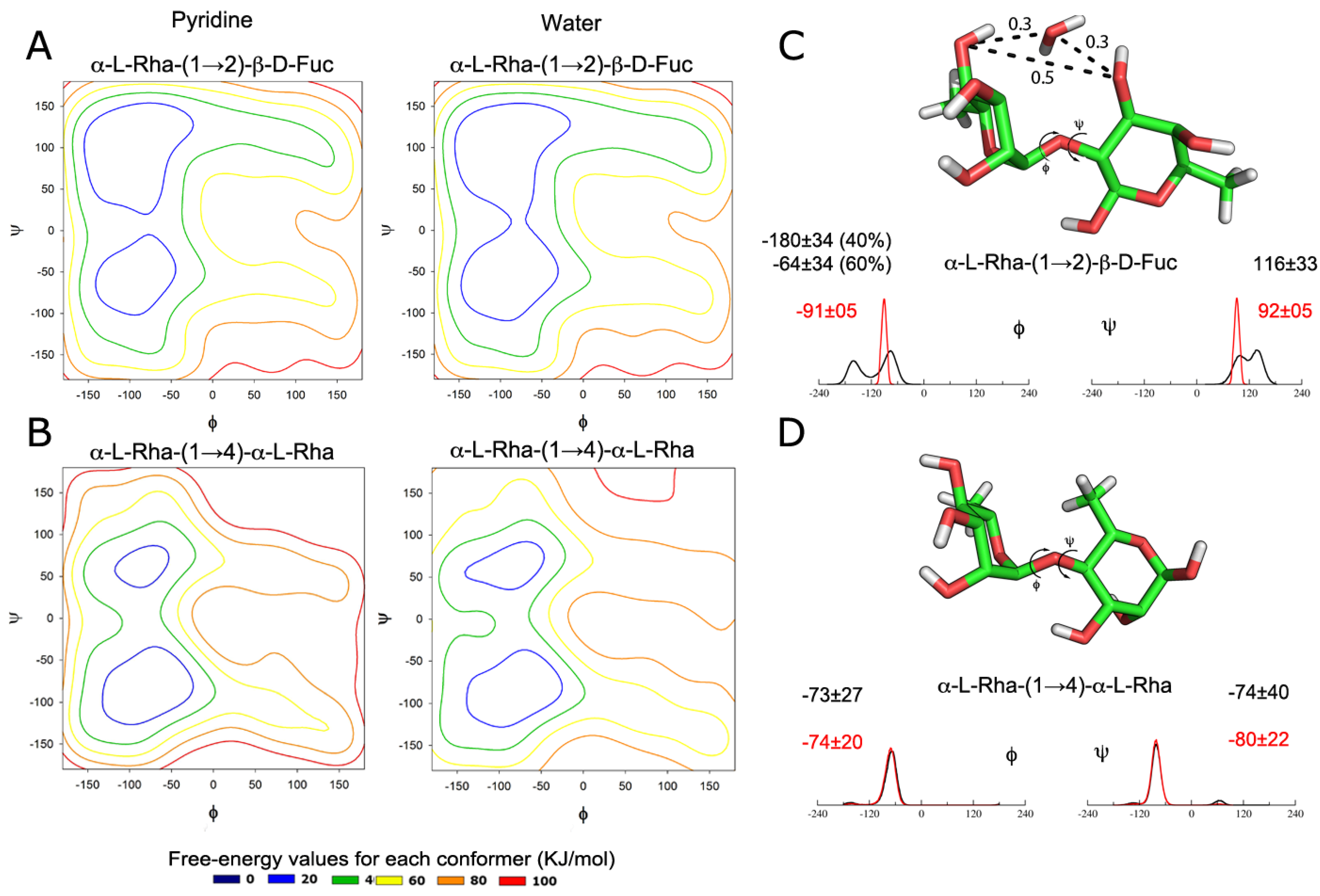
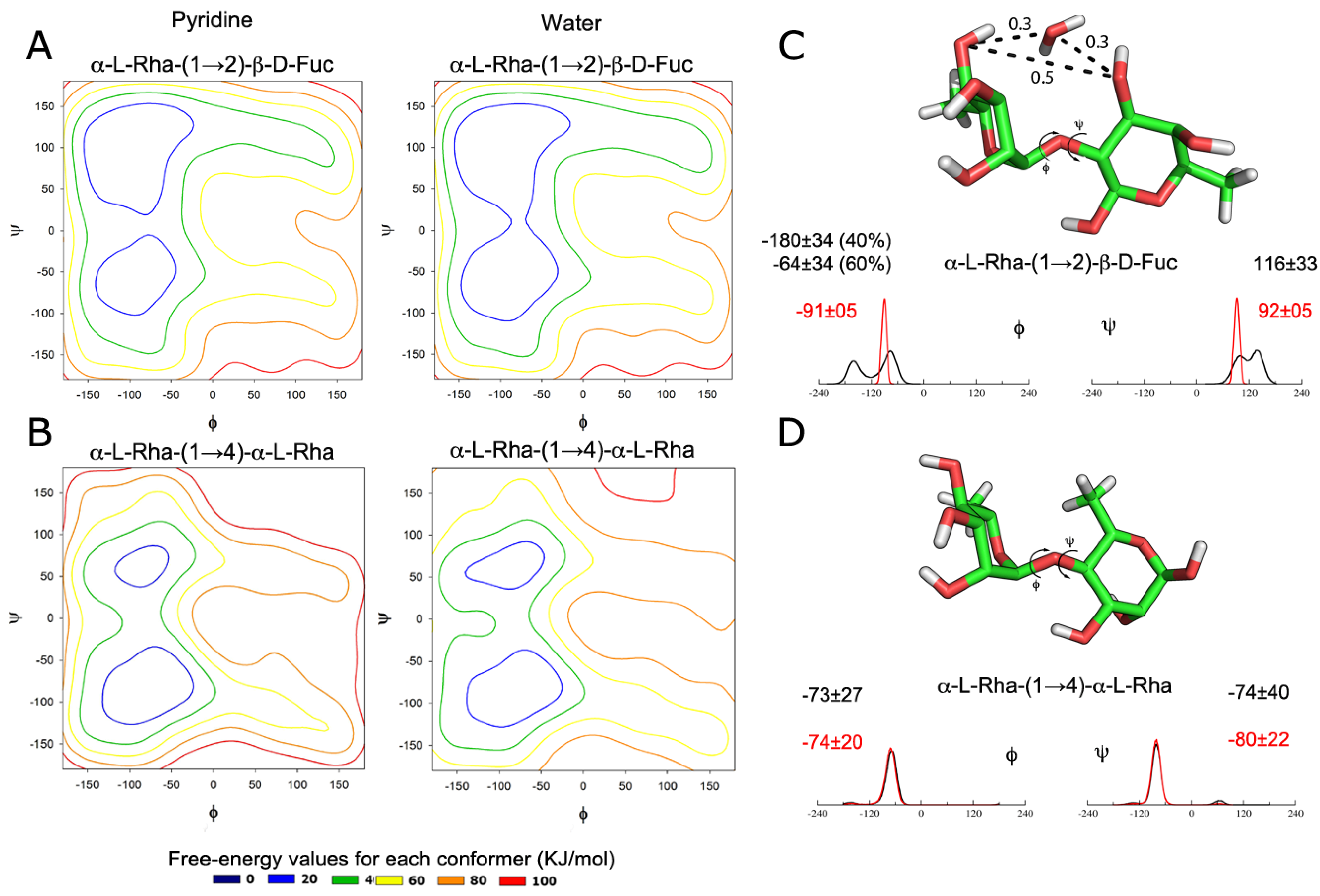
2.1. Dynamics of Isolated Disaccharides
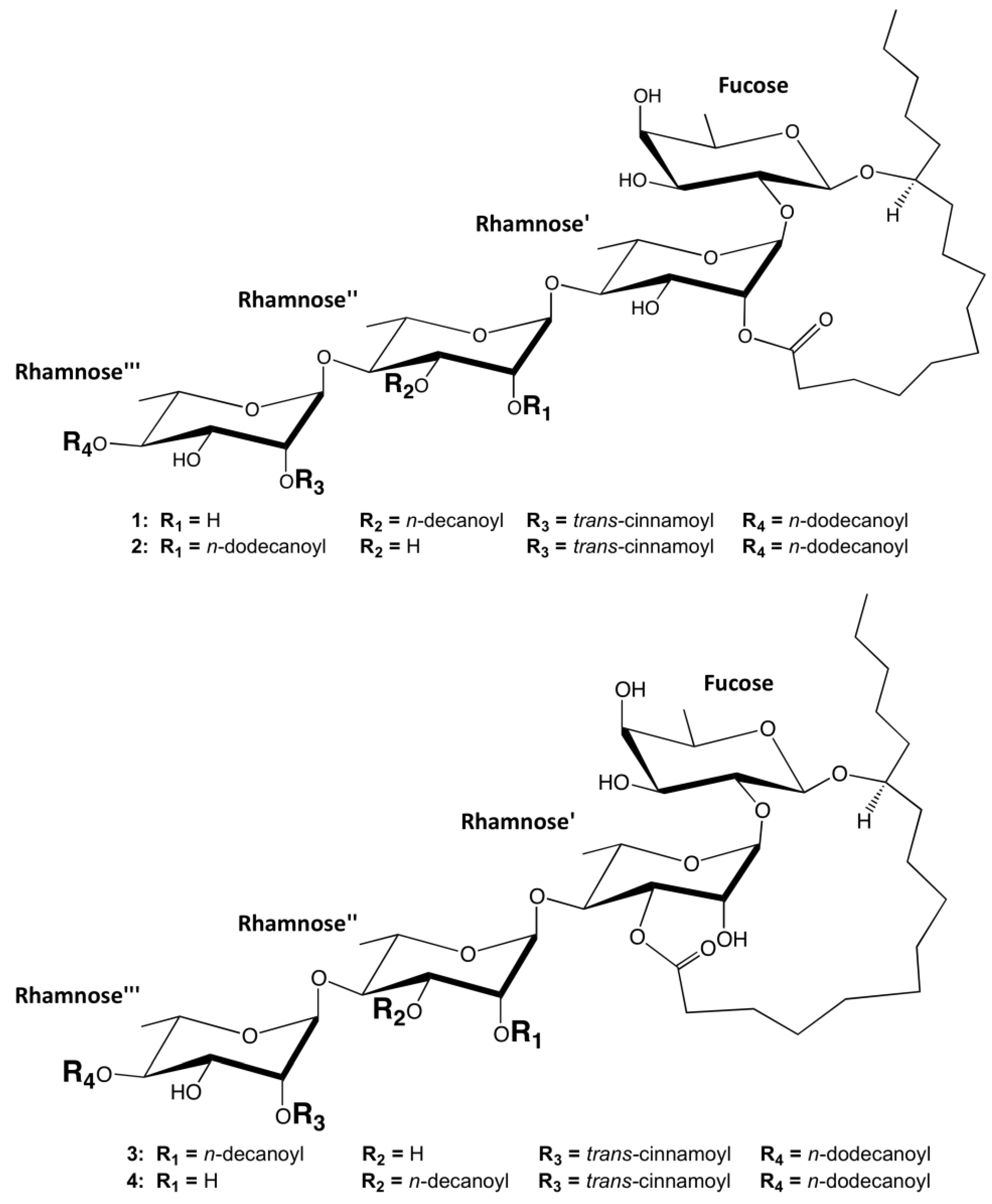
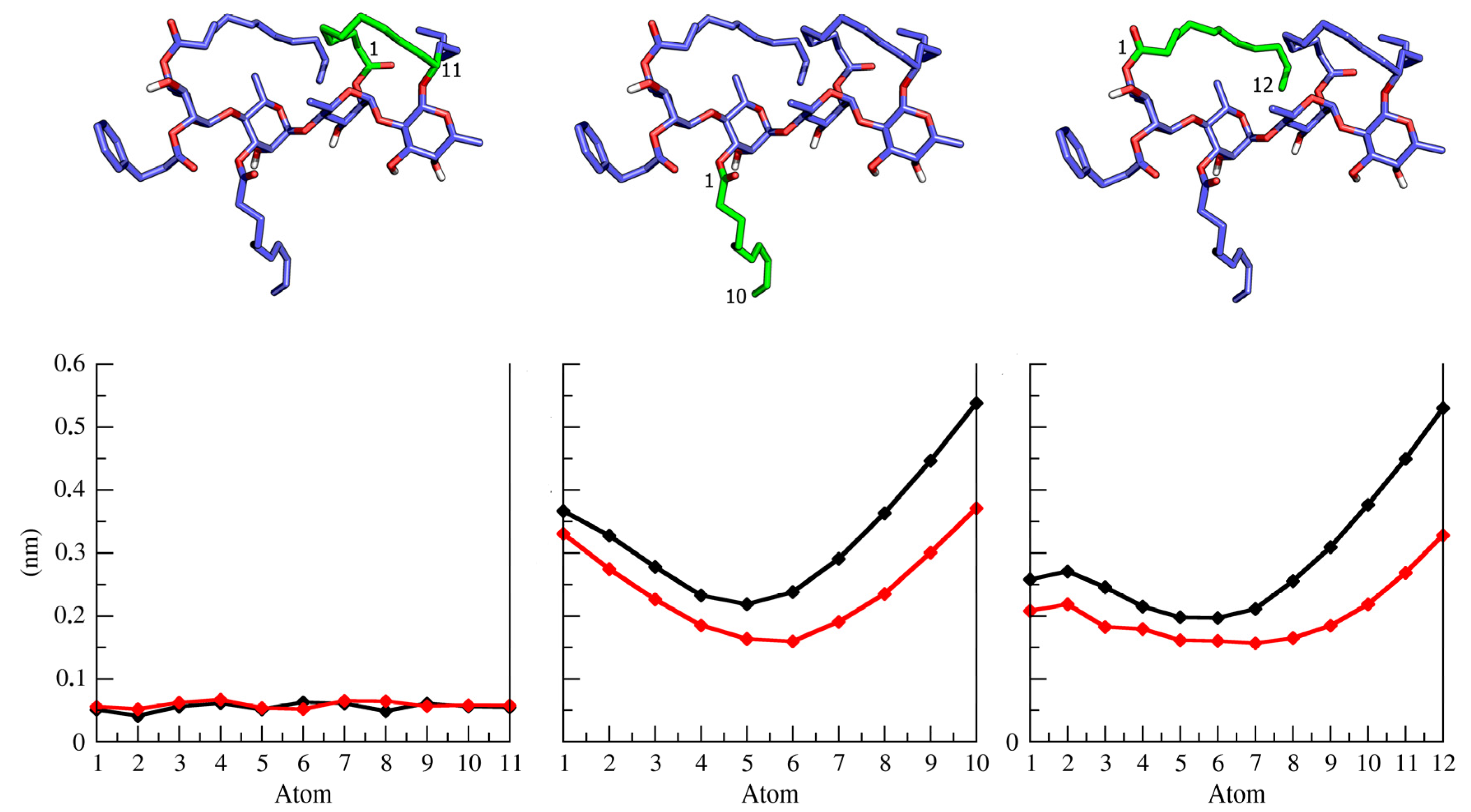
2.2. Ipomotaosides Dynamics

{kind=link}
{kind=link}
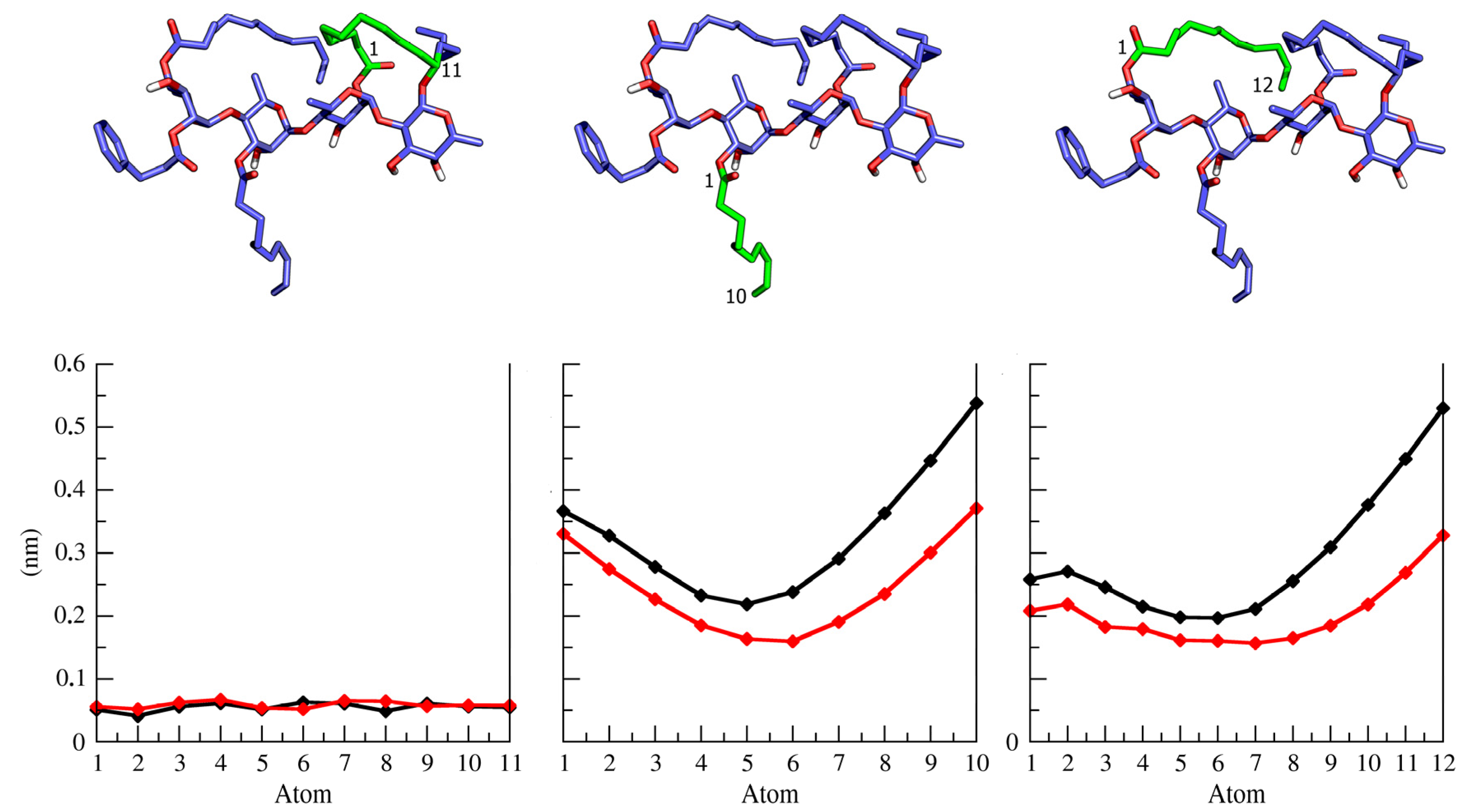{kind=link}
{kind=link}
| Glicosidic Linkage | |||||||
|---|---|---|---|---|---|---|---|
| Compound | Condition | α-l-Rha′-(1→2)-β-d-Fuc | α-l-Rha′′-(1→4)-α-l-Rha′ | α-l-Rha′′′-(1→4)-α-l-Rha′′ | |||
| Φ | Ψ | Φ | Ψ | Φ | Ψ | ||
| Ipomotaoside 1 | MD in pyridine | −171 ± 29 (42%) | 79 ± 40 (37%) | −75 ± 22 | −85 ± 14 | −71 ± 11 | −81 ± 10 |
| −73 ± 28 (58%) | −51 ± 29 (63%) | - | - | - | - | ||
| MD in water | −163 ± 15 | 86 ± 19 | −74 ± 22 | −87 ± 21 | −71 ± 15 | −82 ± 12 | |
| Ipomotaoside 2 | MD in pyridine | −165 ± 14 | 82 ± 15 | −76 ± 14 | −83 ± 11 | −76 ± 14 | −83 ± 11 |
| MD in water | −71 ± 26 (14%) | −51 ± 13 (14%) | −76 ± 42 | −87 ± 19 | −78 ± 40 | −88 ± 18 | |
| −170 ± 31 (86%) | 81 ± 56 (86%) | - | - | - | - | ||
| Ipomotaoside 3 | MD in pyridine | −183 ± 25 (47%) | 76 ± 27 (47%) | −74 ± 11 | −82 ± 10 | −75 ± 14 | −82 ± 11 |
| −91 ± 29 (53%) | −35 ± 37 (53%) | - | - | - | - | ||
| MD in water | −173 ± 10 | 87 ± 11 | −72 ± 26 | −82 ± 13 | −77 ± 37 | −86 ± 17 | |
| Ipomotaoside 4 | MD in pyridine | −171 ± 25 (43%) | 83 ± 26 (43%) | −71 ± 39 | −85 ± 16 | −74 ± 11 | −84 ± 10 |
| −85 ± 25 (57%) | −31 ± 40 (57%) | - | - | - | - | ||
| MD in water | −173 ± 09 | 87 ± 09 | −97 ± 33 (34%) | −83 ± 20 (34%) | −80 ± 26 | −84 ± 15 | |
| - | - | −185 ± 18 (66%) | −150 ± 18 (66%) | - | - | ||
| Ipomotaoside | Proton 1 | Proton 2 | Average of Interproton distance from MD (Å) | Ipomotaoside | Proton 1 | Proton 2 | Average of Interproton distance from MD (Å) |
|---|---|---|---|---|---|---|---|
| 1 | Fuc-(H1) | Fuc-(H5) | 2.4 ± 0.2 | 3 | Fuc-(H1) | Fuc-(H5) | 2.4 ± 0.2 |
| Rha'-(H1) | Fuc-(H1) | 2.5 ± 0.7 | Rha'-(H1) | Fuc-(H1) | 2.5 ± 0.7 | ||
| Rha'-(H1) | Fuc-(H2) | 3.2 ± 0.5 | Rha'-(H1) | Fuc-(H2) | 3.2 ± 0.4 | ||
| Rha'-(H1) | Fuc-(H3) | 3.4 ± 0.8 | Rha'-(H1) | Fuc-(H3) | 3.6 ± 0.8 | ||
| Rha'-(H1) | Fuc-(H4) | 5.5 ± 0.5 | Rha'-(H5) | Fuc-(H1) | 4.9 ± 0.3 | ||
| Rha''-(H1) | Rha'-(H3) | 3.4 ± 0.3 | Rha''-(H1) | Rha'-(H2) | 4.7 ± 0.2 | ||
| Rha''-(H1) | Rha'-(H4) | 2.5 ± 0.3 | Rha''-(H1) | Rha'-(H4) | 2.5 ± 0.2 | ||
| Rha'''-(H1) | Rha''-(H4) | 2.6 ± 0.3 | Rha''-(H1) | Rha'-(H5) | 4.4 ± 0.1 | ||
| 2 | Fuc-(H1) | Fuc-(H5) | 2.4 ± 0.2 | Rha'''-(H1) | Rha''-(H3) | 3.3 ± 0.2 | |
| Rha'-(H1) | Fuc-(H1) | 3.3 ± 0.3 | Rha'''-(H1) | Rha''-(H4) | 2.5 ± 0.3 | ||
| Rha'-(H1) | Fuc-(H2) | 2.6 ± 0.3 | Rha'''-(H1) | Rha''-(H5) | 4.4 ± 0.1 | ||
| Rha'-(H1) | Fuc-(H3) | 4.4 ± 0.1 | 4 | Fuc-(H1) | Fuc-(H5) | 2.4 ± 0.2 | |
| Rha'-(H2) | Fuc-(H2) | 2.2 ± 0.3 | Rha'-(H1) | Fuc-(H1) | 2.5 ± 0.7 | ||
| Rha'-(H2) | Fuc-(H3) | 4.7 ± 0.3 | Rha'-(H1) | Fuc-(H2) | 3.1 ± 0.5 | ||
| Rha''-(H1) | Rha'-(H3) | 3.4 ± 0.2 | Rha'-(H1) | Fuc-(H3) | 3.5 ± 0.8 | ||
| Rha''-(H1) | Rha'-(H4) | 2.5 ± 0.3 | Rha''-(H1) | Rha'-(H4) | 2.5 ± 0.3 | ||
| Rha''-(H1) | Rha'-(H5) | 4.4 ± 0.1 | Rha''-(H1) | Rha'-(H5) | 4.3 ± 0.2 | ||
| Rha''-(H2) | Rha'-(H3) | 4.1 ± 0.3 | Rha'''-(H1) | Rha''-(H4) | 2.5 ± 0.3 | ||
| Rha''-(H2) | Rha'-(H4) | 4.3 ± 0.2 | Rha'''-(H1) | Rha''-(H5) | 4.4 ± 0.1 | ||
| Rha'''-(H1) | Rha''-(H3) | 3.4 ± 0.2 | - | - | - | ||
| Rha'''-(H2) | Rha''-(H3) | 4.1 ± 0.3 | - | - | - |
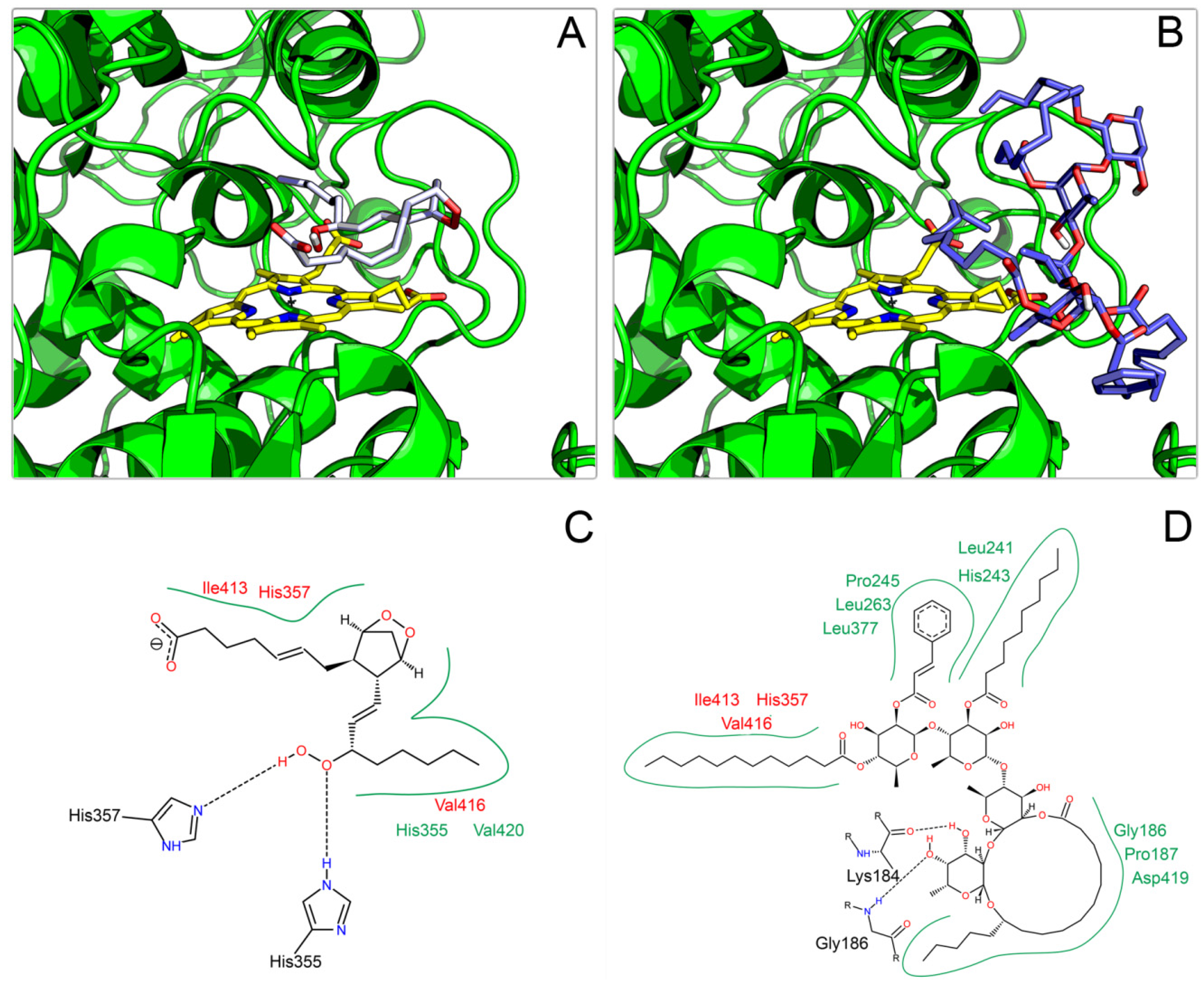
2.3. Docking on COX
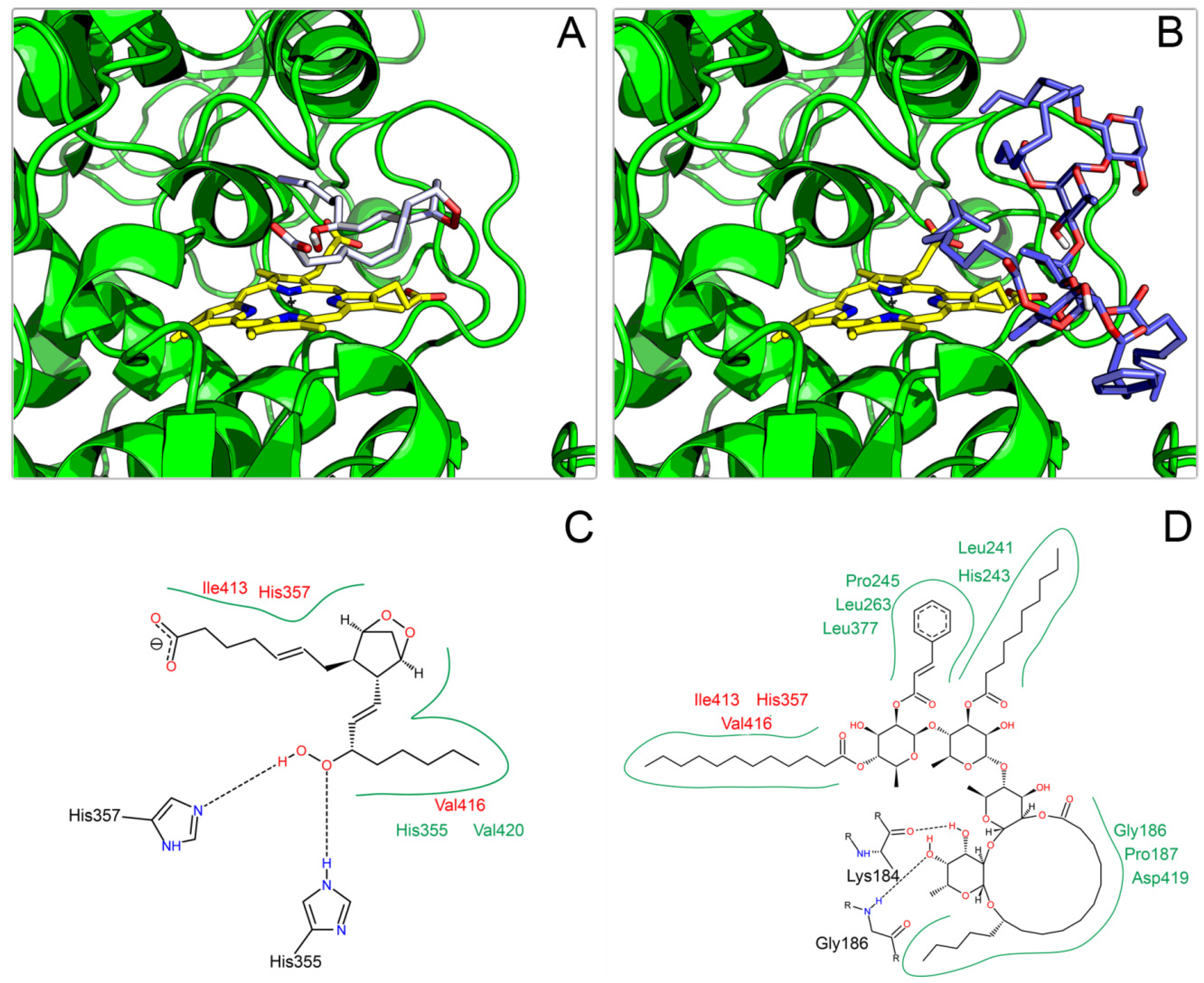
3. Experimental
3.1. Nomenclature, Topologies and Software
- α1→2:Φ = O5 − C1 − O1 − C2’Ψ = C1 − O1 − C2’ − C1’
- α1→4:Φ = O5 − C1 − O1 − C4’Ψ = C1 − O1 − C4’− C3’
3.2. Topology Construction
3.3. Metadynamics
3.4. MD Simulations
3.5. ROESY Signals
3.6. Docking Procedures
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yoshikawa, K.; Yagi, C.; Hama, H.; Tanaka, M.; Arihara, S.; Hashimoto, T. Ipomotaosides A-D, resin glycosides from the aerial parts of Ipomoea batatas and their inhibitory activity against COX-1 and COX-2. J. Nat. Prod. 2010, 73, 1763–1766. [Google Scholar] [CrossRef]
- Li, S.Z. Min Dynasty, Compendium of Materia Medica; Medical Publishing House: Beijing, China, 1999; p. 1501. [Google Scholar]
- Corona-Castañeda, B.; Pereda-Miranda, R. Morning glory resin glycosides as modulators of antibiotic activity in multidrug-resistant Gram-negative bacteria. Planta Med. 2012, 78, 128–131. [Google Scholar] [CrossRef]
- Figueroa-González, G.; Jacobo-Herrera, N.; Zentella-Dehesa, A.; Pereda-Miranda, R. Reversal of multidrug resistance by morning glory resin glycosides in human breast cancer cells. J. Nat. Prod. 2012, 75, 93–97. [Google Scholar] [CrossRef]
- Pereda-Miranda, R.; Rosas-Ramírez, D.; Castañeda-Gómez, J. Resin glycosides from the morning glory family. In Progress in the Chemistry of Natural Products; Kinghorn, D.A., Falk, H., Kobayashi, J., Eds.; Springer: New York, NY, USA, 2010; Volume 92, pp. 77–153. [Google Scholar]
- Smith, W.L.; Garavito, R.M.; DeWitt, D.L. Prostaglandin Endoperoxide H Synthases (Cyclooxygenases)-1 and -2. J. Biol. Chem. 1996, 271, 33157–33160. [Google Scholar] [CrossRef]
- Rencurosi, A.; Mitchell, E.P.; Cioci, G.; Pérez, S.; Pereda-Miranda, R.; Imberty, A. Crystal Structure of Tricolorin A: Molecular Rationale for the Biological Properties of Resin Glycosides Found in Some Mexican Herbal Remedies. Angew. Chem. Int. Ed. 2004, 43, 5918–5922. [Google Scholar] [CrossRef]
- Woods, R.J. Computational carbohydrate chemistry: What theoretical methods can tell us. Glycoconj. J. 1998, 15, 209–216. [Google Scholar] [CrossRef]
- Dwek, R.A. Glycobiology: Toward Understanding the Function of Sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef]
- Pérez, S.; Mulloy, B. Prospects for Glycoinformatics. Curr. Opin. Struct. Biol. 2005, 15, 517–524. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Fernandes, C.L.; Verli, H. GROMOS96 43a1 performance on the characterization of glycoprotein conformational ensembles through molecular dynamics simulations. Carbohydr. Res. 2009, 344, 491–500. [Google Scholar] [CrossRef]
- Pedebos, C.; Pol-Fachin, L.; Verli, H. Unrestrained conformational characterization of Stenocereus eruca saponins in aqueous and nonaqueous solvents. J. Nat. Prod. 2012, 75, 1196–1200. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Serrato, R.V.; Verli, H. Solution conformation and dynamics of exopolysaccharides from Burkholderia species. Carbohydr. Res. 2010, 345, 1922–1931. [Google Scholar] [CrossRef]
- Castro, M.O.; Pomin, V.H.; Santos, L.L.; Vilela-Silva, A.C.E.S.; Hirohashi, N.; Pol-Fachin, L.; Verli, H.; Mourao, P.A.S. A Unique 2-Sulfated β-Galactan from the Egg Jelly of the Sea Urchin Glyptocidaris crenularis Conformation Flexibility Versus Induction of the Sperm Acrosome Reaction. J. Biol. Chem. 2009, 284, 18790–18800. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Verli, H. Effects of glycosylation on heparin binding and antithrombin activation by heparin. Carbohydr. Res. 2008, 343, 1435–1445. [Google Scholar] [CrossRef]
- Jiang, Z.; Geyer, A.; Schmitd, R.R. The Macrolidic Glycolipid Calonyctin A, a Plant Growth Regulator: Synthesis, Structural Assignment, and Conformational Analysis in Micellar Solution. Angew. Chem. Int. Ed. 1995, 34, 2520–2524. [Google Scholar] [CrossRef]
- Stierand, K.; Maaß, P.; Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef]
- Horton, D. Nomenclature of carbohydrates. Pure Appl. Chem. 1996, 68, 1919–2008. [Google Scholar]
- Schaftenaar, G.; Noordik, J.H.J. Molden: A pre- and post-processing program for molecular and electronic structures. Comput.-Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Schuettelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. 2004, D60, 1355–1363. [Google Scholar]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific LCC: San Carlos, CA, USA, 2002. [Google Scholar]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Scott, W.R.P.; Hünenberger, P.H.; Tironi, I.G.; Mark, A.E.; Billeter, S.R.; Fennen, J.; Torda, A.E.; Huber, T.; Krüger, P.; van Gunsteren, W.F. The GROMOS biomolecular simulation program package. J. Phys. Chem. A 1999, 103, 3596–3607. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raitieri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Verli, H.; Guimarães, J.A.A. Molecular dynamics simulation of a decasaccharide fragment of heparin in aqueous solution. Carbohydr. Res. 2004, 339, 281–290. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. A General Definition of Ring Puckering Coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P.J. The missing term in effective pair potentials. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comp. Chem. 2009, 16, 2785–2791. [Google Scholar]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arantes, P.R.; Sachett, L.G.; Graebin, C.S.; Verli, H. Conformational Characterization of Ipomotaosides and Their Recognition by COX-1 and 2. Molecules 2014, 19, 5421-5433. https://doi.org/10.3390/molecules19045421
Arantes PR, Sachett LG, Graebin CS, Verli H. Conformational Characterization of Ipomotaosides and Their Recognition by COX-1 and 2. Molecules. 2014; 19(4):5421-5433. https://doi.org/10.3390/molecules19045421
Chicago/Turabian StyleArantes, Pablo R., Liana G. Sachett, Cedric S. Graebin, and Hugo Verli. 2014. "Conformational Characterization of Ipomotaosides and Their Recognition by COX-1 and 2" Molecules 19, no. 4: 5421-5433. https://doi.org/10.3390/molecules19045421
APA StyleArantes, P. R., Sachett, L. G., Graebin, C. S., & Verli, H. (2014). Conformational Characterization of Ipomotaosides and Their Recognition by COX-1 and 2. Molecules, 19(4), 5421-5433. https://doi.org/10.3390/molecules19045421