RutheniumII Complexes bearing Fused Polycyclic Ligands: From Fundamental Aspects to Potential Applications

Abstract
:1. Introduction
2. Mononuclear RutheniumII Complexes
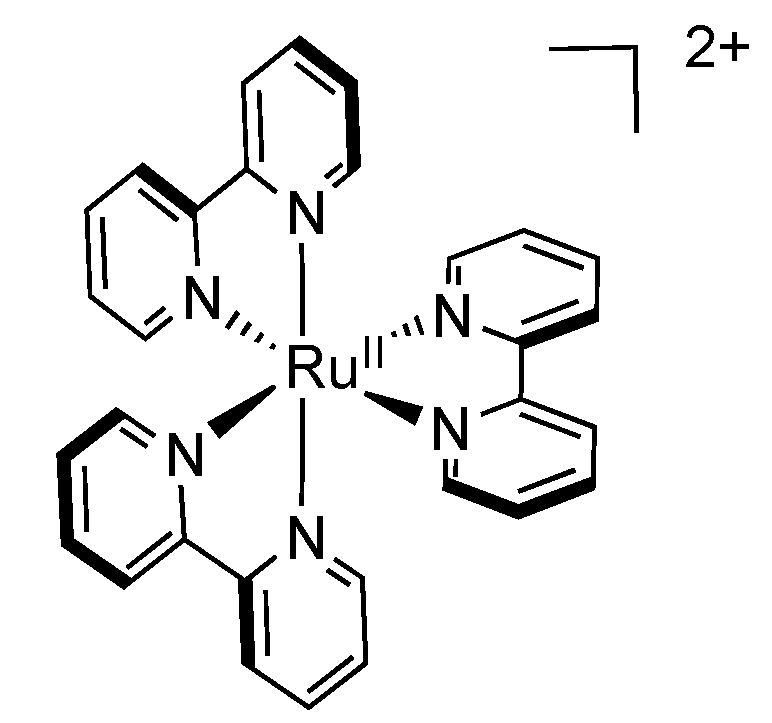
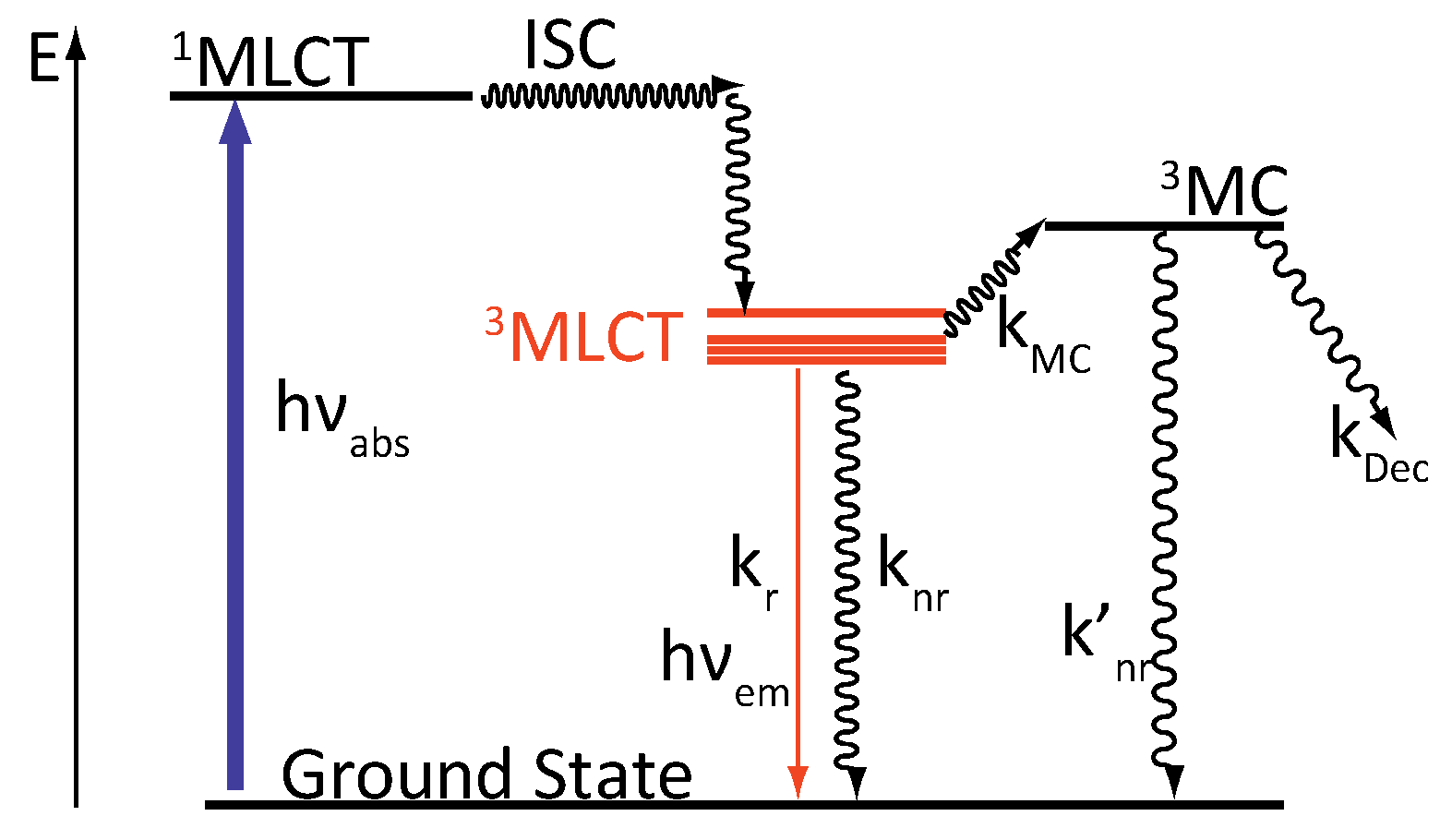
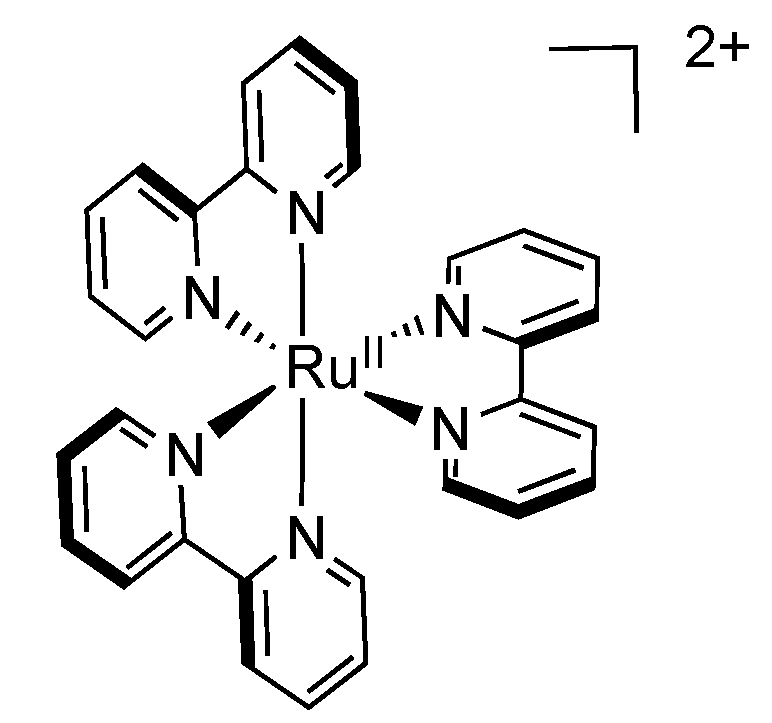
2.1. [Ru(bpy)3]2+

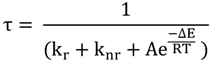
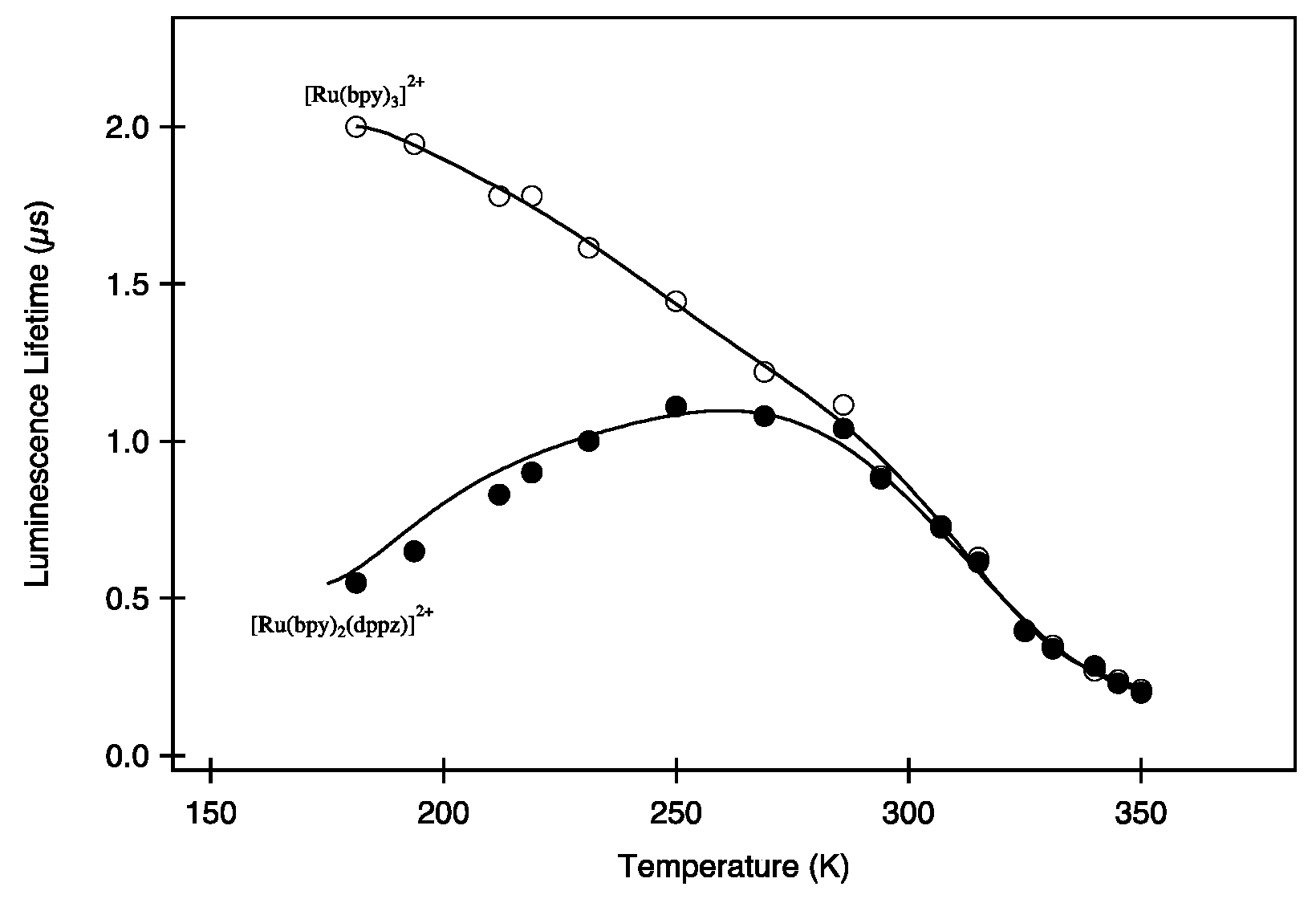
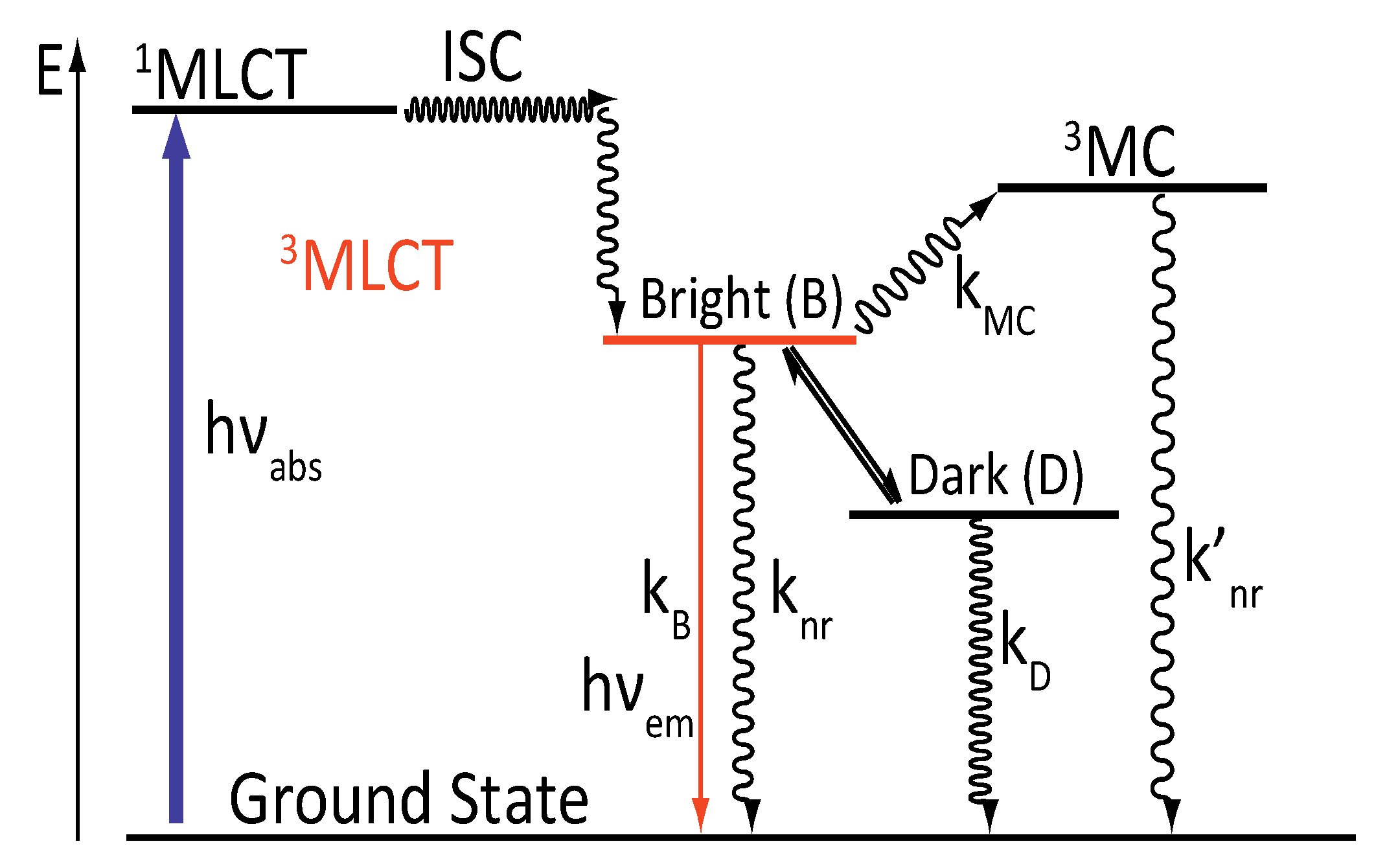
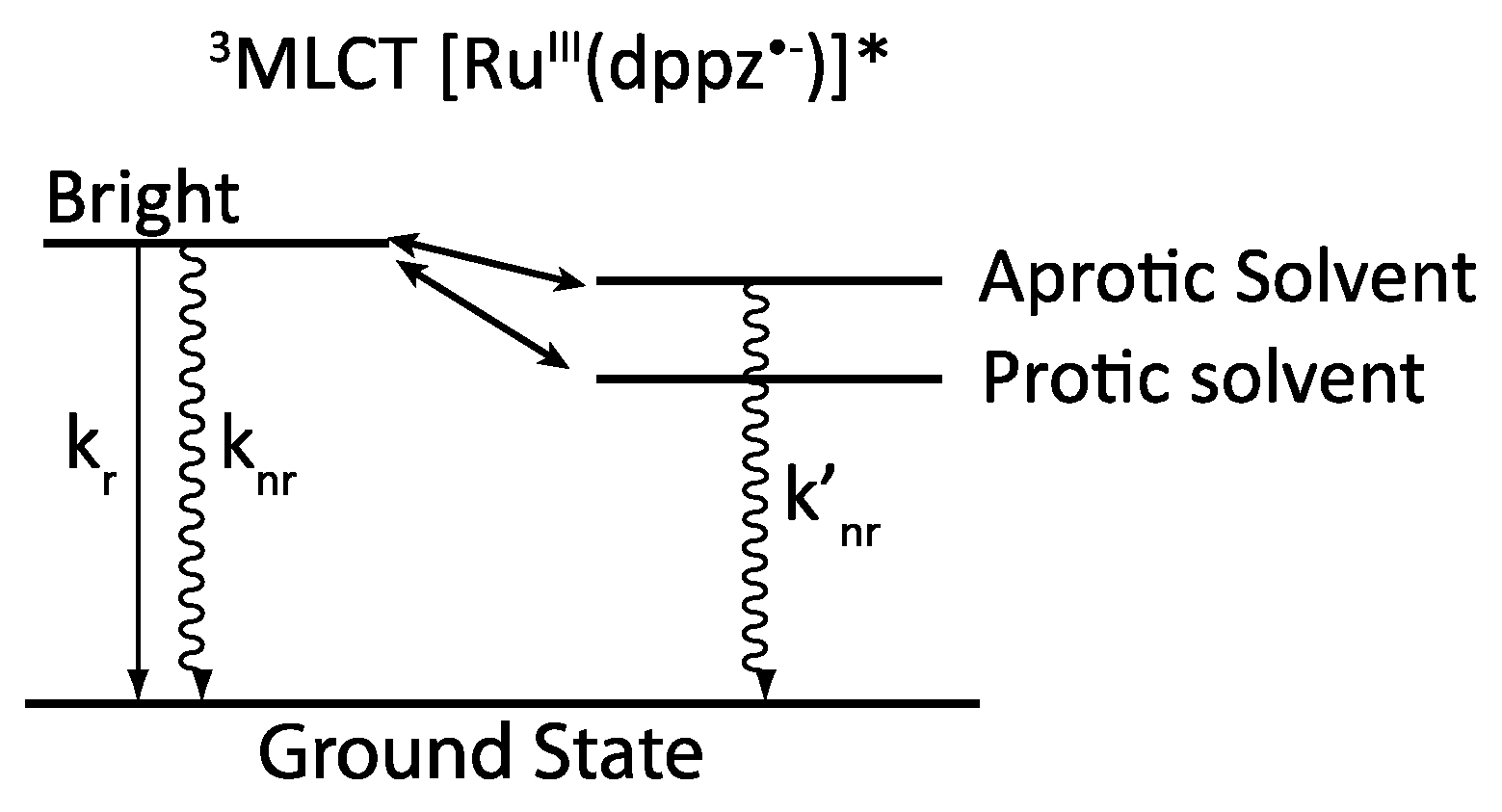
2.2. [Ru(bpy/phen)2(DPPZ)]2+

- -
- A 3MC state higher in energy
- -
- A luminescent 3MLCT “bright state” corresponding to a charge transfer from the ruthenium center to the 1,10-phenanthroline moiety of the DPPZ ligand.
- -
- A “dark state”, non-luminescent and lower in energy, that corresponds to the phenazine moiety of DPPZ.

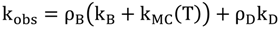
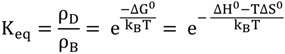
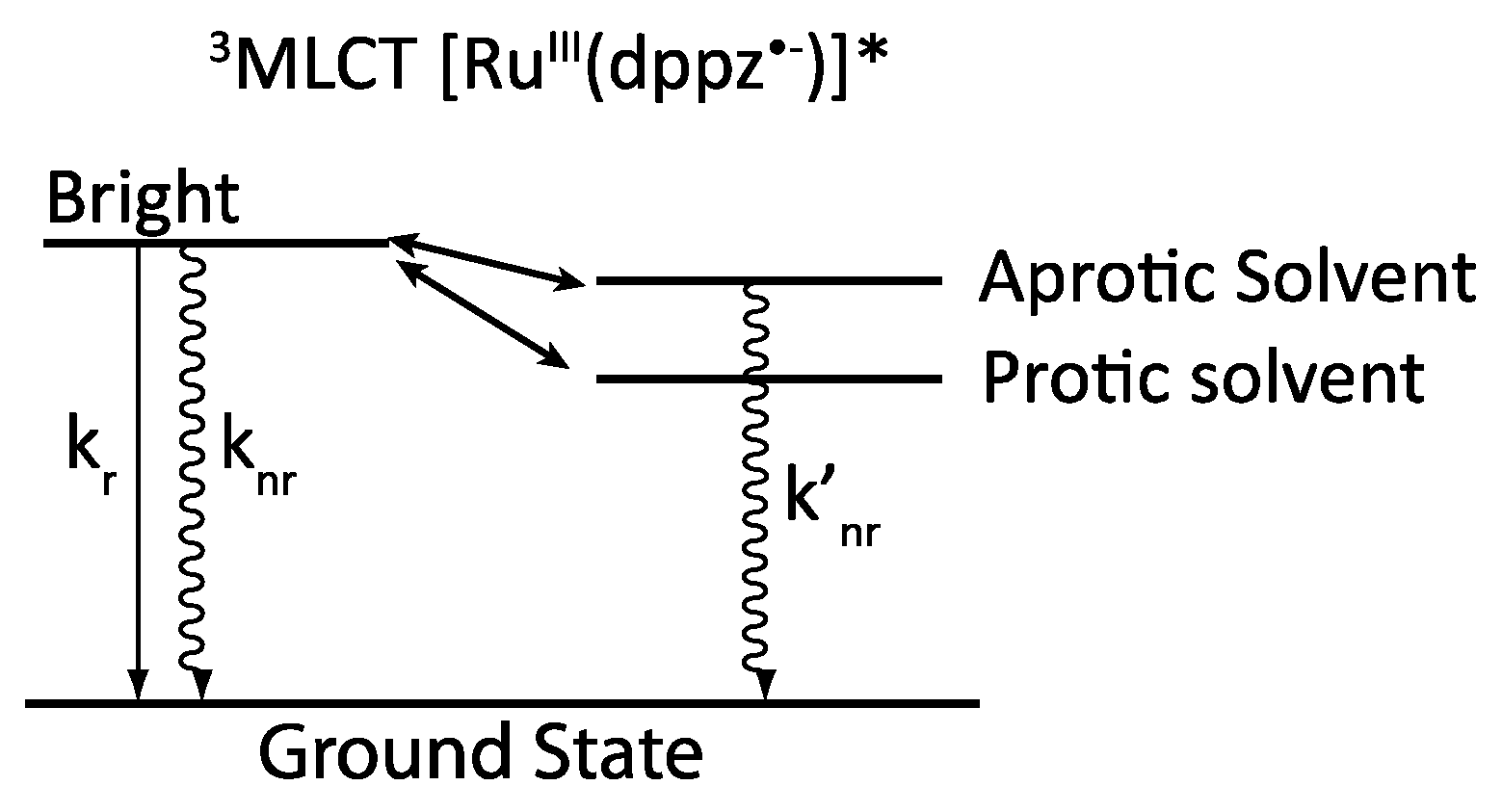

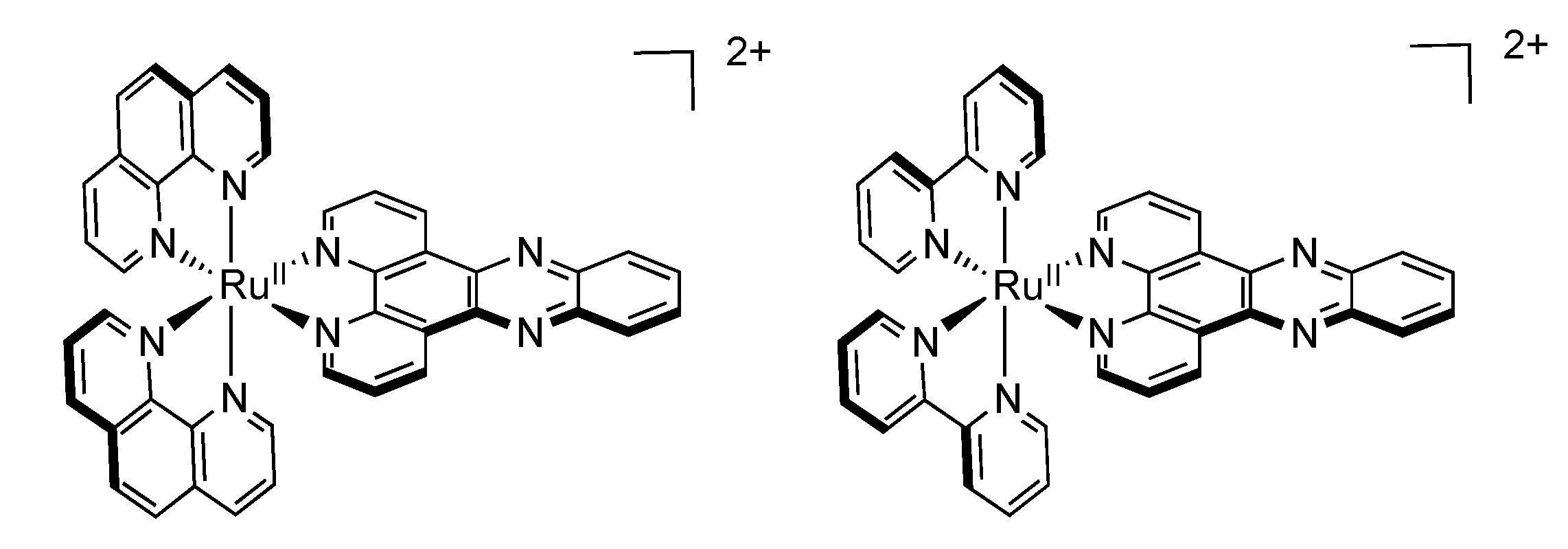
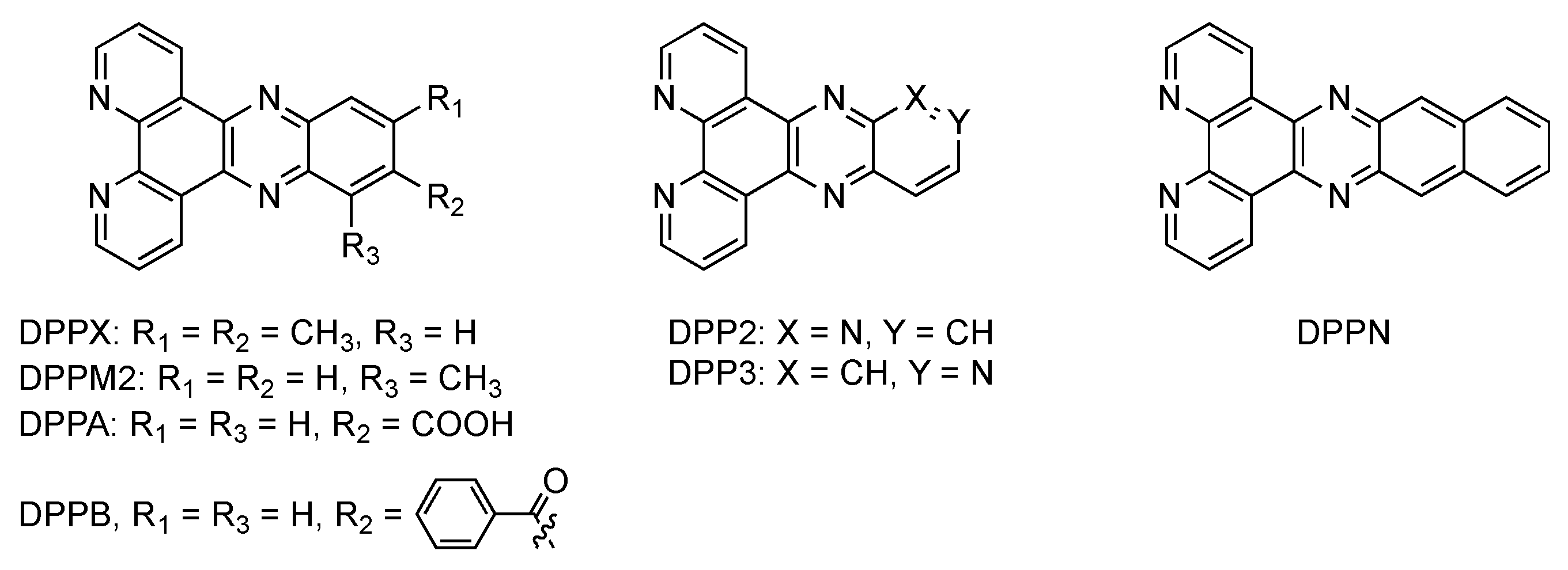
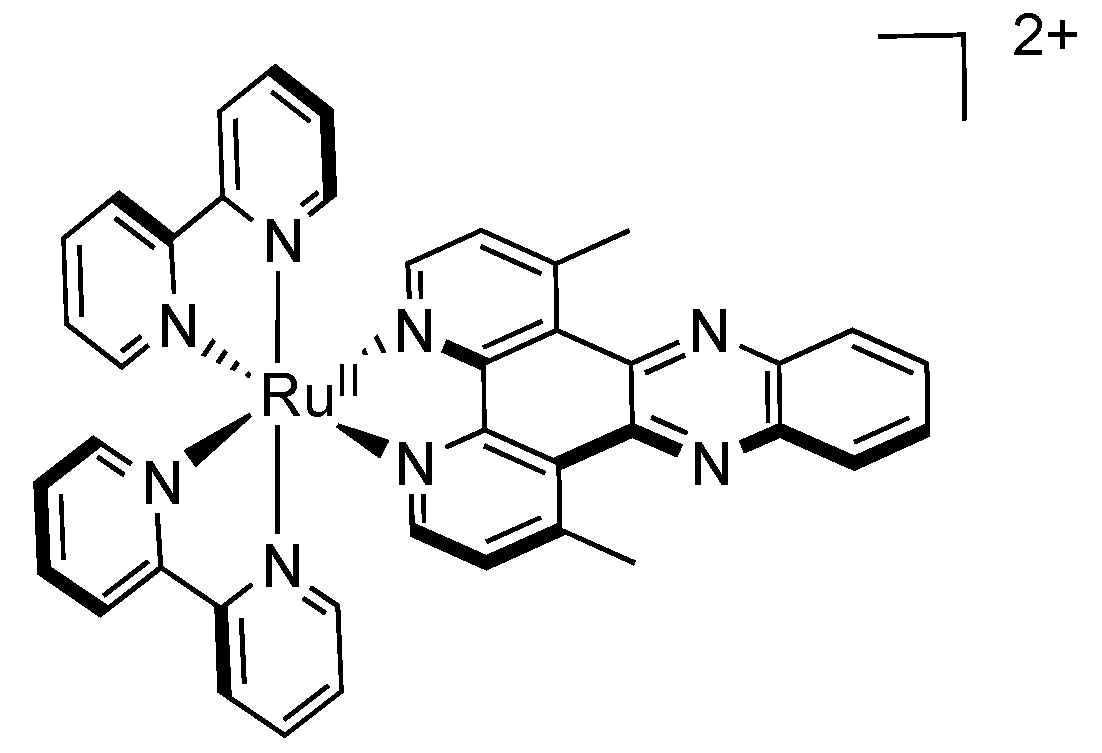
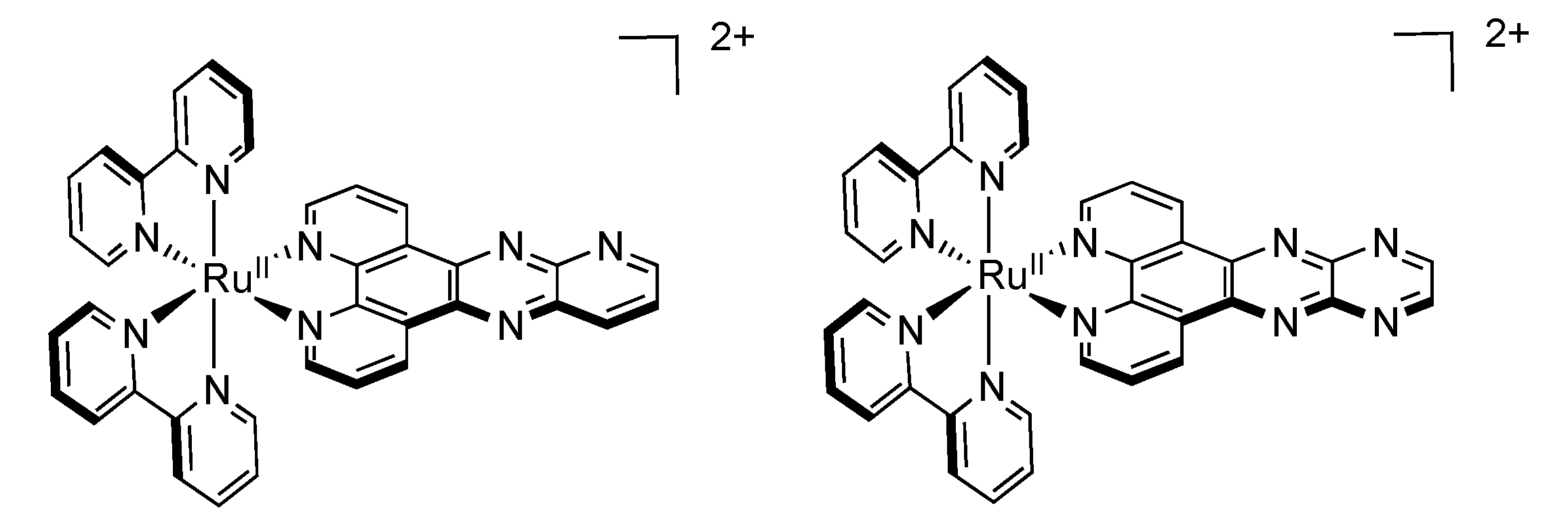
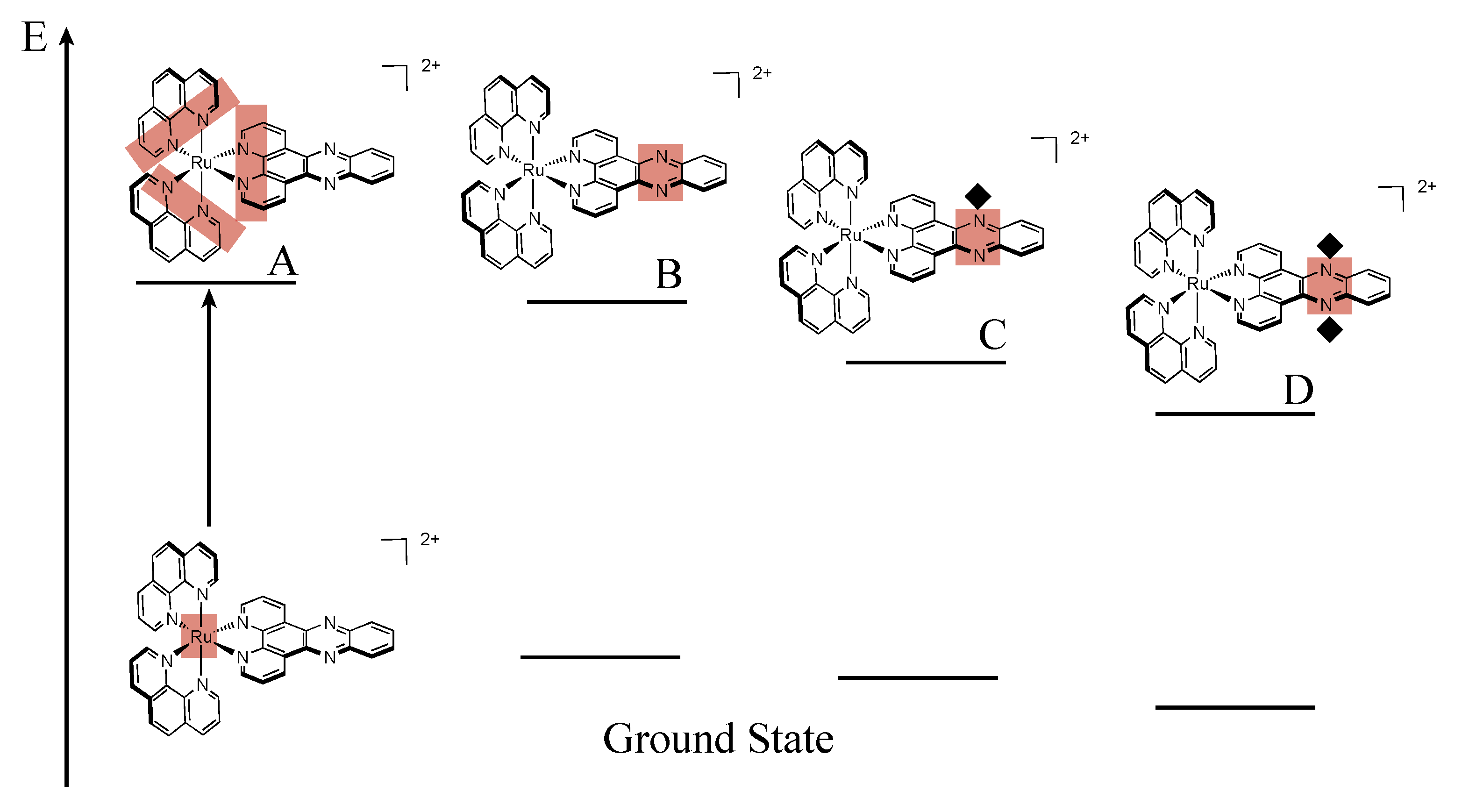
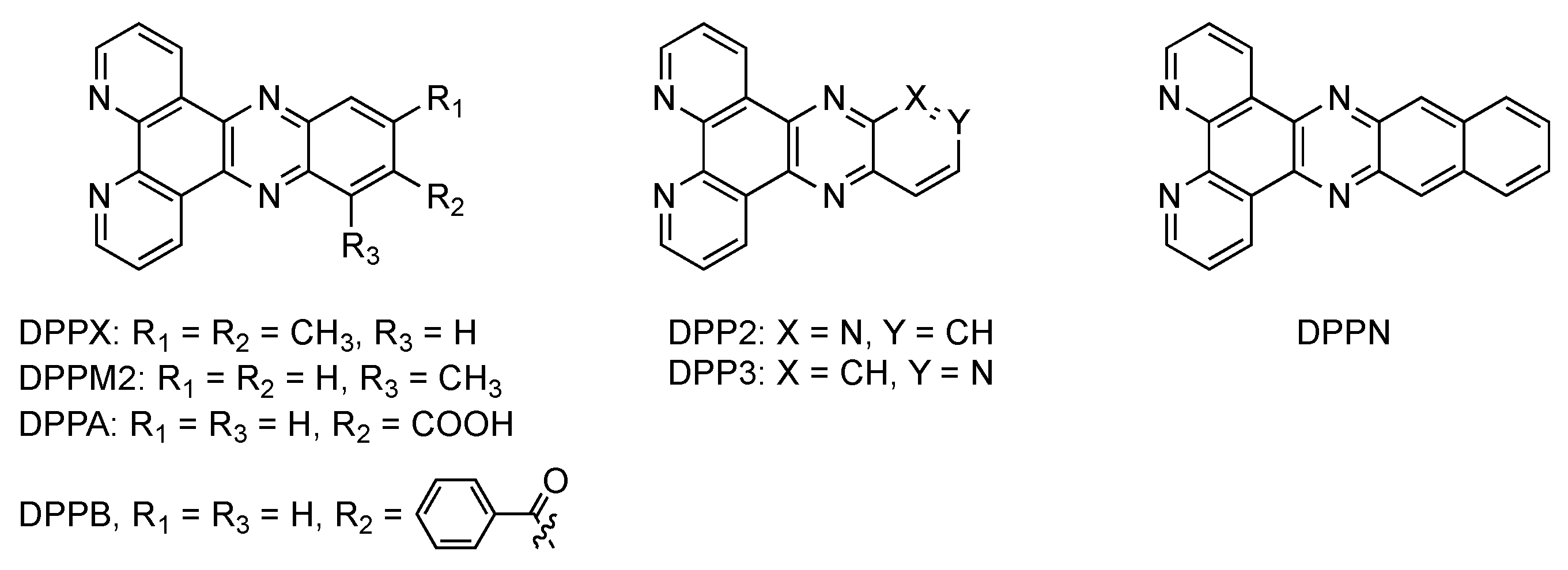
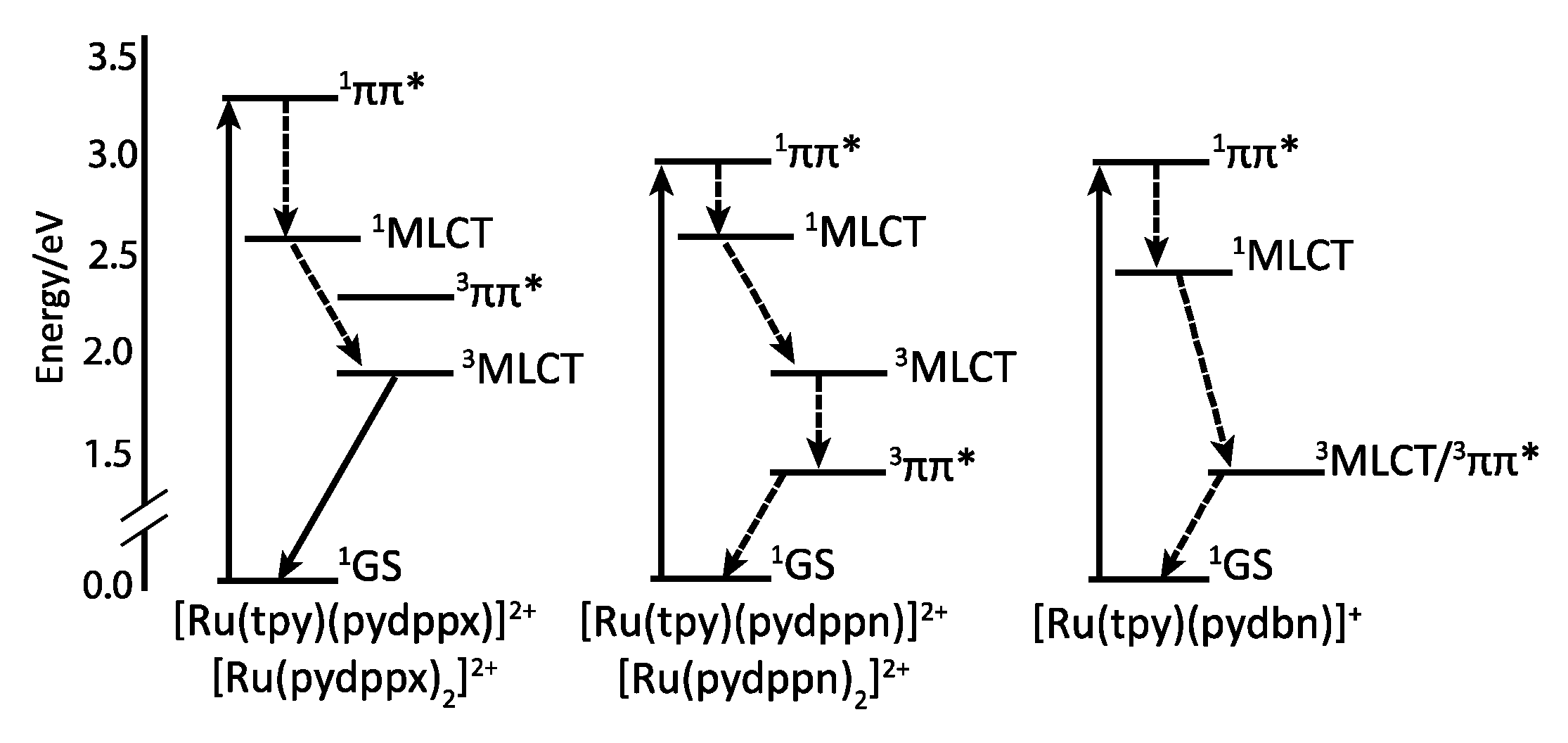
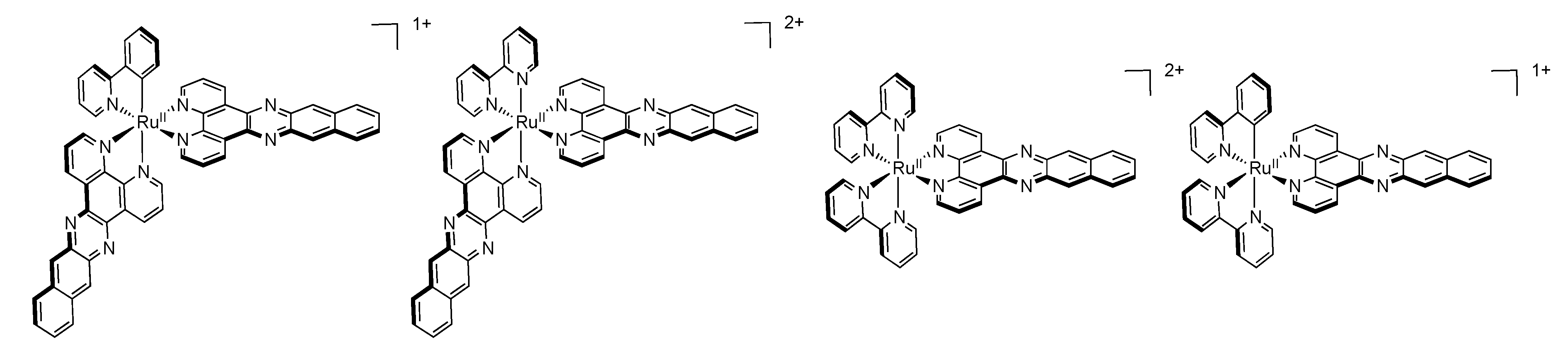
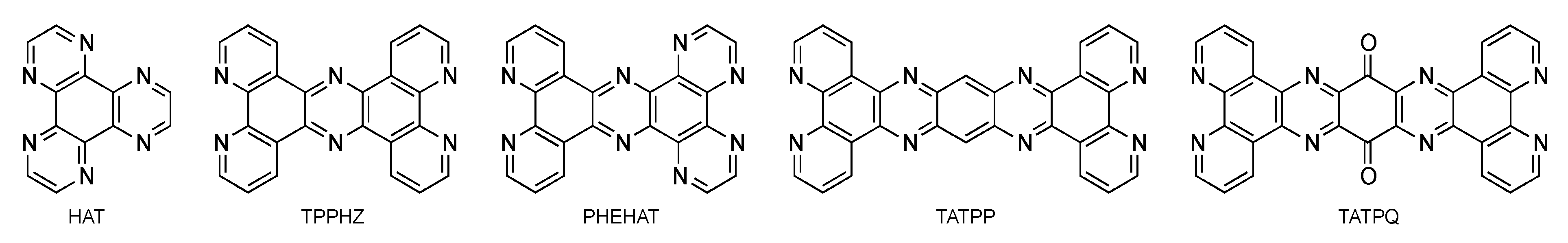
2.3. RutheniumII Complexes bearing DPPZ Analogues

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
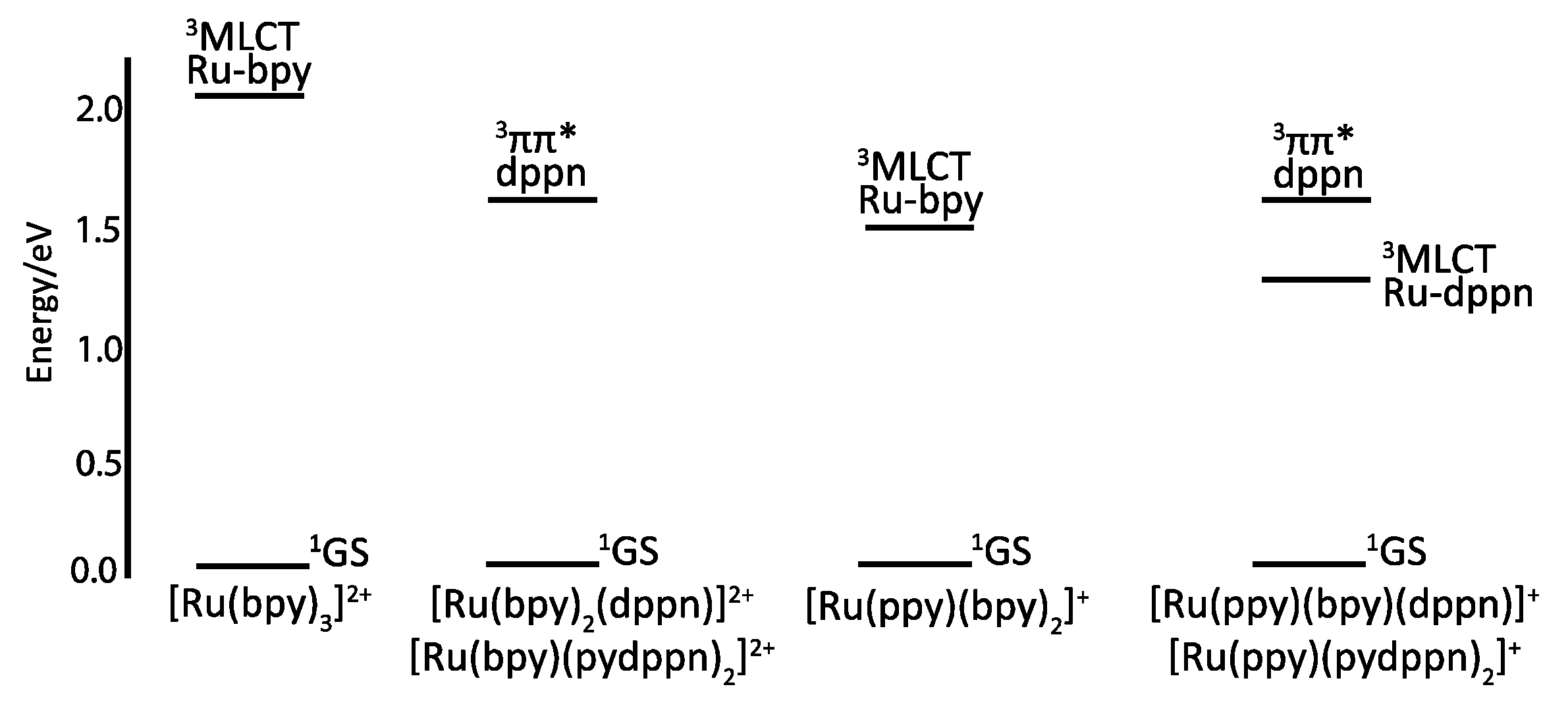{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | pKa*(app) |
|---|---|
| [Ru(TAP)3]2+ | 3.5 |
| [Ru(TAP)2(bpy)]2+ | 4 |
| [Ru(TAP)(bpy)2]2+ | 3.1 |
| [Ru(bpy)2(dpqp)]2+ | 2.1 |
| [Ru(bpz)3]2+ | 3.30 |
| [Ru(bpz)2(bpm)]2+ | 3.4 |
| [Ru(bpm)2(bpz)]2+ | 3.5 |
| [Ru(bpz)2(bpy)]2+ | 3.4 |
| [Ru(bpy)(bpz)(bpm)]2+ | 3.1 |
| [Ru(bpy)2(bpz)]2+ | 2.8 |
| [Ru(bpm)3]2+ | 2.35 |
| [Ru(bpm)2(bpy)]2+ | 2.25 |
| [Ru(bpy)2(bpm)]2+ | 1.90 |


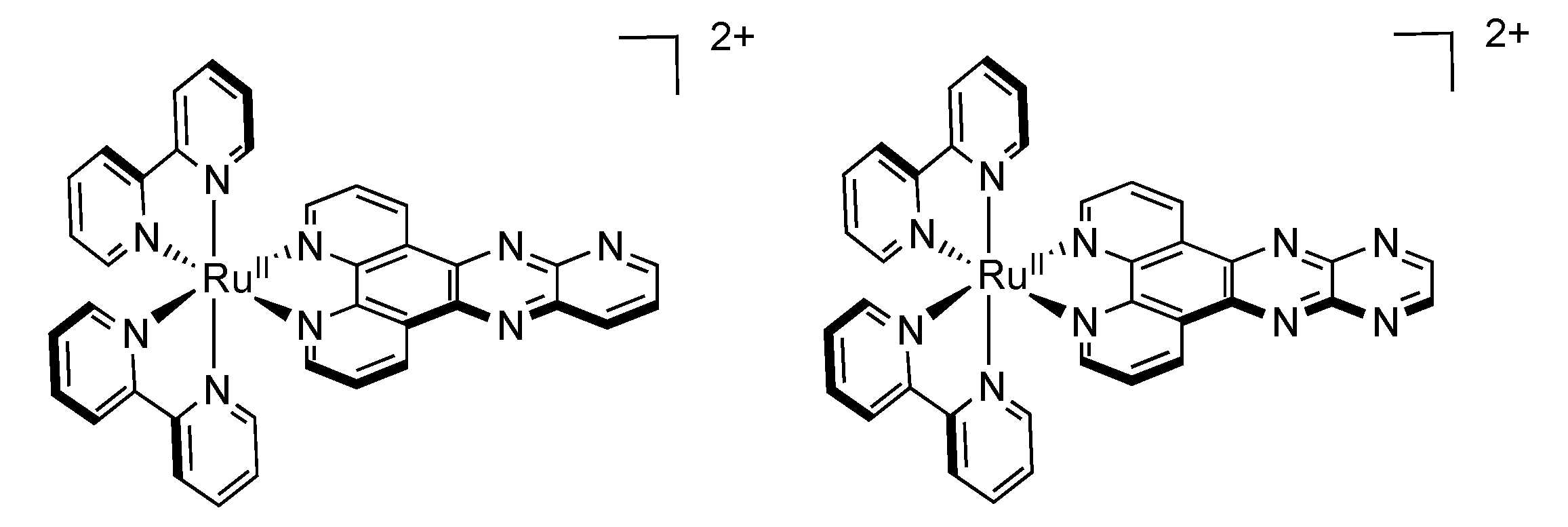
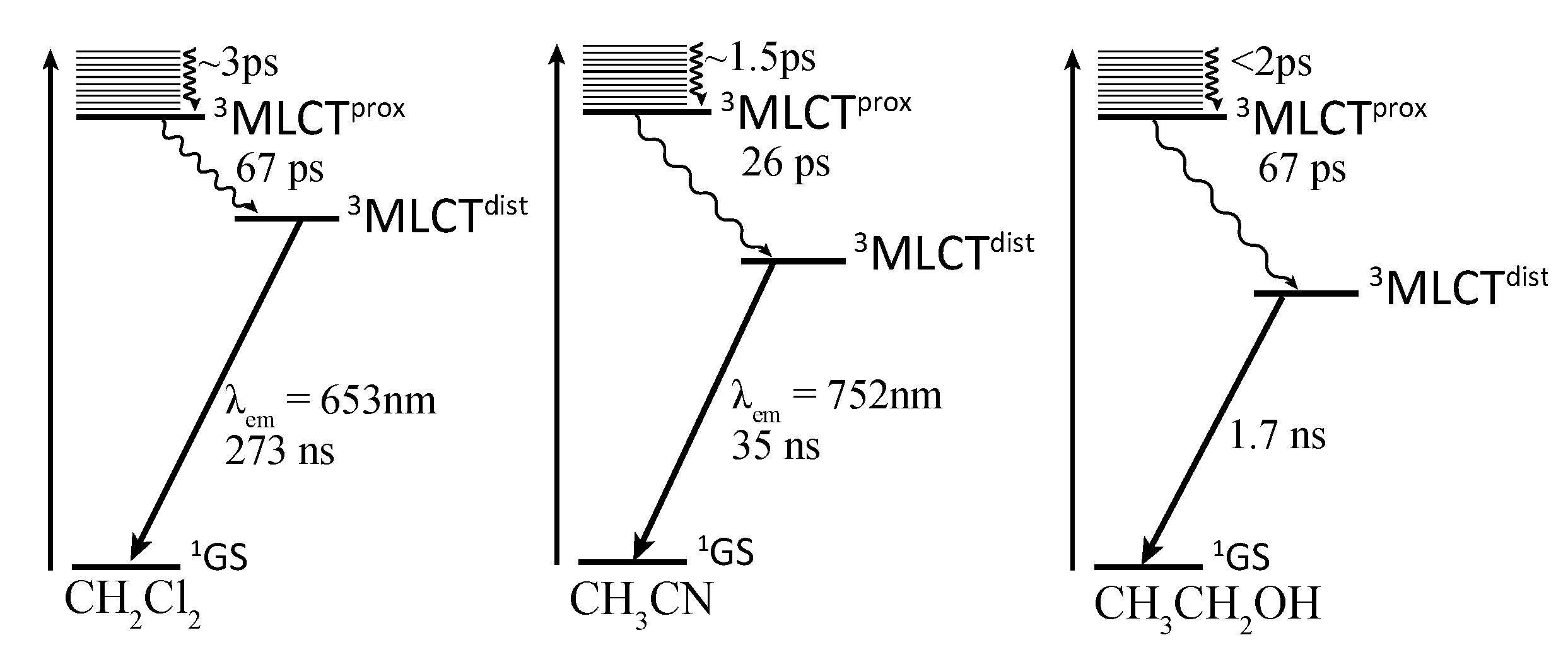
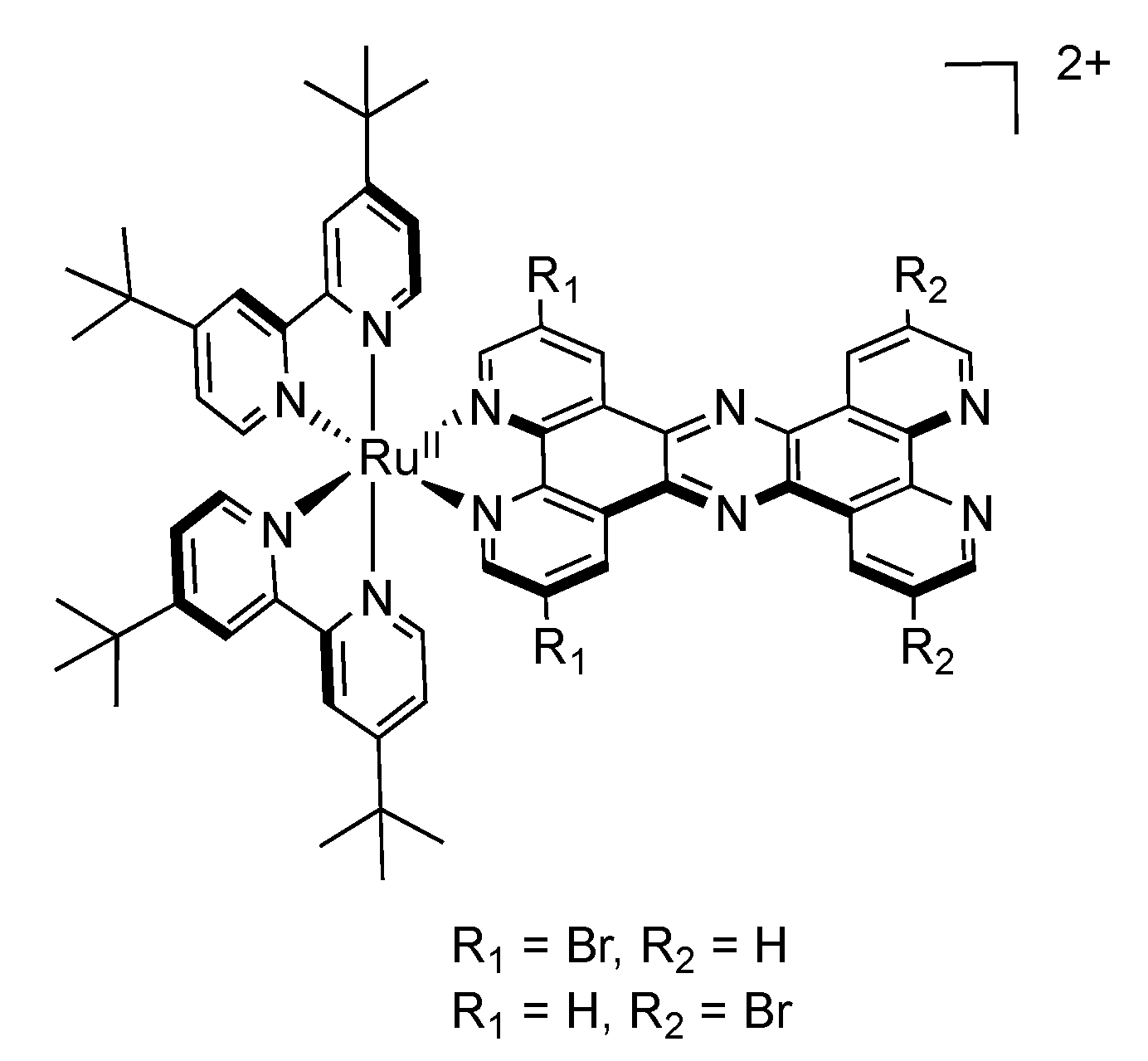
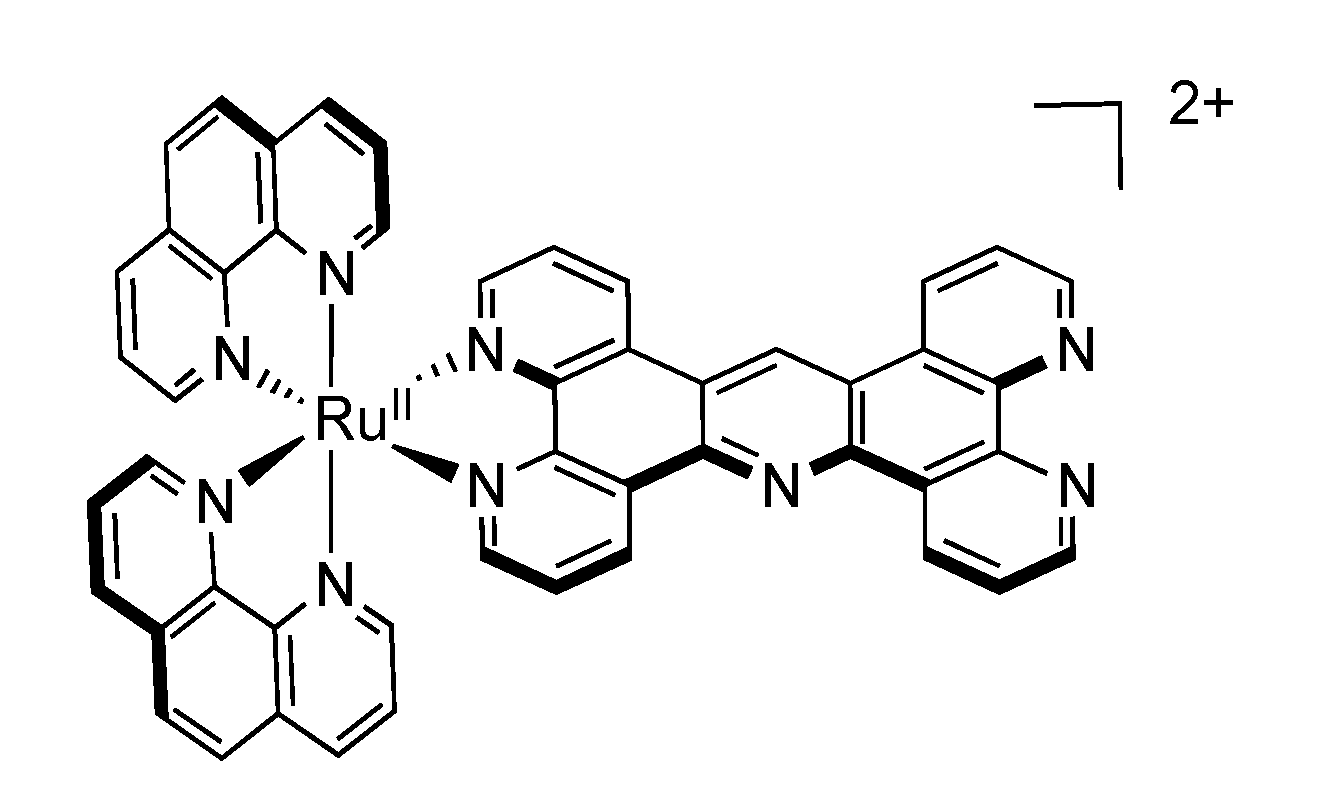
2.4. RutheniumII Complexes bearing Trischelating DPPZ Analogues
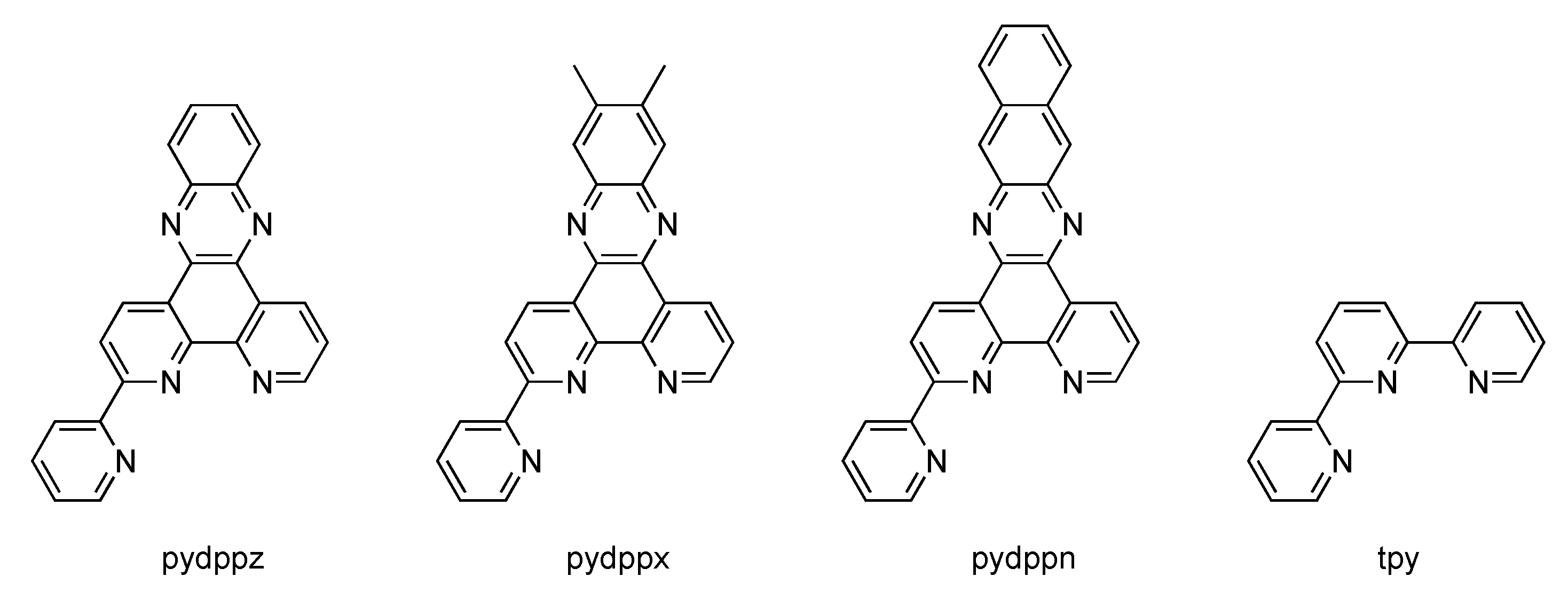

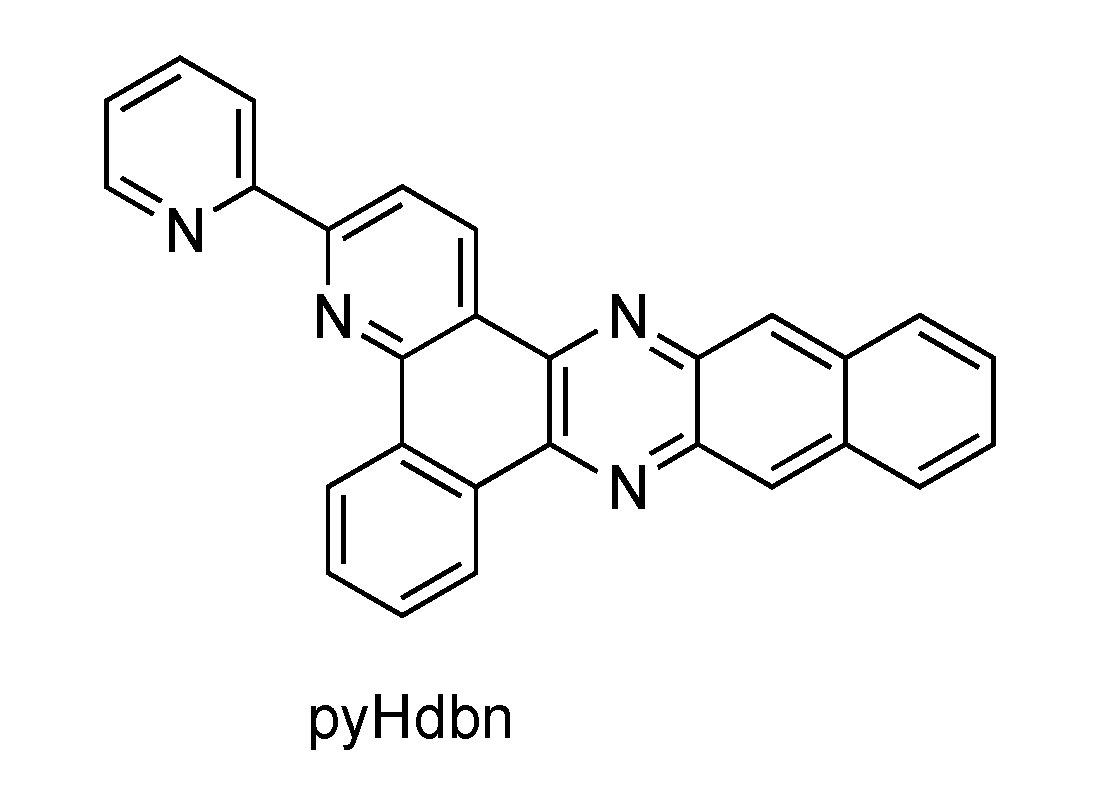
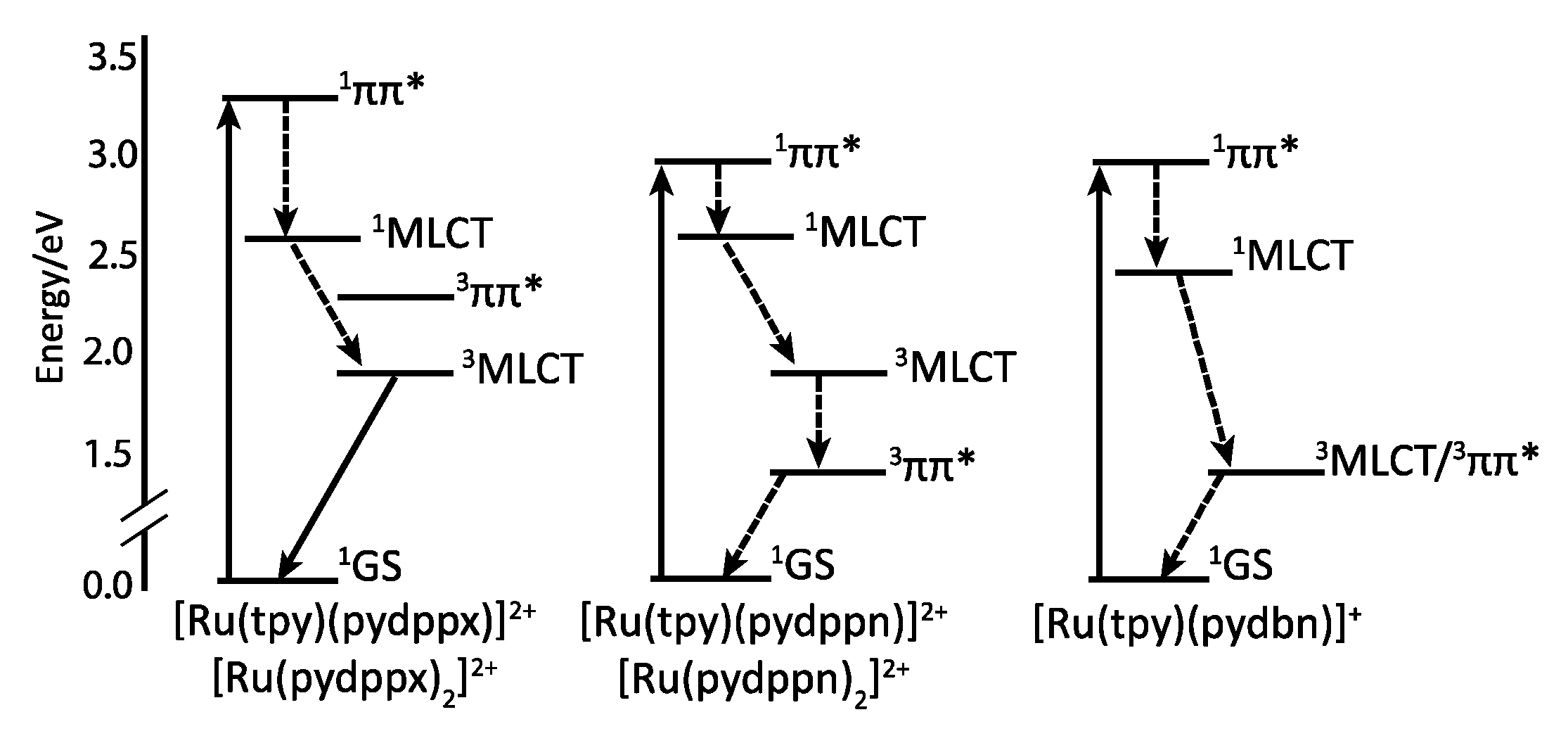
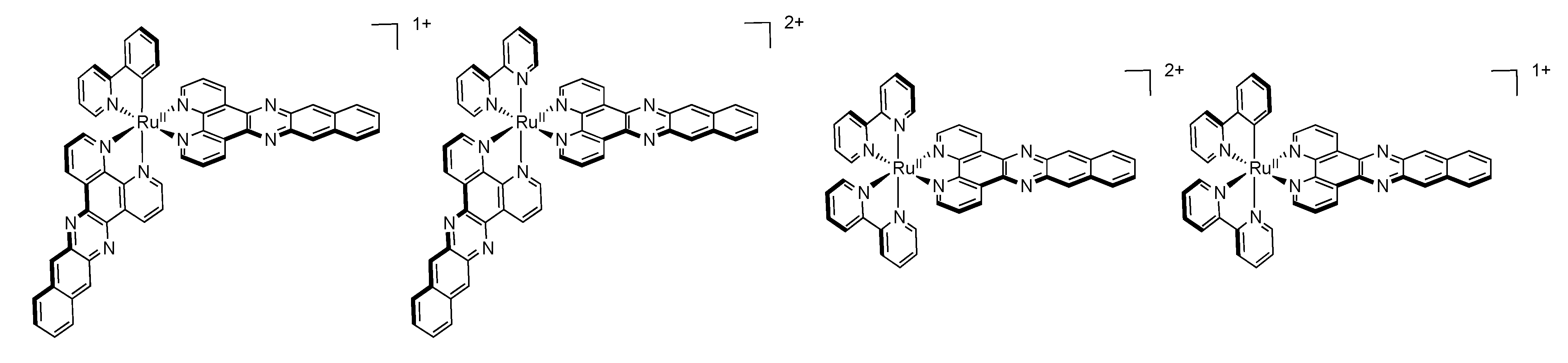

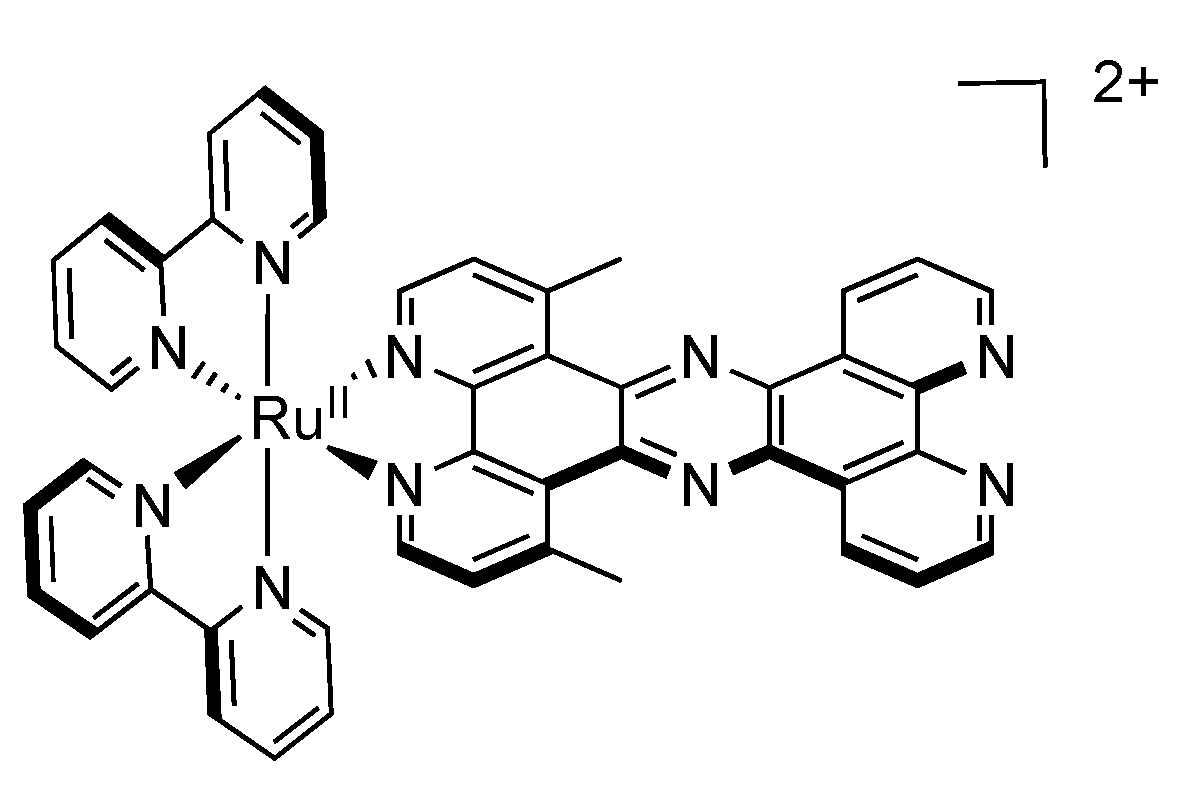
2.5. Mononuclear RutheniumII-TPPHZ Complexes
| Complex | Oxidation | Reduction |
|---|---|---|
| [Ru(bpy)3]2+ [91] | +1.27 | −1.31 −1.50 −1.77 |
| [Ru(phen)3]2+ [141] | +1.27 | −1.35 −1.52 |
| [Os(bpy)3]2+ [124,142] | +0.78 | −1.30 −1.48 −1.78 |
| [Ru(phen)2(TPPHZ)]2+ [137] a | +1.34 | −1.00 −1.38 −1.69 |
| [Ru(bpy)2(TPPHZ)]2+ [124] | +1.33 | −0.87 −1.33 −1.51 −1.73 |
| [Os(bpy)2(TPPHZ)]2+ [124] | +0.88 | −0.87 −1.24 −1.53 −1.79 |
| [Ru(bpy)2(TPPHZ)Ru(bpy)2]4+ [124] | +1.34 (2) | −0.71 −1.31 (2) −1.51 (2) −1.72 |
| [Os(bpy)2(TPPHZ)Os(bpy)2]4+ [124] | +0.89 (2) | −0.70 −1.21 (2) −1.44 (2) |
| [Ru(bpy)2(TPPHZ)Os(bpy)2]4+ [124] | + 0.89 +1.33 | −0.70 −1.27 (2) −1.48 (2) −1.66 −2.06 |
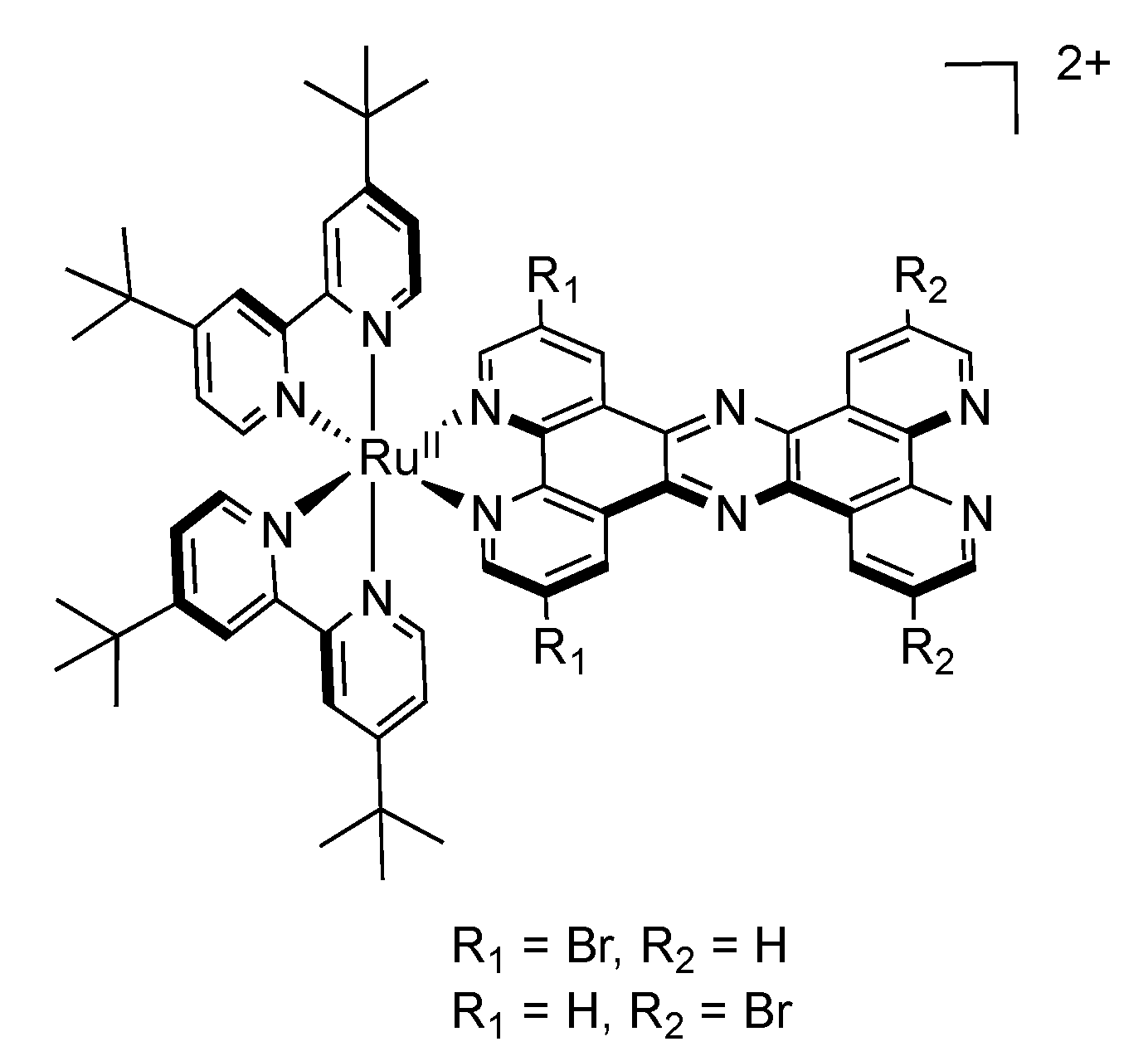
| Complex | λem/nm (CH2Cl2) | λem/nm (CH3CN) | (τ/ns)
CH2Cl2 | (τ/ns)
CH3CN | Oxidation | Reduction |
|---|---|---|---|---|---|---|
| [Ru(tbbpy)2(TPPHZ)]2+ R1 = R2 = H | 629 | 638 | 404.7 | 150.3 | +0.83 | −1.39 −1.88 −2.05 −2.26 |
| [Ru(tbbpy)2(TPPHZ)]2+ R1 = Br, R2 = H | 650 | 661 | 560.3 | 218.3 | +0.89 | −1.18 −1.87 −2.03 −2.18 |
| [Ru(tbbpy)2(TPPHZ)]2+ R1 = H R2 = Br | 633 | 645 | 390.0 | 92.1 | +0.91 | −1.25 −1.89 −2.06 −2.25 |
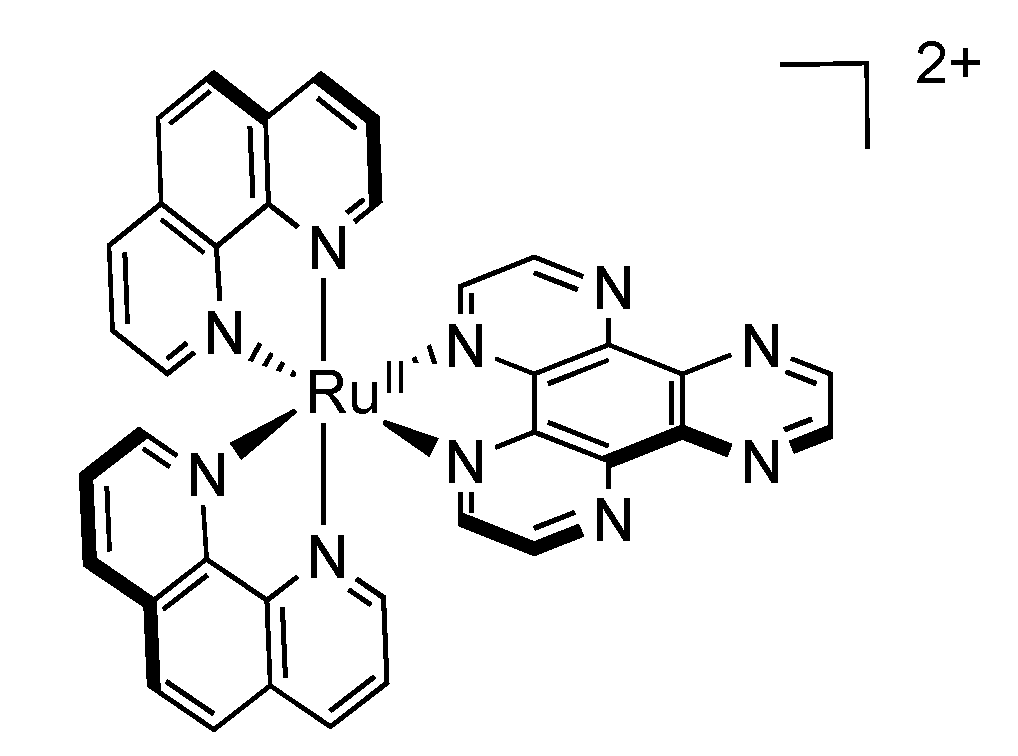
2.6. Mononuclear RutheniumII-TPAC Complexes
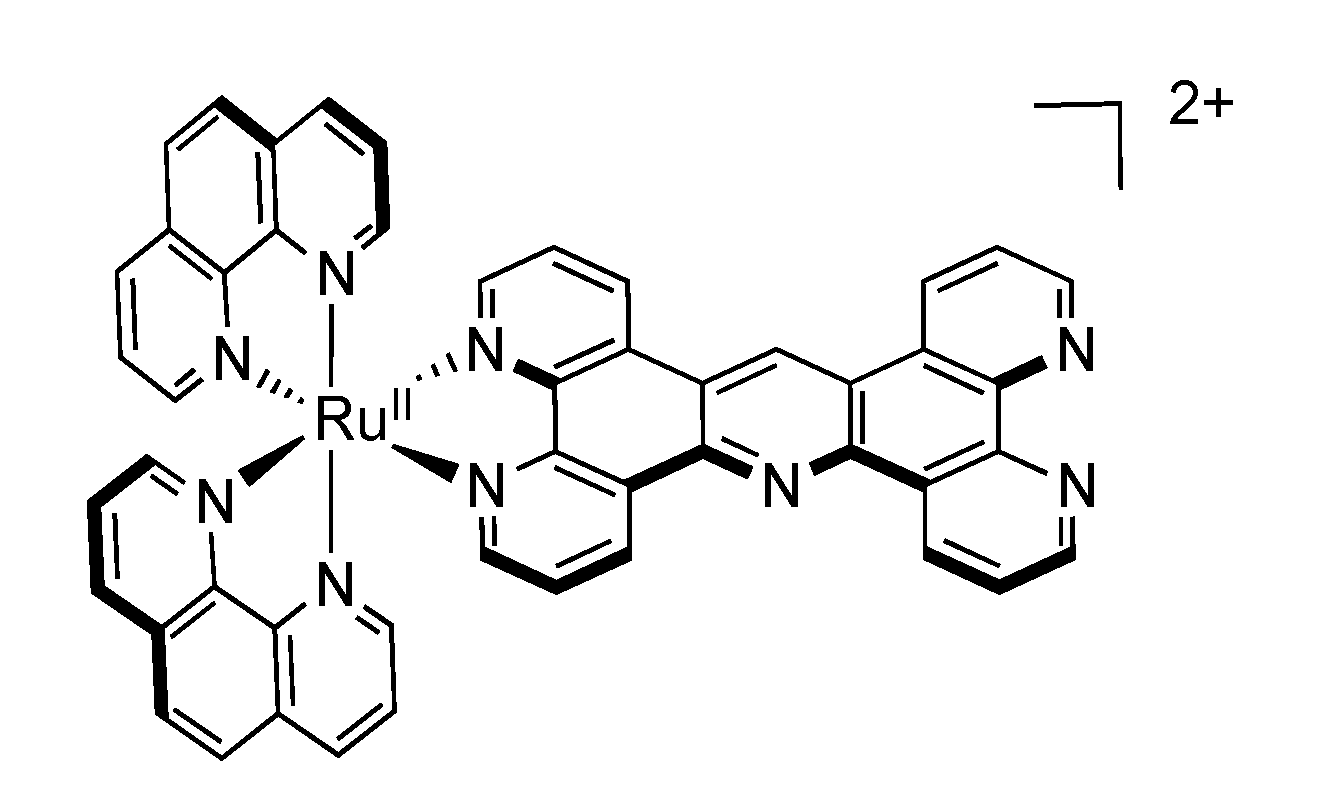
2.7. Mononuclear RutheniumII-PHEHAT Complexes



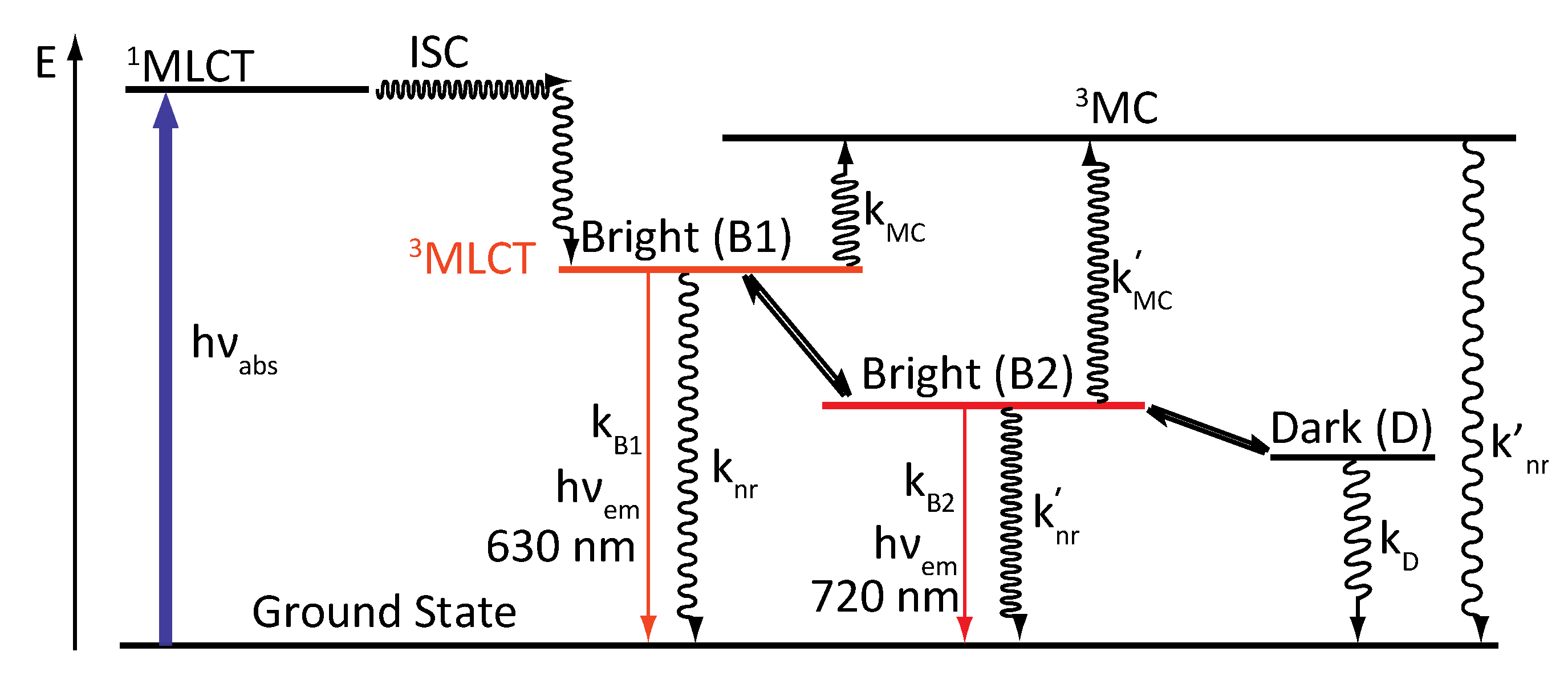
- -
- B1, populated at high temperature should correspond to a 3MLCT state where the electron is located on the phen part of the ligand, whose characteristics are close to those of [Ru(phen)3]2+.
- -
- B2, lower in energy, populated at low temperature, should correspond to a 3MLCT state where the electron is located on the “HAT” moiety of the ligand. This excited state presents similarities with the complex [Ru(phen)2(HAT)]2+.
| Complex | λem/nm CH3CN | τ/ns CH3CN deaerated | λem/nm H2O | τ/ns H2O deaerated |
|---|---|---|---|---|
| [Ru(bpy)3]2+ [88] | 620 | 855 | 626 | 630 |
| [Ru(phen)3]2+ [153] | 604 | 460 | 604 | 920 |
| [Ru(bpy)2(DPPZ)]2+ [107] | 631 | 750 | --- | --- |
| [Ru(phen)2(DPPZ)]2+ [95] | 607 | 663 | --- | --- |
| [Ru(bpy)2(dpqp)]2+ [107] | 618 | 921 | 617 | 582 |
| [Ru(TAP)2(DPPZ)]2+ [115] | 621 | 636 | 1090 | |
| [Ru(phen)2(TPPHZ)]2+ [137] | 625 | 1250 | ||
| [Ru(phen)2(TPAC)]2+ [111] | 608 | 253 | 613 | 839 |
| [Ru(TAP)2(TPAC)]2+ [111] | 624 | 1127 | 640 | 952 |
| [Ru(phen)2(HAT)]2+ [151] | 696 | 776 | 732 | 137 |
| [Ru(phen)2(PHEHAT)]2+ [151] | 662 | 262 | --- | --- |
| [Ru(phen)2(HATPHE]2+ [151] | 692 | 666 | 730 | 130 |
3. Binuclear Complexes
3.1. Binuclear TPPHZ Complexes

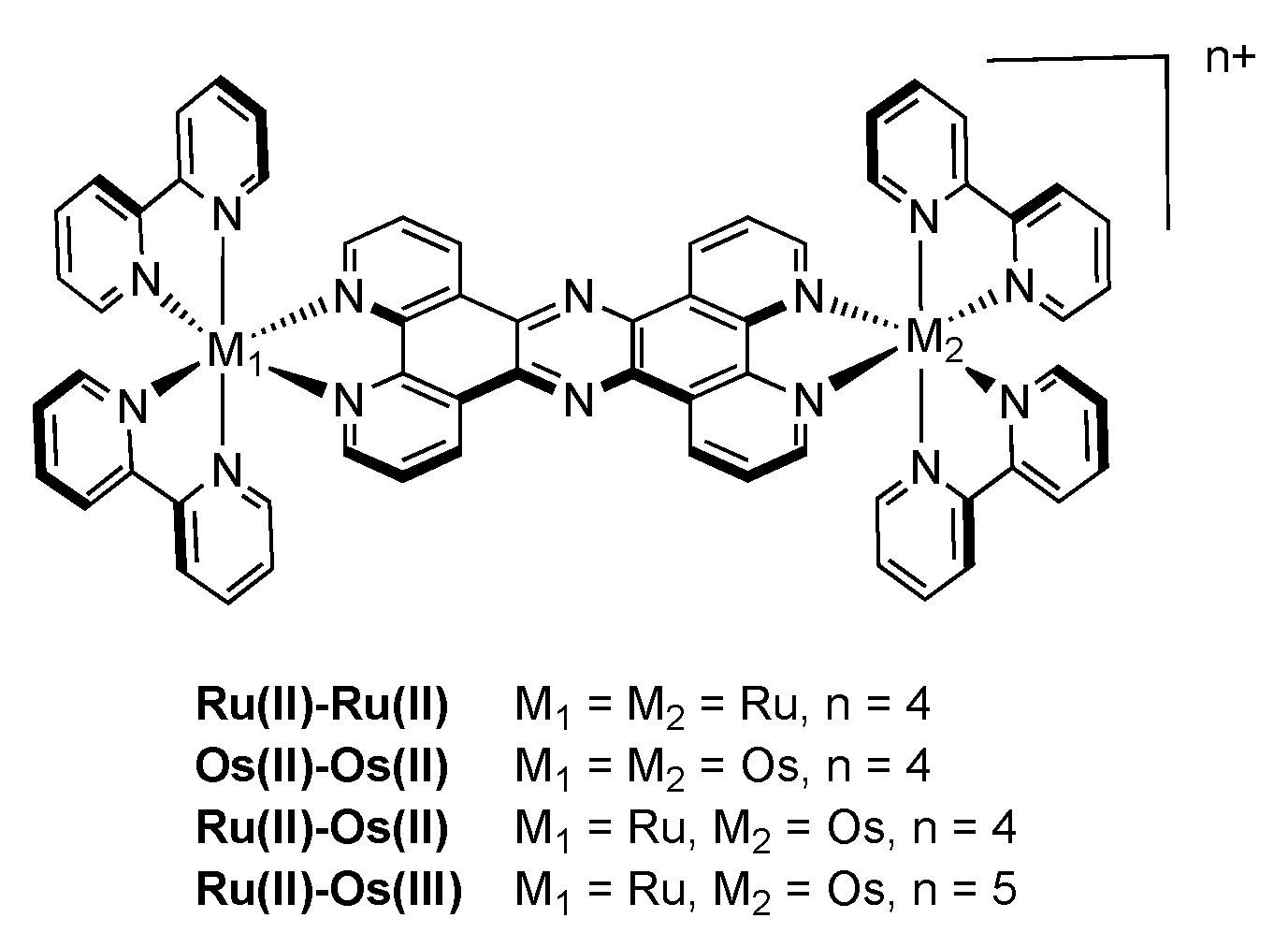
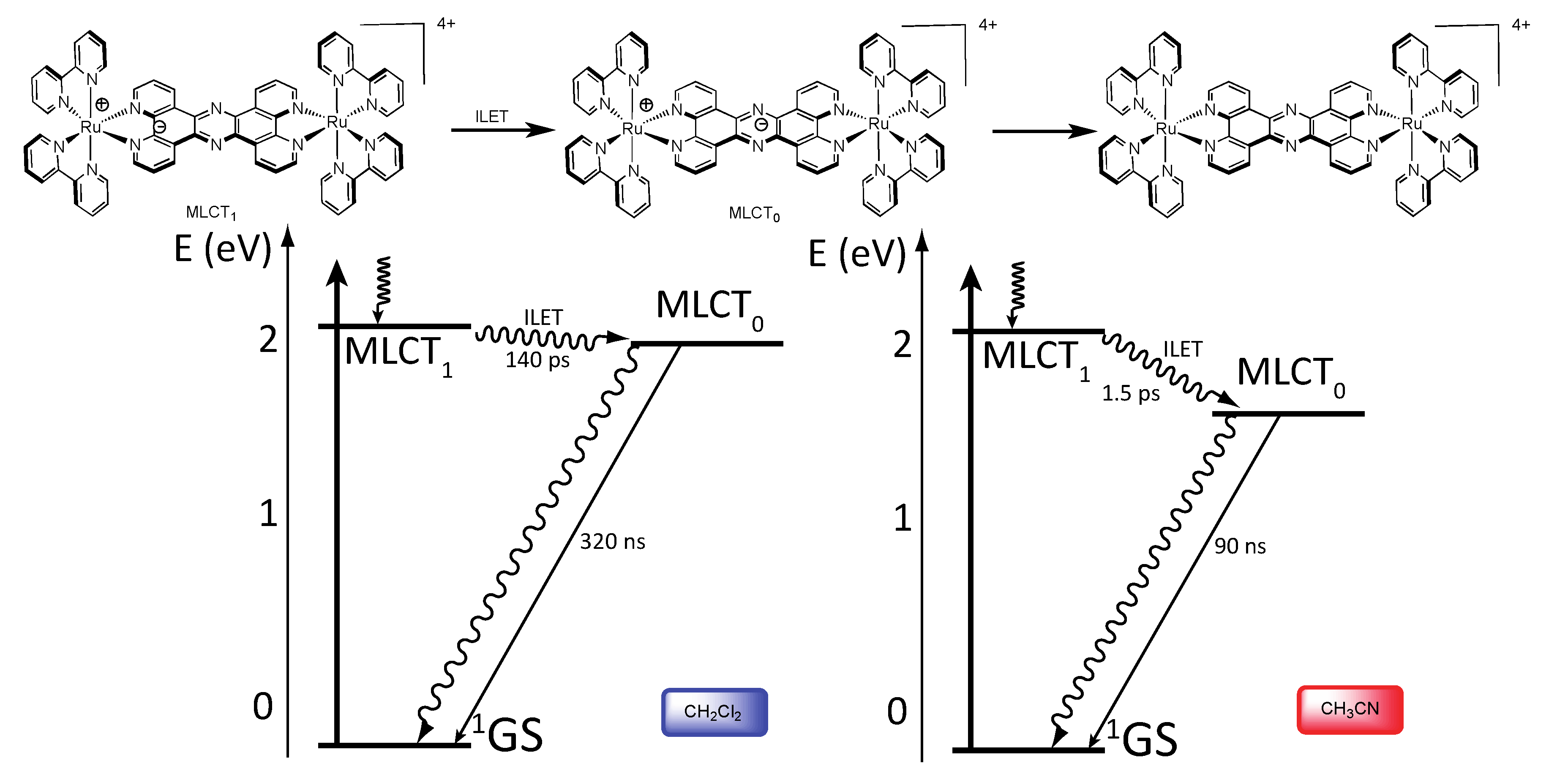
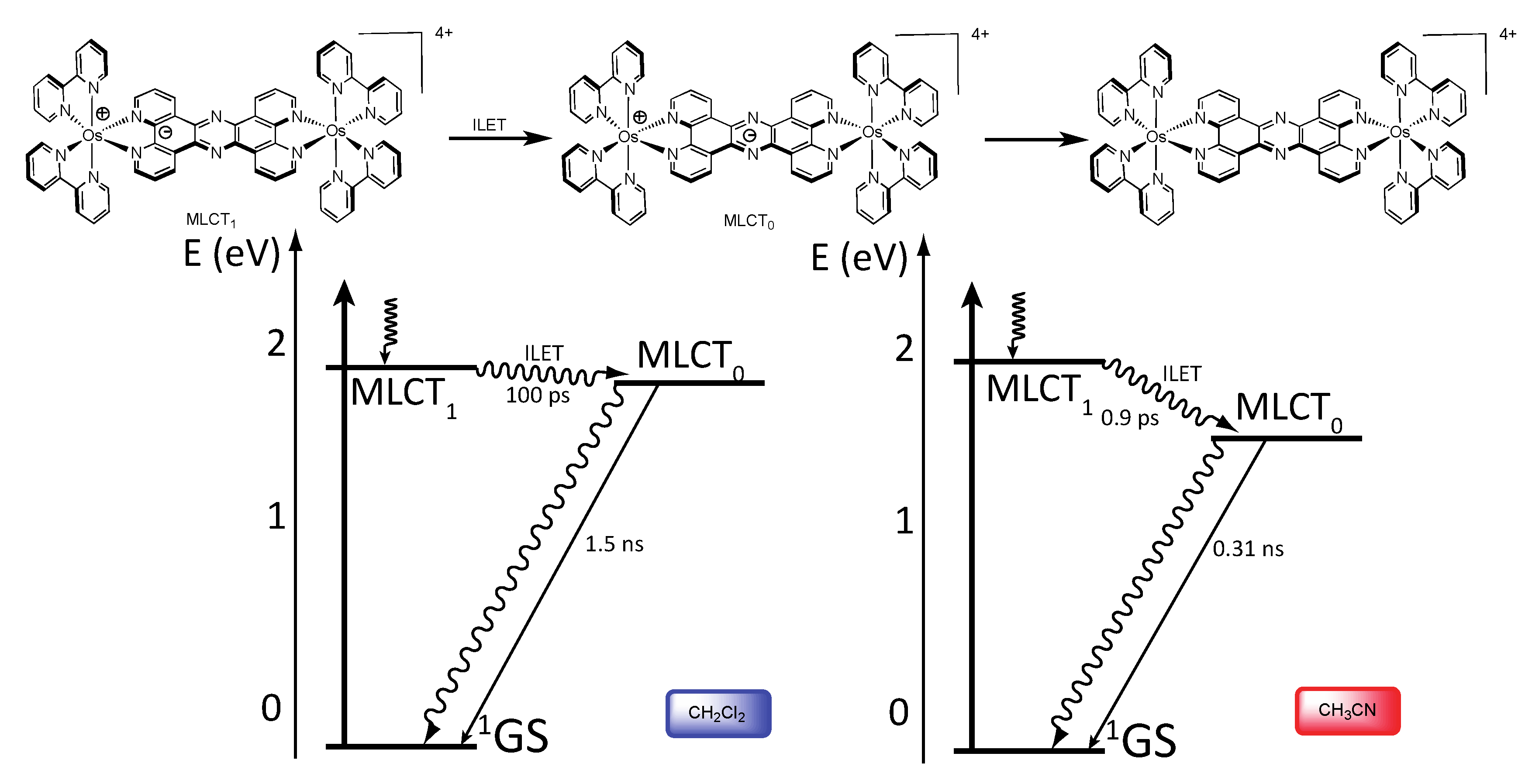
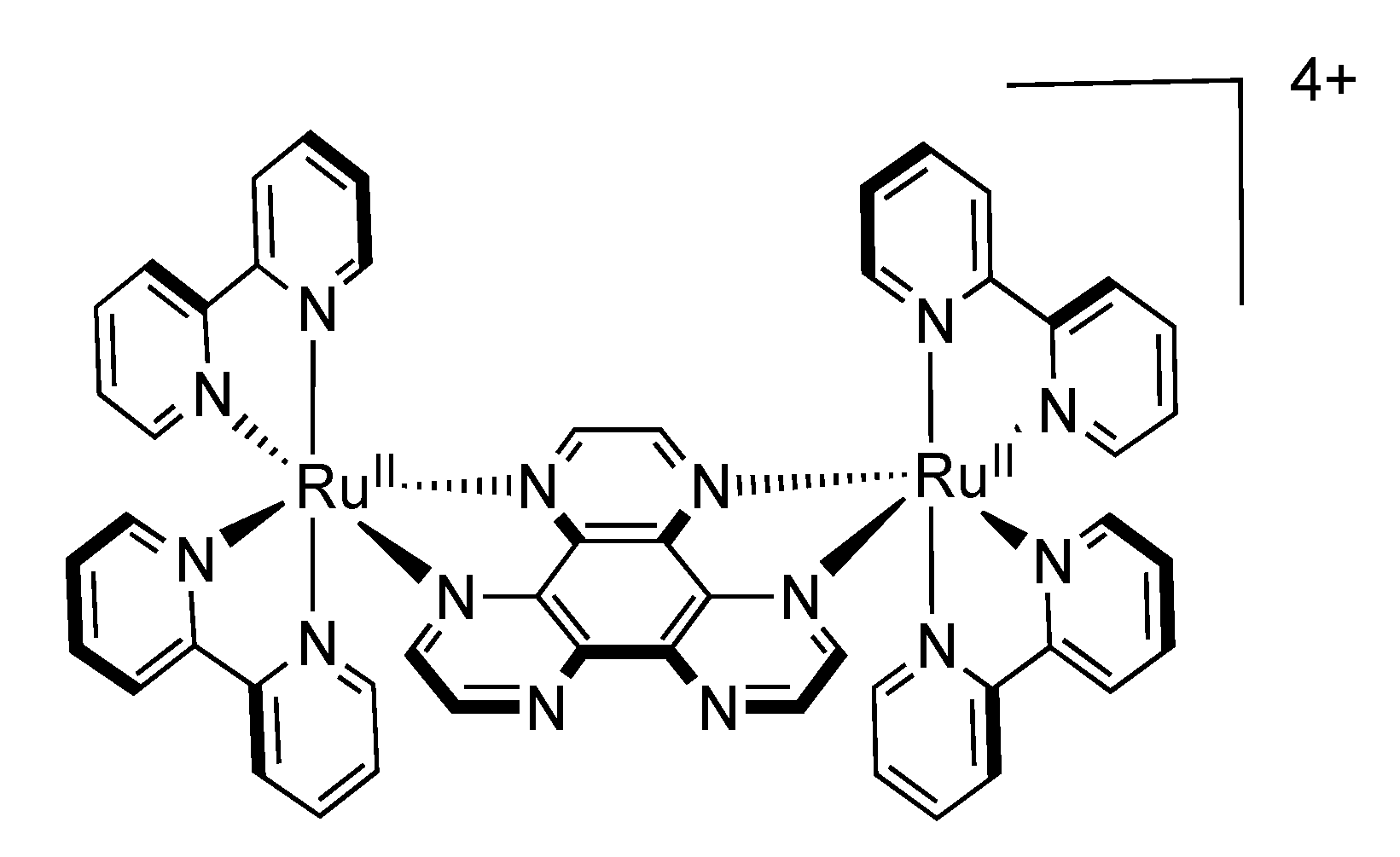
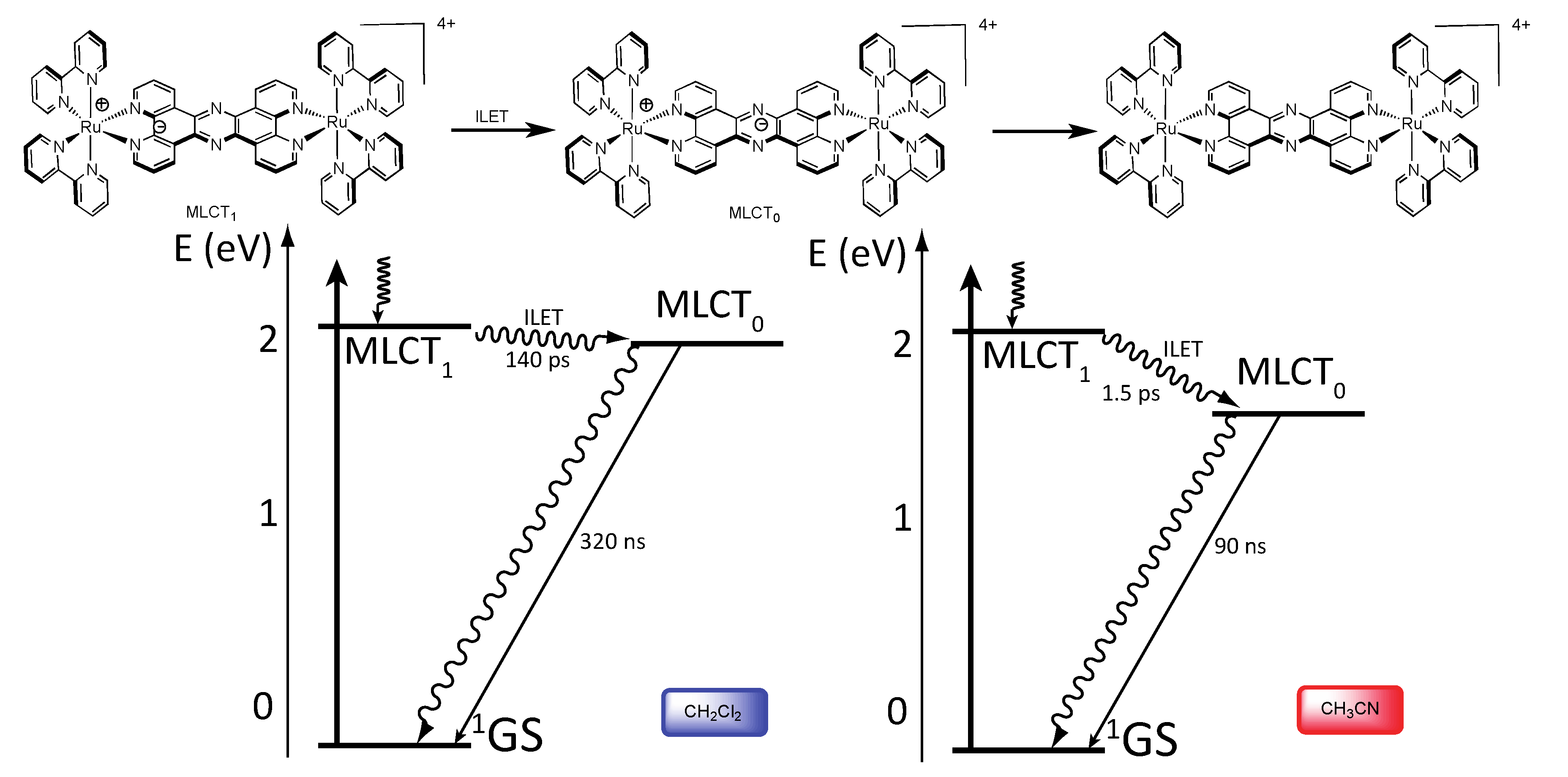
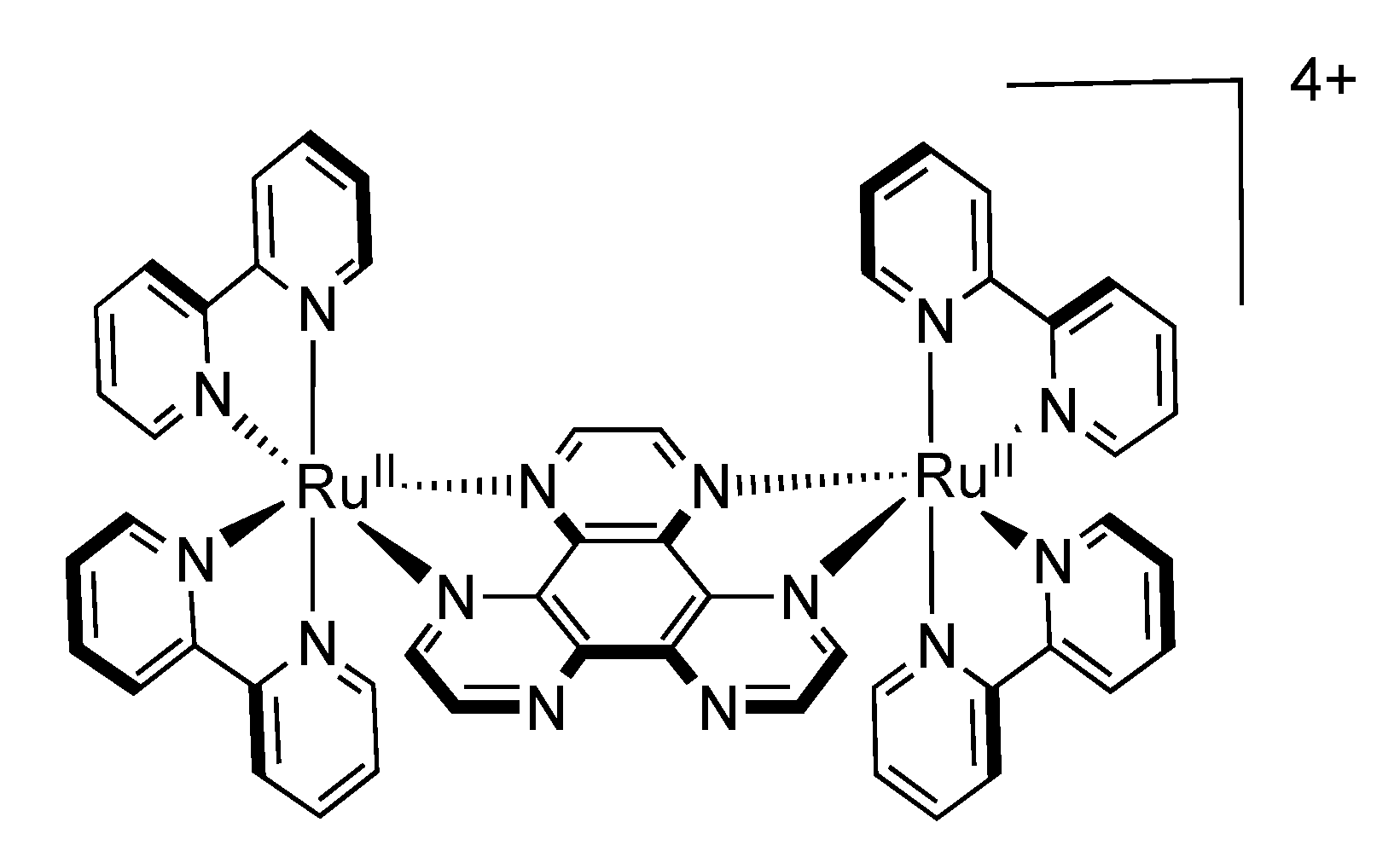
3.1.1. RuII-RuII and OsII-OsII Binuclear Complexes
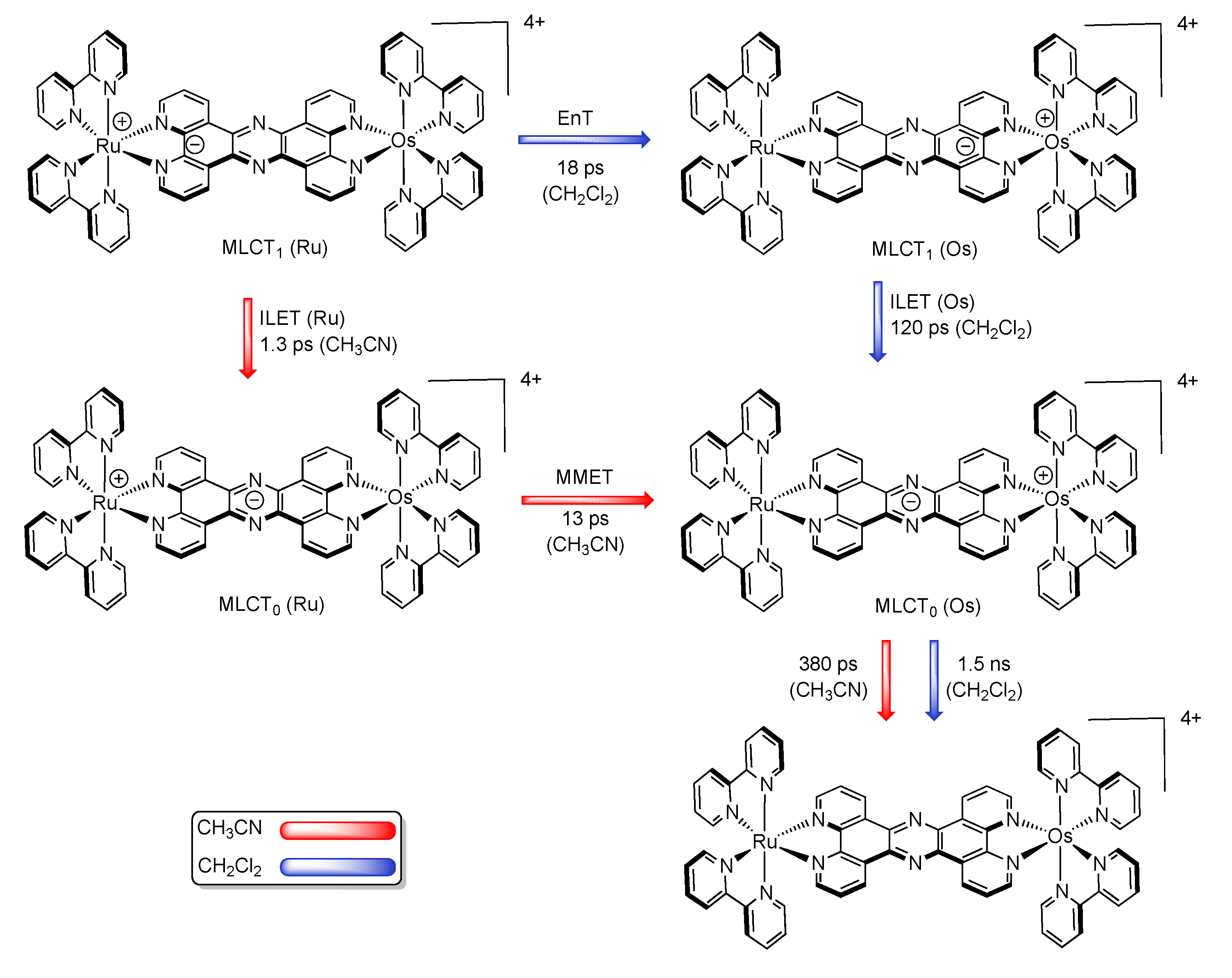
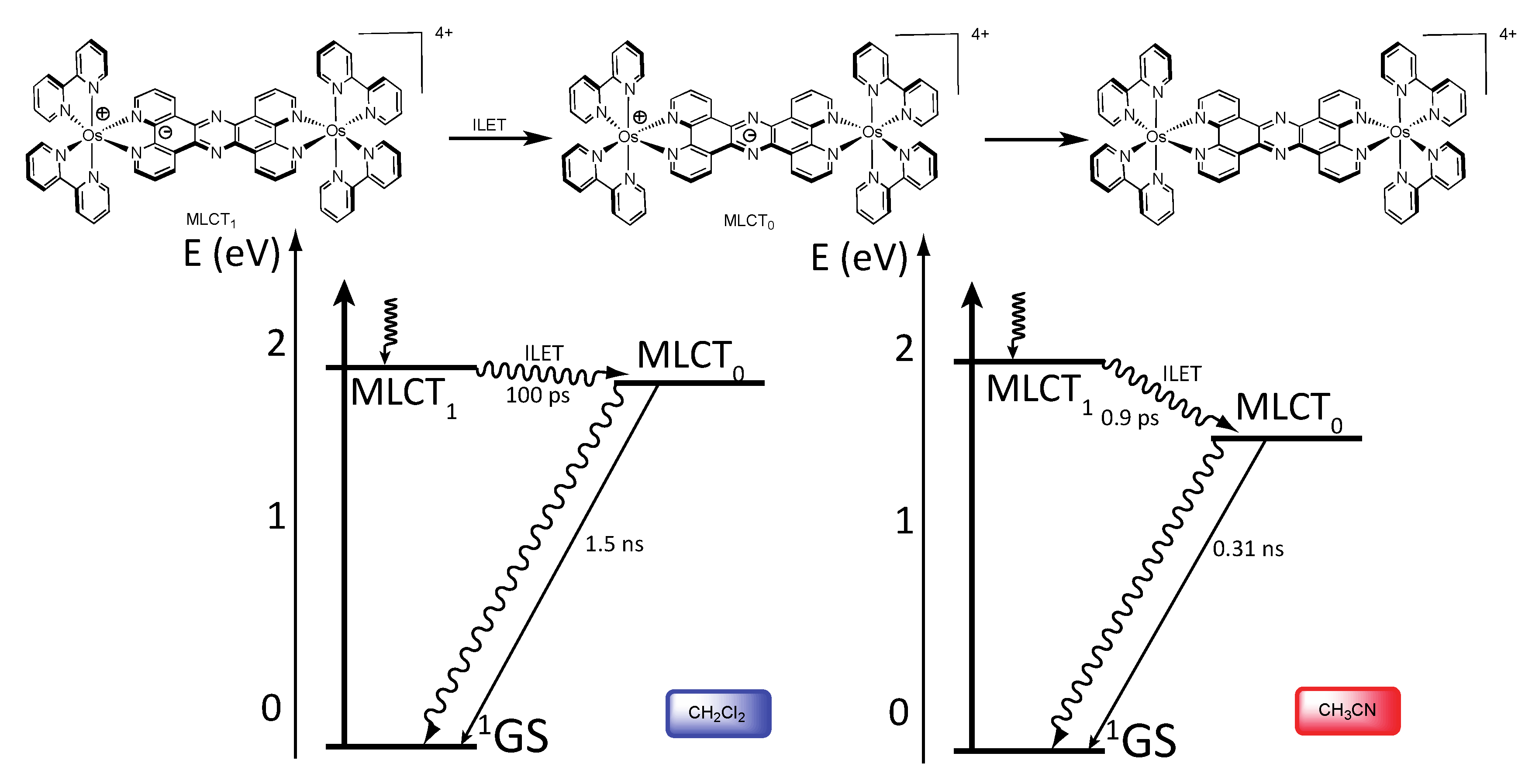
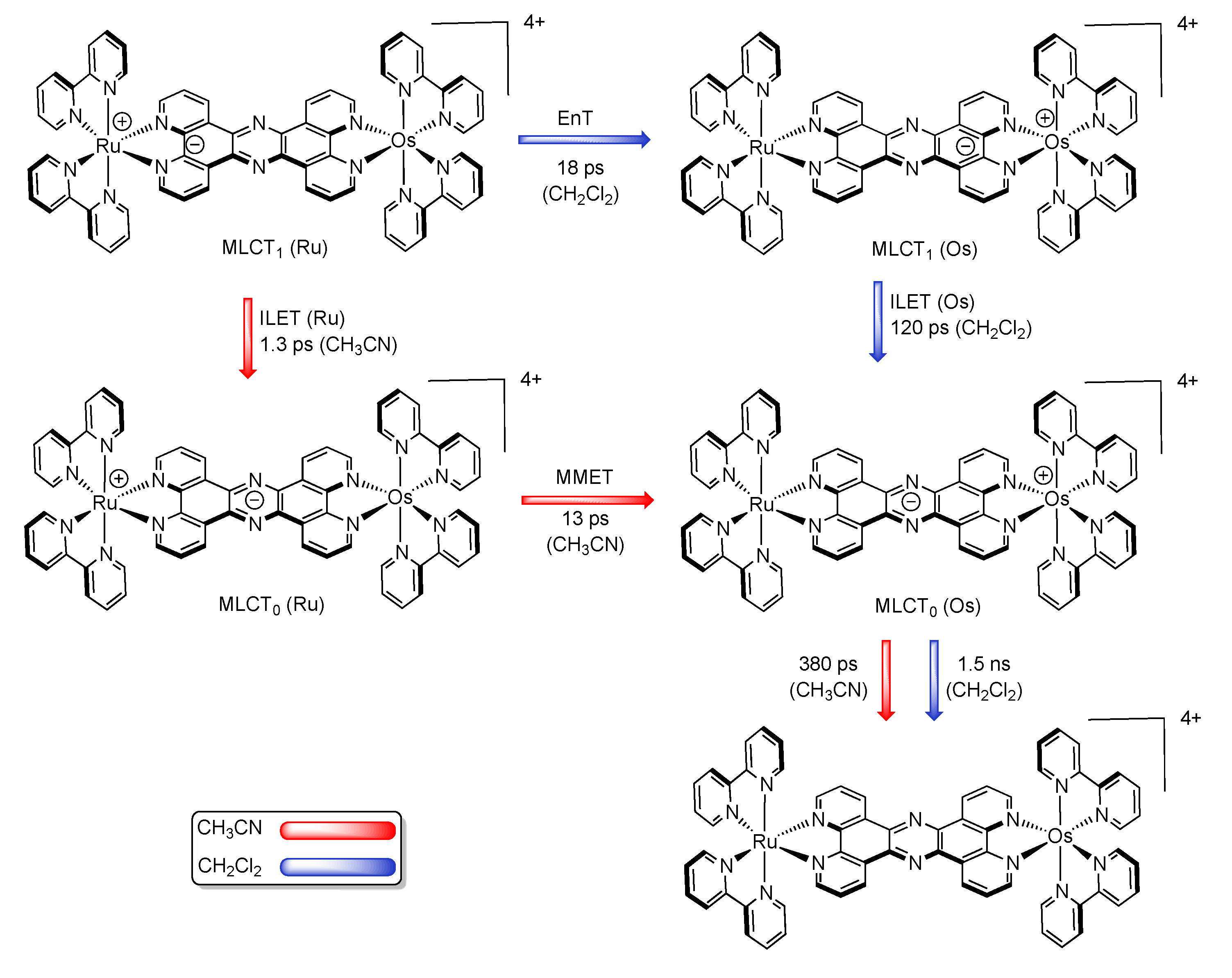
3.1.2. RuII-OsII Binuclear Complexes
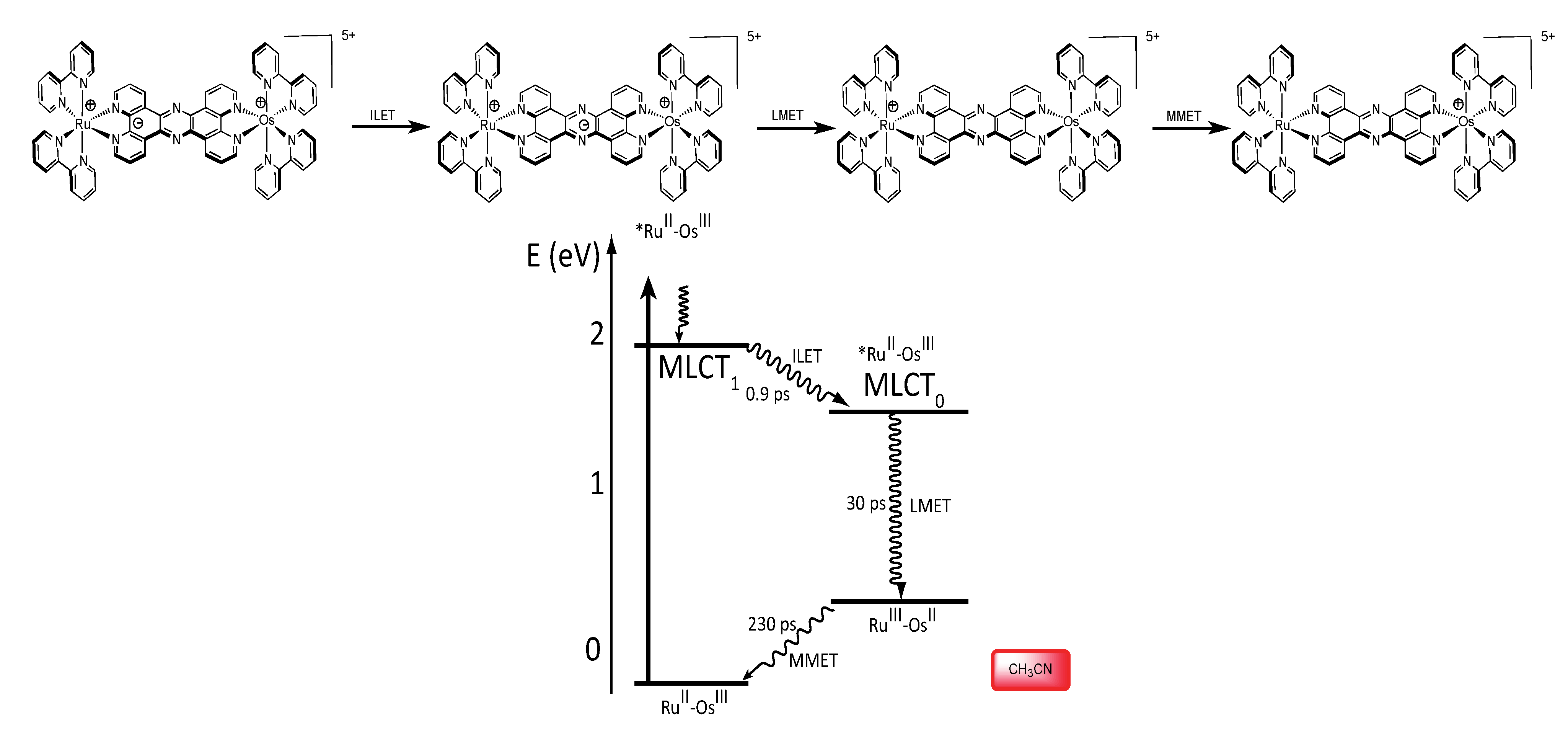
3.1.3. RuII-OsIII Binuclear Complexes
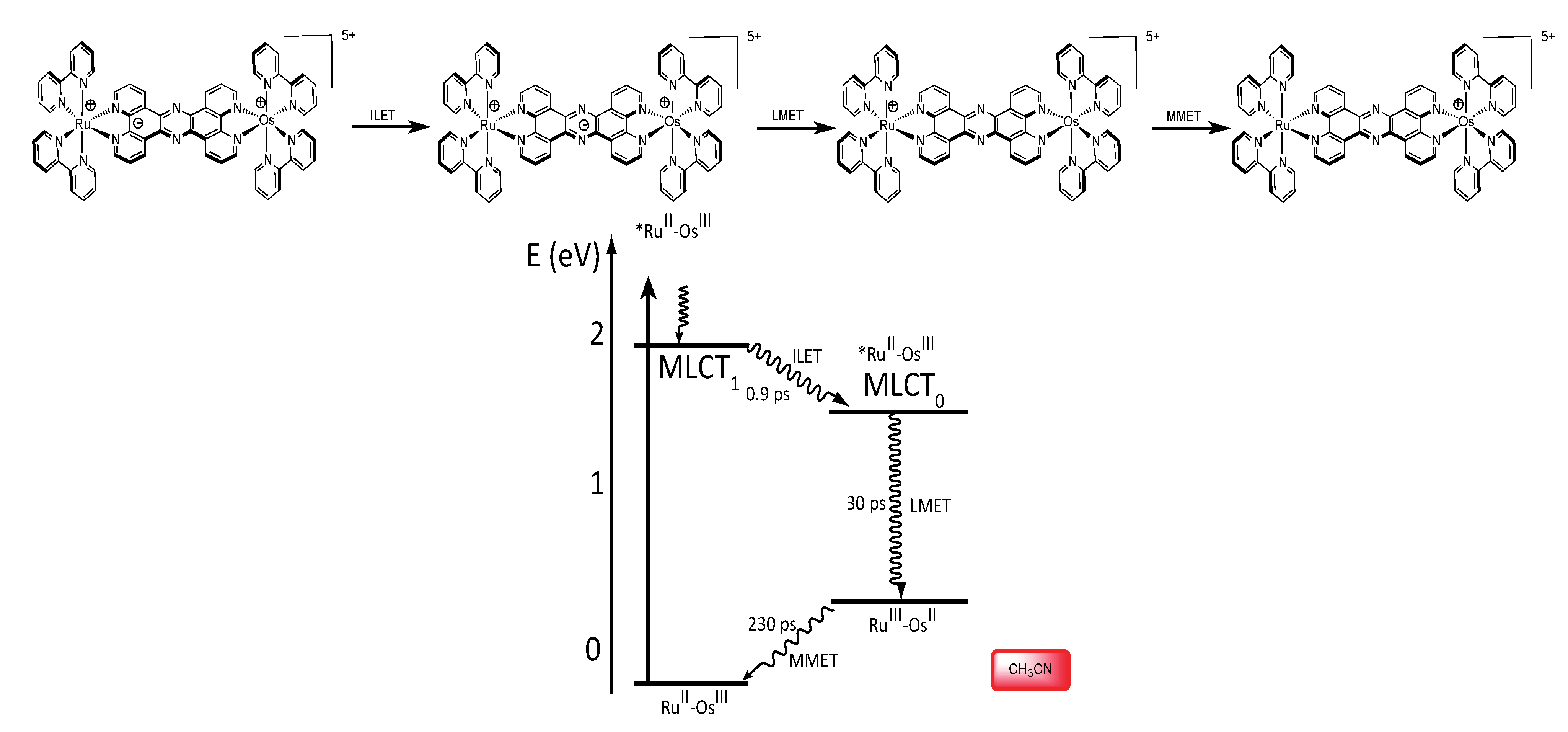
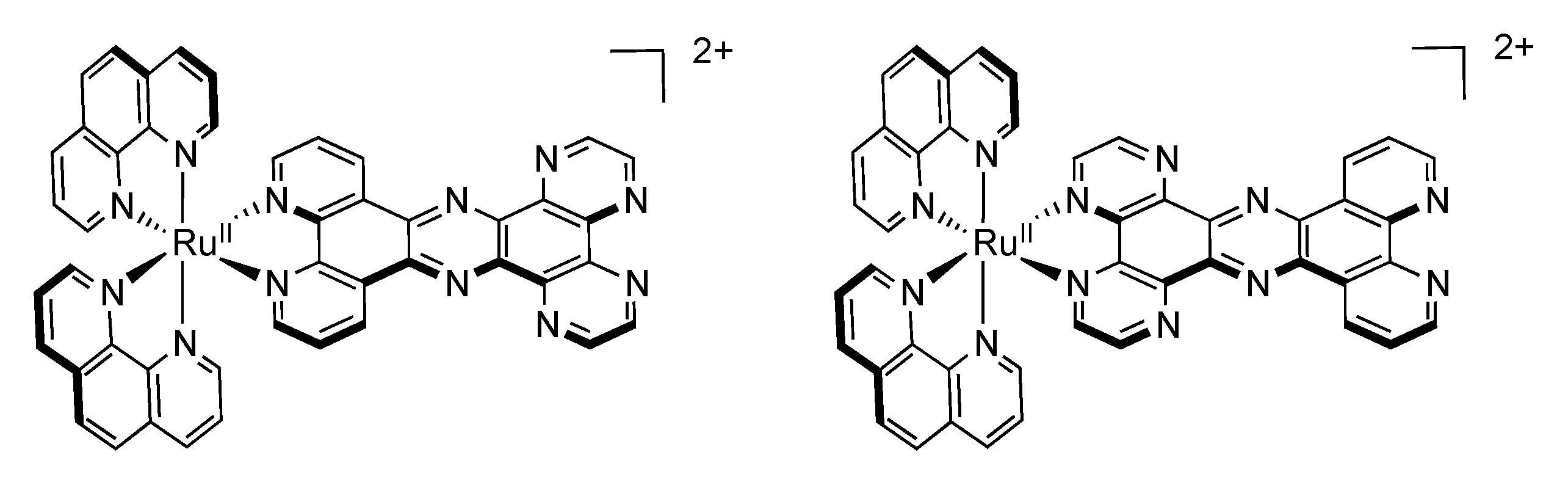
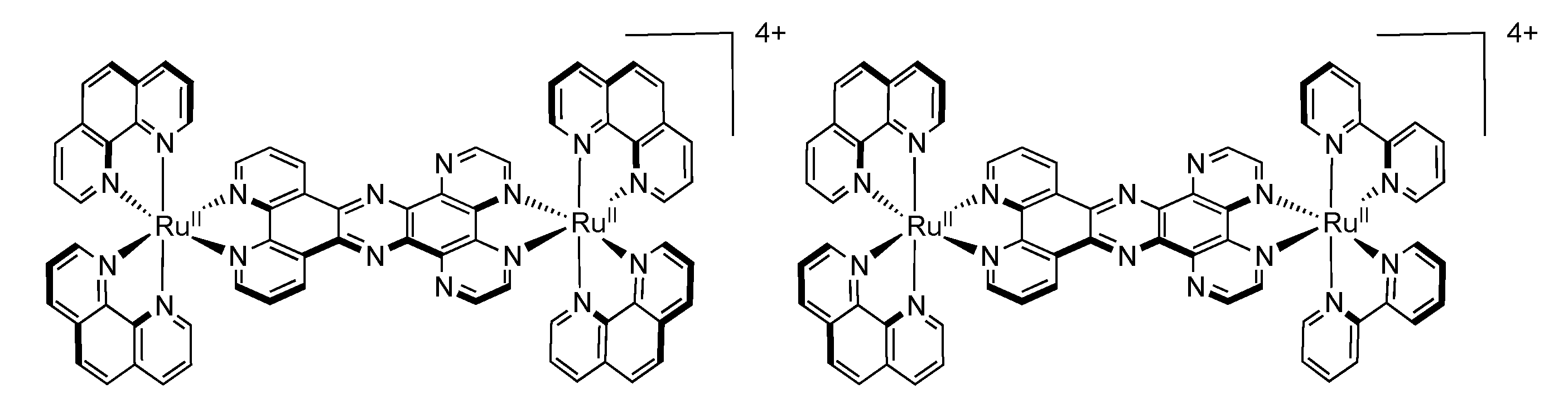
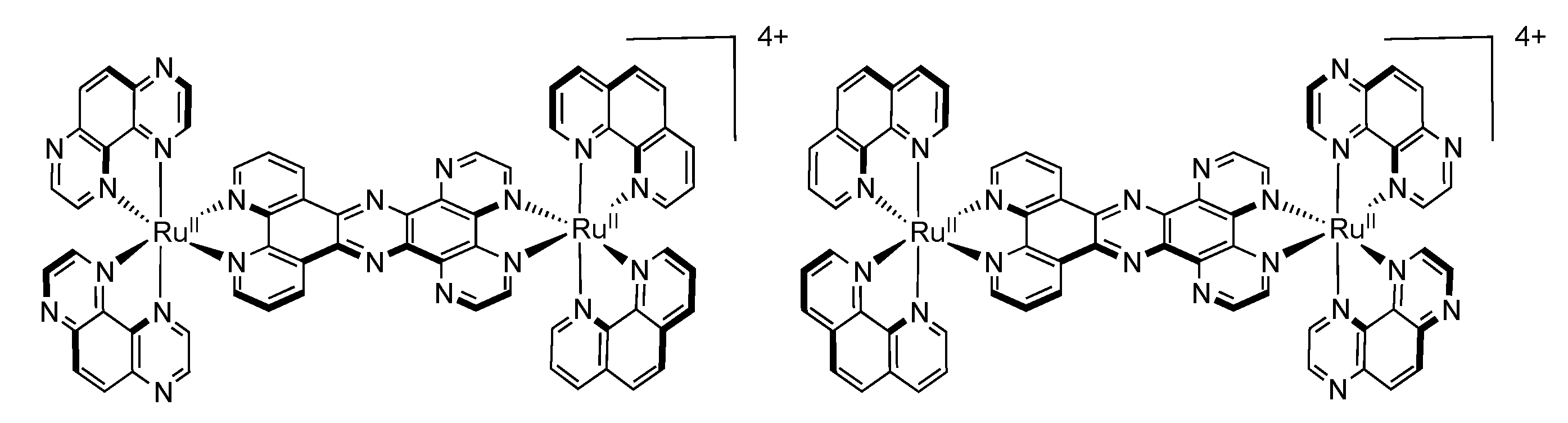
3.2. Binuclear HAT Complexes
| Complex | λem/nm H2O | (τ/ns) H2O | λem/nm CH3CN | (τ/ns) CH3CN |
|---|---|---|---|---|
| [Ru(bpy)2(HAT)]2+ | 742 | 104 | 703 | 620 |
| [Ru(phen)2(HAT)]2+ | 732 | 137 | 698 | 817 |
| [Ru(bpy)(TAP)(HAT)]2+ | 668 | 601 | 647 | 1764 |
| [Ru(bpy)(HAT)2]2+ | 661 | 666 | 642 | 1810 |
| Ru(TAP)2(HAT)]2+ | 608 | 315 | 595 | 102 |
| [Ru(bpy)(HAT)2]2+ | 600 | 226 | 591 | 99 |
| [Ru(HAT)3]2+ | 596 | 145 | 587 | 89 |
| Complex | Oxidation | Reduction |
|---|---|---|
| [Ru(bpy)2(HAT)]2+ | +1.56 | −0.84 −1.43 −1.63 |
| [Ru(phen)2(HAT)]2+ | +1.53 | −0.86 −1.42 −1.69 |
| {[Ru(bpy)2]2(HAT)}4+ | +1.53 +1.78 | −0.49 −1.06 a |
| {[Ru(phen)2]2(HAT)}4+ | +1.52 +1.78 | −0.49 −1.07 a |
| {[Ru(bpy)2]3(HAT)}4+ | +1.61 +1.87 +2.12 | −0.25 −0.58 −1.07 |
| {[Ru(phen)2]3(HAT)}4+ | +1.61 +1.88 +2.16 | −0.30 −0.64 −1.12 |
3.3. Binuclear PHEHAT Complexes
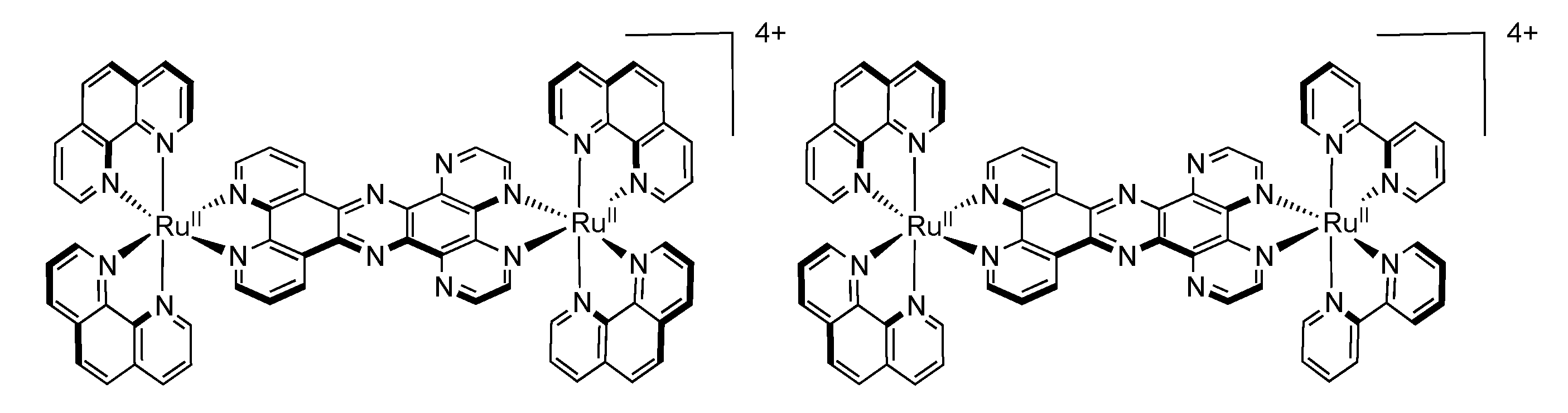
| Complex | Oxidation | Reduction |
|---|---|---|
| [Ru(phen)3]2+ [17] | +1.27 | −1.35 −1.52 |
| [Ru(TAP)3]2+ [17] | +1.94 | −0.75 −0.88 −1.10 −1.60 −1.80 |
| [Ru(phen)2(HAT)]2+ [150] | +1.53 | −0.86 −1.42 −1.69 |
| [Ru(TAP)2(HAT)]2+ [150] | +2.02 | −0.68 −0.86 −1.08 |
| [Ru(phen)2(PHEHAT)]2+ [145] | +1.35 | −1.00 a −1.25 |
| [Ru(phen)2(HATPHE]2+ [145,151] | +1.56 | −0.83 −1.01 |
| [Ru(TAP)2(PHEHAT)]2+ [145] | +1.80 | −0.75 |
| [Ru(phen)2(PHEHAT)Ru(TAP)2]4+ [145] | +1.39 +2.10 a | −0.52 −0.76 |
| [Ru(phen)2(PHEHAT)Ru(phen)2]4+ [157] | +1.34 +1.55 | −0.68 −1.06 |
| [Ru(TAP)2(PHEHAT)Ru(phen)2]4+ [145] | +1.50 +1.86 | −0.57 −0.79 |
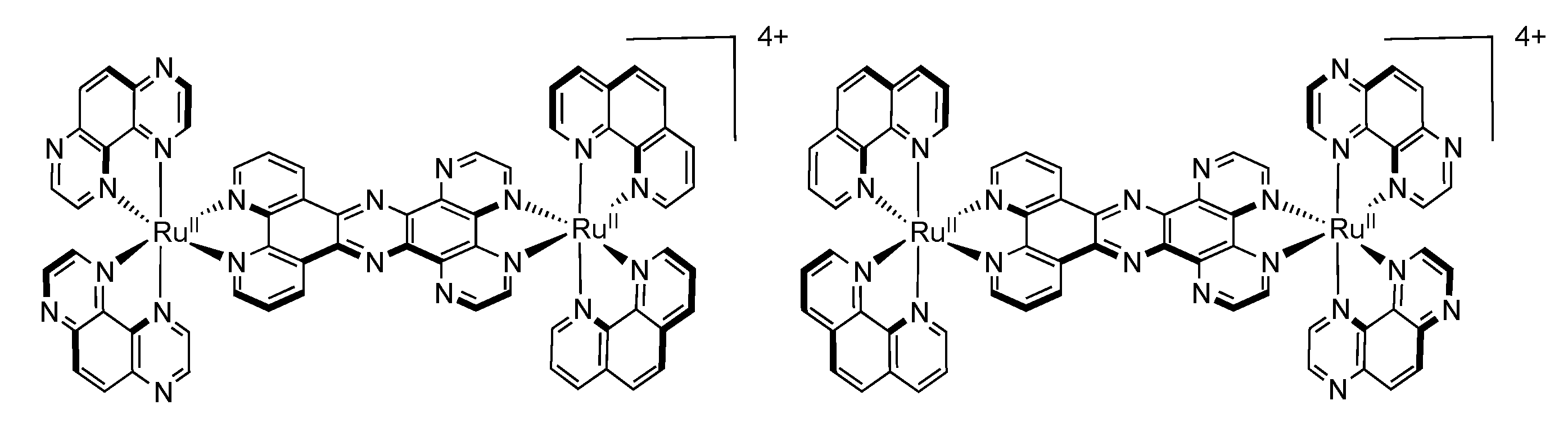
3.4. Binuclear TPAC Complexes
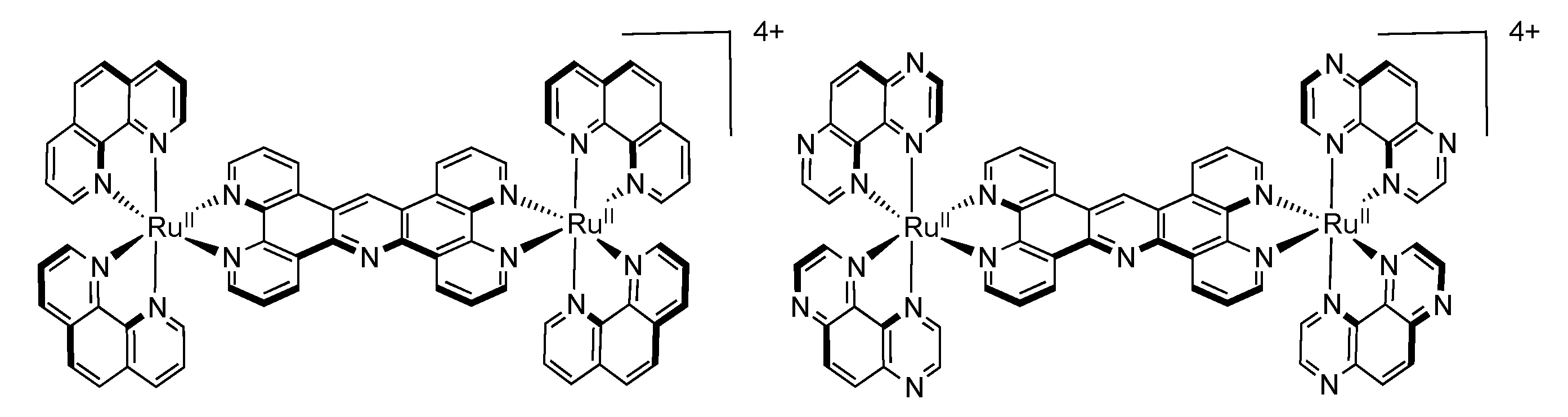
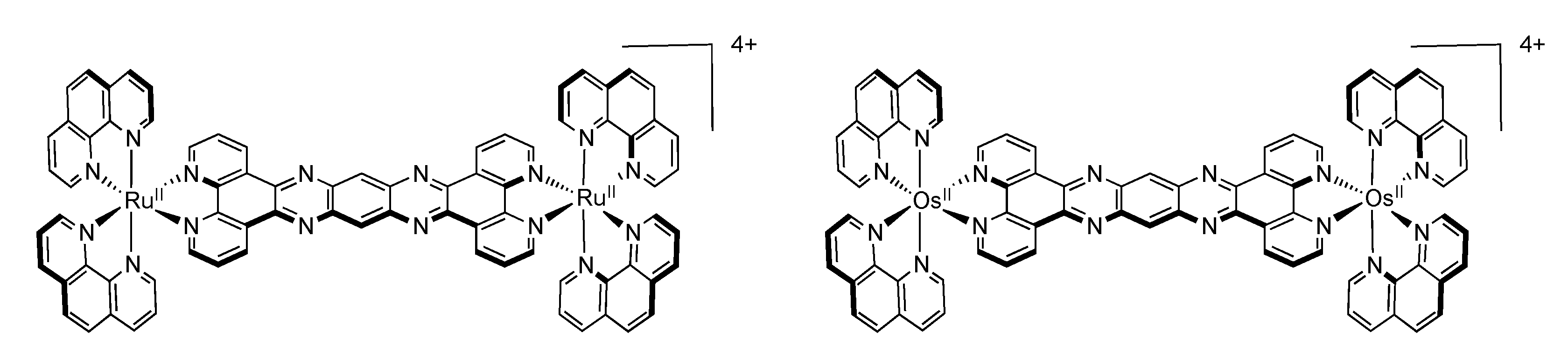
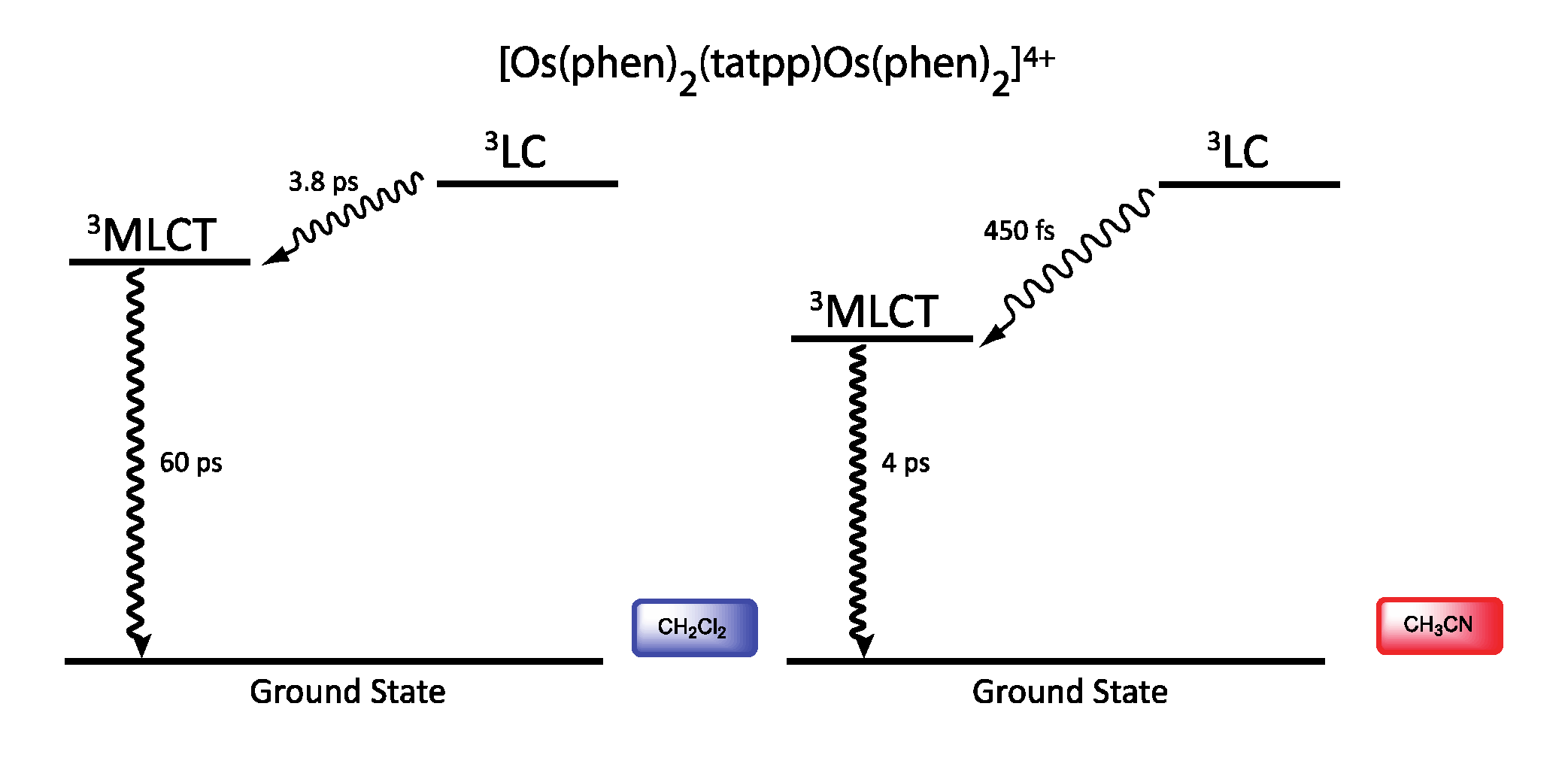
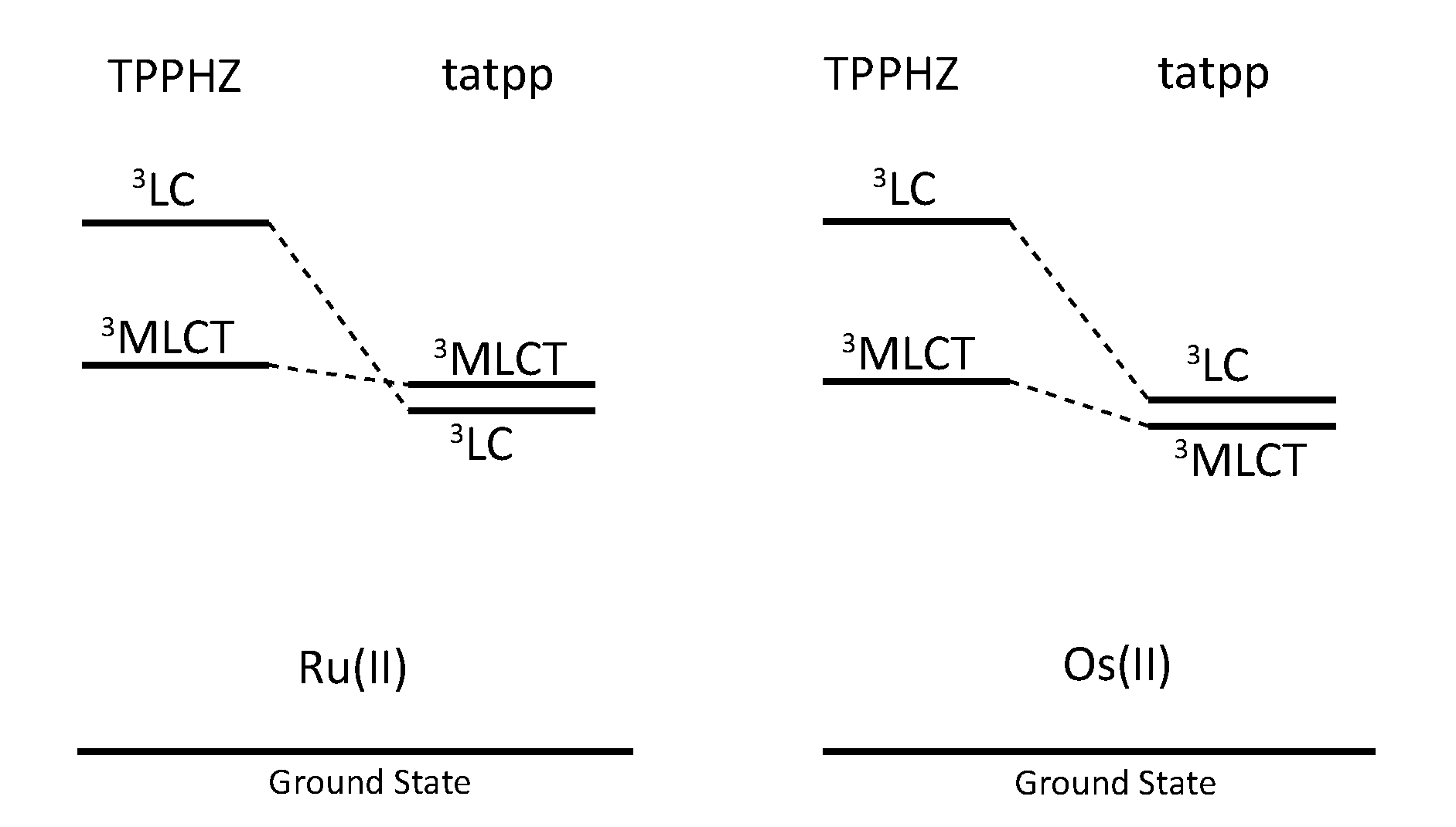
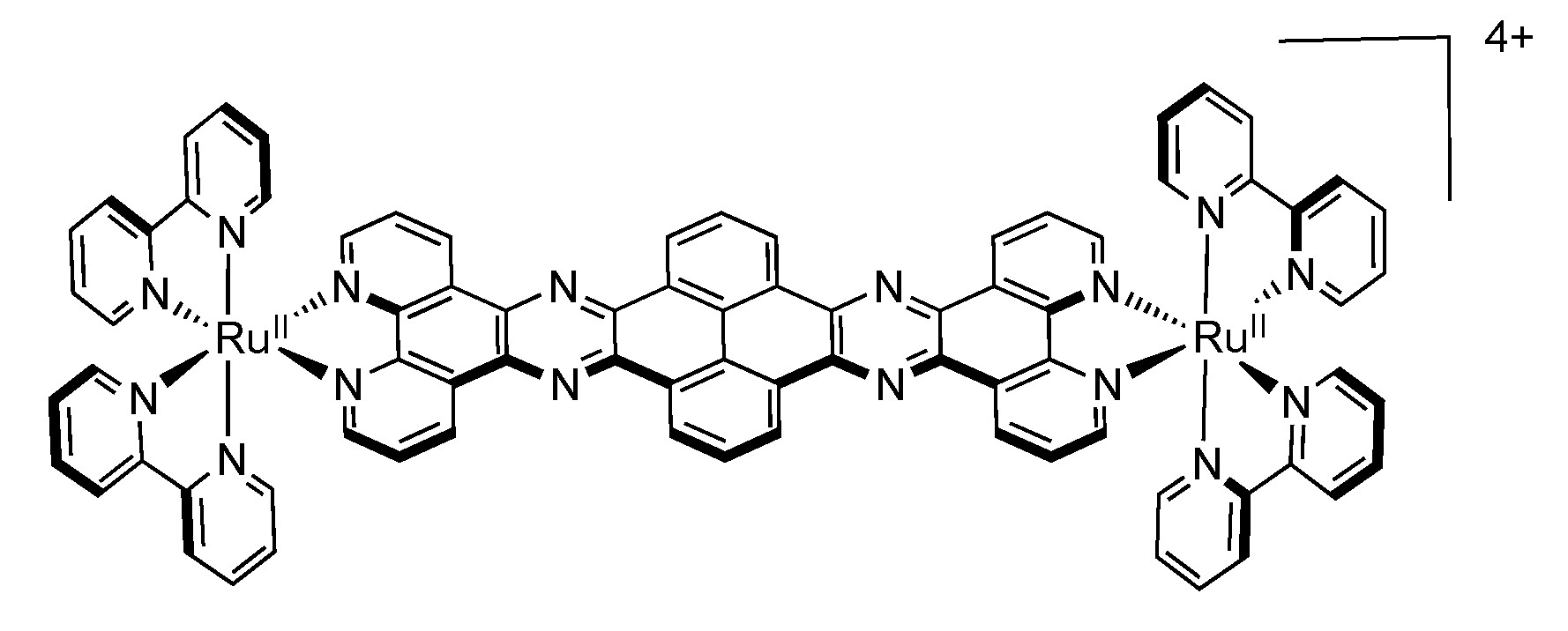
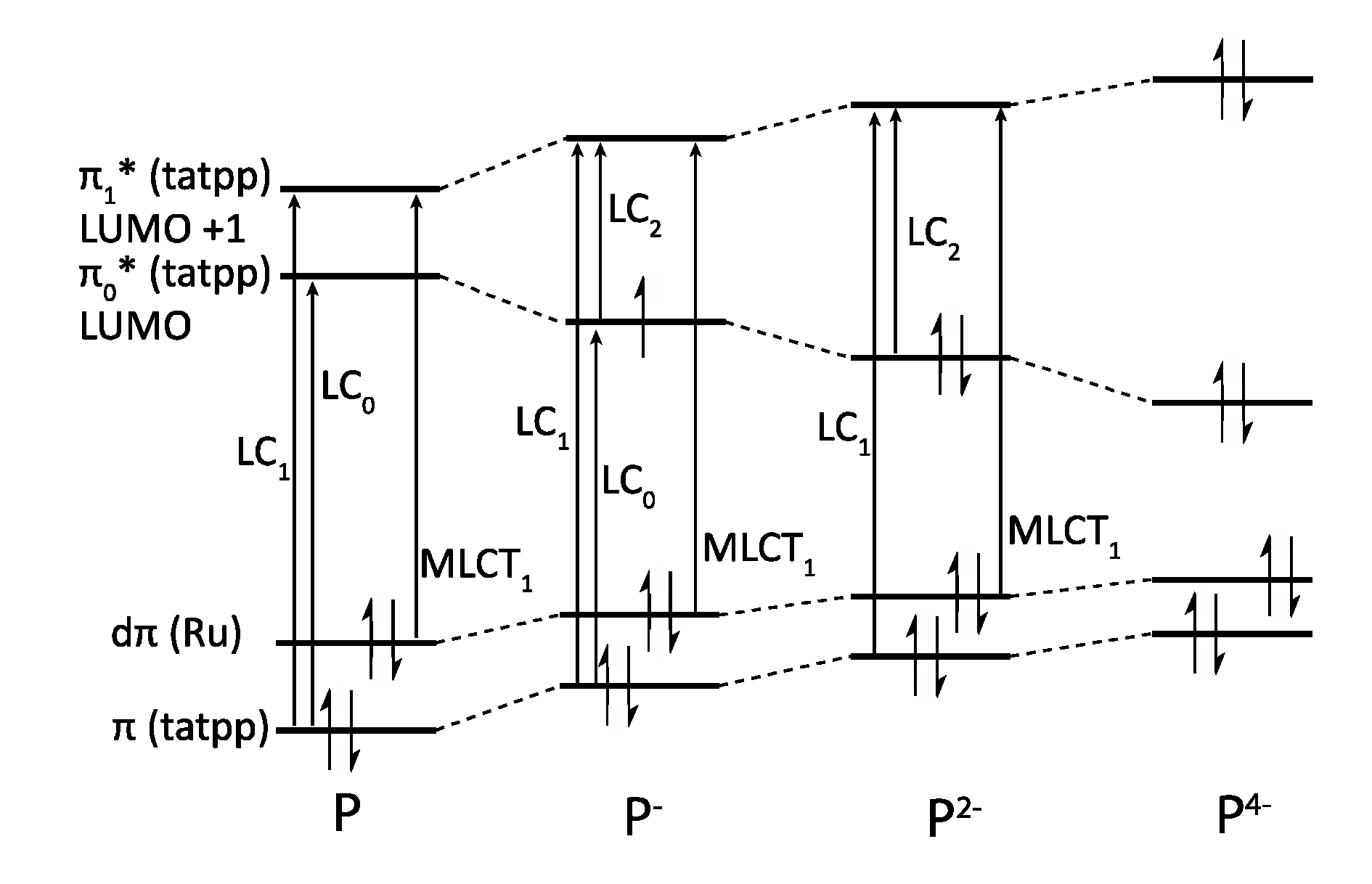
3.5. Binuclear TATPP Complexes

| Complex | Oxidation | Reduction |
|---|---|---|
| [Ru(bpy)2(TPPHZ)Ru(bpy)2]4+ a [124] | +1.34 (2) | −0.71−1.31 (2) −1.51 (2) −1.72 |
| [Os(bpy)2(TPPHZ)Os(bpy)2]4+ a [124] | +0.89 (2) | −0.70 −1.21 (2) −1.44 (2) |
| [Ru(bpy)2(TPPHZ)Os(bpy)2]4+ a [124] | + 0.89 +1.33 | −1.27 (2) −1.48 (2) −1.66 −2.06 |
| [Ru(phen)2(PHEHAT)Ru(bpy)2]4+ [157] | +1.34 +1.55 | −0.68 −1.07 |
| [Ru(TAP)2(PHEHAT)Ru(phen)2]4+ [145] | +1.50 +1.86 | −0.57 −0.79 |
| [Ru(phen)2(PHEHAT)Ru(TAP)2]4+ [145] | +1.39 +2.10 | −0.52 −0.76 |
| [Ru(phen)2(PHEHAT)Ru(phen)2]4+ [157] | +1.34 +1.55 | −0.68 −1.06 |
| [Ru(phen)2(TPAC)Ru(phen)2]4+ [145] | +1.31 (2) | −1.10 −1.32 (2) −1.57 (2) |
| [Ru(TAP)2(TPAC)Ru(TAP)2]4+ [145] | +1.76 (2) | −0.76 (2) −0.92 (2) −1.26 (1) |
| [Ru(phen)2(tatpp)Ru(phen)2]4+ [161] | +1.32 (2) | −0.26 −0.75 −1.32 (2) |
| [Os(phen)2(tatpp)Os(phen)2]4+ [161] | +0.85 (2) | −0.07 −0.24 −1.27 (2) |
| Complex | Oxidation | Reduction |
|---|---|---|
| [Ru(bpy)3]2+ [91] | +1.28 | −1.35 −1.55 −1.79 |
| [Ru(phen)2(TPPHZ)Ru(phen)2]4+ [137] | +1.34 (2) | −0.78 −1.36 −1.52 |
| [Ru(phen)2(tatpp)Ru(phen)2]4+ [161] | +1.32 (2) | −0.26 −0.75 −1.32 (2) |
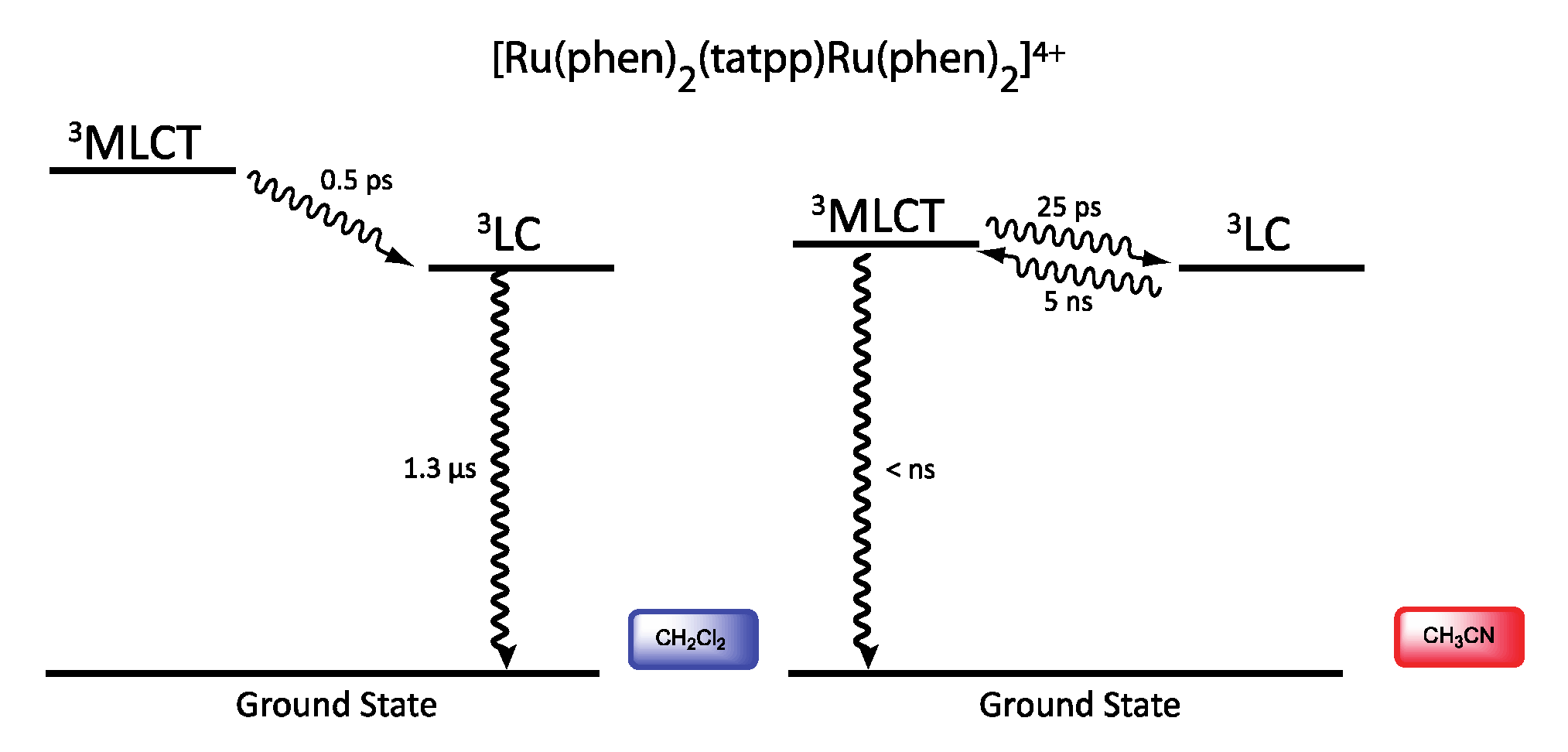
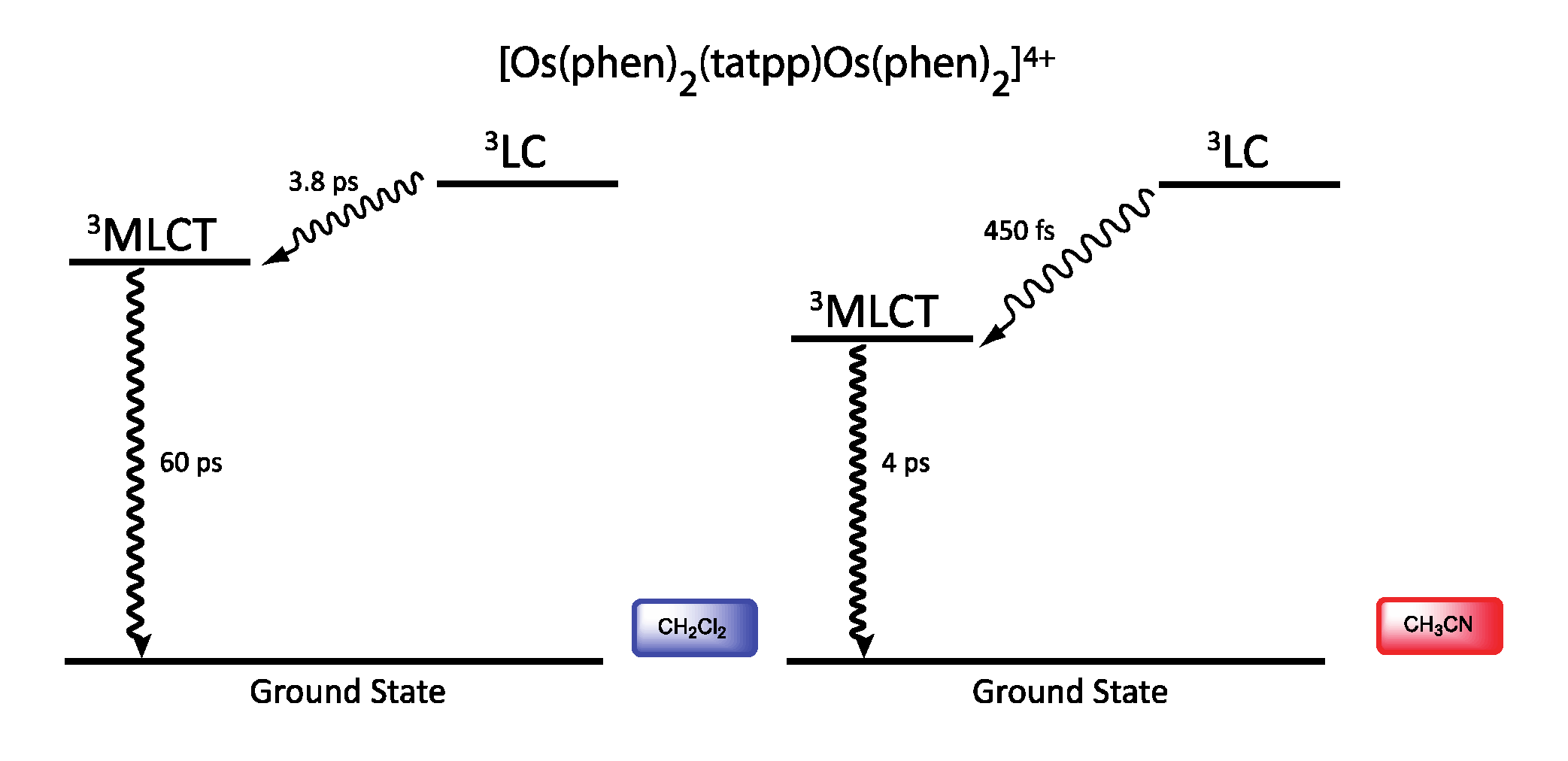
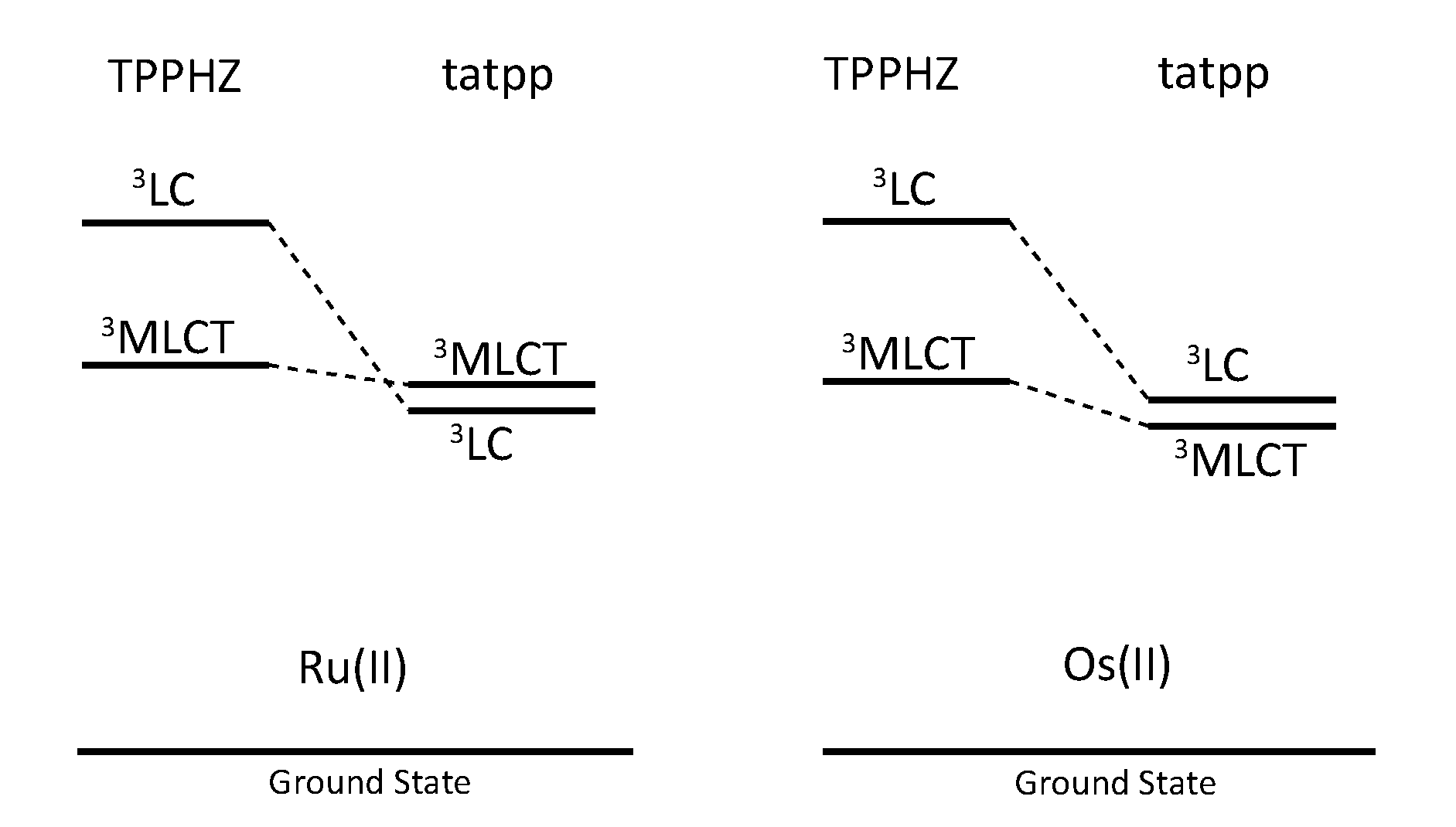
| Complex | Oxidation, V vs. SCE | Reduction, V vs. SCE |
|---|---|---|
| [Ru(phen)2(HAT)]2+ [150] | +1.53 | −0.86 −1.42 −1.69 |
| [Ru(phen)2(TPPHZ)]2+ [137] | +1.34 | −1.00 −1.38 −1.69 |
| [Ru(phen)2(PHEHAT)]2+ [128] | +1.35 | −0.84 −1.25 |
| [Ru(phen)2(TPAC)]2+ [145] | +1.33 | −1.15 −1.25 −1.35 |
| [Ru(phen)2(tatpp)]2+ [127] | +1.33 | −0.30 −0.83 −1.38 |
| [(phen)2Ru(TPPHZ)Ru(phen)2]4+ [137] | +1.34 (2) | −0.78 −1.36(2) −1.52 |
| [(phen)2Ru(HAT)Ru(phen)2]4+ [150] | +1.52 +1.78 | −0.49 −1.07 |
| [(phen)2Ru(TPAC)Ru(phen)2]4+ [145] | +1.31 (2) | −1.10 −1.32 (2) −1.57 (2) |
| [(phen)2Ru(PHEHAT)Ru(phen)2]4+ [157] | +1.34 +1.55 | −0.68 −1.06 |
| [(phen)2Ru(PHEHAT)Ru(bpy)2]4+ [157] | +1.34 +1.55 | −0.68 −1.07 |
| [Ru(phen)2(tatpp)Ru(phen)2]4+ [161] | +1.32 (2) | −0.26 −0.75 −1.32 (2) |
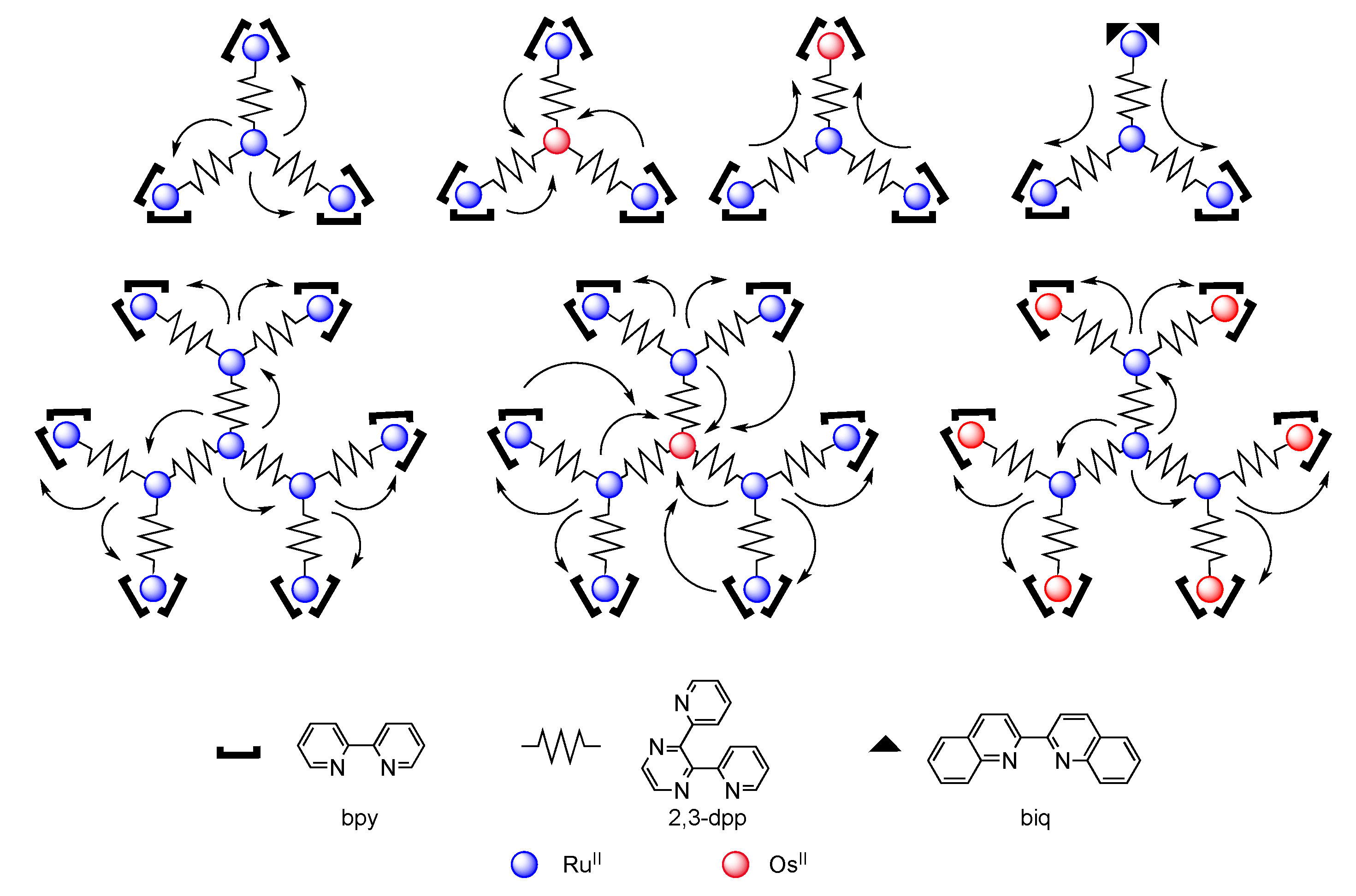
4. Polynuclear Complexes
4.1. Polynuclear Complexes based on 2,3-dpp Ligand
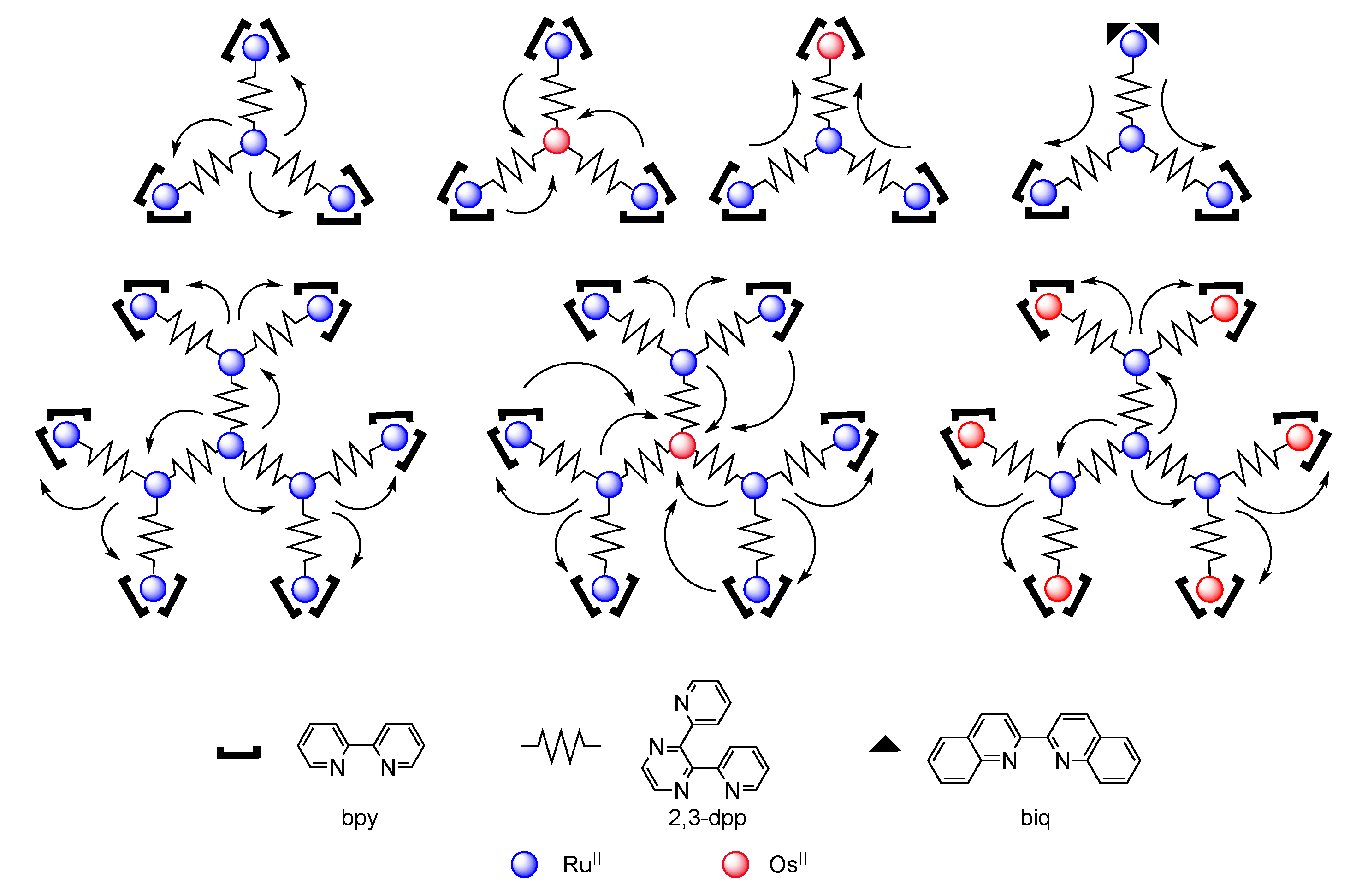
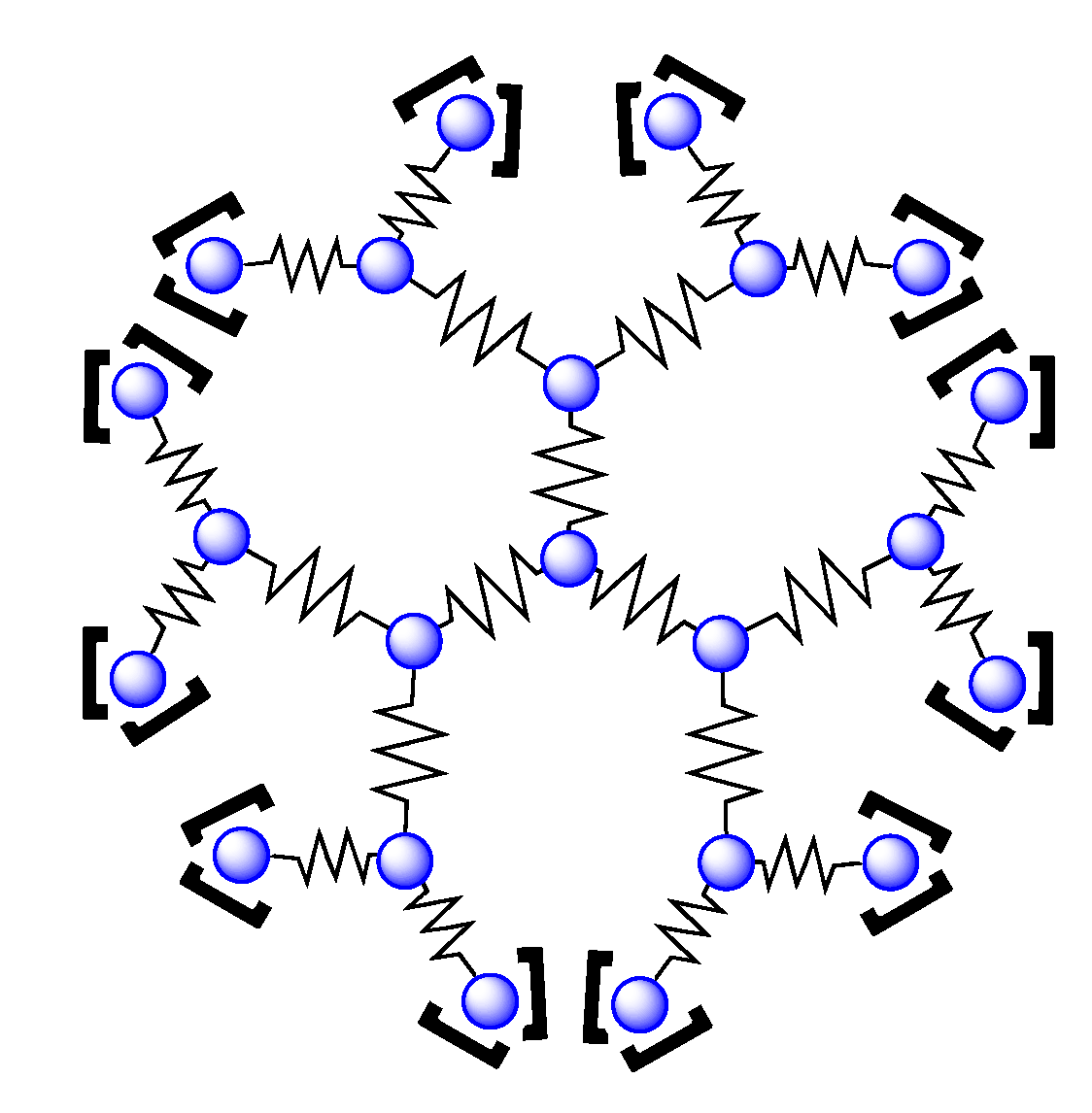
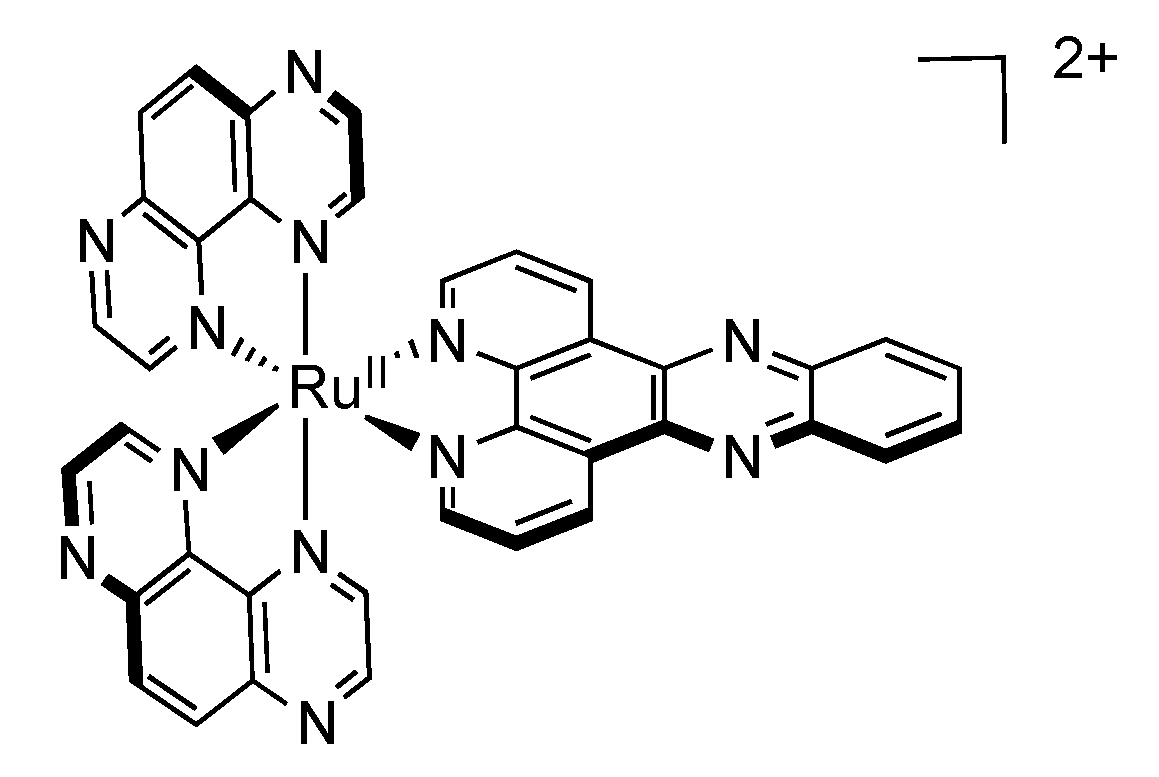
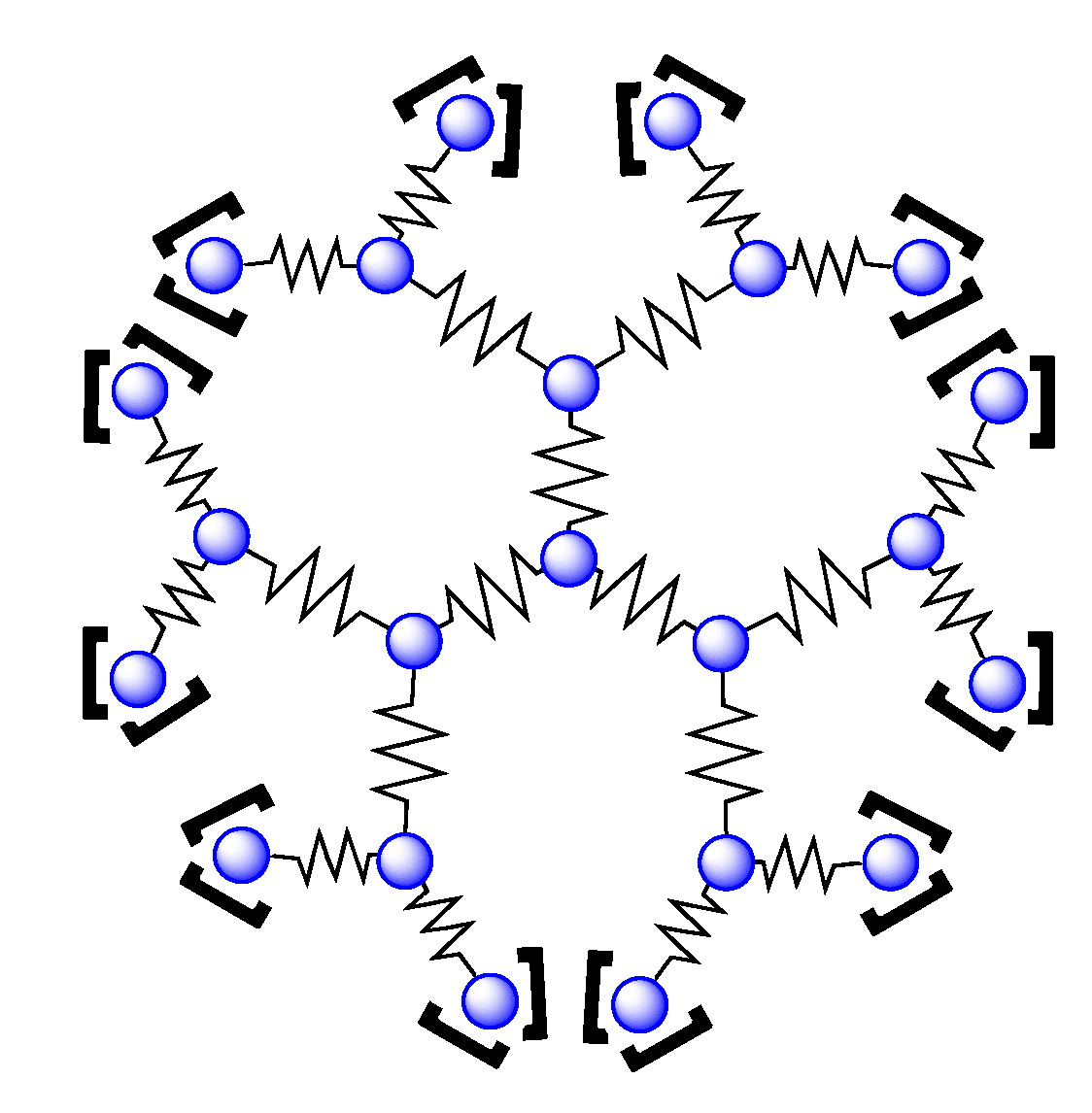
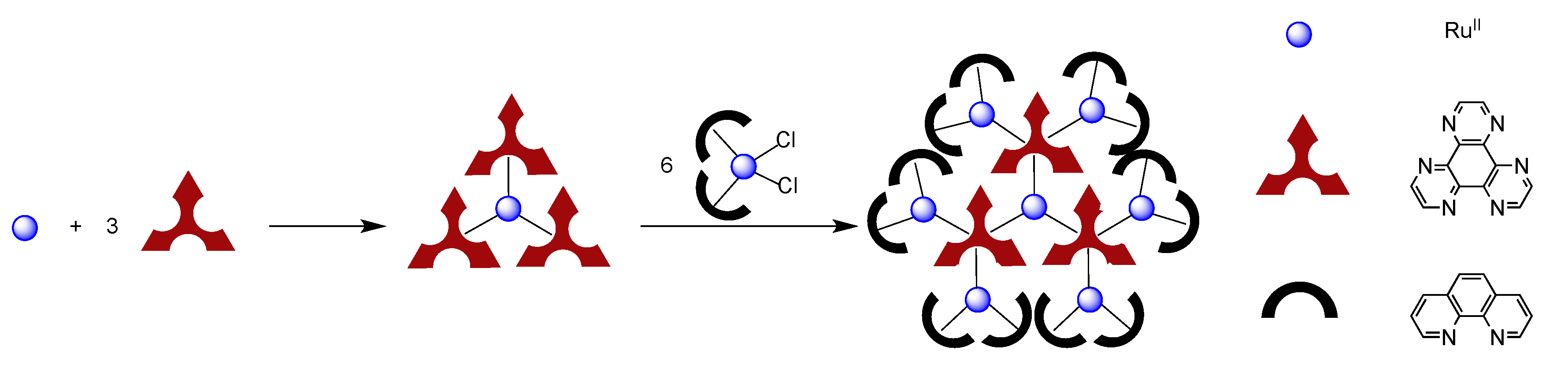
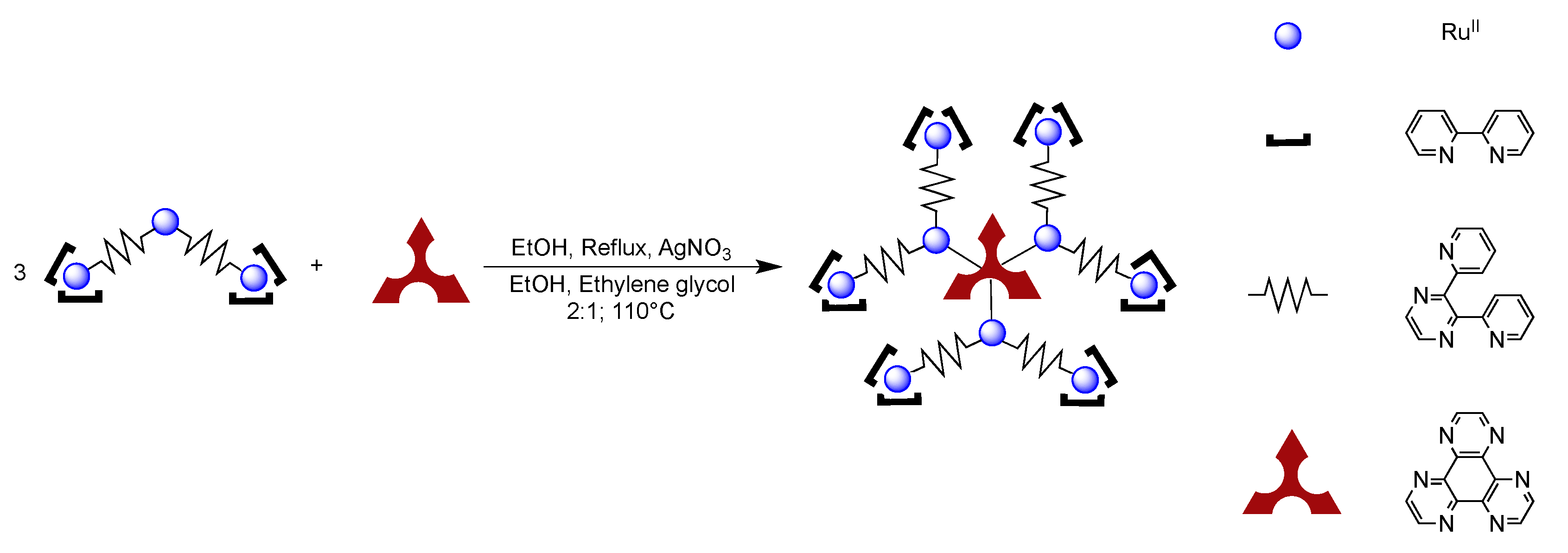
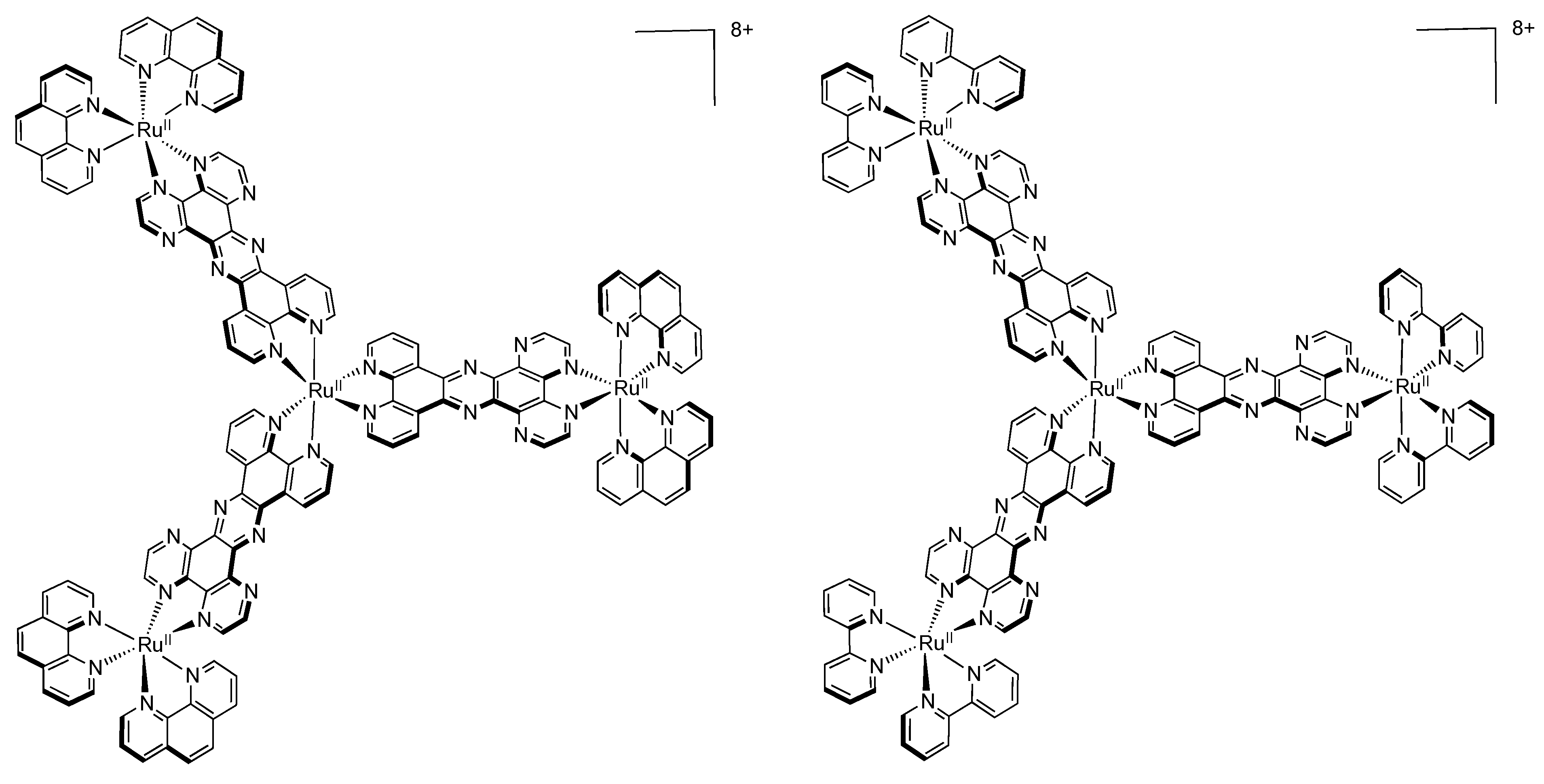
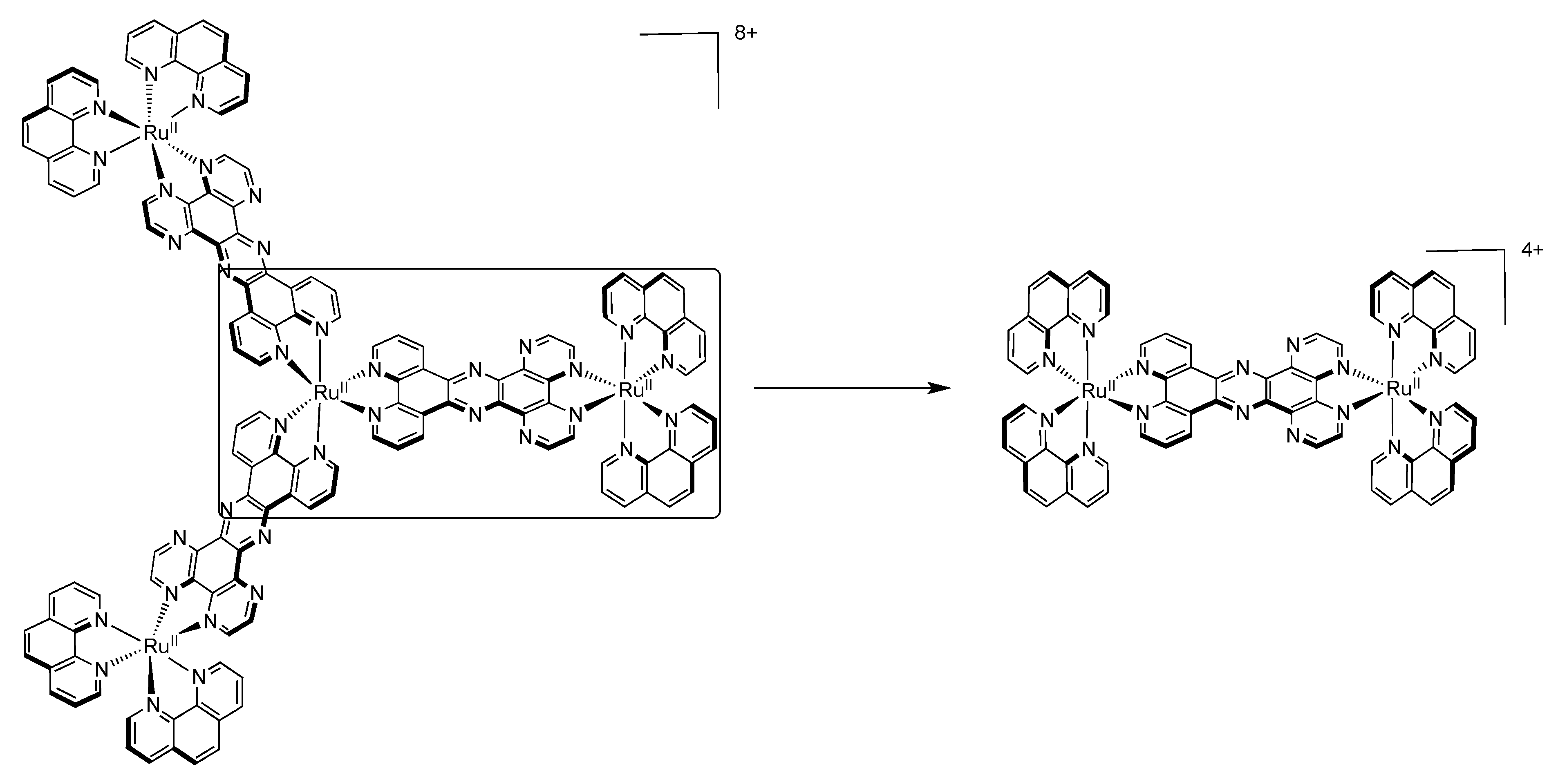
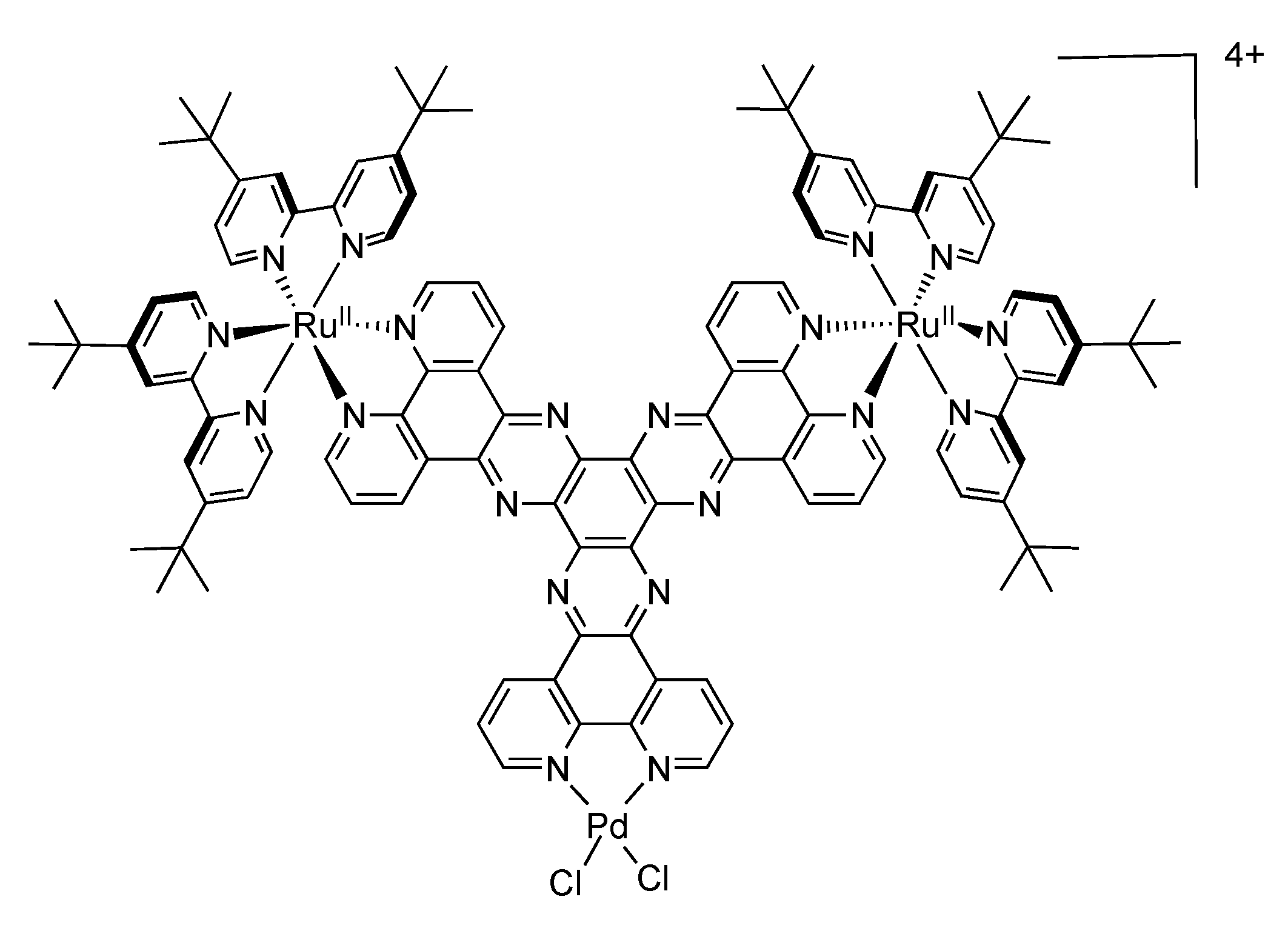
4.2. Polynuclear Complexes based on the HAT Ligand


| Complex | Oxidation, V vs. SCE | Reduction, V vs. SCE |
|---|---|---|
| [HAT{Ru[2,3-dpp)Ru(bpy)2]2}3]18+ | +1.53(6) +2.13 a | −0.56 −0.68 b |
| [Cl2Ru{(2,3-dpp)Ru(bpy)2}2]4+ | +0.82(1) +1.57(2) | −0.72(1) −0.88(1) |
| [HAT{Ru(bpy)2}3]6+ | +1.61(1) +1.87(1) +2.12(1) | −0.25(1) −0.58(1) |
| [Ru{(2,3-dpp)Ru(bpy)2}3]8+ | +1.53(3) | −0.56(1) −0.63(1) |
| [Ru{(2,3-dpp)Ru[(µ-2,3-dpp)Ru(bpy)2]2}3]20+ | +1.53(6) +2.11 +2.44(3) c |
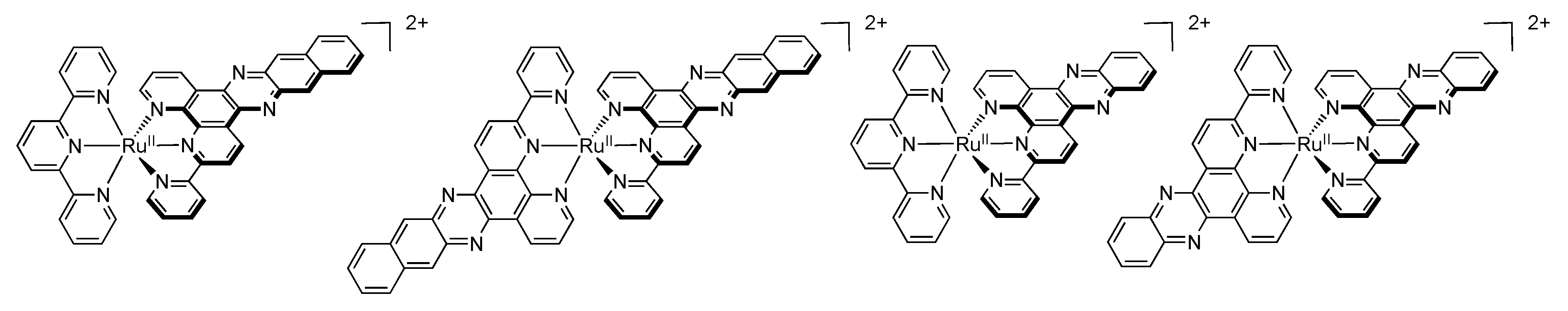
4.3. Polynuclear Complexes based on the TPPHZ Ligand
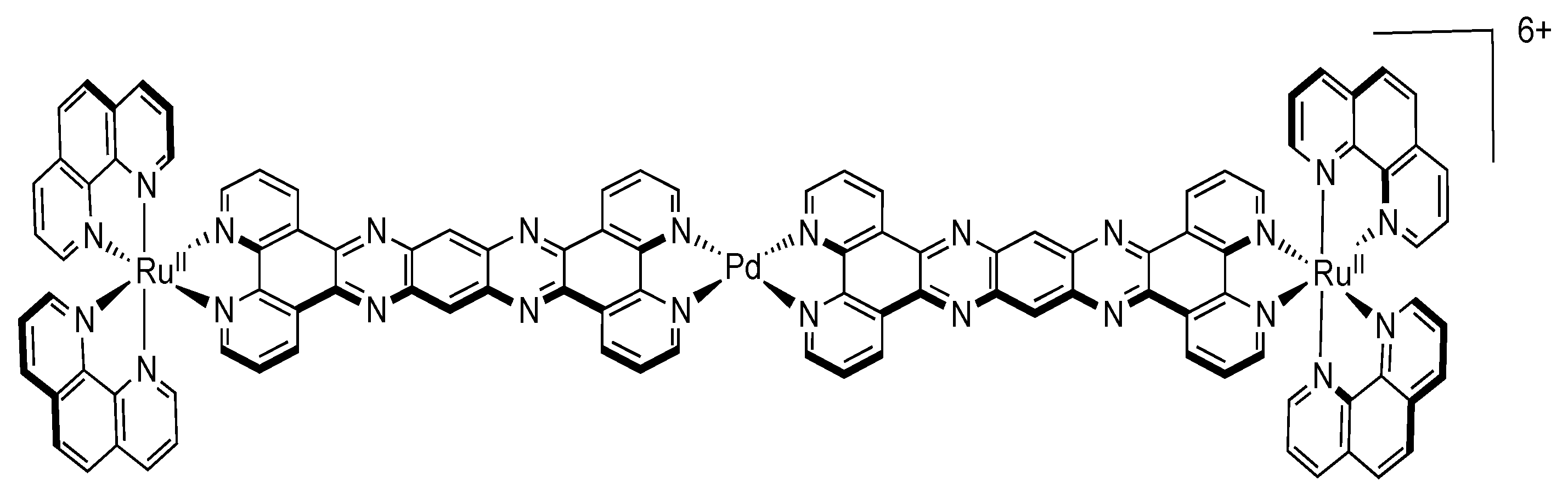
4.4. Polynuclear Complexes based on the PHEHAT and TPAC Ligands

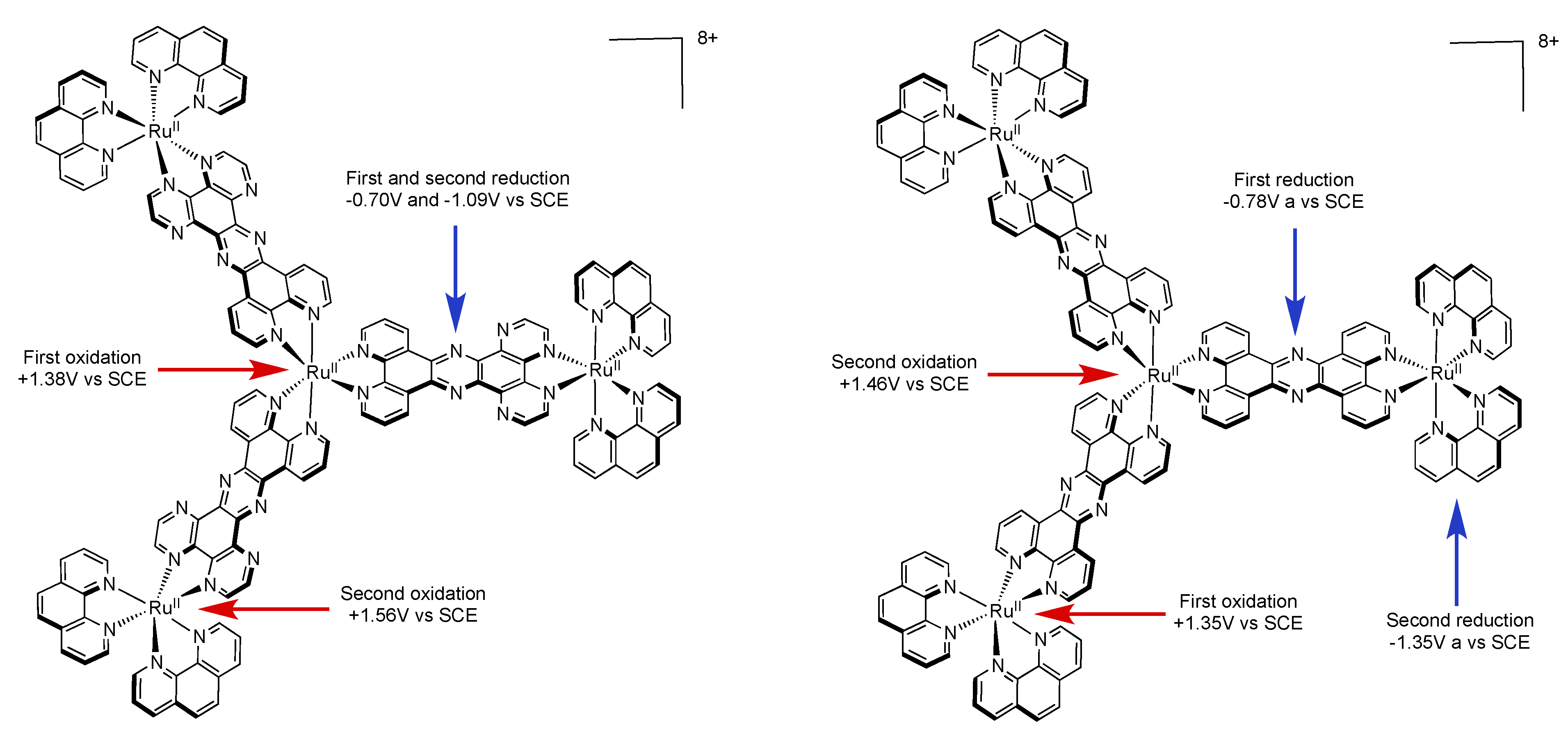
| Complex | Oxidation, V vs. SCE | Reduction, V vs. SCE |
|---|---|---|
| [Ru(phen)3]2+ [141] | +1.27(1) | −1.35(1) −1.52(1) |
| [Ru(phen)2(TPPHZ)]2+ [137] | +1.34(1) | −1.00(1) −1.38(1) −1.69(1) |
| [Ru(phen)2(PHEHAT)]2+ [145] | +1.35 | −0.84 −1.25 |
| [(phen)2Ru(TPPHZ)Ru(phen)2]4+ [137] | +1.34(2) | −0.78(1) −1.36(2) −1.52 |
| [(phen)2Ru(PHEHAT)Ru(phen)2]4+ [157] | +1.34(1) +1.55(1) | −0.68(1) −1.06(1) |
| [(phen)2Ru(TPPHZ)Ru(bpy)2]4+ [137] | +1.34(1) +1.55(1) | −0.68(1) −1.07(1) |
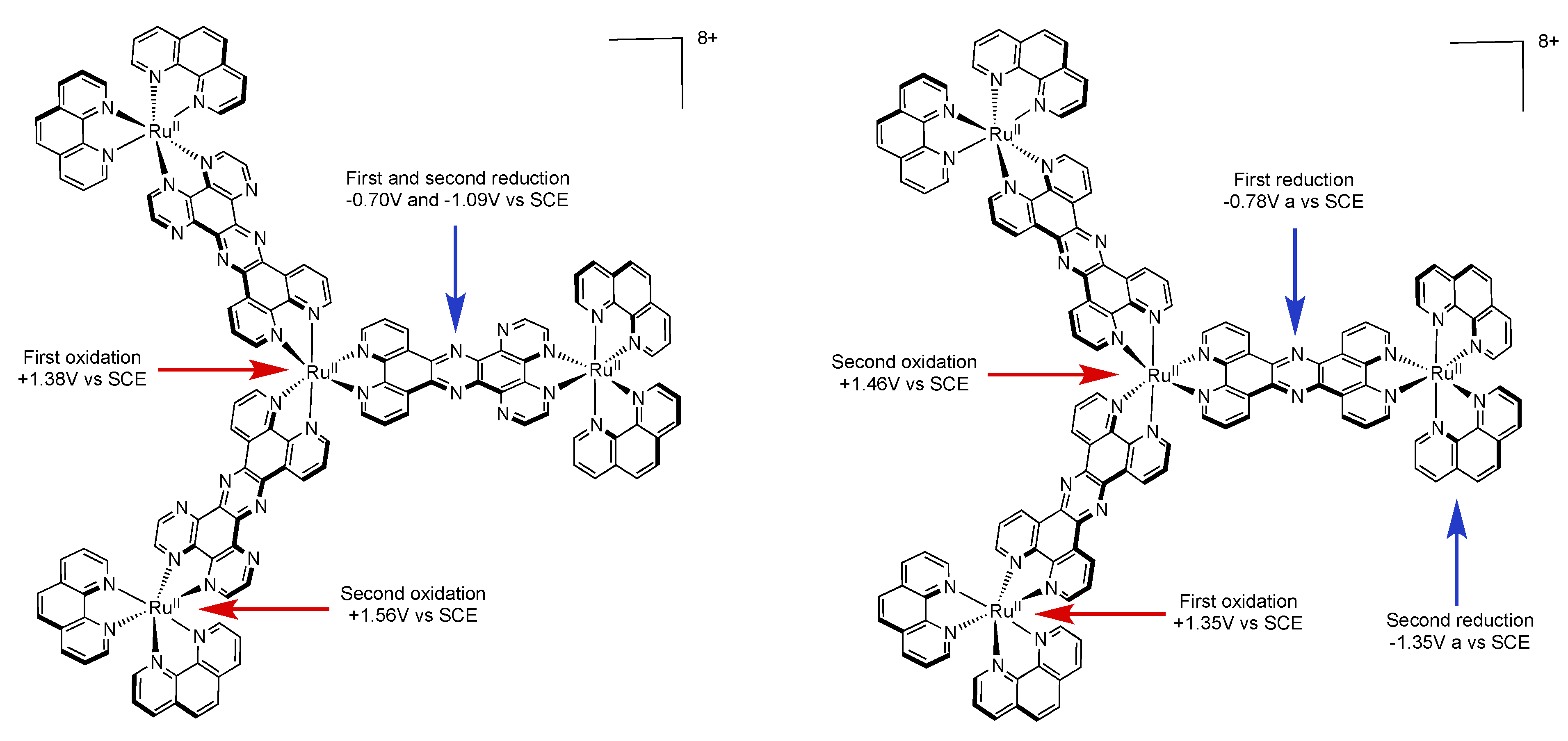
| {Ru[(TPPHZ)Ru(phen)2]3}8+ [137] | +1.35(3) +1.46(1) | −0.78(3) −1.35 (3) −1.54 |
| {Ru[(PHEHAT)Ru(phen)2]3}8+ [157] | +1.38(1) +1.56(3) | −0.70(3) −1.09(3) |
| {Ru[(PHEHAT)Ru(bpy)2]3}8+ [157] | +1.34(1) +1.54(3) | −0.69(3) −1.07(3) |
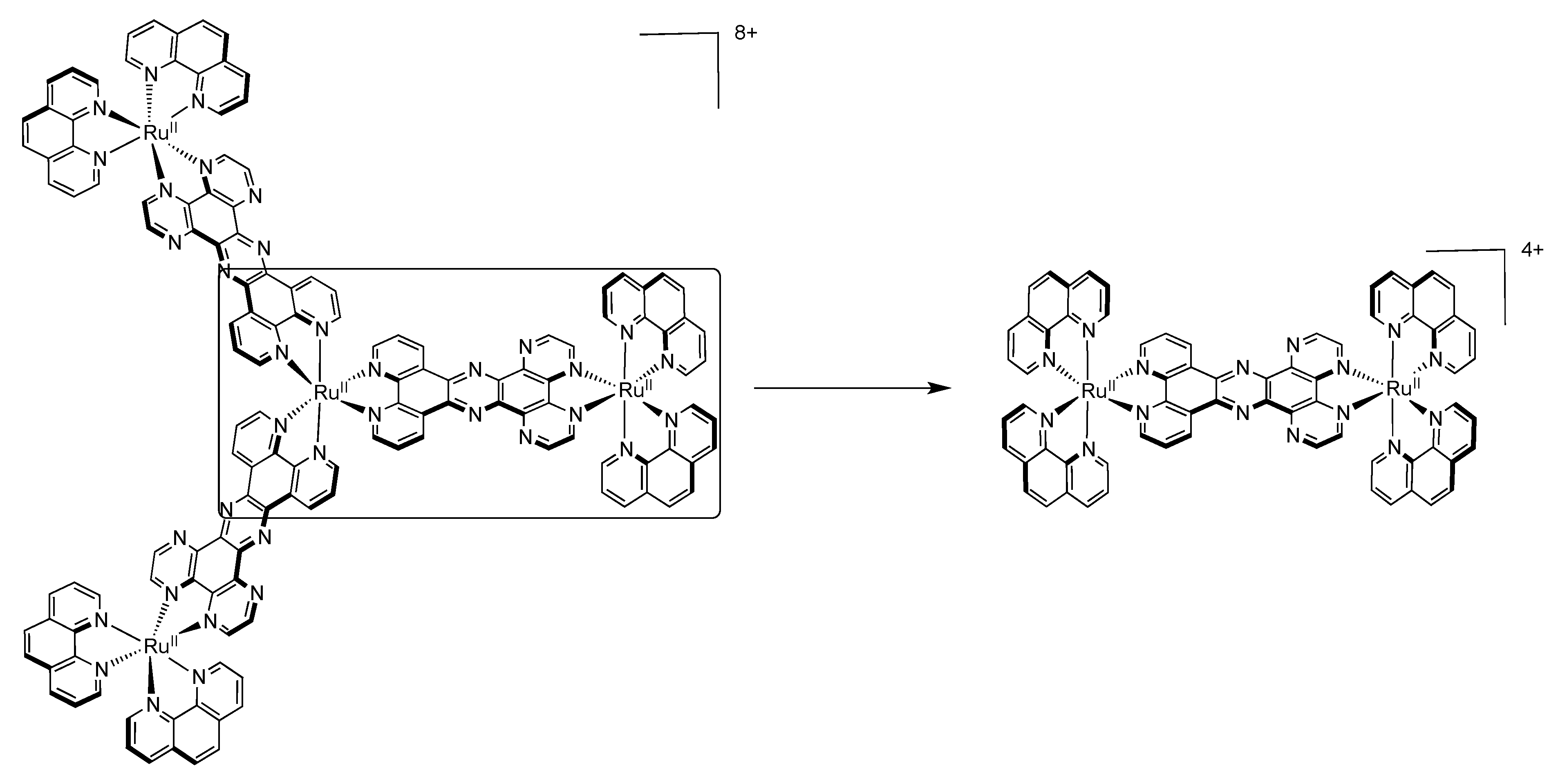
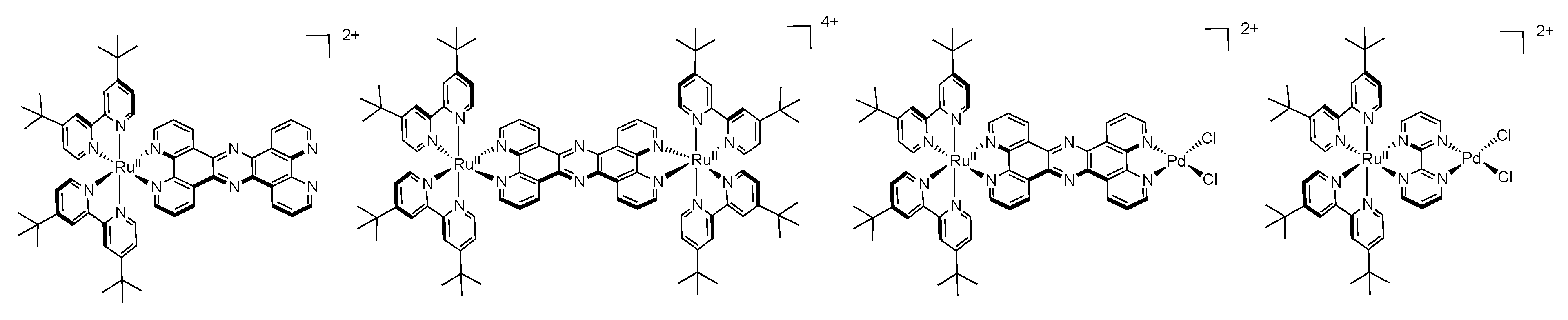
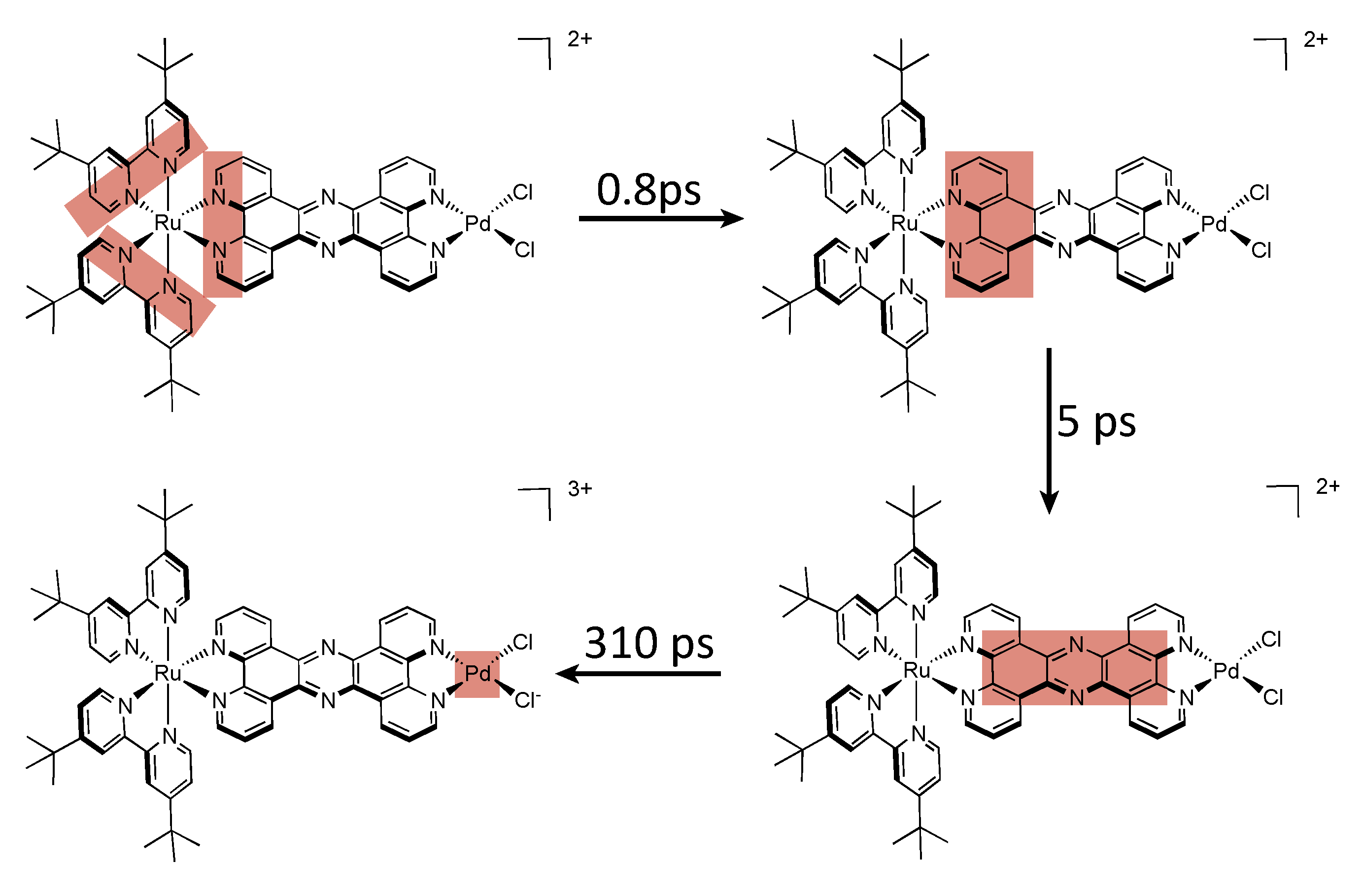
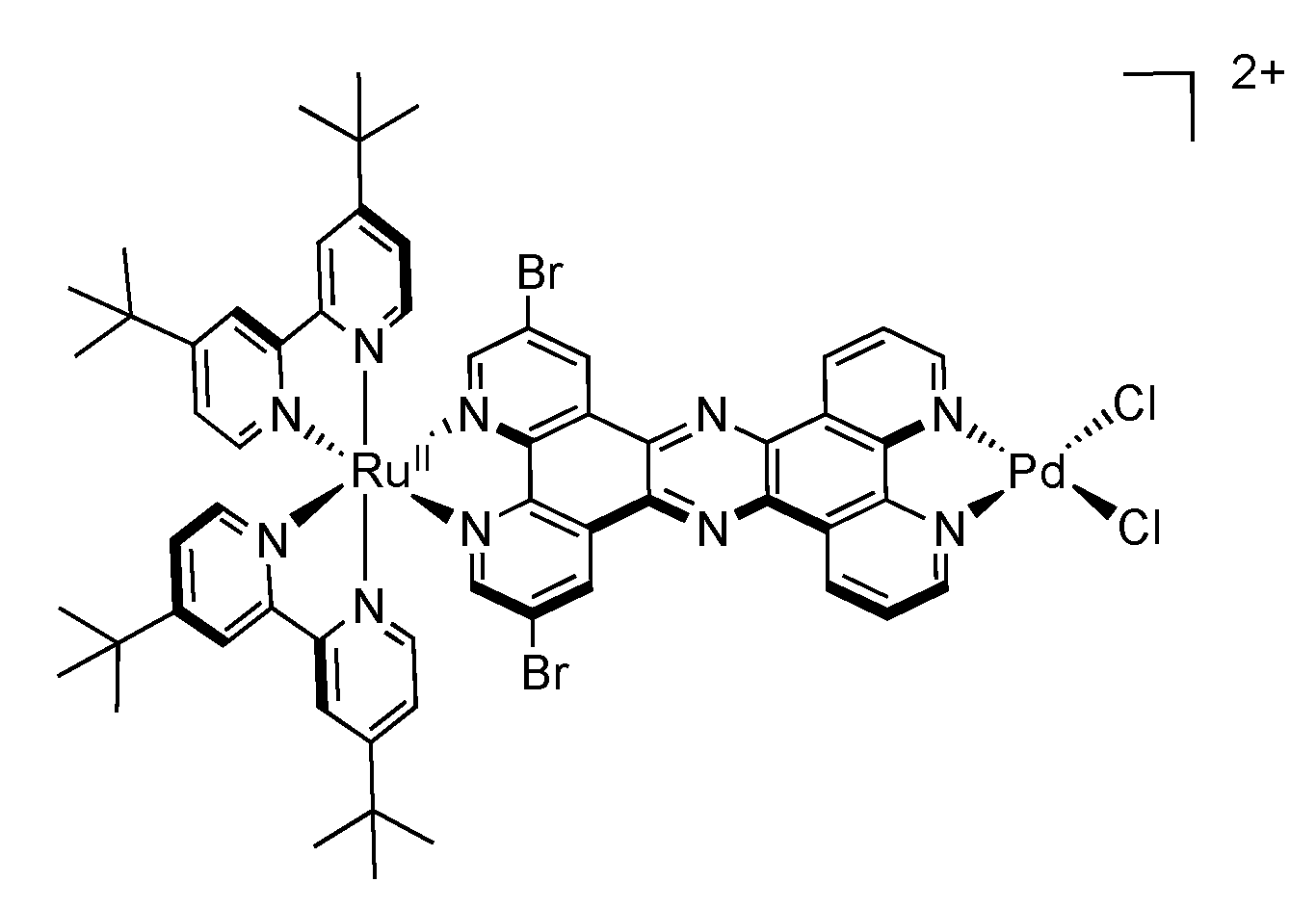
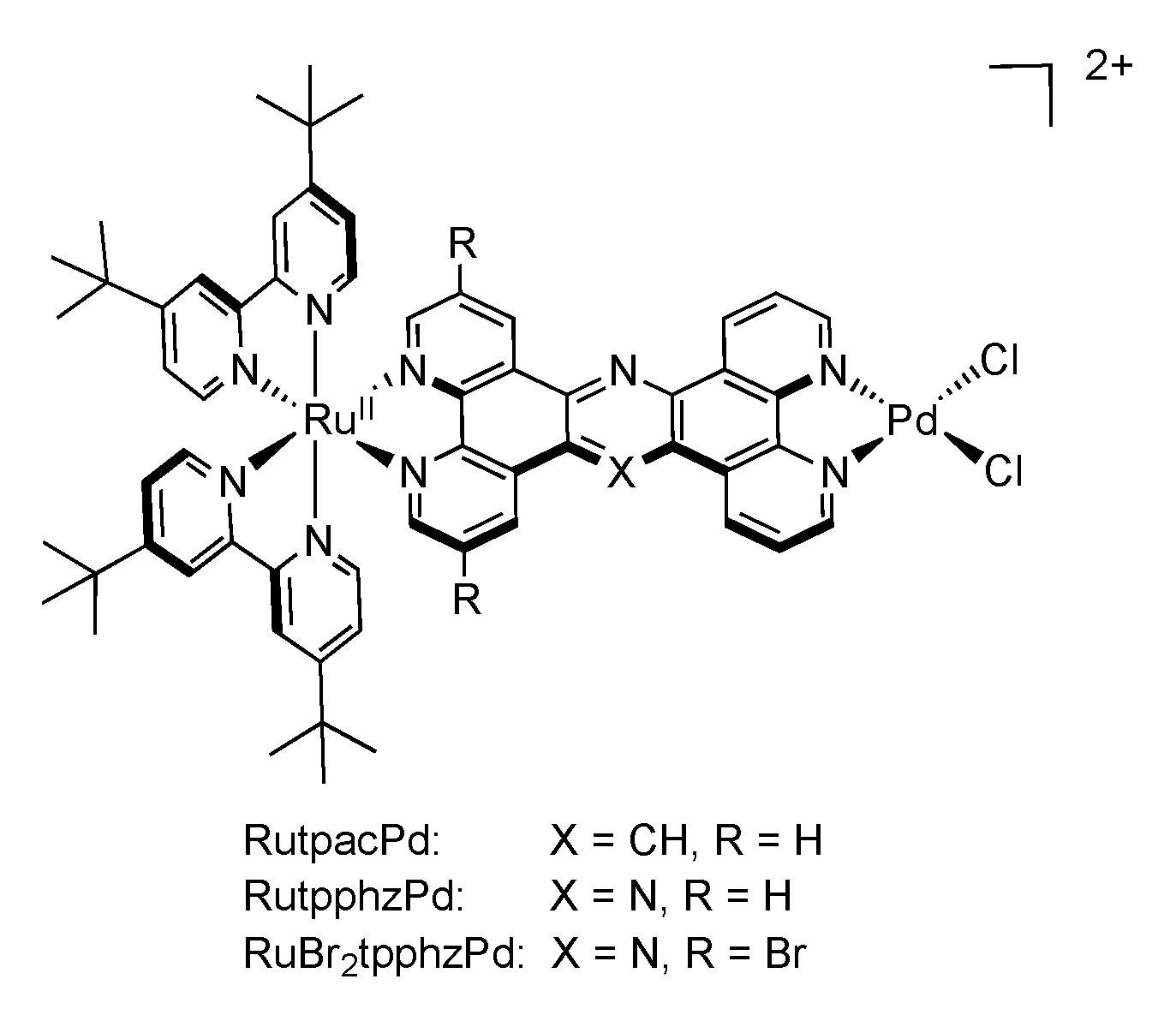
5. Water Splitting and Hydrogen Production
5.1. Complexes based on Ligands with Extended Aromaticity as Catalysts for Hydrogen Production

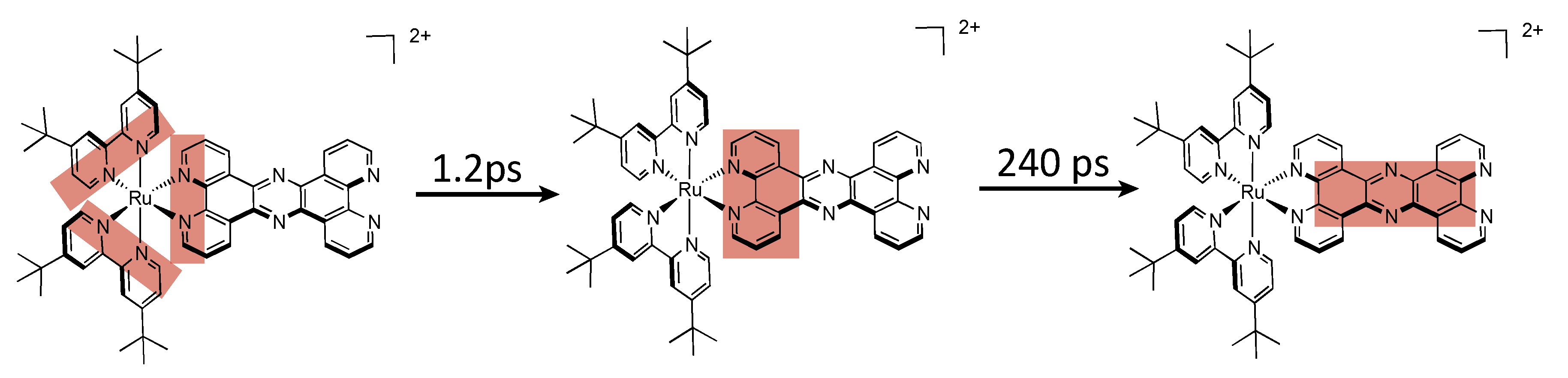
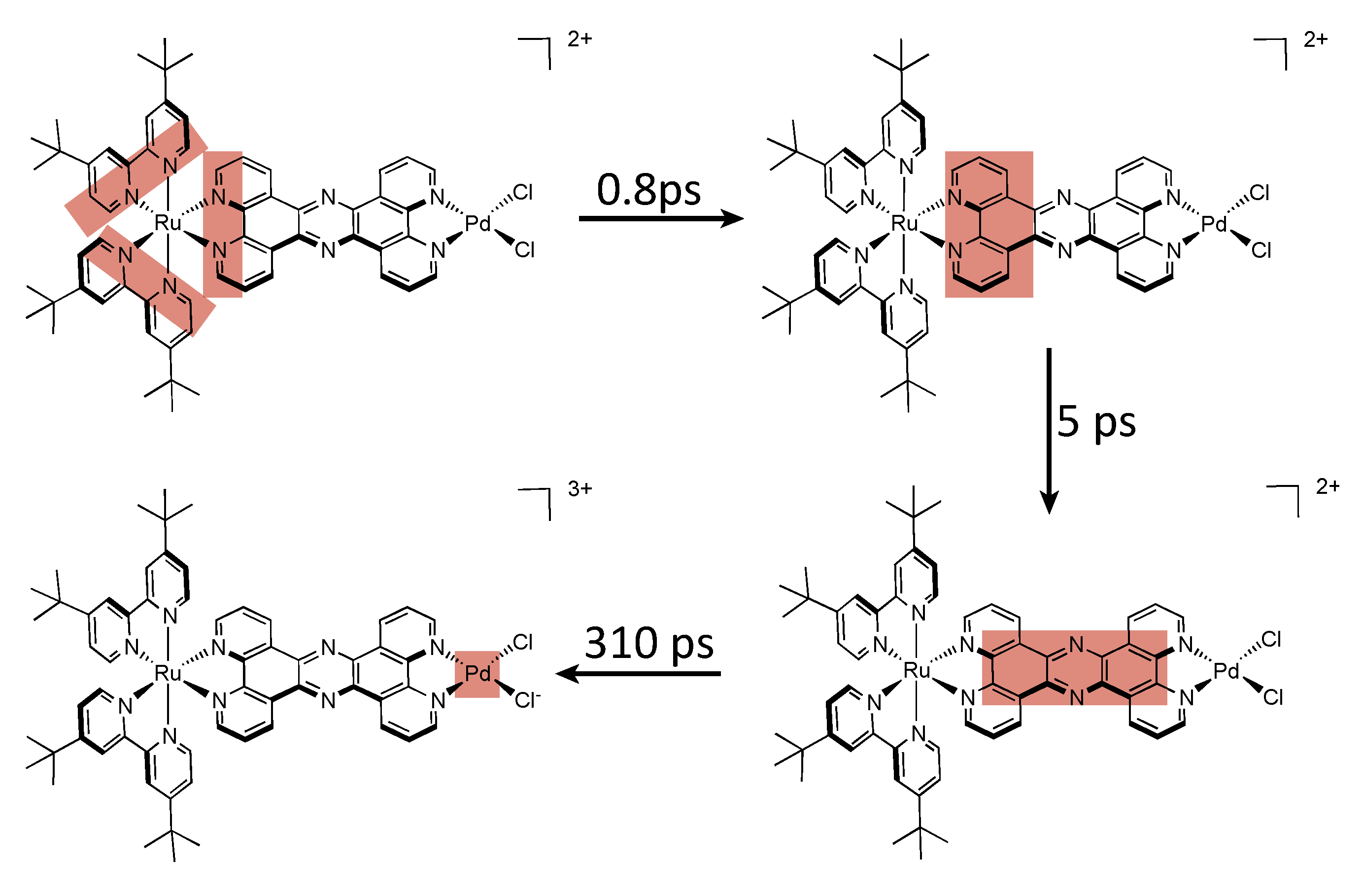
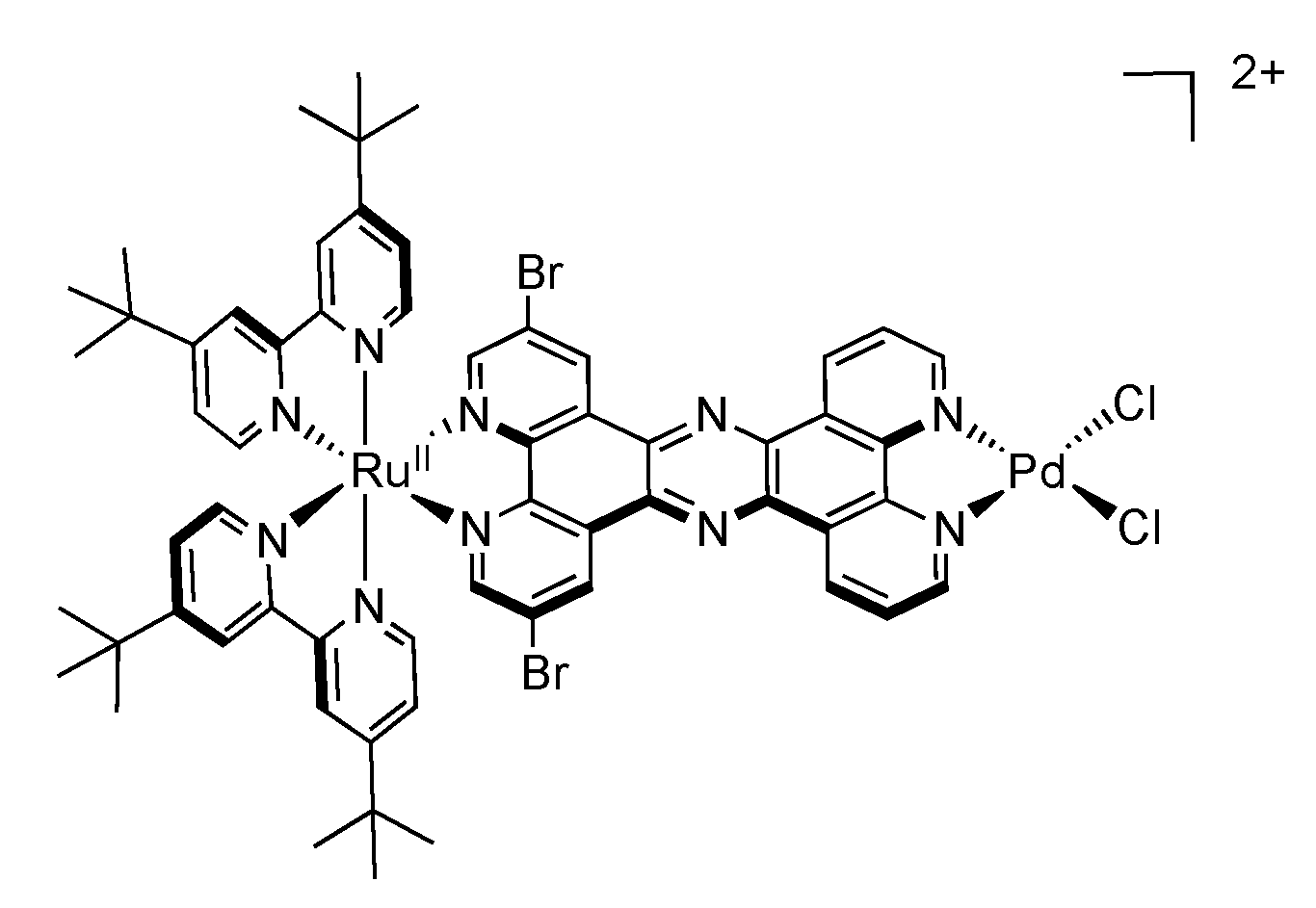
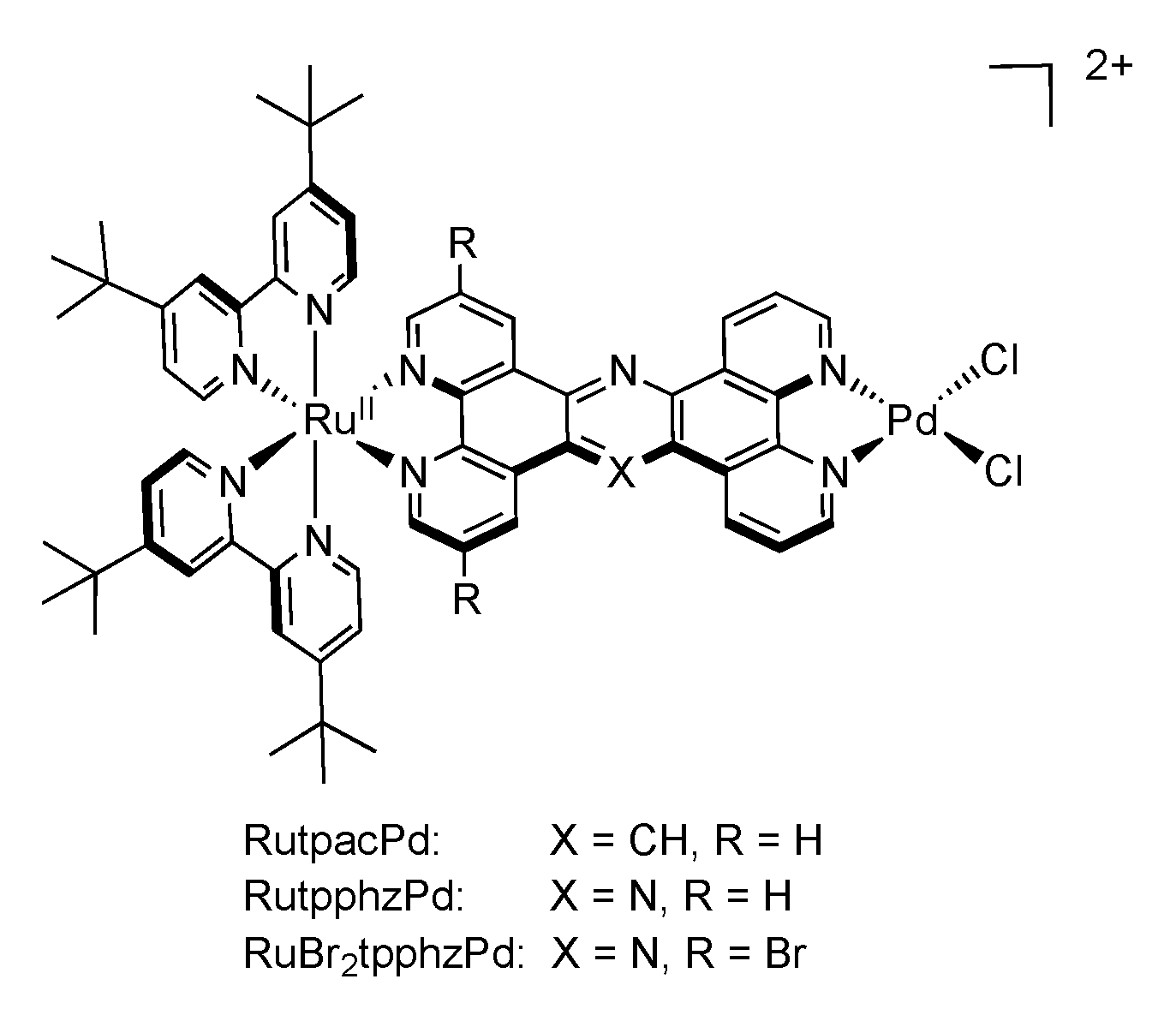
| Complex | λabs/nm | λem/nm | τ/ns | Solvent (catalysis) | Donor | TON (time (h)) |
|---|---|---|---|---|---|---|
| [Ru(tbbpy)2(TPAC)PdCl2]2+ | 475 | 617 | 180 | ACN + 10% H2O | TEA | 138.7 (18) |
| [Ru(tbbpy)2(TPPHZ)PdCl2]2+ | 445 | 650 | 27 | ACN + 15% H2O | TEA | 238.3 (18) |
| [Ru(tbbpy)2(Br2TPPHZ)PdCl2]2+ | 484 | 675 | 84 | ACN + 7.1% H2O | TEA | 94.2 (18) |
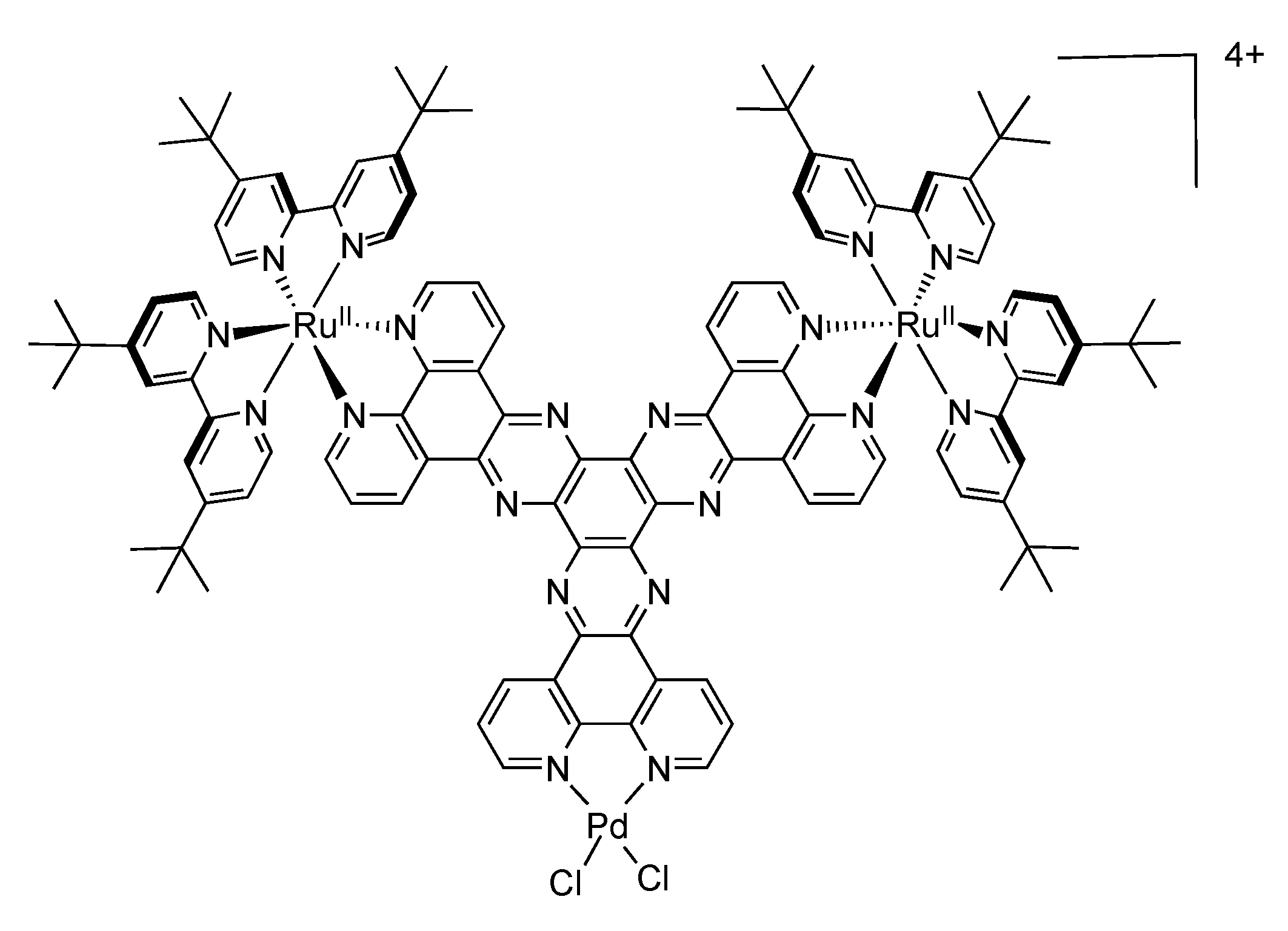
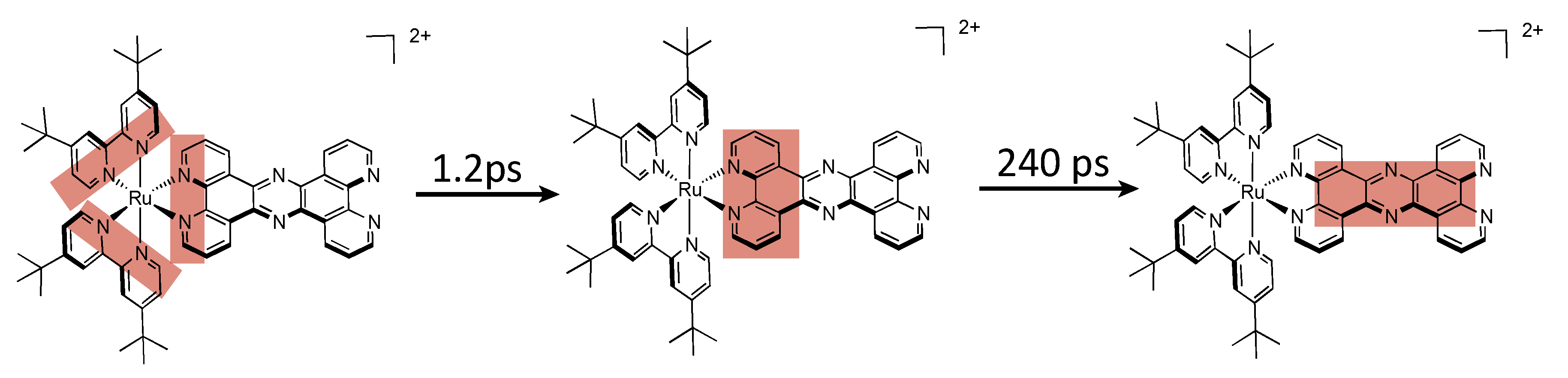
5.2. Development of Ligands with More Extended π-Systems
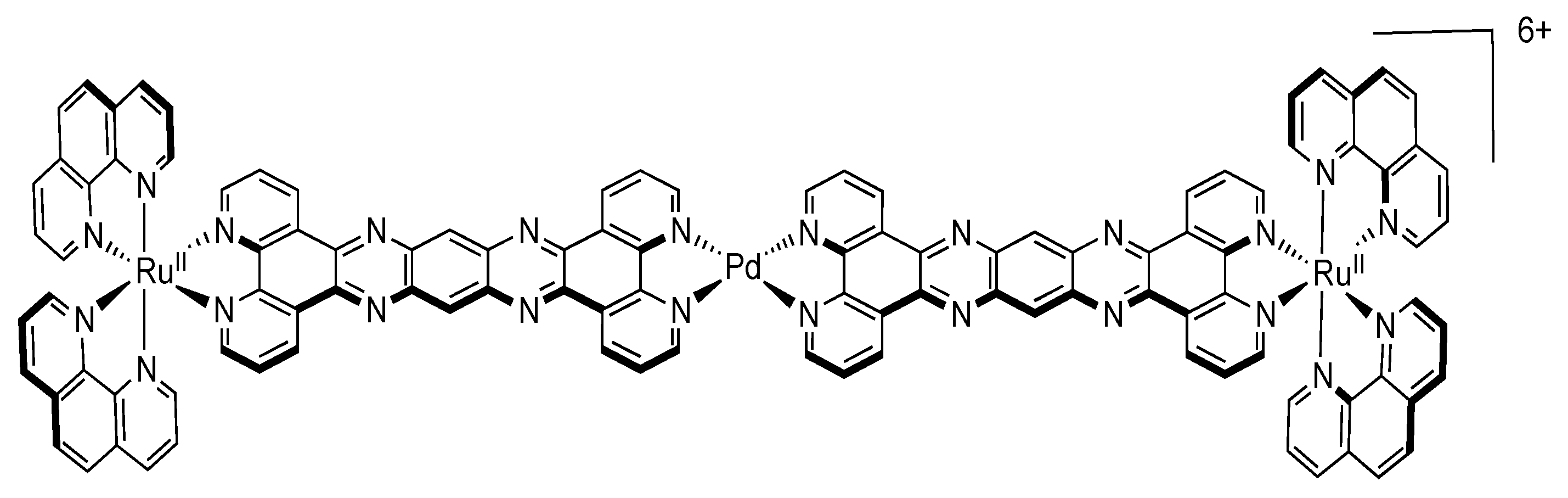


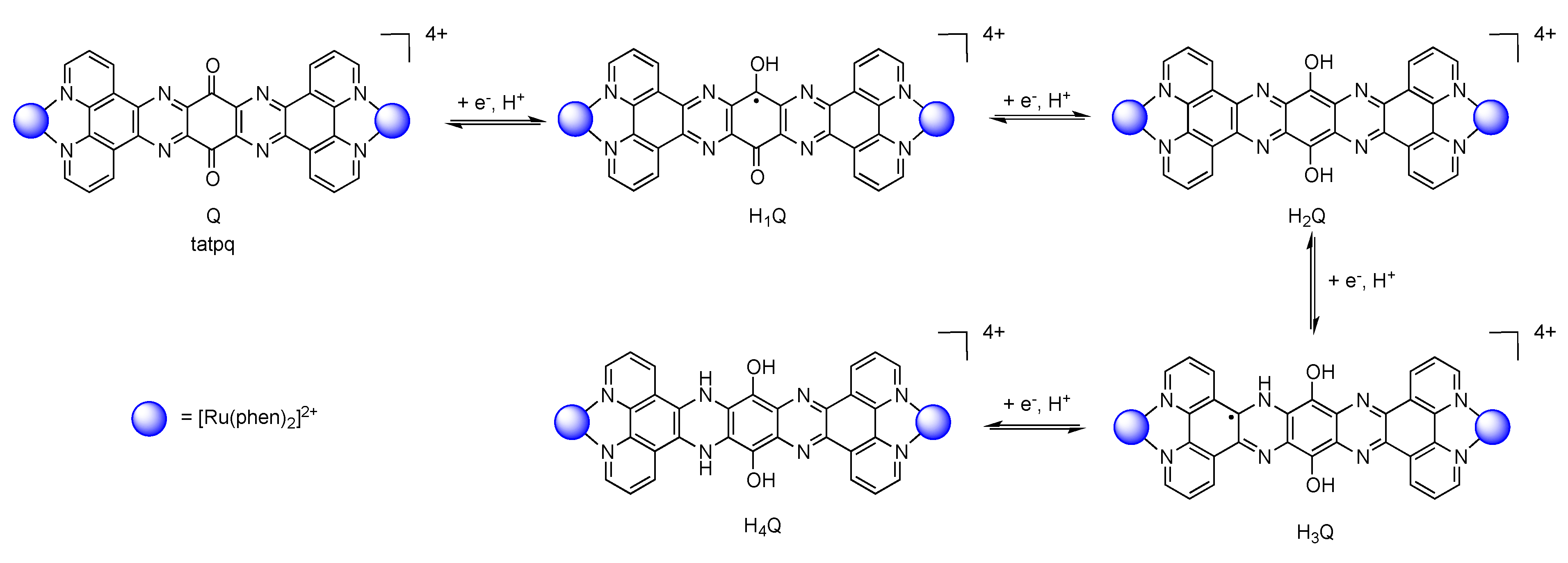
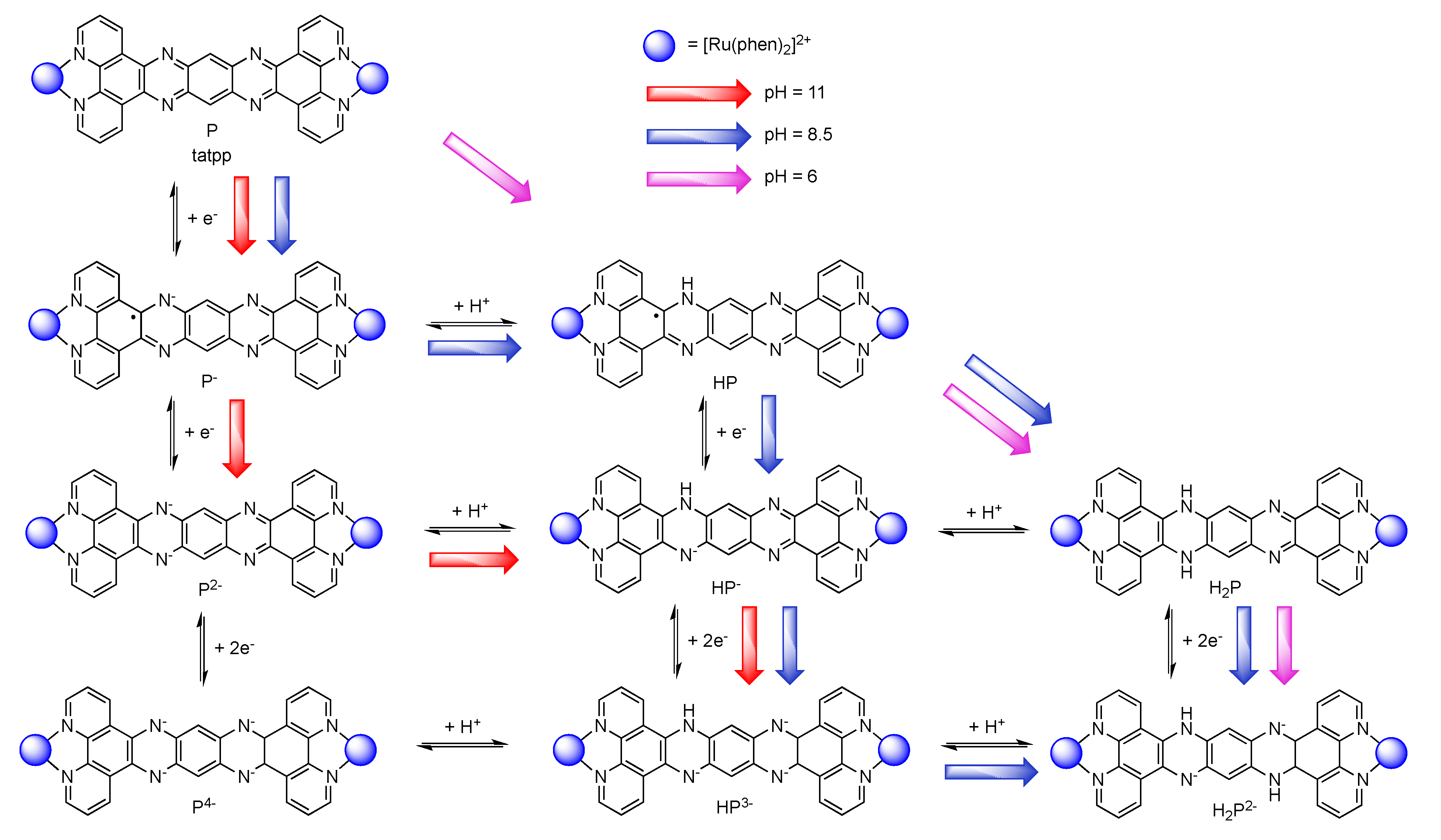
5.3. Complexes based on Ligands with Extended Aromaticity for Electron Photo-Accumulation
| Complex | Oxidation | Reduction |
|---|---|---|
| [Ru(phen)2(tatpp)Ru(phen)2]4+ [161] | +1.32 (2) | −0.26 −0.75 −1.32 (2) |
| [Ru(phen)2(tatpq)Ru(phen)2]4+ [129] | +1.37 (2) | −0.23 −0.60 |
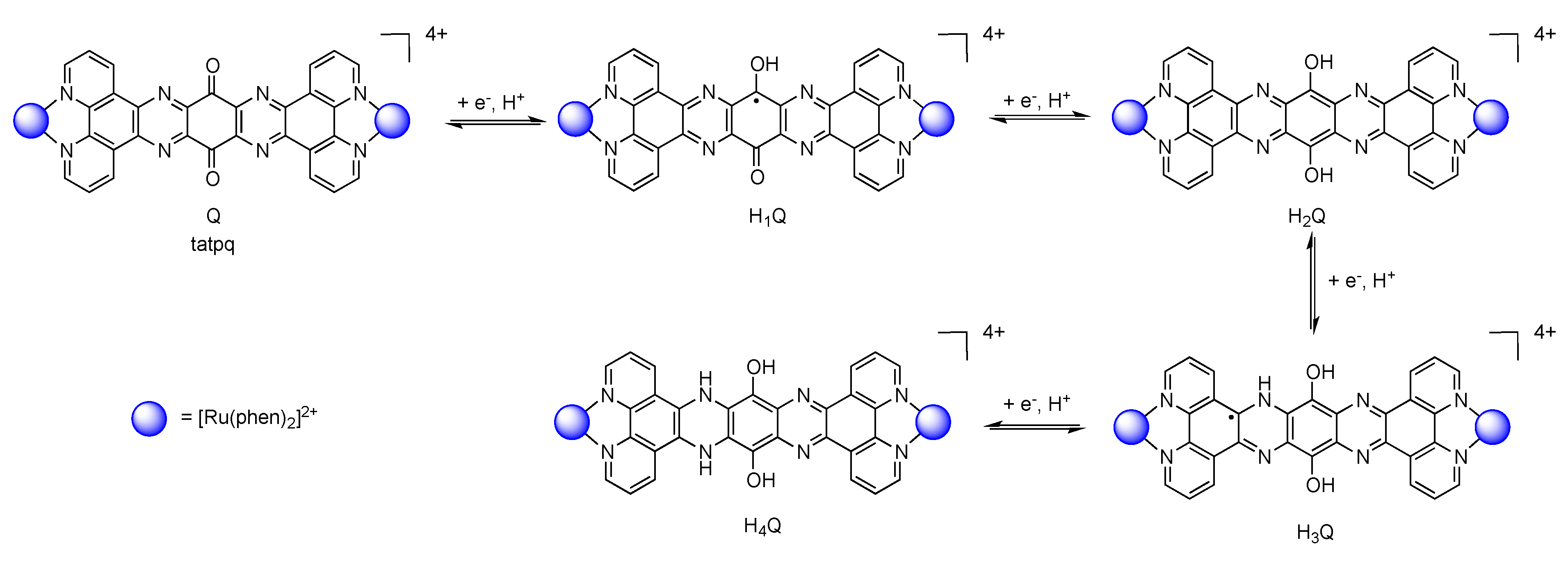
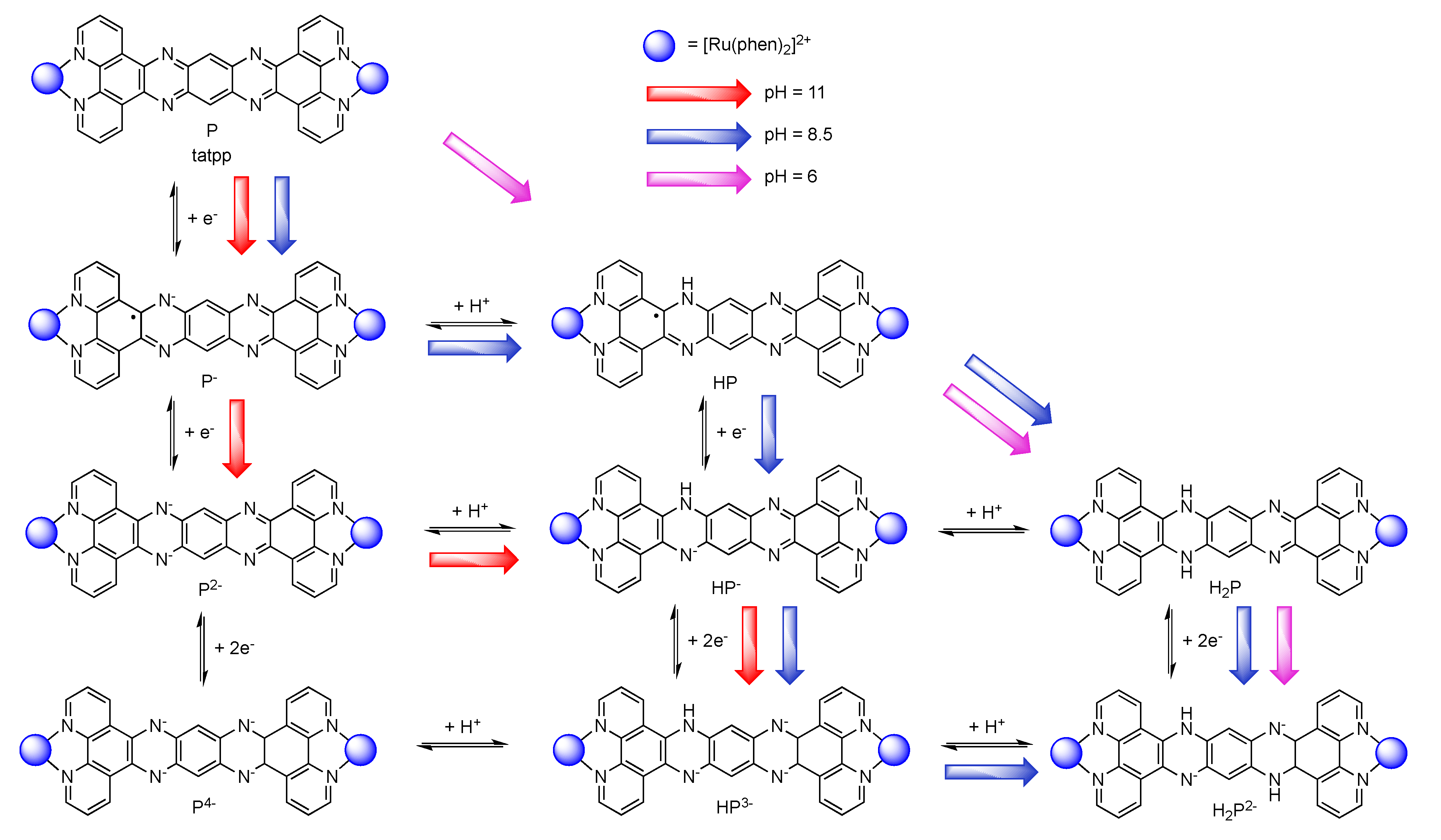

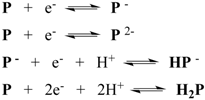
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Barton, J.K.; Danishefsky, A.; Goldberg, J.M. Tris(phenanthroline)ruthenium(II): Stereoselectivity in binding to DNA. J. Am. Chem. Soc. 1984, 106, 2172–2176. [Google Scholar] [CrossRef]
- Barton, J.K.; Goldberg, J.M.; Kumar, C.V.; Turro, N.J. Binding modes and base specificity of tris(phenanthroline)ruthenium(II) enantiomers with nucleic acids: Tuning the stereoselectivity. J. Am. Chem. Soc. 1986, 108, 2081–2088. [Google Scholar] [CrossRef]
- Pyle, A.M.; Barton, J.K. Probing nucleic acids with transition metal complexes. Prog. Inorg. Chem. 1990, 38, 413–475. [Google Scholar] [CrossRef]
- Moucheron, C.; Kirsch-De Mesmaeker, A.; Kelly, J.M. Photophysics and Photochemistry of Metal Polypyridyl and Related Complexes with Nucleic Acids. In Less Common Metals In Proteins And Nucleic Acid Probes; Richmond, J.P., Ed.; Springer Berlin: Heidelberg, Germany, 1998; pp. 163–216. [Google Scholar]
- Metcalfe, C.; Thomas, J.A. Kinetically inert transition metal complexes that reversibly bind to DNA. Chem. Soc. Rev. 2003, 32, 215–224. [Google Scholar] [CrossRef]
- Vos, J.G.; Kelly, J.M. Ruthenium polypyridyl chemistry; from basic research to applications and back again. J. Chem. Soc. Dalton Trans. 2006, 41, 4869–4883. [Google Scholar]
- Gill, M.R.; Thomas, J.A. Ruthenium(II) polypyridyl complexes and DNA – from structural probes to cellular imaging and therapeutics. Chem. Soc. Rev. 2012, 41, 3179–3192. [Google Scholar] [CrossRef]
- Lo, K.K.-W.; Choi, A.W.-T.; Law, W.H.-T. Applications of luminescent inorganic and organometallic transition metal complexes as biomolecular and cellular probes. J. Chem. Soc. Dalton Trans. 2012, 41, 6021–6047. [Google Scholar] [CrossRef]
- Nordén, B.; Lincoln, P.; Åkerman, B.; Tuite, E. DNA Interactions with Substitution-Inert Transition Metal Ion Complexes. In Metal Ions in Biological Systems; Sigel, A., Sigel, H., Eds.; Marcel Dekker: New York, NY, USA, 1996; Volume 33, pp. 177–252. [Google Scholar]
- Erkkila, K.E.; Odom, D.T.; Barton, J.K. Recognition and reaction of metallointercalators with DNA. Chem. Rev. 1999, 99, 2777–2795. [Google Scholar] [CrossRef]
- Hartshorn, R.M.; Barton, J.K. Novel dipyridophenazine complexes of ruthenium(II): Exploring luminescent reporters of DNA. J. Am. Chem. Soc. 1992, 114, 5919–5925. [Google Scholar] [CrossRef]
- Gill, M.R.; Garcia-Lara, J.; Foster, S.J.; Smythe, C.; Battaglia, G.; Thomas, J.A. A ruthenium(II) polypyridyl complex for direct imaging of DNA structure in living cells. Nat. Chem. 2009, 1, 662–667. [Google Scholar] [CrossRef]
- Tan, L.-F.; Chao, H.; Liu, Y.-J.; Li, H.; Sun, B.; Ji, L.-N. DNA-binding and photocleavage studies of [Ru(phen)2(NMIP)]2+. Inorg. Chim. Acta 2005, 358, 2191–2198. [Google Scholar] [CrossRef]
- Xu, L.-C.; Shi, S.; Li, J.; Liao, S.-Y.; Zheng, K.-C.; Ji, L.-N. A combined computational and experimental study on DNA-photocleavage of Ru(II) polypyridyl complexes [Ru(bpy)2(L)]2+ (L = pip, o-mopip and p-mopip). J. Chem. Soc. Dalton Trans. 2008, 291–301. [Google Scholar]
- Gao, F.; Chao, H.; Ji, L.-N. DNA binding, photocleavage, and topoisomerase inhibition of functionalized ruthenium(II)-polypyridine complexes. Chem. Biodivers. 2008, 5, 1962–1979. [Google Scholar] [CrossRef]
- Sun, Y.; Joyce, L.E.; Dickson, N.M.; Turro, C. Efficient DNA photocleavage by [Ru(bpy)2(dppn)]2+ with visible light. Chem. Commun. 2010, 46, 2426–2428. [Google Scholar] [CrossRef]
- Moucheron, C.; Kirsch-De Mesmaeker, A.; Kelly, J.M. Photoreactions of ruthenium(II) and osmium(II) complexes with deoxyribonucleic acid (DNA). J. Photochem. Photobiol. B 1997, 40, 91–106. [Google Scholar] [CrossRef]
- Marcélis, L.; Ghesquière, J.; Garnir, K.; Kirsch-De Mesmaeker, A.; Moucheron, C. Photo-oxidizing RuII complexes and light: Targeting biomolecules via photoadditions. Coord. Chem. Rev. 2012, 256, 1569–1582. [Google Scholar] [CrossRef]
- Ghesquière, J.; le Gac, S.; Marcélis, L.; Moucheron, C.; Kirsch-De Mesmaeker, A. What does the future hold for photo-oxidizing RuII complexes with polyazaaromatic ligands in medicinal chemistry? Curr. Top. Med. Chem. 2012, 12, 185–196. [Google Scholar] [CrossRef]
- Marcélis, L.; Moucheron, C.; Kirsch-De Mesmaeker, A. Ru-TAP complexes and DNA: From photo-induced electron transfer to gene photo-silencing in living cells. Phil. Trans. R. Soc. A 2013, 371, 20120131. [Google Scholar] [CrossRef]
- Chow, C.S.; Barton, J.K. Transition metal complexes as probes of nucleic acids. Meth. Enzymol. 1992, 212, 219–242. [Google Scholar] [CrossRef]
- Long, E.C.; Barton, J.K. On demonstrating DNA intercalation. Acc. Chem. Res. 1990, 23, 271–273. [Google Scholar] [CrossRef]
- Chambron, J.-C.; Sauvage, J.-P.; Amouyal, E.; Koffi, P. Ru(bipy)2(dipyridophenazine)2+: A complex with a long range directed charge transfer excited state. Nouv. J. Chim. 1985, 9, 527–529. [Google Scholar]
- Friedman, A.E.; Chambron, J.-C.; Sauvage, J.-P.; Turro, N.J.; Barton, J.K. A molecular light switch for DNA: Ru(bpy)2(DPPZ)2+. J. Am. Chem. Soc. 1990, 112, 4960–4962. [Google Scholar] [CrossRef]
- Dickeson, J.E.; Summers, L.A. Derivatives of 1,10-phenanthroline-5,6-quinone. Aust. J. Chem. 1970, 23, 1023–1027. [Google Scholar] [CrossRef]
- Friedman, A.E.; Kumar, C.V.; Turro, N.J.; Barton, J.K. Luminescence of ruthenium(II) polypyridyls: Evidence for intercalative binding to Z-DNA. Nucleic Acids Res. 1991, 19, 2595–2602. [Google Scholar]
- Jenkins, Y.; Friedman, A.E.; Turro, N.J.; Barton, J.K. Characterization of dipyridophenazine complexes of ruthenium(II): The light switch effect as a function of nucleic acid sequence and conformation. Biochemistry 1992, 31, 10809–10816. [Google Scholar] [CrossRef]
- Hiort, C.; Lincoln, P.; Nordén, B. DNA binding of .DELTA.- and .LAMBDA.-[Ru(phen)2DPPZ]2+. J. Am. Chem. Soc. 1993, 115, 3448–3454. [Google Scholar] [CrossRef]
- Lincoln, P.; Broo, A.; Nordén, B. Diastereomeric DNA-binding geometries of intercalated ruthenium(II) trichelates probed by linear dichroism: [Ru(phen)2DPPZ]2+ and [Ru(phen)2BDPPZ]2+. J. Am. Chem. Soc. 1996, 118, 2644–2653. [Google Scholar] [CrossRef]
- Tuite, E.; Lincoln, P.; Nordén, B. Photophysical evidence that ∆- and Λ-[Ru(phen)2(DPPZ)]2+ intercalate DNA from the minor groove. J. Am. Chem. Soc. 1997, 119, 239–240. [Google Scholar] [CrossRef]
- Liu, Y.; Chouai, A.; Degtyareva, N.N.; Lutterman, D.A.; Dunbar, K.R.; Turro, C. Chemical control of the DNA light switch: Cycling the switch on and off. J. Am. Chem. Soc. 2005, 127, 10796–10797. [Google Scholar] [CrossRef]
- Önfelt, B.; Olofsson, J.; Lincoln, P.; Nordén, B. Picosecond and steady-state emission of [Ru(phen)2DPPZ]2+ in glycerol: Anomalous temperature dependence. J. Phys. Chem. A 2003, 107, 1000–1009. [Google Scholar] [CrossRef]
- Coates, C.G.; Olofsson, J.; Coletti, M.; McGarvey, J.J.; Önfelt, B.; Lincoln, P.; Nordén, B.; Tuite, E.; Matousek, P.; Parker, A.W. Picosecond time-resolved resonance Raman probing of the light-switch states of [Ru(phen)2DPPZ]2+. J. Phys. Chem. B 2001, 105, 12653–12664. [Google Scholar] [CrossRef]
- Elias, B.; Creely, C.; Doorley, G.W.; Feeney, M.M.; Moucheron, C.; Kirsch-De Mesmaeker, A.; Dyer, J.; Grills, D.C.; George, M.W.; Matousek, P.; et al. Photooxidation of guanine by a ruthenium dipyridophenazine complex intercalated in a double-stranded polynucleotide monitored directly by picosecond visible and infrared transient absorption spectroscopy. Chem. Eur. J. 2008, 14, 369–375. [Google Scholar] [CrossRef]
- Smith, J.A.; George, M.W.; Kelly, J.M. Transient spectroscopy of dipyridophenazine metal complexes which undergo photo-induced electron transfer with DNA. Coord. Chem. Rev. 2011, 255, 2666–2675. [Google Scholar] [CrossRef]
- Chen, W.; Turro, C.; Friedman, L.A.; Barton, J.K.; Turro, N.J. Resonance Raman investigation of Ru(phen)2(DPPZ)2+ and related complexes in water and in the presence of DNA. J. Phys. Chem. B 1997, 101, 6995–7000. [Google Scholar] [CrossRef]
- McGarvey, J.J.; Callaghan, P.; Coates, C.G.; Schoonover, J.R.; Kelly, J.M.; Jacquet, L.; Gordon, K.C. Comment on “resonance Raman investigation of [Ru(phen)2(DPPZ)2+] and related complexes in water and in the presence of DNA”. J. Phys. Chem. B 1998, 102, 5941–5942. [Google Scholar] [CrossRef]
- Coates, C.G.; Jacquet, L.; McGarvey, J.J.; Bell, S.E.J.; Al-Obaidi, A.H.R.; Kelly, J.M. Resonance Raman probing of the interaction between dipyridophenazine complexes of Ru(II) and DNA. J. Am. Chem. Soc. 1997, 119, 7130–7136. [Google Scholar]
- Hall, J.P.; O'Sullivan, K.; Naseer, A.; Smith, J.A.; Kelly, J.M.; Cardin, C.J. Structure determination of an intercalating ruthenium dipyridophenazine complex which kinks DNA by semiintercalation of a tetraazaphenanthrene ligand. Proc. Natl. Acad. Sci. USA 2011, 108, 17610–17614. [Google Scholar] [CrossRef]
- Niyazi, H.; Hall, J.P.; O'Sullivan, K.; Winter, G.; Sorensen, T.; Kelly, J.M.; Cardin, C.J. Crystal structures of Λ-[Ru(phen)2(DPPZ)]2+ with oligonucleotides containing TA/TA and AT/AT steps show two intercalation modes. Nat. Chem. 2012, 4, 621–628. [Google Scholar] [CrossRef]
- Song, H.; Kaiser, J.T.; Barton, J.K. Crystal structure of ∆-[Ru(bpy)2DPPZ]2+ bound to mismatched DNA reveals side-by-side metalloinsertion and intercalation. Nat. Chem. 2012, 4, 615–620. [Google Scholar] [CrossRef]
- Hall, J.P.; Cook, D.; Ruiz Morte, S.; McIntyre, P.; Buchner, K.; Beer, H.; Cardin, D.J.; Brazier, J.A.; Winter, G.; Kelly, J.M.; et al. X-ray crystal structure of rac-[Ru(phen)2DPPZ]2+ with d(ATGCAT)2 shows enantiomer orientations and water ordering. J. Am. Chem. Soc. 2013, 135, 12652–12659. [Google Scholar] [CrossRef]
- Ardhammar, M.; Lincoln, P.; Nordén, B. Ligand substituents of ruthenium dipyridophenazine complexes sensitively determine orientation in liposome membrane. J. Phys. Chem. B 2001, 105, 11363–11368. [Google Scholar] [CrossRef]
- Svensson, F.R.; Li, M.; Nordén, B.; Lincoln, P. Luminescent dipyridophenazine-ruthenium probes for liposome membranes. J. Phys. Chem. B 2008, 112, 10969–10975. [Google Scholar] [CrossRef]
- Rajendiran, V.; Palaniandavar, M.; Periasamy, V.S.; Akbarsha, M.A. [Ru(phen)2(DPPZ)]2+ as an efficient optical probe for staining nuclear components. J. Inorg. Biochem. 2010, 104, 217–220. [Google Scholar] [CrossRef]
- Yam, V.W.-W.; Lee, V.W.-M.; Ke, F.; Siu, K.-W.M. Synthesis, photophysics, and electrochemistry of ruthenium(II) polypyridine complexes with crown ether pendants. Inorg. Chem. 1997, 36, 2124–2129. [Google Scholar] [CrossRef]
- Cheng, F.; Tang, N. A new family of dinuclear Ru (II) polypyridyl complexes containing dibenzo-14-crown-4. Inorg. Chem. Commun. 2008, 11, 939–942. [Google Scholar] [CrossRef]
- Cheng, F.; Sun, Y.; Wu, W.; Tang, N. Synthesis, photophysical, and electrochemical properties of three novel Ru (II) polypyridyl complexes containig dibenzo-18-crown-6. Inorg. Chem. Commun. 2008, 11, 687–690. [Google Scholar] [CrossRef]
- Shang, X.-F.; Lin, D.-K. Phenanthroline complexes bearing diamide-anion recognition sites. Transit. Met. Chem. 2007, 32, 38–41. [Google Scholar] [CrossRef]
- Lin, T.-P.; Chen, C.-Y.; Wen, Y.-S.; Sun, S.-S. Synthesis, photophysical, and anion-sensing properties of quinoxalinebis(sulfonamide) functionalized receptors and their metal complexes. Inorg. Chem. 2007, 46, 9201–9212. [Google Scholar] [CrossRef]
- Shang, X.; Li, X.; Xi, N.; Zhai, Y.; Zhang, J.; Xu, X. Theory and experiment: Recognition properties of chemosensor containing ruthenium(II) system in water solution. Sens. Actuators B Chem. 2011, 160, 1112–1119. [Google Scholar] [CrossRef]
- Shang, X.; Li, J.; Lin, H.; Jiang, P.; Cai, Z.-C.; Lin, H.-K. Anion recognition and sensing of ruthenium(II) and cobalt(II) sulfonamido complexes. J. Chem. Soc. Dalton Trans. 2009, 2096–2102. [Google Scholar]
- nflet, B.; Lincoln, P.; Nordén, B. Enantioselective DNA threading dynamics by phenazine-linked [Ru(phen)2DPPZ]2+ dimers. J. Am. Chem. Soc. 2001, 123, 3630–3637. [Google Scholar] [CrossRef]
- Lincoln, P.; Nordén, B. Binuclear ruthenium(II) phenanthroline compounds with extreme binding affinity for DNA. Chem. Commun. 1996, 2145–2146. [Google Scholar] [CrossRef]
- Wilhelmsson, L.M.; Westerlund, F.; Lincoln, P.; Nordén, B. DNA-binding of semirigid binuclear ruthenium complex ∆,∆-[µ-(11,11’-biDPPZ)(phen)4Ru2]4+: Extremely slow intercalation kinetics. J. Am. Chem. Soc. 2002, 124, 12092–12093. [Google Scholar] [CrossRef]
- Campagna, S.; Serroni, S.; Puntoriero, F.; di Pietro, C.; Ricevuto, V. Electron Transfer in Chemistry; Balzani, V., Ed.; VCH-Wiley: Weinheim, Germany, 2001; p. 186. [Google Scholar]
- Sommovigo, M.; Denti, G.; Serroni, S.; Campagna, S.; Mingazzini, C.; Mariotti, C.; Juris, A. Polynuclear polypyridine complexes incorporating Ru(II), Os(II), and Pt(II): Decanuclear dendrimeric antennas. Inorg. Chem. 2001, 40, 3318–3323. [Google Scholar] [CrossRef]
- Campagna, S.; Serroni, S.; Puntoriero, F.; Loiseau, F.; de Cola, L.; Kleverlaan, C.J.; Becher, J.; Sorensen, A.P.; Hascoat, P.; Thorup, N. Coupling of metal-based light harvesting antennas and electron-donor subunits: Trinuclear ruthenium(II) complexes containing tetrathiafulvalene-substituted polypyridine ligands. Chem. Eur. J. 2002, 8, 4461–4469. [Google Scholar] [CrossRef]
- Serroni, S.; Campagna, S.; Puntoriero, F.; Loiseau, F.; Ricevuto, V.; Passalacqua, R.; Galletta, M. Dendrimers made of Ru(II) and Os(II) polypyridine subunits as artificial light-harvesting antennae. C. R. Chim. 2003, 6, 883–893. [Google Scholar] [CrossRef]
- Puntoriero, F.; Serroni, S.; Galletta, M.; Juris, A.; Licciardello, A.; Chiorboli, C.; Campagna, S.; Scandola, F. A new heptanuclear dendritic ruthenium(II) complex featuring photoinduced energy transfer across high-energy subunits. ChemPhysChem 2005, 6, 129–138. [Google Scholar] [CrossRef]
- Balzani, V.; Ceroni, P.; Juris, A.; Venturi, M.; Campagna, S.; Puntoriero, F.; Serroni, S. Dendrimers based on photoactive metal complexes. Recent advances. Coord. Chem. Rev. 2001, 219–221, 545–572. [Google Scholar]
- Serroni, S.; Campagna, S.; Puntoriero, F.; di Pietro, C.; McClenaghan, N.D.; Loiseau, F. Dendrimers based on ruthenium(II) and osmium(II) polypyridine complexes and the approach of using complexes as ligands and complexes as metals. Chem. Soc. Rev. 2001, 30, 367–375. [Google Scholar] [CrossRef]
- Konduri, R.; Ye, H.; MacDonnell, F.M.; Serroni, S.; Campagna, S.; Rajeshwar, K. Ruthenium photocatalysts capable of reversibly storing up to four electrons in a single acceptor ligand: A step closer to artificial photosynthesis. Angew. Chem. Int. Ed. 2002, 41, 3185–3187. [Google Scholar] [CrossRef]
- Puntoriero, F.; Nastasi, F.; Cavazzini, M.; Quici, S.; Campagna, S. Coupling synthetic antenna and electron donor species: A tetranuclear mixed-metal Os(II)-Ru(II) dendrimer containing six phenothiazine donor subunits at the periphery. Coord. Chem. Rev. 2007, 251, 536–545. [Google Scholar] [CrossRef]
- Pellegrin, Y.; Odobel, F. Molecular devices featuring sequential photoinduced charge separations for the storage of multiple redox equivalents. Coord. Chem. Rev. 2011, 255, 2578–2593. [Google Scholar] [CrossRef]
- Balzani, V.; Bergamini, G.; Ceroni, P. From the photochemistry of coordination compounds to light-powered nanoscale devices and machines. Coord. Chem. Rev. 2008, 252, 2456–2469. [Google Scholar] [CrossRef]
- Fantacci, S.; de Angelis, F. A computational approach to the electronic and optical properties of Ru(II) and Ir(III) polypyridyl complexes: Applications to DSC, OLED and NLO. Coord. Chem. Rev. 2011, 255, 2704–2726. [Google Scholar] [CrossRef]
- Coe, B.J. Developing iron and ruthenium complexes for potential nonlinear optical applications. Coord. Chem. Rev. 2013, 257, 1438–1458. [Google Scholar] [CrossRef]
- Campagna, S.; Puntoriero, F.; Nastasi, F.; Bergamini, G.; Balzani, V. Photochemistry and photophysics of coordination compounds: Ruthenium. Top. Curr. Chem. 2007, 280, 117–214. [Google Scholar] [CrossRef]
- Puntoriero, F.; Sartorel, A.; Orlandi, M.; la Ganga, G.; Serroni, S.; Bonchio, M.; Scandola, F.; Campagna, S. Photoinduced water oxidation using dendrimeric Ru(II) complexes as photosensitizers. Coord. Chem. Rev. 2011, 255, 2594–2601. [Google Scholar] [CrossRef]
- Manbeck, G.F.; Brewer, K.J. Photoinitiated electron collection in polyazine chromophores coupled to water reduction catalysts for solar H2 production. Coord. Chem. Rev. 2013, 257, 1660–1675. [Google Scholar] [CrossRef]
- Burstall, F.H. Optical activity dependent on coordinated bivalent ruthenium. J. Chem. Soc. 1936, 173–175. [Google Scholar] [CrossRef]
- Brandt, W.W.; Paris, J.P. Charge transfer luminescence of a ruthenium(II) chelate. J. Am. Chem. Soc. 1959, 81, 5001–5002. [Google Scholar] [CrossRef]
- Harrigan, R.W.; Hager, G.D.; Crosby, G.A. Evidence for multiple-state emission from ruthenium(II) complexes. Chem. Phys. Lett. 1973, 21, 487–490. [Google Scholar] [CrossRef]
- Demas, J.N.; Taylor, D.G. On the « intersystem crossing » yields in ruthenium(II) and osmium(II) photosensitizers. Inorg. Chem. 1979, 18, 3177–3179. [Google Scholar] [CrossRef]
- Durham, B.; Caspar, J.V.; Nagle, J.K.; Meyer, T.J. Photochemistry of Ru(bpy)32+. J. Am. Chem. Soc. 1982, 75, 4803–4810. [Google Scholar]
- Barrigelletti, F.; Juris, A.; Balzani, V.; Belser, P.; Von Zelewsky, A. Excited-state properties of complexes of the Ru(diimine)32+ family. Inorg. Chem. 1983, 22, 3335–3886. [Google Scholar] [CrossRef]
- Kober, E.M.; Meyer, T.J. An electronic structural mode for the emitting MLCT excited states of Ru(bpy)32+ and Os(bpy)32+. Inorg. Chem. 1984, 23, 3877–3886. [Google Scholar] [CrossRef]
- Yersin, H.; Gallhuber, E. On the lowest excited states of [Ru(bpy)3](PF6)2 single crystals. J. Am. Chem. Soc. 1984, 106, 6582–6586. [Google Scholar] [CrossRef]
- Meyer, T.J. Photochemistry of metal coordination complexes: Metal to ligand charge transfer excited states. Pure Appl. Chem. 1986, 58, 1193–1206. [Google Scholar] [CrossRef]
- Damrauer, N.H.; Cerullo, G.; Yeh, A.; Boussie, R.R.; Shank, C.V.; McCusker, J.K. Femtosecond dynamics of excited-state evolution in [Ru(bpy)3]2+. Science 1997, 275, 54–57. [Google Scholar] [CrossRef]
- Braun, D.; Huber, J.; Wudy, J.; Schmidt, J.; Yersin, H. Electron delocalization and localization in mixed-ligand [Ru(LL)n(LL’)3-n]2+ complexes. J. Chem. Phys. 1994, 98, 8044–8049. [Google Scholar] [CrossRef]
- Malone, R.A.; Kelley, D.F. Interligand electron transfer and transition state dynamics in Ru(II)trisbipyridine. J. Phys. Chem. 1991, 95, 8970–8976. [Google Scholar] [CrossRef]
- Cooley, L.F.; Bergquist, P.; Kelley, D.F. Determination of excition hopping rates in ruthenium(II) tris(bipyridine) complexes by picosecond polarized absorption spectroscopy. J. Am. Chem. Soc. 1990, 112, 2612–2617. [Google Scholar] [CrossRef]
- Kober, E.M.; Sullivan, B.P.; Meyer, T.J. Solvent dependence of metal-to-ligand charge transfer transitions. Evidence for initial electron localization in MLCT excited states of 2,2’-bipyridine complexes of ruthenium(II) and osmium(II). Inorg. Chem. 1984, 23, 2098–2104. [Google Scholar] [CrossRef]
- Barqawi, K.R.; Llobet, A.; Meyer, T.J. Synthetic design of MLCT excited states. Ligand-substituted, mono-2,2'-bipyridine complexes of Ru (II). J. Am. Chem. Soc. 1988, 110, 7751–7759. [Google Scholar] [CrossRef]
- Demas, J.N.; Crosby, G.A. Quantum efficiencies on transition metal complexes. II. Charge-transfer luminescence. J. Am. Chem. Soc. 1971, 93, 2841–2847. [Google Scholar] [CrossRef]
- Caspar, J.V.; Meyer, T.J. Photochemistry of Ru(bpy)32+. Solvent effects. Inorg. Chem. 1983, 105, 5583–5590. [Google Scholar]
- Van Houten, J.; Watts, R.J. Photochemistry of tris(2,2'-bipyridyl)ruthenium(II) in aqueous solutions. Inorg. Chem. 1978, 17, 3381–3385. [Google Scholar] [CrossRef]
- Van Houten, J.; Watts, R.J. Temperature dependence of the photophysical and photochemical properties of the tris(2,2’-bipyridyl)ruthenium(II) ion in aqueous solution. J. Am. Chem. Soc. 1976, 98, 4853–4858. [Google Scholar] [CrossRef]
- Rillema, D.P.; Allen, G.; Meyer, T.J.; Conrad, D. Redox properties of ruthenium(II) tris chelate complexes containing the ligands 2,2'-bipyrazine, 2,2'-bipyridine, and 2,2'-bipyrimidine. Inorg. Chem. 1983, 22, 1617–1622. [Google Scholar] [CrossRef]
- Makuta, T.; Fukazawa, N.; Murata, K.; Inagaki, A.; Akita, M.; Tanaka, S.; Koshihara, S.; Onda, K. Infrared vibrational spectroscopy of [Ru(bpy)2(bpm)]2+ and [Ru(bpy)3]2+ in the excited triplet state. Inorg. Chem. 2014, 53, 2481–2490. [Google Scholar] [CrossRef]
- Bhasikuttan, A.C.; Suzuki, M.; Nakashima, S.; Okada, T. Ultrafast fluorescence detection in tris(2,2’-bipyridine)ruthenium(II) complex in solution: Relaxation dynamics involving higher excited states. J. Am. Chem. Soc. 2002, 124, 8389–8405. [Google Scholar] [CrossRef]
- Turro, C.; Bossmann, S.H.; Jenkins, Y.; Barton, J.K.; Turro, N.J. Proton transfer quenching of the MLCT excited state of Ru(phen)2DPPZ2+ in homogeneous solution and bound to DNA. J. Am. Chem. Soc. 1995, 117, 9026–9032. [Google Scholar] [CrossRef]
- Nair, R.B.; Cullum, B.M.; Murphy, C.J. Optical properties of [Ru(phen)2DPPZ]2+ as a function of nonaqueous environment. Inorg. Chem. 1997, 36, 962–965. [Google Scholar] [CrossRef]
- Olson, E.J.C.; Hu, D.; Hörnman, A.; Jonkman, A.M.; Arkin, M.R.; Stemp, E.D.A.; Barton, J.K.; Barbara, P.F. First observation of the key intermediate in the “light-switch” mechanism of [Ru(phen)2DPPZ]2+. J. Am. Chem. Soc. 1997, 119, 11458–11467. [Google Scholar] [CrossRef]
- Brennaman, M.K.; Alstrum-Acevedo, J.H.; Fleming, C.N.; Jang, P.; Meyer, T.J.; Papanikolas, J.M. Turning the [Ru(bpy)2DPPZ]2+ light-switch on and off with temperature. J. Am. Chem. Soc. 2002, 124, 15094–15098. [Google Scholar] [CrossRef]
- Brennaman, M.K.; Meyer, T.J.; Papanikolas, J.M. [Ru(bpy)2DPPZ]2+ light-switch mechanism in protic solvents as studied through temperature-dependent lifetime measurements. J. Phys. Chem. A 2004, 108, 9939–9944. [Google Scholar]
- Olofsson, J.; Wilhelmsson, L.M.; Lincoln, P. Effects of methyl substitution on radiative and solvent quenching rate constants of [Ru(phen)2DPPZ] in polyol solvents and bound to DNA. J. Am. Chem. Soc. 2004, 126, 15458–15465. [Google Scholar] [CrossRef]
- Olofsson, J.; Önfelt, B.; Lincoln, P. Three-state light switch of [Ru(phen)2DPPZ]2+: Distinct excited-state species with two, one or no hydrogen bonds from solvent. J. Phys. Chem. A 2004, 108, 4391–4398. [Google Scholar] [CrossRef]
- McKinley, A.; Lincoln, P.; Tuite, E.M. Environmental effects on the photophysics of transition metal complexs with dipyrido[2,3-a:3',2'-c]phenazine (dppz) and related ligands. Coord. Chem. Rev. 2011, 255, 2676–2692. [Google Scholar] [CrossRef]
- Ling, L.-S.; He, Z.-K.; Song, G.-W.; Zeng, Y.E.; Wang, C.; Bai, C.-L.; Che, X.-D.; Shen, P. High sensitive determination of DNA by use of molecular “light switch” complex of Ru(phen)2(dppx)2+. Anal. Chim. Acta 2001, 436, 207–214. [Google Scholar] [CrossRef]
- Sun, Y.; Lutterman, D.A.; Turro, C. Role of electronic structure on DNA light-switch behavior of Ru(II) intercalators. Inorg. Chem. 2008, 47, 6427–6434. [Google Scholar] [CrossRef]
- Komatsuzaki, N.; Katoh, R.; Himeda, Y.; Sugihara, H.; Arakawa, H.; Kasuga, K. Structure and photochemical properties of ruthenium complexes having dimethyl-substituted DPPZ or TPPHZ as a ligand. J. Chem. Soc. Dalton Trans. 2000, 3053–3054. [Google Scholar]
- Lin, C.T.; Boettcher, W.; Chou, M.; Creutz, C.; Sutin, N. Mechanism of the quenching of the emission of substituted polypyridineruthenium(II) complexes by iron(III), chromium(III), and europium(III) ions. J. Am. Chem. Soc. 1976, 98, 6536–6544. [Google Scholar] [CrossRef]
- Mabrouk, P.A.; Wrighton, M.S. Resonance Raman spectroscopy of the lowest excited state of derivatives of tris(2,2'-bipyridine)ruthenium(II): Substituent effects on electron localization in mixed-ligand complexes. Inorg. Chem. 1986, 25, 526–531. [Google Scholar] [CrossRef]
- Sun, Y.; Collins, S.N.; Joyce, L.E.; Turro, C. Unusual photophysical properties of a ruthenium(II) complex related to [Ru(bpy)2(DPPZ)]2+. Inorg. Chem. 2010, 49, 4257–4262. [Google Scholar] [CrossRef]
- Kirsch-De Mesmaeker, A.; Jacquet, L.; Nasielski, J. Ruthenium(II) complexes of 1,4,5,8-tetraazaphenanthrene (TAP) and 2,2’-bipyridine (bpy). Ground- and excited-state basicities of Ru2+ (bpy)n(TAP)3-n (n = 0, 1, 2): Their luminescence quenching by organic buffers. Inorg. Chem. 1988, 27, 4451–4458. [Google Scholar] [CrossRef]
- Sun, H.; Hoffman, M.Z. Protonation of the excited states of ruthenium(II) complexes containing 2,2'-bipyridine, 2,2'-bipyrazine, and 2,2'-bipyrimidine ligands in aqueous solution. J. Phys. Chem. 1993, 97, 5014–5018. [Google Scholar] [CrossRef]
- Crutchley, R.J.; Kress, N.; Lever, A.B.P. Protonation equilibria in excited-state tris(bipyrazine)ruthenium(II). J. Am. Chem. Soc. 1983, 105, 1170–1178. [Google Scholar] [CrossRef]
- Herman, L.; Elias, B.; Pierard, F.; Moucheron, C.; Kirsch-De Mesmaeker, A. Effects of protonation on the spectroscopic properties of tetrapyridoacridine (TPAC) mono- and dinuclear Ru(II) complexes in their ground and 3MLCT excited states. J. Phys. Chem. A 2007, 111, 9756–9763. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, Y.; Turro, C. Ultrafast dynamics of the low-lying 3MLCT states of [Ru(bpy)2(dppp2]2+. J. Am. Chem. Soc. 2010, 132, 5594–5595. [Google Scholar] [CrossRef]
- Sun, Y.; Turro, C. Highly solvent dependent luminescence from [Ru(bpy)n(dppp2)3-n]2+ (n = 0–2). Inorg. Chem. 2010, 49, 5025–5032. [Google Scholar] [CrossRef]
- Coates, C.G.; Callaghan, P.; McGarvey, J.J.; Kelly, J.M.; Jacquet, L.; Kirsch-De Mesmaeker, A. Spectroscopic studies of structurally similar DNA-binding ruthenium(II) complexes containing the dipyridophenazine ligand. J. Mol. Struct. 2001, 598, 15–25. [Google Scholar] [CrossRef]
- Ortmans, I.; Elias, B.; Kelly, J.M.; Moucheron, C.; Kirsch-De Mesmaeker, A. [Ru(TAP)2(DPPZ)]2+: A DNA intercalating complex, which luminesces strongly in water and undergoes photo-induced proton-coupled electron transfer with guanosine-5'-monophosphate. J. Chem. Soc. Dalton Trans. 2004, 668–676. [Google Scholar]
- Liu, Y.; Hammitt, R.; Lutterman, D.A.; Joyce, L.E.; Thummel, R.P.; Turro, C. Ru(II) complexes of new tridentade ligands: Unexpected high yield of sensitized 1O2. Inorg. Chem. 2009, 48, 375–385. [Google Scholar] [CrossRef]
- Foxon, S.P.; Alamiry, M.A.H.; Walker, M.G.; Meijer, A.J.H.M.; Sazanovich, I.V.; Weinstein, J.A.; Thomas, J.A. Photophysical properties and singlet oxygen production by ruthenium(II) complexes of benzo[i]dipyrido[3,2-a:2';3'-c]phenazine: Spectroscopic and TD-DFT study. J. Phys. Chem. A 2009, 113, 12754–12762. [Google Scholar] [CrossRef]
- Sun, Y.; El Ojaimi, M.; Hammitt, R.; Thummel, R.P.; Turro, C. Effect of ligands with extended π-system on the photophysical properties of Ru (II) complexes. J. Phys. Chem. B. 2010, 114, 14664–14670. [Google Scholar] [CrossRef]
- Peña, B.; Leed, N.A.; Dunbar, K.R.; Turro, C. Excited state dynamics of two new Ru (II) cyclometallated dyes: Relation to cells for solar energy conversion and comparison to conventional systems. J. Phys. Chem. C 2012, 116, 22186–22195. [Google Scholar] [CrossRef]
- Kohne, B.; Praefcke, K. Eine neue und einfache Synthese des Dipyrazino[2,3-f:2',3'-h]-chinoxalin-Ringsystems. Liebigs Ann. Chem. 1985, 3, 522–528. [Google Scholar] [CrossRef]
- Rogers, D.Z. Improved synthesis of 1,4,5,8,9,12-hexaazatriphenylene. J. Org. Chem. 1986, 51, 3904–3905. [Google Scholar] [CrossRef]
- Nasielski-Hinkens, R.; Benedek-Vamos, M.; Maetens, D.; Nasielski, J. A new heterocyclic ligand for transition metals: 1,4,5,8,9,12-hexaazatriphenylene and its chromium carbonyl complexes. J. Organomet. Chem. 1981, 217, 179–182. [Google Scholar] [CrossRef]
- Bolger, J.; Gourdon, A.; Ishow, E.; Launay, J.-P. Stepwise syntheses of mono- and di-nuclear ruthenium tpphz complexes [(bpy)2Ru(tpphz)]2+ and [(bpy)2Ru(tpphz)Ru(bpy)2]4+ {tpphz = tetrapyrido[3,2-a:2',3'-c:3'',2''-h:2''',3'''-j]phenazine}. J. Chem. Soc. Chem. Commun. 1995, 1799–1800. [Google Scholar]
- Bolger, J.; Gourdon, A.; Ishow, E.; Launay, J.-P. Mononuclear and binuclear tetrapyrido[3,2-a:2',3'-c:3'',2''-h:2''',3'''-j]phenazine (tpphz) ruthenium and osmium complexes. Inorg. Chem. 1996, 35, 2937–2944. [Google Scholar] [CrossRef]
- Moucheron, C.; Kirsch-De Mesmaeker, A.; Choua, S. Photophysics of Ru(phen)2(PHEHAT)2+: A novel “light switch” for DNA and photo-oxidant for mononucleotides. Inorg. Chem. 1997, 36, 584–592. [Google Scholar] [CrossRef]
- Wärnmark, K.; Thomas, J.A.; Heyke, O.; Lehn, J.-M. Stereoisomerically controlled inorganic architectures: Synthesis of enantio-and diastereo-merically pure ruthenium-palladium molecular rods from enantiopure building blocks. Chem. Commun. 1996, 701–702. [Google Scholar]
- Majewski, M.B.; de Tacconi, N.R.; MacDonnell, F.M.; Wolf, M.O. Long-lived, directional photoinduced charge separation in RuII complexes bearing laminate polypyridyl ligands. Chem. Eur. J. 2013, 19, 8331–8341. [Google Scholar] [CrossRef]
- De Tacconi, N.R.; Lezna, R.O.; Chitakunye, R.; MacDonnell, F.M. Electroreduction of the ruthenium complex [(bpy)2Ru(tatpp)]Cl2 in water: Insights on the mechanism of multielectron reduction and protonation of the tatpp acceptor ligand as a function of pH. Inorg. Chem. 2008, 47, 8847–8858. [Google Scholar] [CrossRef]
- Kim, M.-J.; Konduri, R.; Ye, H.; MacDonnell, F.M.; Puntoriero, F.; Serroni, S.; Campagna, S.; Holder, T.; Kinsel, G.; Rajeshwar, K. Dinuclear ruthenium(II) polypyridyl complexes containing large, redox-active, aromatic bridging ligands: Synthesis, characterization, and intramolecular quenching of MLCT excited states. Inorg. Chem. 2002, 41, 2471–2476. [Google Scholar] [CrossRef]
- Wilson, T.; Williamson, M.P.; Thomas, J.A. Differentiating quadruplexes: Binding preferences of a luminescent dinuclear ruthenium(II) complex with four-stranded DNA structures. Org. Biomol. Chem. 2010, 8, 2617–2621. [Google Scholar] [CrossRef]
- Baggaler, E.; Gill, M.R.; Green, N.H.; Turton, D.; Sazanovich, I.V.; Botchway, S.W.; Smythe, C.; Haycock, J.W.; Weinstein, J.A.; Thomas, J.A. Dinuclear ruthenium(II) complexes as two-photon, time-resolved emission microscopy probes for cellular DNA. Angew. Chem. Int. Ed. 2014, 126, 3435–3439. [Google Scholar] [CrossRef]
- Gill, M.R.; Derrat, H.; Smythe, C.G.W.; Battaglia, G.; Thomas, J.A. Ruthenium(II) metallo-intercalators: DNA imaging and cytotoxicity. ChemBioChem 2011, 12, 877–880. [Google Scholar] [CrossRef]
- Knapp, R.; Schott, A.; Rehahn, M. A novel synthetic strategy toward soluble, well-defined ruthenium(II) coordination polymers. Macromolecules 1996, 29, 478–480. [Google Scholar] [CrossRef]
- Kelch, S.; Rehahn, M. High-molecular-weight ruthenium(II) coordination polymers: Synthesis and solution properties. Macromolecules 1997, 30, 6185–6193. [Google Scholar] [CrossRef]
- Kelch, S.; Rehahn, M. Soluble ruthenium(II) coordination polymers bearing bulky side groups. Macromolecules 1998, 31, 4102–4106. [Google Scholar] [CrossRef]
- Ishow, E.; Gourdon, A.; Launay, J.-P.; Lecante, P.; Verelst, M.; Chiorboli, C.; Scandola, F.; Bignozzi, C.-A. Tetranuclear tetrapyrido[3,2-a:2',3'-c:3'',2''-h:2''',3'''-j]phenazine ruthenium complex: Synthesis, wide-angle X-ray scattering, and photophysical studies. Inorg. Chem. 1998, 37, 3603–3609. [Google Scholar] [CrossRef]
- Campagna, S.; Serroni, S.; Bodige, S.; MacDonnell, F.M. Absorption spectra, photophysical properties, and redox behavior of stereochemically pure dendritic ruthenium(II) tetramers and related dinuclear and mononuclear complexes. Inorg. Chem. 1999, 38, 692–701. [Google Scholar] [CrossRef]
- Kim, M.-J.; MacDonnell, F.M.; Gimon-Kinsel, M.E.; Du Bois, T.; Asgharian, N.; Griener, J.C. Global chirality in rigid decametallic ruthenium dendrimers. Angew. Chem. Int. Ed. 2000, 39, 615–619. [Google Scholar] [CrossRef]
- Rau, S.; Schäfer, B.; Gleich, D.; Anders, E.; Rudolph, M.; Friedrich, M.; Görls, H.; Henry, W.; Vos, J.G. A supramolecular photocatalyst for the production of hydrogen and the selective hydrogenation of tolane. Angew. Chem. Int. Ed. 2006, 45, 6215–6218. [Google Scholar] [CrossRef]
- Tschierlei, S.; Karnahl, M.; Presselt, M.; Dietzek, B.; Guthmuller, J.; González, L.; Schimtt, M.; Rau, S.; Popp, J. Photochemical fate: The first step determines efficiency of H2 formation with a supramolecular photocatalyst. Angew. Chem. Int. Ed. 2010, 49, 3981–3984. [Google Scholar] [CrossRef]
- Barigelletti, F.; Juris, A.; Balzani, V.; Belser, P.; Von Zelewsky, A. Influence of the ligand structure on the electrochemical and spectroscopic properties of ruthenium(II)-polypyridine complexes. Inorg. Chem. 1987, 26, 4115–4119. [Google Scholar] [CrossRef]
- Richter, M.M.; Brewer, K.J. Synthesis and characterization of osmium(II) complexes incorporationg polypyridyl bridging ligands. Inorg. Chim. Acta 1991, 180, 125–131. [Google Scholar] [CrossRef]
- Karnahl, M.; Tschierlei, S.; Kuhnt, C.; Dietzek, B.; Schmitt, M.; Popp, J.; Schwalbe, M.; Krieck, S.; Görls, H.; Heinemann, F.W.; Rau, S. Synthesis and characterization of regioselective substituted tetrapyridophenazine ligands and their Ru(II) complexes. J. Chem. Soc. Dalton Trans. 2010, 39, 2359–2370. [Google Scholar] [CrossRef]
- Demeunynck, M.; Moucheron, C.; Kirsch-De Mesmaeker, A. Tetrapyrido[3,2-a:2',3'-c:3'',2''-h:2''',3'''-j]acridine (tpac): A new extended polycyclic bis-phenanthroline ligand. Tetrahedron Lett. 2002, 43, 261–264. [Google Scholar] [CrossRef]
- Elias, B.; Herman, L.; Moucheron, C.; Kirsch-De Mesmaeker, A. Dinuclear RuIIPHEHAT and –TPAC complexes: Effects of the second RuII center on their spectroelectrochemical properties. Inorg. Chem. 2007, 46, 4979–4988. [Google Scholar] [CrossRef]
- Rochester, C.H. Organic Chemistry, A Series of Monographs; Academic Press: New York, NY, USA, 1970; Volume 7. [Google Scholar]
- Kirsch-De Mesmaeker, A.; Jacquet, L.; Masschelein, A.; Vanhecke, F.; Heremans, K. Resonance Raman spectra and spectroelectrochemical properties of mono- and polymetallic ruthenium complexes with 1,4,5,8,9,12-hexaazatriphenylene. Inorg. Chem. 1989, 28, 2465–2470. [Google Scholar] [CrossRef]
- De Buyl, F.; Kirsch-De Mesmaeker, A.; Tossi, A.; Kelly, J.M. Medium dependence of the spectroscopic and photophysical properties of Ru(bpy)2(HAT)2+. The effect of solvent, pH and binding to polyelectrolytes. J. Photochem. Photobiol. A 1991, 60, 27–45. [Google Scholar] [CrossRef]
- Lecomte, J.-P.; Kirsch-De Mesmaeker, A.; Orellana, G. Photophysics of polyazaaromatic ruthenium(II) complexes interacting with DNA. J. Phys. Chem. 1994, 98, 5382–5388. [Google Scholar] [CrossRef]
- Jacquet, L.; Kirsch-De Mesmaeker, A. Spectroelectrochemical characteristics and photophysics of a series of Ru (II) complexes with 1,4,5,8,9,12-hexaazatriphenylene: Effects of polycomplexation. J. Chem. Soc. Faraday Trans. 1992, 88, 2471–2480. [Google Scholar] [CrossRef]
- Boisdenghien, A.; Moucheron, C.; Kirsch-De Mesmaeker, A. [Ru(phen)2(PHEHAT)]2+ and [Ru(phen)2(HATPHE)]2+: Two ruthenium(II) complexes with the same ligands but different photophysics and spectroelectrochemistry. Inorg. Chem. 2005, 44, 7678–7685. [Google Scholar] [CrossRef]
- Pourtois, G.; Beljonne, D.; Moucheron, C.; Schumm, S.; Kirsch-De Mesmaeker, A.; Lazzaroni, R.; Brédas, J.-L. Photophysical properties of ruthenium(II) polyazaaromatic compounds: A theoretical insight. J. Am. Chem. Soc. 2004, 126, 683–692. [Google Scholar] [CrossRef]
- Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Ru(II) polypyridine complexes: Photophysics, photochemistry, electrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988, 85–277. [Google Scholar]
- Chiorboli, C.; Bignozzi, C.A.; Scandola, F.; Ishow, E.; Gourdon, A.; Launay, J.-P. Photophysics of dinuclear Ru(II) and Os(II) complexes based on the tetrapyrido[3,2-a:2',3'-c:3'',2''-h:2'''-3'''-j]phenazine (tpphz) bridging ligand. Inorg. Chem. 1999, 38, 2402–2140. [Google Scholar] [CrossRef]
- Chiorboli, C.; Rodgers, M.A.; Scandola, F. Ultrafast processes in bimetallic dyads with extended aromatic bridges. Energy and electron transfer pathways in tetrapyridophenazine-bridged complexes. J. Am. Chem. Soc. 2003, 125, 483–491. [Google Scholar] [CrossRef]
- Ali, Md. M.; MacDonnell, F.M. Topospecific self-assembly of mixed-metal molecular hexagons with diameters of 5.5 nm using chiral control. J. Am. Chem. Soc. 2000, 122, 11527–11528. [Google Scholar] [CrossRef]
- Leveque, J.; Elias, B.; Moucheron, C.; Kirsch-De Mesmaeker, A. Dendritic tetranuclear Ru(II) complexes based on the nonsymmetrical PHEHAT bridging ligand and their building blocks: Synthesis, characterization, and electrochemical and photophysical properties. Inorg. Chem. 2005, 44, 393–400. [Google Scholar] [CrossRef]
- Kuciauska, D.; Liddell, P.A.; Lin, S.; Johnson, T.E.; Weghorn, S.J.; Lindsey, J.S.; Moore, A.L.; Moore, T.A.; Gust, D. An artificial photosynthetic antenna-reaction center complex. J. Am. Chem. Soc. 1999, 121, 8604–8614. [Google Scholar] [CrossRef]
- De Cola, L.; Belser, P. Photoinduced energy and electron transfer processes in rigidly bridged dinuclear Ru/Os complexes. Coord. Chem. Rev. 1998, 177, 301–346. [Google Scholar] [CrossRef]
- Schanze, K.S.; Walters, K.A. Organic and Inorganic Photochemistry; Ramamurthy, V., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1998; Volume 2, pp. 275–127. [Google Scholar]
- Chiorboli, C.; Fracasso, S.; Ravaglia, M.; Scandola, F.; Campagna, S.; Wouters, K.L.; Konduri, R.; MacDonnell, F.M. Primary photoinduced processes in bimetallic dyads with extended aromatic bridges. Tetraazatetrapyridopentacene complexes of ruthenium(II) and osmium(II). Inorg. Chem. 2005, 44, 8368–8378. [Google Scholar] [CrossRef]
- Marcaccio, M.; Paolucci, F.; Paradisi, C.; Roffia, S.; Fontanesi, C.; Yellowlees, L.J.; Serroni, S.; Campagna, S.; Denti, G.; Balzani, V. Electrochemistry of multicomponent systems. Redox series comprising up to 26 reversible reduction processes in polynuclear ruthenium(II) bipyridine-type complexes. J. Am. Chem. Soc. 1999, 121, 10081–10091. [Google Scholar] [CrossRef]
- Denti, G.; Campagna, S.; Serroni, S.; Ciano, M.; Balzani, V. Decanuclear homo- and heterometallic polypyridine complexes: Syntheses, absorption spectra, luminescence, electrochemical oxidation, and intercomponent energy transfer. J. Am. Chem. Soc. 1992, 114, 2944–2950. [Google Scholar] [CrossRef]
- Denti, G.; Serroni, S. Hexanuclear polypyridine complexes containing different metals, bridging ligands and/or terminal ligands. Absorption spectra, electrochemical oxidation, luminescence properties and intercomponent energy transfer. Inorg. Chim. Acta 1992, 507–512. [Google Scholar] [CrossRef]
- Denti, G.; Serroni, S.; Campagna, S.; Ricevuto, V.; Balzani, V. Made-to-order control of the direction of electronic energy transfer in tetranuclear luminescent metal complexes. Coord. Chem. Rev. 1991, 111, 227–236. [Google Scholar] [CrossRef]
- Venturi, M.; Serroni, S.; Juris, A.; Denti, G.; Campagna, S.; Balzani, V. Designing dendrimers based on transition-metal complexes. Light-harvesting properties and predetermined redox patterns. Acc. Chem. Res. 1998, 31, 26–34. [Google Scholar] [CrossRef]
- Campagna, S.; Denti, G.; Serroni, S.; Juris, A.; Venturi, M.; Ricevuto, V.; Balzani, V. Dendrimers of nanometer size based on metal complexes: Luminescent and redox-active polynuclear metal complexes containing up to twenty-two metal centers. Chem. Eur. J. 1995, 1, 211–221. [Google Scholar] [CrossRef]
- Moucheron, C.; Kirsch-De Mesmaeker, A.; Dupont-Gervais, A.; Leize, E.; Van Dorsselaer, A. Synthesis and characterization by electrospray mass spectrometry of a novel dendritic heptanuclear complex of ruthenium(II). J. Am. Chem. Soc. 1996, 118, 12834–12835. [Google Scholar] [CrossRef]
- Latterini, L.; Pourtois, G.; Moucheron, C.; Lazzaroni, R.; Brédas, J.-L.; Kirsch-De Mesmaeker, A.; de Schryver, F.C. STM imaging of a heptanuclear ruthenium(II) dendrimer, mono-add layer on graphite. Chem. Eur. J. 2000, 6, 1331–1336. [Google Scholar] [CrossRef]
- Latterini, L.; Schweitzer, G.; De Schryver, F.C.; Moucheron, C.; Kirsch-De Mesmaeker, A. Femtosecond transient dynamics of a heptametallic HAT-ruthenium(II) complex. A photophysical study. Chem. Phys. Lett. 1997, 281, 267–271. [Google Scholar] [CrossRef]
- Leveque, J.; Moucheron, C.; Kirsch-De Mesmaeker, A.; Loiseau, F.; Serroni, S.; Puntoriero, F.; Campagna, S.; Nierengarten, H.; van Dorsselaer, A. A mixed-bridging ligand nonanuclear Ru(II) dendrimer containing a tris-chelating core. Synthesis and redox properties. Chem. Commun. 2004, 878–879. [Google Scholar]
- Kimura, M.; Sugihara, Y.; Muto, T.; Hanabusa, K.; Shirai, H.; Kobayashi, N. Dendritic metallophthalocyanines – synthesis, electrochemical properties, and catalytic activities. Chem. Eur. J. 1999, 5, 3495–3500. [Google Scholar] [CrossRef]
- Pollak, K.W.; Leon, J.W.; Fréchet, J.M.J.; Maskus, M.; Abruña, H.D. Effects of dendrimer generation on site isolation of core moieties: Electrochemical and fluorescence quenching studies with metalloporphyrin core dendrimers. Chem. Mater. 1998, 10, 30–38. [Google Scholar] [CrossRef]
- Gorman, C.B.; Smith, J.C.; Hager, M.W.; Parkhurst, B.L.; Sierzputowska-Gracz, H.; Haney, C.A. Molecular structure-property relationships for electron-transfer rate attenuation in redox-active core dendrimers. J. Am. Chem. Soc. 1999, 121, 9958–9966. [Google Scholar] [CrossRef]
- Ceroni, P.; Paolucci, F.; Paradisi, C.; Juris, A.; Roffia, S.; Serroni, S.; Campagna, S.; Bard, A.J. Dinuclear and dendritic polynuclear ruthenium(II) and osmium(II) polypyridine complexes: Electrochemistry at very positive potentials in liquid SO2. J. Am. Chem. Soc. 1998, 120, 5480–5487. [Google Scholar] [CrossRef]
- Sprintschnik, G.; Sprintschnik, H.W.; Kirsch, P.P.; Whitten, D.G. Photochemical cleavage of water: A system for solar energy conversion using monolayer-bound transition metal complexes. J. Am. Chem. Soc. 1976, 98, 2337–2338. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Grätzel, M. Cyclic cleavage of water into H2 and O2 by visible light with coupled redox catalysts. Angew. Chem. Int. Ed. 1979, 18, 701–702. [Google Scholar] [CrossRef]
- Lehn, J.-M.; Sauvage, J.-P. Chemical storage of light energy-catalytic generation of hydrogen by visible-light or sunlight-irradiation of neutral aqueous-solutions. Nouv. J. Chim. 1977, 1, 449–451. [Google Scholar]
- Gilbert, J.A.; Eggleston, D.S.; Murphy, W.R.; Geselowitz, D.A.; Gersten, S.W.; Hodgson, D.J.; Meyer, T.J. Structure and redox properties of the water-oxidation catalyst [(bpy)2(OH2)RuORu(OH2)(bpy)2]2+. J. Am. Chem. Soc. 1985, 107, 3855–3864. [Google Scholar] [CrossRef]
- Gersten, S.W.; Samuels, G.J.; Meyer, T.J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982, 104, 4029–4030. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Rotzinger, F.P.; Comte, P.; Grätzel, M. Spontaneous oxidation of water to oxygen by the mixed-valence µ-oxo ruthenium dimer L2(H2O)RuIII-O-RuIV(OH)L2 (L = 2,2’-bipyridyl-5,5’-dicarboxylic acid). J. Chem. Soc. Chem. Commun. 1988, 872–874. [Google Scholar]
- Nallas, G.N.A.; Jones, S.W.; Brewer, K.J. bipyrimidine-bridged mixed-metal trimetallic complexes of ruthenium(II) with rhodium(III) or iridium(III), {[(bpy)2Ru(bpm)]2MCl2}5+. Inorg. Chem. 1996, 35, 6974–6980. [Google Scholar] [CrossRef]
- Molnar, S.M.; Nallas, G.; Bridgewater, J.S.; Brewer, K.J. Photoinitiated electron collection in a mixed-metal trimetallic complex of the form {[(bpy)2Ru(dpb)}2IrCl2}(PF6)5 (bpy = 2,2’-bipyridine and dpb = 2,3-bis(2-pyridyl)benzoquinoxaline). J. Am. Chem. Soc. 1994, 116, 5206–5210. [Google Scholar] [CrossRef]
- Heyduk, A.F.; Nocera, D.G. Hydrogen produced from hydrohalic acid solutions by a two-electron mixed-valence photocatalyst. Science 2001, 293, 1639–1641. [Google Scholar] [CrossRef]
- Kanan, M.W.; Nocera, D.G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef]
- Dincā, M.; Surendranath, Y.; Nocera, D.G. A Nickel-Borate oxygen evolving catalyst that functions under benign conditions. Proc. Natl. Acad. Sci. USA 2010, 107, 10337–10341. [Google Scholar] [CrossRef]
- Bediako, D.K.; Surendranath, Y.; Nocera, D.G. Mechanistic studies of the oxygen evolution reaction mediated by a Nickel-Borate thin film electrocatalyst. J. Am. Chem. Soc. 2013, 135, 3662–3674. [Google Scholar] [CrossRef]
- Nocera, D.G. The artificial leaf. Acc. Chem. Res. 2012, 45, 767–776. [Google Scholar] [CrossRef]
- Du, P.; Eisenberg, R. Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: Recent progress and future challenges. Energy Environ. Sci. 2012, 5, 6012–6021. [Google Scholar] [CrossRef]
- La Ganga, G.; Puntoriero, F.; Campagna, S.; Bazzan, I.; Berardi, S.; Bonchio, M.; Sartorel, A.; Natali, M.; Scandola, F. Light-driven water oxidation with a molecular tetra-cobalt(III) cubane cluster. Faraday Discuss. 2012, 155, 177–190. [Google Scholar] [CrossRef]
- McCool, N.S.; Robinson, D.M.; Sheats, J.E.; Dismukes, G.C. A Co4O4 « cubane » water oxidation catalyst inspired by photosynthesis. J. Am. Chem. Soc. 2011, 133, 11446–11449. [Google Scholar] [CrossRef]
- Berardi, S.; la Ganga, G.; Natali, M.; Bazzan, I.; Puntoriero, F.; Sartorel, A.; Scandola, F.; Campagna, S.; Bonchio, M. Photocatalytic water oxidation: Tuning light-induced electron transfer by molecular Co4O4 cores. J. Am. Chem. Soc. 2012, 134, 11104–11107. [Google Scholar] [CrossRef]
- Gao, Y.; Ding, X.; Lio, J.; Wang, L.; Lu, Z.; Li, L.; Sun, L. Visible light driven water splitting in a molecular device with unprecedentedly high photocurrent density. J. Am. Chem. Soc. 2013, 135, 4219–4222. [Google Scholar] [CrossRef]
- Zhang, B.; Li, F.; Yu, F.; Wang, X.; Zhou, X.; Li, H.; Jiang, Y.; Sun., L. Electrochemical and photoelectrochemical water oxidation by supported cobalt-oxo cubanes. ACS Catal. 2014, 4, 804–809. [Google Scholar] [CrossRef]
- Duan, L.; Xu, Y.; Zhang, P.; Wang, M.; Sun, L. Visible light-driven water oxidation by a molecular ruthenium catalyst in homogeneous system. Inorg. Chem. 2010, 49, 209–215. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Tsai, M.-K.; Muckerman, J.T.; Meyer, T.J. Mechanism of water oxidation by single-site ruthenium complex catalysts. J. Am. Chem. Soc. 2010, 132, 1545–1557. [Google Scholar] [CrossRef]
- Staehle, R.; Ton, L.; Wang, L.; Duan, L.; Fischer, A.; Ahlquist, M.S.G.; Sun, L.; Rau, S. Water oxidation catalyzed by mononuclear ruthenium complexes with a 2,2'-bipyridine-6,6'-dicarboxylate (bda) ligand: How ligand environment influences the catalytic behavior. Inorg. Chem. 2014, 53, 1307–1319. [Google Scholar] [CrossRef]
- Duan, L.; Fischer, A.; Xu, Y.; Sun., L. Isolated seven-coordinate Ru(IV) dimer complex with [HOHOH]- bridging ligand as an intermediate for catalytic water oxidation. J. Am. Chem. Soc. 2009, 131, 10397–10399. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, F.; Zhang, B.; Li, X.; Wang, X.; Huang, F.; Sun, L. Promoting the activity of catalysts for the oxidation of water with bridged dinuclear ruthenium complexes. Angew. Chem. Int. Ed. 2013, 125, 3482–3485. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Jurss, J.W.; Templeton, J.L.; Meyer, T.J. Mediator-assisted water oxidation by ruthenium “blue dimer” cis,cis-[(bpy)2(H2O)RuORu(OH2)(bpy)2]4+. Proc. Natl. Acad. Sci. USA 2008, 105, 17632–17635. [Google Scholar] [CrossRef]
- Liu, F.; Concepcion, J.J.; Jurss, J.W.; Cardolaccia, T.; Templeton, J.L.; Meyer, T.J. Mechanisms of water oxidation from blue dimer to photosystem II. Inorg. Chem. 2008, 47, 1727–1752. [Google Scholar] [CrossRef]
- Ozawa, H.; Sakai, K. Homogeneous catalysis of platinum(II) complexes in photochemical hydrogen production from water. Coord. Chem. Rev. 2007, 251, 2753–2766. [Google Scholar] [CrossRef]
- Ozawa, H.; Haga, M.-A.; Sakai, K. A photo-hydrogen-evolving molecular device driving visible-light-induced EDTA-reduction of water into molecular hydrogen. J. Am. Chem. Soc. 2006, 128, 4926–4927. [Google Scholar] [CrossRef]
- Duan, L.; Bozoglian, F.; Mandal, S.; Steart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef]
- Stoll., T.; Genneri, M.; Fortage, J.; Castillo, C.E.; Rebarz, M.; Sliwa, M.; Poizat, O.; Odobel, F.; Deronzier, A.; Collomb, M.-N. An efficient RuII-RhIII-RuII polypyridyl photocatalyst for visible-light-driven hydrogen production in aqueous solution. Angew. Chem. Int. Ed. 2014, 53, 1654–1658. [Google Scholar] [CrossRef]
- Tschierlei, S.; Presselt, M.; Kuhnt, C.; Yartsev, A.; Pascher, T.; Sundström, V.; Karnahl, M.; Schwalbe, M.; Schäfer, B.; Rau, S.; et al. Photophysics of an intramolecular hydrogen-evolving Ru-Pd photocatalyst. Chem. Eur. J. 2009, 15, 7678–7688. [Google Scholar] [CrossRef]
- Inagaki, A.; Akita, M. Visible-light promoted bimetallic catalysis. Coord. Chem. Rev. 2010, 254, 1220–1239. [Google Scholar] [CrossRef]
- Karnahl, M.; Kuhnt, C.; Ma, F.; Yartsev, A.; Schmitt, M.; Dietzek, B.; Rau, S.; Popp, J. Tuning of photocatalytic hydrogen production and photoinduced intramolecular electron transfer rates by regioselective bridging ligand substitution. ChemPhysChem 2011, 12, 2101–2109. [Google Scholar] [CrossRef]
- Karnahl, M.; Kuhnt, C.; Heinemann, F.W.; Schmitt, M.; Rau, S.; Popp, J.; Dietzek, B. Synthesis and photophysics of a novel photocatalyst for hydrogen production based on a tetrapyridoacridine bridging ligand. Chem. Phys. 2012, 393, 65–73. [Google Scholar] [CrossRef]
- Wärnmark, K.; Heyke, O.; Thomas, J.A.; Lehn, J.-M. Stereoisomerically controlled inorganic architectures: Synthesis of extended enantio-and diastereo-merically pure tris-ruthenium disks from enantiopure building blocks. Chem. Commun. 1996, 2603–2604. [Google Scholar]
- Pilz, T.D.; Rockstroh, N.; Rau, S. Synthesis and characterization of heterooligonuclear ruthenium complexes with tri(phenanthrolino)hexaazatriphenylene ligands. J. Coord. Chem. 2010, 63, 2727–2742. [Google Scholar] [CrossRef]
- Ishow, E.; Gourdon, A.; Launay, J.-P. Observation of supramolecular π-π dimerization of a dinuclear ruthenium complex by 1H NMR and ESMS. Chem. Commun. 1998, 1909–1910. [Google Scholar] [CrossRef]
- Ishow, E.; Gourdon, A.; Launay, J.-P.; Chiorboli, C.; Scandola, F. Synthesis, mass spectrometry, and spectroscopic properties of a dinuclear ruthenium complex comprising a 20 Å long fully aromatic bridging ligand. Inorg. Chem. 1999, 38, 1504–1510. [Google Scholar] [CrossRef]
- Wouters, K.L.; de Tacconi, N.R.; Konduri, R.; Lezna, R.O.; MacDonnell, F.M. Driving multi-electron reactions with photons: Dinuclear ruthenium complexes capable of stepwise and concerted multi-electron reduction. Photosynth. Res. 2006, 87, 41–55. [Google Scholar] [CrossRef]
- Konduri, R.; de Tacconi, N.R.; Rajeshwar, K.; MacDonnell, F.M. Multielectron photoreduction of a bridged ruthenium dimer, [(phen)2Ru(tatpp)Ru(phen)2][PF6]4: Aqueous reactivity and chemical and spectroelectrochemical identification of the photoproducts. J. Am. Chem. Soc. 2004, 126, 11621–11629. [Google Scholar] [CrossRef]
- De Tacconi, N.R.; Lezna, R.O.; Konduri, R.; Ongeri, F.; Rajeshwar, K.; MacDonnell, F.M. Influence of pH on the photochemical and electrochemical reduction on the dinuclear ruthenium complex, [(phen)2Ru(tatpp)Ru(phen)2]Cl4, in water: Proton-coupled sequential and concerted multi-electron reduction. Chem. Eur. J. 2005, 11, 4327–4339. [Google Scholar] [CrossRef]
- MacDonnell, F.M.; Ali, M.D.M.; Kim, M.-J. Robust chiral nanostructures: Global chirality in supramolecules constructed from enantiopure ruthenium(II) trisdiimine building blocks. Comments Inorg. Chem. 2000, 22, 203–225. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Troian-Gautier, L.; Moucheron, C. RutheniumII Complexes bearing Fused Polycyclic Ligands: From Fundamental Aspects to Potential Applications. Molecules 2014, 19, 5028-5087. https://doi.org/10.3390/molecules19045028
Troian-Gautier L, Moucheron C. RutheniumII Complexes bearing Fused Polycyclic Ligands: From Fundamental Aspects to Potential Applications. Molecules. 2014; 19(4):5028-5087. https://doi.org/10.3390/molecules19045028
Chicago/Turabian StyleTroian-Gautier, Ludovic, and Cécile Moucheron. 2014. "RutheniumII Complexes bearing Fused Polycyclic Ligands: From Fundamental Aspects to Potential Applications" Molecules 19, no. 4: 5028-5087. https://doi.org/10.3390/molecules19045028
APA StyleTroian-Gautier, L., & Moucheron, C. (2014). RutheniumII Complexes bearing Fused Polycyclic Ligands: From Fundamental Aspects to Potential Applications. Molecules, 19(4), 5028-5087. https://doi.org/10.3390/molecules19045028