Anti-Inflammatory Triterpenoids from the Stems of Microtropis Fokienensis

 ,
,  and
and 
Abstract
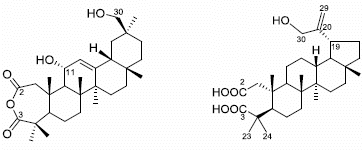
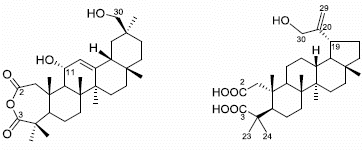
:
1. Introduction
2. Results and Discussion
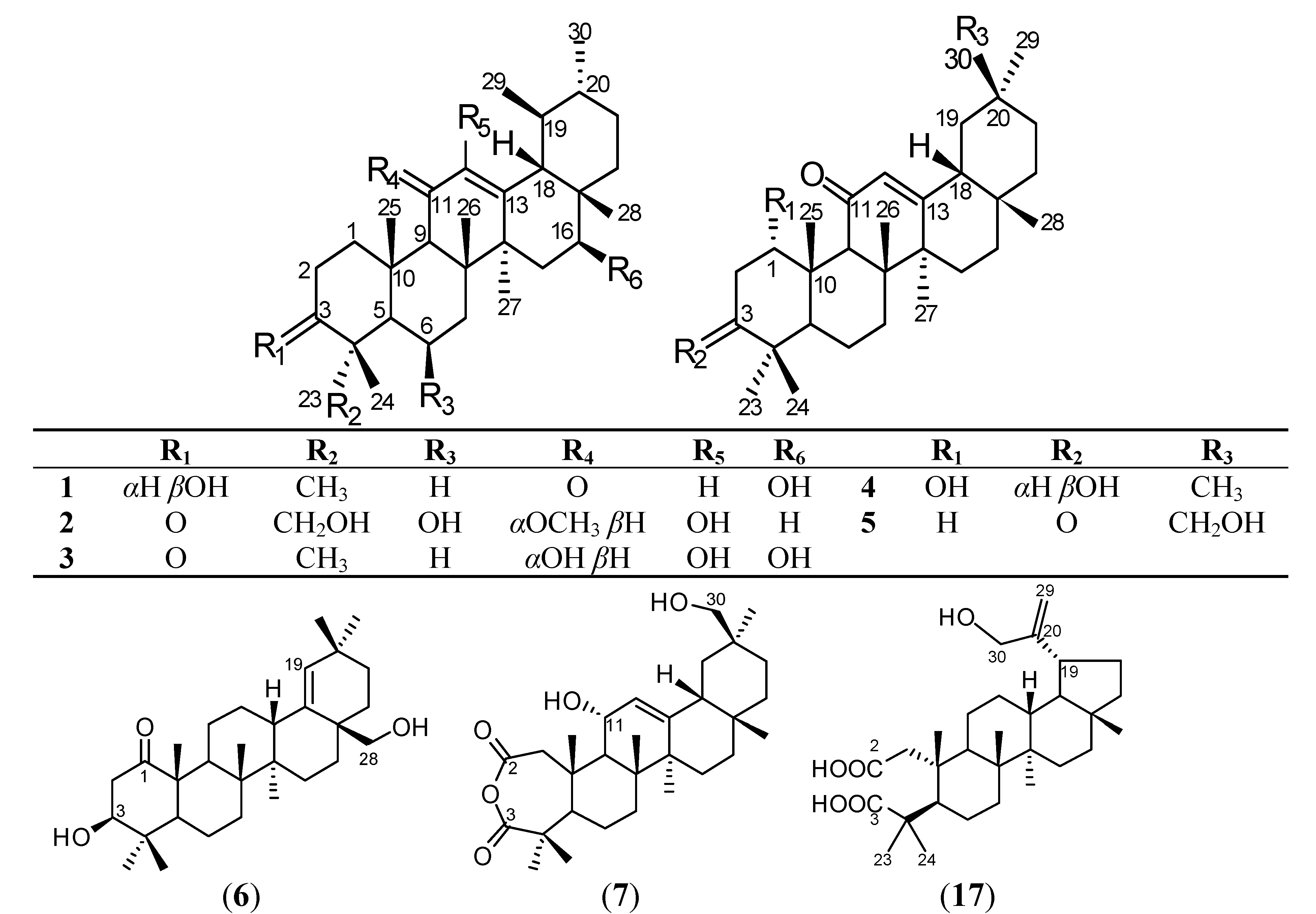

{kind=link}
{kind=link}
{kind=link}
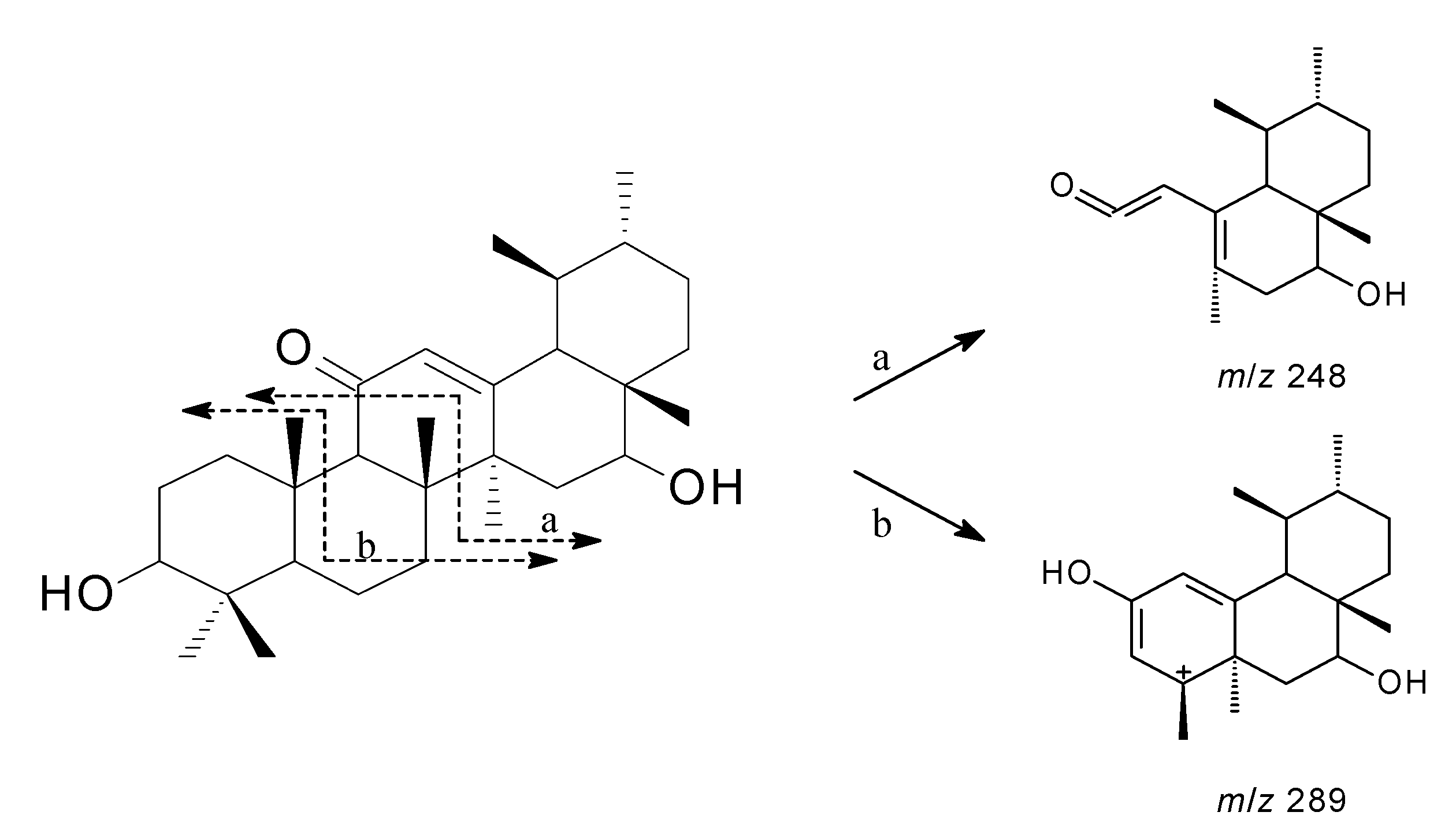{kind=link}
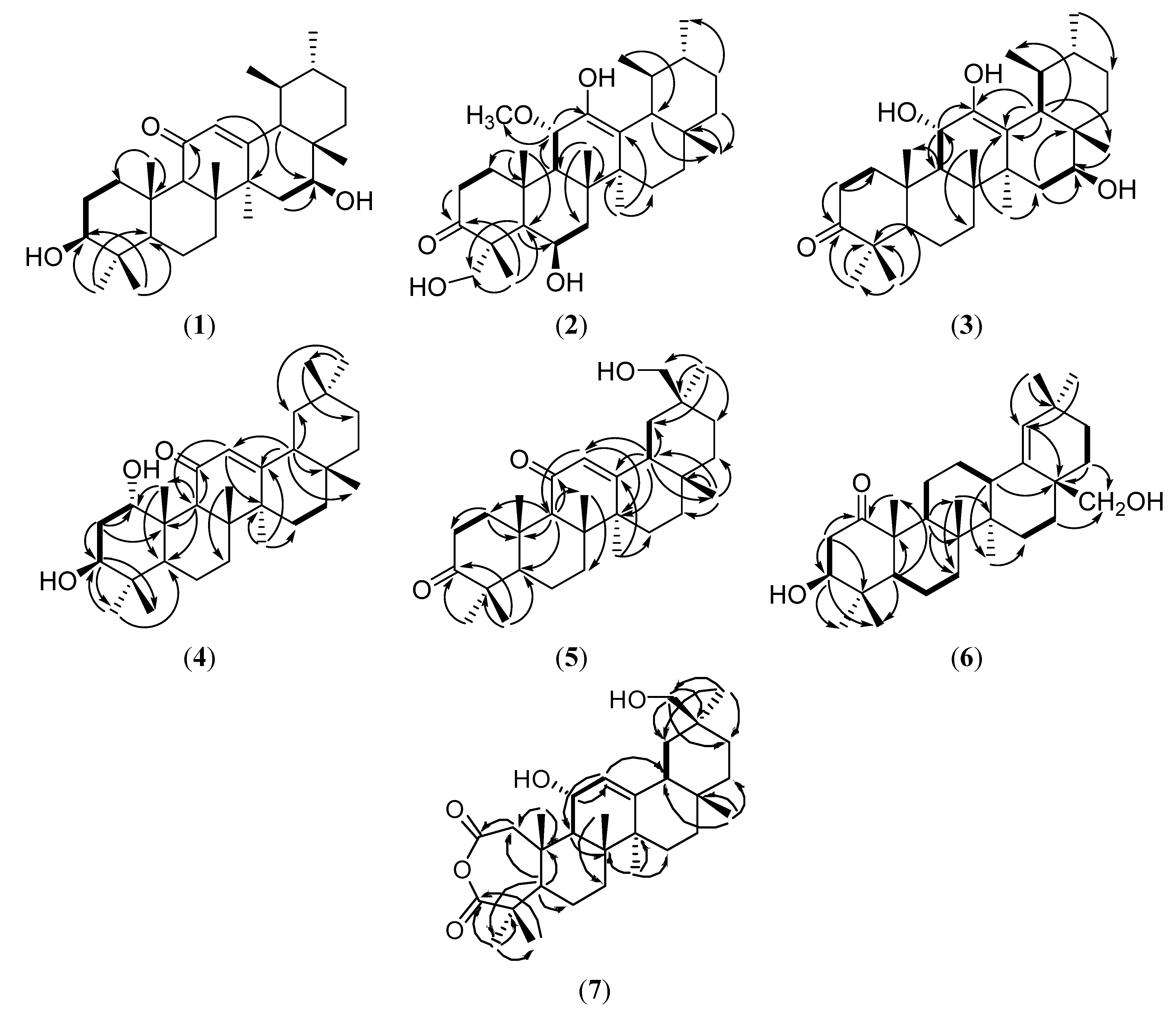{kind=link}
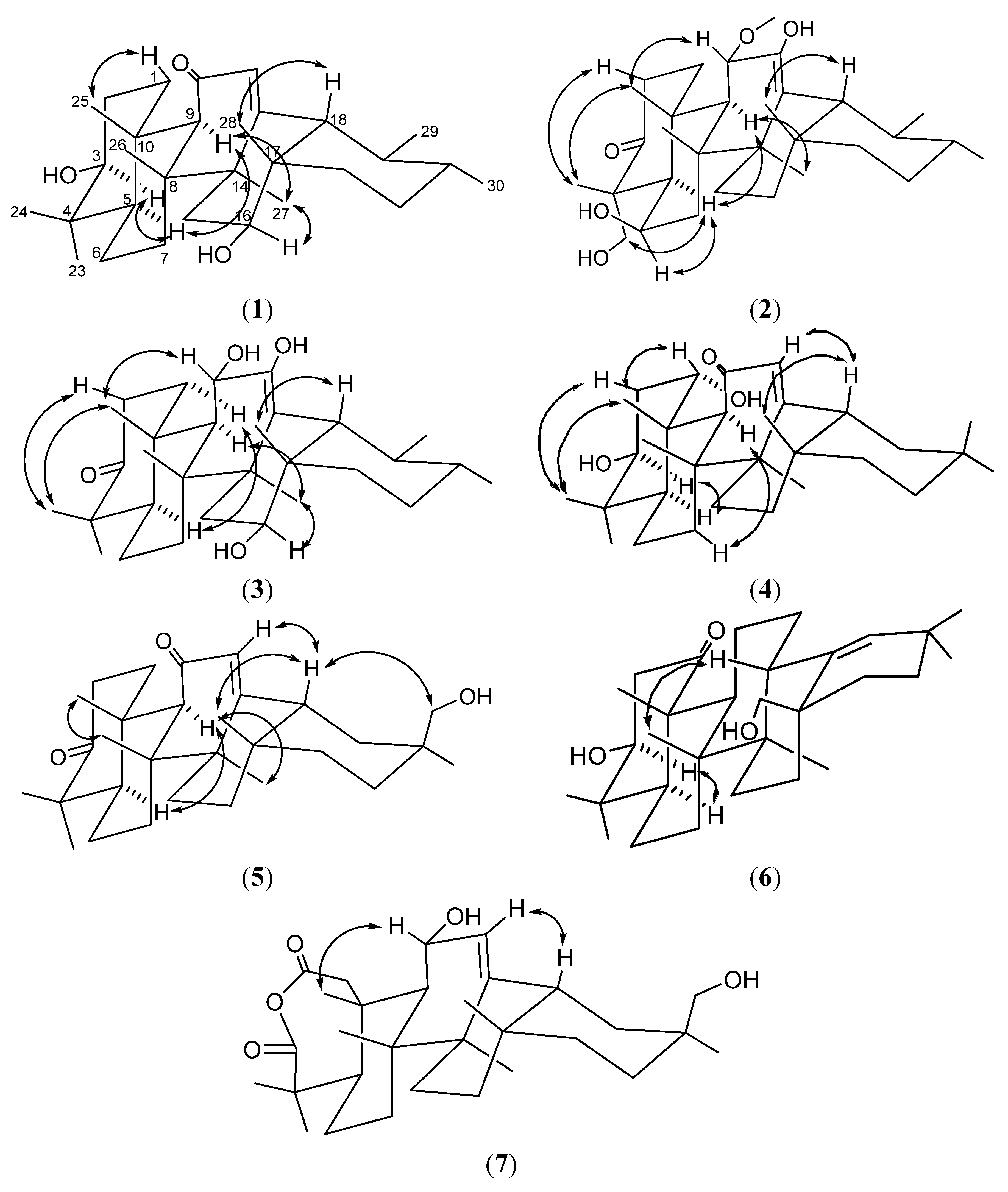
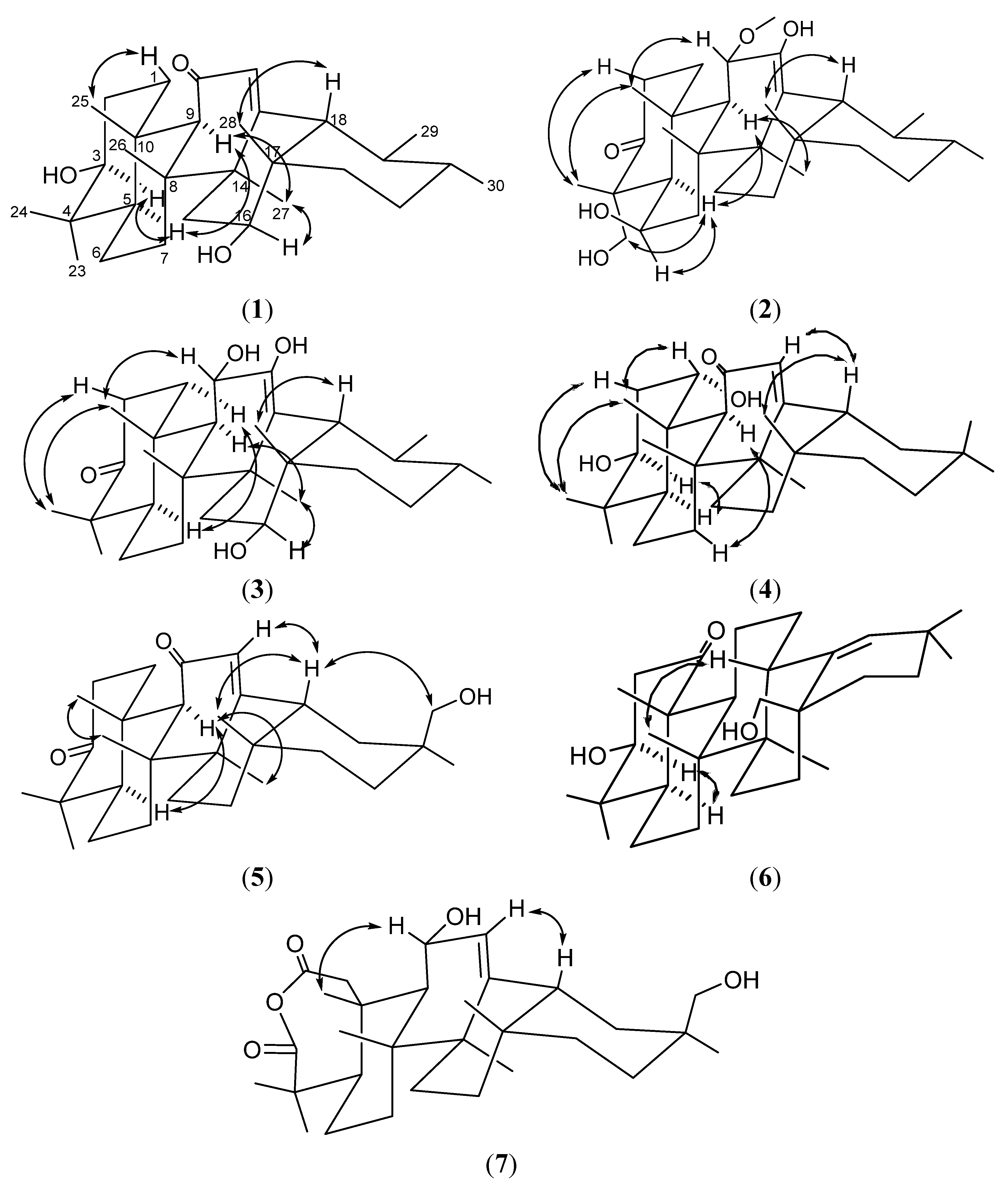{kind=link}
| No. | 1 | 2 | 3 a | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | 1.20 (m, α) 3.14 (dt, 13.6, 3.6, β) | 1.80 (m, α) 2.84 (m, β) | 2.04 (m, α) 3.04 (m, β) | 5.37 (s) | 1.55 (m, α) 3.20 (m, β) | 3.14 (d, 17.6) 3.33 (d, 17.6) | |
| 2 | 1.88 (m, α) 2.04 (m, β) | 2.52 (m, α) 2.91 (m, β) | 2.55 (m, α) 2.62 (m, β) | 2.33 (dt, 13.6, 3.6, α) 2.48 (dd, 13.6, 12.0, β) | 2.46 (m, α) 2.69 (m, β) | 2.74 (dd, 11.6, 4.8, α) 3.50 (dd, 12.0, 11.6, β) | |
| 3 | 3.50 (m, α) | 4.43 (dd, 12.0, 3.6, α) | 3.75 (dd, 12.0, 4.8, α) | ||||
| 5 | 0.87 (m) | 2.58 (brs) | 1.59 (m) | 1.71 | 1.41 (m) | 0.98 (dd, 11.2, 2.4) | 1.96 (m) |
| 6 | 1.56 (2H, m) | 4.94 (brs, α) | 1.44 (2H, m) | 1.61 (m) 1.72 (m) | 1.43 (2H) | 1.50 (m) 1.58 (m) | 1.58 (m) 1.67 (m) |
| 7 | 1.71 (m, α) 1.40 (m, β) | 1.98 (m, α) 1.86 (m, β) | 1.66 (m, α) 1.46 (m, β) | 1.74 (m, α) 1.35 (m, β) | 1.30 (m) 1.60 (m) | 1.26 (dd, 12.0, 3.2) 1.40 (m) | 1.33 (m) 1.73 (m) |
| 9 | 2.55 (s) | 2.36 (d, 10.0) | 2.19 (d, 10.0) | 3.87 (s) | 2.55 (s) | 2.27 (dd, 12.0, 2.0) | 2.19 (d, 11.2) |
| 11 | 4.82 (d, 10.0, β) | 4.58 (d, 9.5, β) | 1.22 (m) 1.93 (m) | 4.91 (dd, 10.8, 2.4) | |||
| 12 | 5.85 (s) | 5.76 (s) | 5.83 (s) | 1.43 (m)1.51 (m) | 5.49 (d, 2.0) | ||
| 13 | 2.40 (m) | ||||||
| 15 | 1.79 (m, α) 2.11 (m, β) | 1.38 (2H, m) | 1.71 (m, α) 2.13 (m, β) | 1.07 1.75 | 1.09 (m) 1.72 (m) | 1.13 (m) 1.89 (m) | 0.95 (m) 1.74 (m) |
| 16 | 4.59 (ddd, 11.0, 5.2, 4.4) | 1.43 (2H, m) | 4.63 (m, α) | 1.97 (td, 13.6, 4.4, α) 0.83 (m, β) | 2.10 (td, 13.6, 4.4, α) 0.92 (dt, 13.6, 2.0, β) | 1.32 (m) 2.21 (m) | 0.89 (m) 2.04 (td, 13.2, 4.0) |
| 18 | 1.79 (d, 11.2) | 2.90 (m) | 3.10 (d, 11.5) | 2.11 (dd, 13.6, 4.0) | 2.30 (dd, 13.4, 4.4) | 2.21 (dd, 10.8, 4.4) | |
| 19 | 1.55 (m) | 1.81 (m) | 1.68 (m) | 0.79 (m) 1.53 (m) | 1.52 (m) 1.69 (m) | 5.11 (s) | 1.60 (m) 1.75 (m) |
| 20 | 0.91 (m) | 2.52 (m) | 1.80 (m) | ||||
| 21 | 1.46 (m, 2H) | 1.38 (2H, m) | 1.51 (m) 1.59 (m) | 1.06 (m) 1.34 (m) | 1.40 (m) 1.69 (m) | 1.38 (m) 1.70 (m) | 1.32 (m) 1.70 (m) |
| 22 | 1.13 (d, 3.2, α) 2.57 (dt, 13.6, 3.2, β) | 0.80 (m) 1.96 (m) | 1.29 (m) 2.68 (brd, 13.0) | 1.22 (m) 1.43 (m) | 1.28 (m) 1.56 (m) | 1.39 (m) 2.36 (m) | 1.32 (m) 1.70 (m) |
| 23 | 1.26 (s) | 3.93 (d, 10.4) 4.47 (d, 10.4) | 1.20 (s) | 1.38 (s) | 1.08 (s) | 1.18 (s) | 1.40 (s) |
| 24 | 1.09 (s) | 1.70 (s) | 1.13 (s) | 1.18 (s) | 1.18 (s) | 1.22 (s) | 1.47 (s) |
| 25 | 1.38 (s) | 1.92 (s) | 1.27 (s) | 1.46 (s) | 1.35 (s) | 1.29 (s) | 1.39 (s) |
| 26 | 1.27 (s) | 1.84 (s) | 1.30 (s) | 1.22 (s) | 1.12 (s) | 1.04 (s) | 1.01 (s) |
| 27 | 1.47 (s) | 1.31 (s) | 1.54 (s) | 1.36 (s) | 1.37 (s) | 0.94 (s) | 1.27 (s) |
| 28 | 1.06 (s) | 0.98 (s) | 1.26 (s) | 0.85 (s) | 0.85 (s) | 3.81 (d, 10.4) 4.02 (d, 10.4) | 0.86 (s) |
| 29 | 0.80 (d, 6.4) | 1.26 (d, 6.4) | 1.18 (d, 6.5) | 0.81 (s) | 1.17 (s) | 1.06 (s) | 1.16 (s) |
| 30 | 0.91 (d, 6.0) | 0.97 (d, 7.2) | 0.95 (d, 6.5) | 0.83 (s) | 3.74 (d, 10.4) 3.80 (d, 10.4) | 1.07 (s) | 3.74 (d, 10.4) 3.82 (d, 10.4) |
| No. | 1 | 2 | 3 a | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | 39.8 | 40.5 | 42.5 | 72.3 | 40.0 | 212.9 | 49.6 |
| 2 | 28.1 | 36.1 | 35.1 | 35.5 | 34.4 | 45.4 | 170.6 |
| 3 | 77.9 | 216.0 | 217.2 | 72.3 | 215.9 | 79.0 | 182.0 |
| 4 | 39.8 | 54.7 | 48.3 | 40.1 | 47.8 | 40.1 | 45.4 |
| 5 | 55.3 | 49.2 | 56.1 | 47.8 | 55.1 | 54.9 | 55.2 |
| 6 | 18.0 | 67.8 | 20.5 | 17.8 | 19.0 | 18.1 | 20.7 |
| 7 | 33.2 | 41.6 | 34.4 | 32.8 | 32.1 | 34.4 | 33.4 |
| 8 | 45.7 | 42.6 | 43.5 | 45.2 | 45.3 | 40.7 | 41.1 |
| 9 | 61.3 | 46.6 | 53.8 | 53.8 | 61.2 | 42.8 | 45.9 |
| 10 | 37.4 | 37.8 | 38.5 | 42.1 | 37.0 | 53.5 | 38.5 |
| 11 | 199.3 | 77.3 | 70.2 | 200.9 | 198.9 | 24.4 | 74.5 |
| 12 | 130.7 | 145.0 | 148.4 | 128.6 | 128.4 | 26.7 | 121.4 |
| 13 | 163.6 | 116.5 | 113.2 | 169.7 | 170.6 | 39.6 | 149.8 |
| 14 | 45.8 | 41.4 | 44.2 | 44.0 | 43.7 | 43.7 | 43.1 |
| 15 | 37.1 | 27.6 | 37.8 | 26.7 | 26.7 | 27.8 | 25.8 |
| 16 | 64.7 | 28.0 | 65.9 | 26.5 | 26.9 | 31.3 | 27.5 |
| 17 | 39.2 | 33.7 | 39.5 | 32.5 | 32.5 | 32.5 | 32.9 |
| 18 | 60.7 | 47.5 | 49.9 | 47.6 | 47.3 | 140.7 | 47.2 |
| 19 | 39.1 | 41.3 | 41.9 | 45.2 | 40.8 | 132.6 | 41.6 |
| 20 | 39.4 | 40.0 | 40.7 | 31.0 | 36.0 | 32.5 | 36.1 |
| 21 | 30.8 | 31.6 | 31.8 | 34.5 | 29.9 | 33.4 | 30.0 |
| 22 | 35.4 | 42.2 | 36.8 | 36.7 | 36.4 | 31.1 | 36.6 |
| 23 | 28.7 | 66.8 | 27.5 | 28.8 | 21.5 | 28.6 | 28.2 |
| 24 | 16.6 | 20.5 | 22.1 | 16.2 | 26.5 | 16.6 | 23.3 |
| 25 | 17.0 | 17.6 | 16.9 | 18.0 | 15.9 | 16.1 | 17.7 |
| 26 | 18.7 | 20.3 | 18.9 | 19.1 | 18.5 | 17.0 | 17.1 |
| 27 | 21.9 | 24.0 | 26.0 | 23.6 | 23.4 | 15.1 | 25.5 |
| 28 | 23.0 | 28.9 | 23.6 | 28.8 | 28.7 | 63.6 | 28.4 |
| 29 | 17.6 | 17.4 | 17.8 | 33.0 | 28.2 | 31.4 | 28.2 |
| 30 | 21.2 | 21.5 | 22.0 | 23.5 | 65.3 | 29.7 | 65.6 |
| OCH3 | 51.4 |


| Compounds | Superoxide anion | Elastase |
|---|---|---|
| IC50 (μg/mL) a or (Inh %) | IC50 (µg/mL) a or (Inh %) | |
| 4 | 125.9 ± 6.72 b *** | 44.62 ± 2.82 c *** |
| 5 | 19.65 ± 4.23 b ** | 1.53 ± 0.09 |
| 6 | (4.35 ± 6.05) | 3.23 ± 0.24 |
| 7 | 2.10 ± 0.13 | 2.93 ± 0.27 |
| 8 | 0.93 ± 0.20 | 4.39 ± 1.13 |
| 9 | 16.71 ± 3.39 b ** | 5.65 ± 0.26 |
| 10 | 30.27 ± 5.04 b *** | 5.33 ± 0.73 |
| 14 | (−2.09 ± 4.02) | (14.50 ± 7.26) |
| 15 | 2.66 ± 0.78 | 3.23 ± 0.88 |
| 16 | 3.12 ± 0.60 | 4.53 ± 0.39 |
| 17 | 0.06 ± 0.01 | 1.03 ± 0.35 |
| DPI d | 0.43 ± 0.12 | - |
| PMSF d | - | 17.58 ± 5.79 |
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. The Preparation of Human Neutrophils
3.5. The Measurement of Superoxide Generation
3.6. The Measurement of Elastase Release
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flora of Taiwan, 2nd ed.; Huang, T.C. (Ed.) Lungwei Printing Co. Ltd Press: Taipei, Taiwan, 1993; Volume 3, pp. 640–658.
- Chen, J.J.; Chou, T.H.; Duh, C.Y.; Chen, I.S. Cytotoxic dihydroagarofuranoid sesquiterpenes from the stem of Microtropis fokienensis. J. Nat. Prod. 2006, 69, 685–688. [Google Scholar] [CrossRef]
- Koyama, Y.; Matsunami, K.; Otsuka, H.; Shinzato, T.; Takeda, Y. Microtropiosides A–F: Ent-labdane diterpenoid glucosides from the leaves of Microtropis japonica (Celastraceae). Phytochemistry 2010, 71, 675–681. [Google Scholar] [CrossRef]
- Chen, I.H.; Chang, F.R.; Wu, C.C.; Chen, S.L.; Hsieh, P.W.; Yen, H.F.; Du, Y.C.; Wu, Y.C. Cytotoxic triterpenoids from the leaves of Microtropis fokienensis. J. Nat. Prod. 2006, 69, 1543–1546. [Google Scholar] [CrossRef]
- Chen, I.H.; Lu, M.C.; Du, Y.C.; Yen, M.H.; Wu, C.C.; Chen, Y.H.; Hung, C.S.; Chen, S.L.; Chang, F.R.; Wu, Y.C. Cytotoxic triterpenoids from the stems of Microtropis japonica. J. Nat. Prod. 2009, 72, 1231–1236. [Google Scholar] [CrossRef]
- Chen, I.H.; Du, Y.C.; Lu, M.C.; Lin, A.S.; Hsieh, P.W.; Wu, C.C.; Chen, S.L.; Yen, H.F.; Chang, F.R.; Wu, Y.C. Lupane-type triterpenoids from Microtropis fokienensis and Perrottetia arisanensis and the apoptotic effect of 28-Hydroxy-3-oxo-lup-20(29)-en-30-al. J. Nat. Prod. 2008, 71, 1352–1357. [Google Scholar] [CrossRef]
- González, A.G.; Andres, L.S.; Ravelo, A.G.; Luis, J.G.; Bazzocchi, I.L.; West, J. Terpenoids from Salvia mellifera. Phytochemistry 1990, 29, 1691–1693. [Google Scholar] [CrossRef]
- Ikuta, A.; Tomiyasu, H.; Morita, Y.; Yoshimura, K. Ursane- and oleanane-type triterpenes from Ternstroemia gymnanthera Callus Tissues. J. Nat. Prod. 2003, 66, 1051–1054. [Google Scholar] [CrossRef]
- de Souza e Silva, S.R.; Silva, G.D.F.; Barbosa, L.C.A.; Duarte, L.P.; Vieira Filho, S.A. Lupane pentacyclic triterpenes isolated from stems and branches of Maytenus imbricata (Celastraceae). Helv. Chim. Acta 2005, 88, 1102–1109. [Google Scholar] [CrossRef]
- Saimaru, H.; Orihara, Y.; Tansakul, P.; Kang, Y.-H.; Shibuya, M.; Ebizuka, Y. Production of triterpene acids by cell suspension cultures of Olea europaea. Chem. Pharm. Bull. 2007, 55, 784–788. [Google Scholar] [CrossRef]
- Cheng, J.J.; Zhang, L.J.; Cheng, H.L.; Chiou, C.T.; Lee, I.J.; Kuo, Y.H. Cytotoxic hexacyclic triterpene acids from Euscaphis japonica. J. Nat. Prod. 2010, 73, 1655–1658. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Sarkar, A.C.; Talapatra, B. Two pentacyclic triterpenes from Rubia cordifolia. Phytochemistry 1981, 20, 1923–1927. [Google Scholar] [CrossRef]
- Shibata, S.; Takahashi, K.; Yano, S.; Harada, M.; Saito, H.; Tamura, Y.; Kumagai, A.; Hirabayashi, K.; Yamamoto, M.; Nagara, N. Chemical modification of glycyrrhetinic acid in relation to the biological activities. Chem. Pharm. Bull. 1987, 35, 1910–1918. [Google Scholar] [CrossRef]
- Taniguchi, S.; Imayoshi, Y.; Kobayashi, E.; Takamatsu, Y.; Shimura, S.; Yoshidda, T. Production of bioactive triterpenes by Eriobotrya japonica calli. Phytochemistry 2002, 59, 315–323. [Google Scholar] [CrossRef]
- Su, X.; Lawrence, H.; Ganeshapillai, D.; Cruttenden, A.; Purohit, A.; Reed, M.J.; Vickera, N.; Pottera, B.V. L. Novel 18β-glycyrrhetinic acid analogues as potent and selective inhibitors of 11β-hydroxysteroid dehydrogenases. Bioorg. Med. Chem. 2004, 12, 4439–4457. [Google Scholar] [CrossRef]
- Fingolo, C.E.; de S. Santos, T.; Vianna Filho, M.D.M.; Kaplan, M.A.C. Triterpene esters: Natural products from Dorstenia arifolia (Moraceae). Molecules 2013, 18, 4247–4256. [Google Scholar] [CrossRef]
- Toriumi, Y.; Kakuda, R.; Kikuchi, M.; Yaoita, Y.; Kikuchi, M. New Triterpenoids from Gentiana lutea. Chem. Pharm. Bull. 2003, 51, 89–91. [Google Scholar] [CrossRef]
- Morikawa, T.; Oominami, H.; Matsuda, H.; Yoshikawa, M. Four new ursane-type triterpenes, olibanumols K, L, M, and N, from traditional Egyptian medicine olibanum, the Gum-Resin of Boswellia carterii. Chem. Pharm. Bull. 2010, 58, 1541–1544. [Google Scholar] [CrossRef]
- Shirota, O.; Tamemura, T.; Morita, H.; Takeya, K.; Hideji, I. Triterpenes from Brazilian medicinal plant “Chuchuhuasi” (Maytenus krukovii). J. Nat. Prod. 1996, 59, 1072–1075. [Google Scholar] [CrossRef]
- Lahlou, H.E.; Hirai, N.; Tsuda, M.; Ohigashi, H. Triterpene phytoalexins from nectarine fruits. Phytochemistry 1999, 52, 623–629. [Google Scholar] [CrossRef]
- Barnes, R.A.; Pereira, A.L.; Scofield, T.C.V.; Filho, R.B.; Pinto, A.C. A new triterpene from Vellozia compacta. Chem. Pharm. Bull. 1984, 32, 3674–3677. [Google Scholar] [CrossRef]
- González, A.G.; Fraga, B.M.; González, P.; Hernandez, M.G.; Ravelo, A.G. 13C NMR spectra of olean-18-ene derivatives. Phytochemistry 1981, 20, 1919–1921. [Google Scholar] [CrossRef]
- Osorio, A.A.; Muñóz, A.; Torres-Romero, D.; Bedoya, L.M.; Perestelo, N.R.; Jiménez, I.A.; Alcamí, J.; Bazzocchi, I.L. Olean-18-ene triterpenoids from Celastraceae species inhibit HIV replication targeting NF-kB and Sp1 dependent transcription. Eur. J. Med. Chem. 2012, 52, 295–303. [Google Scholar] [CrossRef]
- Kuo, Y.H.; Huang, H.C.; Chiou, W.F.; Shi, L.S.; Wu, T.S.; Wu, Y.C. A Novel Anti-NO triterpene, laxifolone-A, and sesquiterpene pyridine alkaloids from Euonymus laxiflorus. J. Nat. Prod. 2003, 66, 554–557. [Google Scholar] [CrossRef]
- Kagawa, M.; Minami, H.; Nakahara, M.; Takahashi, H.; Takaoka, S.; Fukuyama, Y. Oleanane-type triterpenes from Viburnum awabuki. Phytochemistry 1998, 47, 1101–1105. [Google Scholar] [CrossRef]
- Wang, K.W. A new fatty acid ester of triterpenoid from Celastrus rosthornianus with anti-tumor activities. Nat. Prod. Res. 2007, 21, 669–674. [Google Scholar] [CrossRef]
- Okada, Y.; Omae, A.; Okuyama, T. A New Triterpenoid Isolated from Lagerstronemia speciosa (L.) PERS. Chem. Pharm. Bull. 2003, 51, 452–454. [Google Scholar] [CrossRef]
- Rodrí guez, F.M.; Perestelo, N.R.; Jiménez, I.A.; Bazzocchi, I.L. Friedelanes from Crossopetalum lobatum a new example of a triterpene anhydride. Helv. Chim. Acta 2009, 92, 188–194. [Google Scholar] [CrossRef]
- Shernyukov, A.V.; Mainagashev, I.Y.; Korchagina, D.V.; Gatilov, Y.V.; Salakhutdinov, N.F.; Tolstikov, G.A. Reduction of 2,3-seco-28-oxo-19β,28-epoxy-18α-olean-2,3-dicarboxylic acid and its cyclic anhydride. Chem. Nat. Comp. 2011, 47, 237–242. [Google Scholar] [CrossRef]
- González, A.G.; González, C.M.; Ravelo, A.G. 3-Oxo-28,29-dihydroxyolean-12-ene from Orthosphenia mexicana. J. Nat. Prod. 1986, 49, 148–150. [Google Scholar] [CrossRef]
- Hwang, T.L.; Su, Y.C.; Chang, H.L.; Leu, Y.L.; Chung, P.J.; Kuo, L.M.; Chang, Y.J. Suppression of superoxide anion and elastase release by C18 unsaturated fatty acids in human neutrophils. J. Lipid Res. 2009, 50, 1395–1408. [Google Scholar] [CrossRef]
- Hwang, T.L.; Li, G.L.; Lan, Y.H.; Chia, Y.C.; Hsieh, P.W.; Wu, Y.H.; Wu, Y.C. Potent inhibition of superoxide anion production in activated human neutrophils by isopedicin, a bioactive component of the Chinese medicinal herb Fissistigma oldhamii. Free Radic. Biol. Med. 2009, 46, 520–528. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds in the manuscript are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, I.-H.; Du, Y.-C.; Hwang, T.-L.; Chen, I.-F.; Lan, Y.-H.; Yen, H.-F.; Chang, F.-R.; Wu, Y.-C. Anti-Inflammatory Triterpenoids from the Stems of Microtropis Fokienensis. Molecules 2014, 19, 4608-4623. https://doi.org/10.3390/molecules19044608
Chen I-H, Du Y-C, Hwang T-L, Chen I-F, Lan Y-H, Yen H-F, Chang F-R, Wu Y-C. Anti-Inflammatory Triterpenoids from the Stems of Microtropis Fokienensis. Molecules. 2014; 19(4):4608-4623. https://doi.org/10.3390/molecules19044608
Chicago/Turabian StyleChen, I-Hsiao, Ying-Chi Du, Tsong-Long Hwang, I-Fen Chen, Yu-Hsuan Lan, Hsin-Fu Yen, Fang-Rong Chang, and Yang-Chang Wu. 2014. "Anti-Inflammatory Triterpenoids from the Stems of Microtropis Fokienensis" Molecules 19, no. 4: 4608-4623. https://doi.org/10.3390/molecules19044608