Synthesis of Regiospecifically Fluorinated Conjugated Dienamides

Abstract
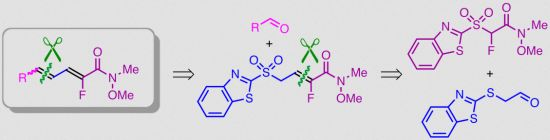
:
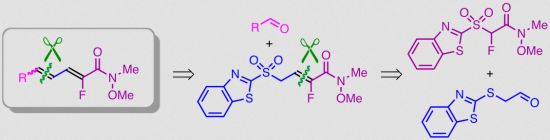
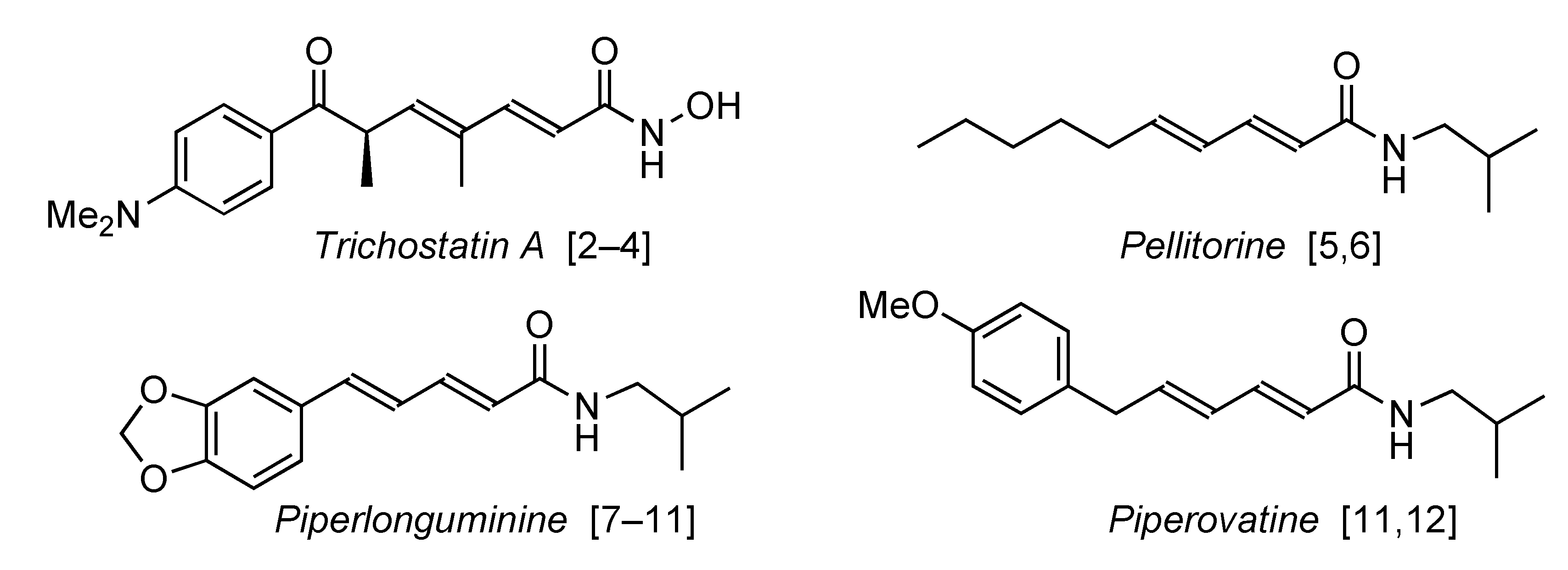
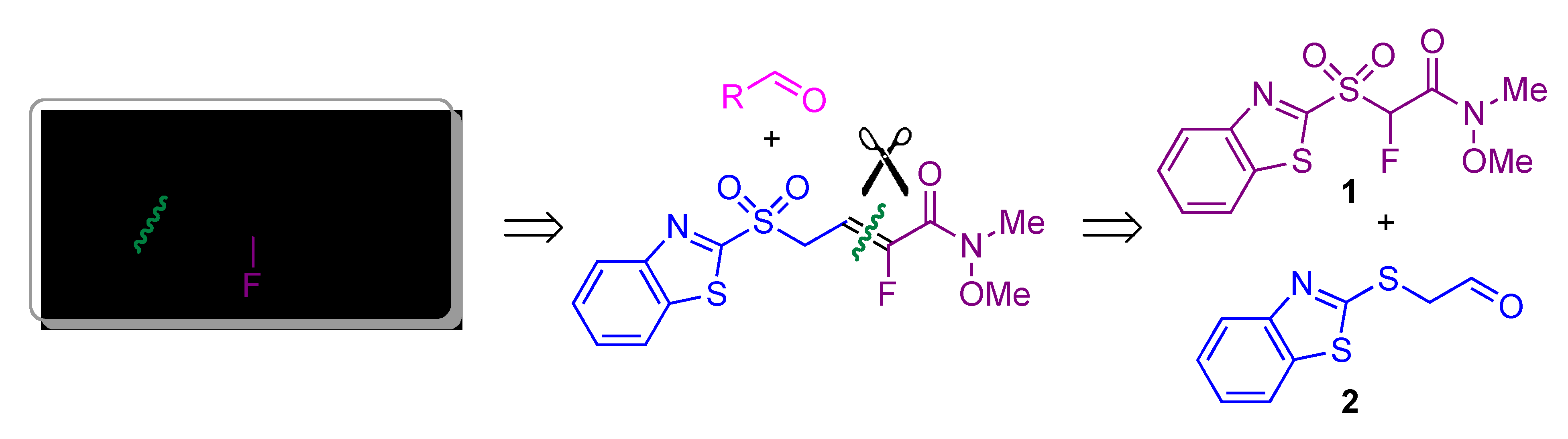
1. Introduction

2. Results and Discussion
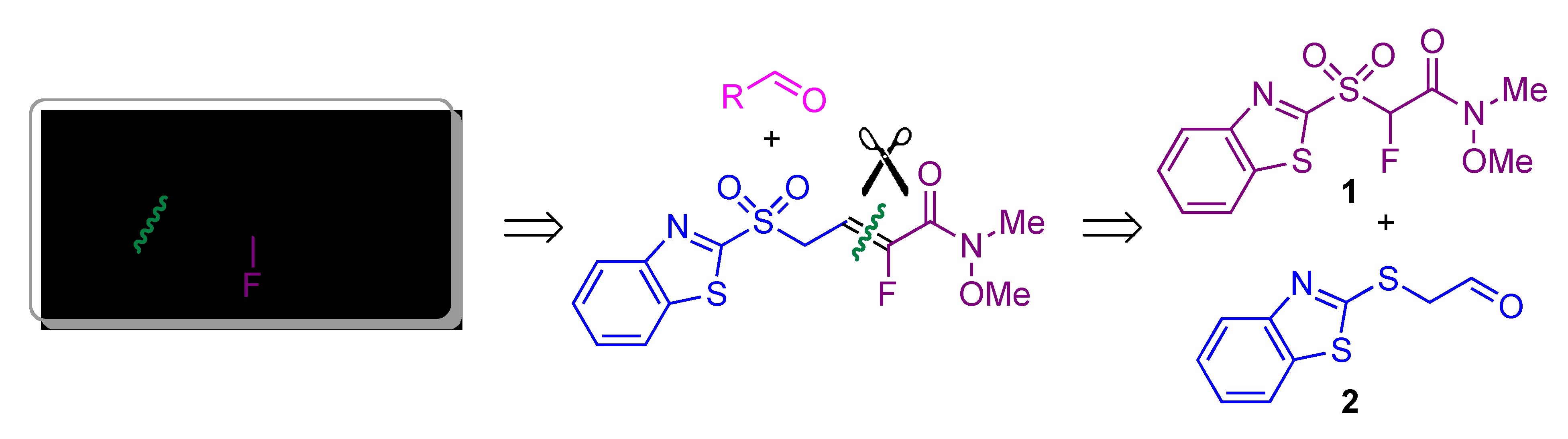


| Entry | Reagent | Solvent | T (°C) | Time | Yield (%) a |
|---|---|---|---|---|---|
| 1 | I2 | acetone | rt | overnight | -- b |
| 2 | CBr4 | CH3CN–H2O 1:3 | 80 | 3 days | -- b |
| 3 | PTSA | THF–H2O 1:1 | rt | 3 days | -- b |
| 4 | HCl (4 M) | acetone | 40 | 24 h | 20 c |
| 5 | HCl (4 M) | acetone | 40 | 4 h | 56 |
| 6 | HCl (12 M) | acetone | 50 | 30 min | 81 |
| 7 | HCl (12 M) | acetone–H2O 10:1 | 50 | 40 min | 84 |
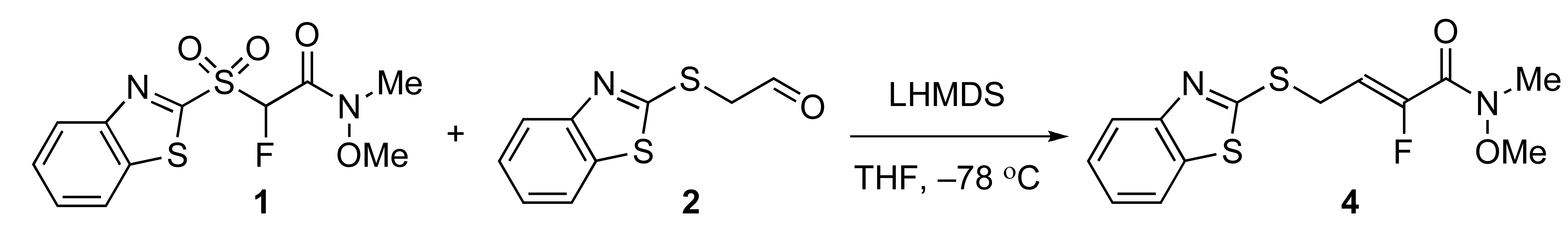
| Entry | Molar Ratio of 1:2:LHMDS | Time | Yield (%) a |
|---|---|---|---|
| 1 | 1.5:1:1.5 | 3 h | 32 |
| 2 | 1:1:2 | 2.5 h | 20 |
| 3 | 1:3:5 | 4 h | 60 |
| 4 | 1:2:3 | 3.5 h | 76 |
| Entry | Base | Solvent | T | Time | % 4E/4Z Ratio a | Yield (%) b |
|---|---|---|---|---|---|---|
| 1 | LHMDS | THF | −78 to 0 °C | overnight | -- | -- c |
| 2 | LHMDS | THF | 0 °C to rt | 12 h | -- | -- c |
| 3 | DBU | THF | rt | 2 h | -- | -- c |
| 4 | DBU | THF | −78 to 0 °C | overnight | 57/43 | 35 |
| 5 | Cs2CO3 | THF | 0 °C | overnight | -- | -- c |
| 6 | DBU | THF | 0 °C | overnight | 43/57 | 55 |
| 7 | Cs2CO3 | CH2Cl2 | 0 °C | overnight | -- | -- c |
| 8 | DBU | CH2Cl2 | 0 °C | overnight | 35/65 | 66 |

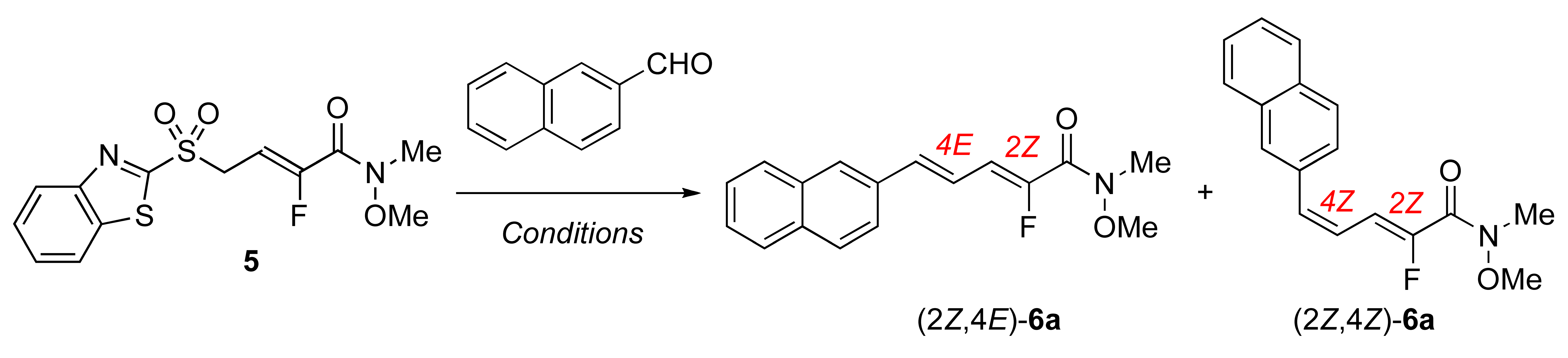
| Entry | RCHO | Product (6a–e); % 4E/4Z Ratio a; Yield (%) b | 19F-NMR Data: c δ (ppm); Mult, J (Hz) |
|---|---|---|---|
| 1 |  | 6a: 35/65; 66 | (4E)-6a: −123.4; d, 30.5 (4Z)-6a: −121.2; d, 30.5 |
| 2 |  | 6b: 23/77; 50 | (4E)-6b: −125.2; d, 33.6 (4Z)-6b: −122.6; d, 33.6 |
| 3 |  | 6c: 40/60; 74 | (4E)-6c: −119.6; d, 30.5 (4Z)-6c: −118.4; d, 30.5 |
| 4 |  | 6d: 10/90; 63 | (4E)-6d: −123.7; d, 30.5 (4Z)-6d: −120.7; d, 33.6 |
| 5 |  | 6e: 15/85; 51 | (4E)-6e: −125.7; d, 30.5 (4Z)-6e: −124.2; d, 33.6 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Isomer Mixture | Time | Product a | Yield (%) b |
|---|---|---|---|---|
| 1 | (2Z,4E/Z)-6a | 3 h | (2Z,4E)-6a | 75 |
| 2 | (2Z,4E/Z)-6b | 3 h | (2Z,4E)-6b | 86 |
| 3 | (2Z,4E/Z)-6c | 1.5 h | (2Z,4E)-6c | 89 |
| 4 | (2Z,4E/Z)-6d | overnight | (2Z,4E)-6d | 92 |
3. Experimental
3.1. General Information
3.2. Synthesis of “Second-Generation” Julia-Kocienski Reagent 5
3.3. Condensation Reactions of Julia-Kocienski Reagent 5
3.4. Isomerization of the (2Z,4E/Z)-Isomer Mixture of 6a–d to the (2Z,4E)-Isomer
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References and Notes
- Nájera, C.; Yus, M. Natural products with polyene amide structures. In Bioactive Natural Products (Part B). Studies in Natural Products Chemistry; Rahman, A., Ed.; Elsevier Science B. V.: Amsterdam, The Netherlands, 2000; Volume 21, pp. 373–455. [Google Scholar]
- Tsuji, N.; Kobayashi, M.; Nagashima, K. A new antifungal antibiotic, trichostatin. J. Antibiot. 1976, 29, 1–6. [Google Scholar] [CrossRef]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 1990, 265, 17174–17179. [Google Scholar]
- Drummond, D.C.; Noble, C.O.; Kirpotin, D.B.; Guo, Z.; Scott, G.K.; Benz, C.C. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 495–528. [Google Scholar] [CrossRef]
- Jacobson, M. The structure of pellitorine. J. Am. Chem. Soc. 1949, 71, 366–367. [Google Scholar] [CrossRef]
- Ee, G.C.L.; Lim, C.M.; Rahmani, M.; Shaari, K.; Bong, C.F.J. Pellitorine, a potential anti-cancer lead compound against HL60 and MTC–7 cell lines and microbial transformation of piperine from Piper nigrum. Molecules 2010, 15, 2398–2404. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef]
- Bezerra, D.P.; Pessoa, C.; de Moraes, M.O.; de Alencar, N.M.N.; Mesquita, R.O.; Lima, M.W.; Alves, A.P.N.N.; Pessoa, O.D.L.; Chaves, J.H.; Silveira, E.R.; et al. In vivo growth inhibition of sarcoma 180 by piperlonguminine, an alkaloid amide from the Piper species. J. Appl. Toxicol. 2008, 28, 599–607. [Google Scholar] [CrossRef]
- Lee, W.; Yoo, H.; Ku, S.-K.; Kim, J.A.; Bae, J.-S. Anticoagulant activities of piperlonguminine in vitro and in vivo. BMP Rep. 2013, 46, 484–489. [Google Scholar] [CrossRef]
- Kim, K.-S.; Kim, J.A.; Eom, S.-Y.; Lee, S.H.; Min, K.R.; Kim, Y. Inhibitory effect of piperlonguminine on melanin production in melanoma B16 cell line by downregulation of tyrosinase expression. Pigment Cell Res. 2006, 19, 90–98. [Google Scholar] [CrossRef]
- Rodrigues Silva, D.; Baroni, S.; Svidzinski, A.E.; Bersani-Amado, C.A.; Cortez, D.A.G. Anti-inflammatory activity of the extract, fractions and amides from the leaves of Piper ovatum Vahl (Piperaceae). J. Ethnopharmacol. 2008, 116, 569–573. [Google Scholar] [CrossRef]
- Makapugay, H.C.; Soejarto, D.D.; Kinghorn, A.D.; Bordas, E. Piperovatine, the tongue-numbing principle of Ottonia frutescens. J. Ethnopharmacol. 1983, 7, 235–238. [Google Scholar] [CrossRef]
- Charrier, C.; Roche, J.; Gesson, J.-P.; Bertrand, P. Biological activities of substituted trichostatic acid derivatives. J. Chem. Sci. 2009, 121, 471–479. [Google Scholar] [CrossRef]
- Welch, J.T. (Ed.) Selective Fluorination in Organic and Bioorganic Chemistry; American Chemical Society: Washington, DC, USA, 1991.
- Kirsch, P. Modern Fluoroorganic Chemistry. Synthesis, Reactivity, Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004. [Google Scholar]
- Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluorine Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Lemal, D.M. Perspective on fluorocarbon chemistry. J. Org. Chem. 2004, 69, 1–11. [Google Scholar] [CrossRef]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef]
- Blakemore, P.R. The modified Julia olefination: alkene synthesis via the condensation of metallated heteroarylalkylsulfones with carbonyl compounds. J. Chem. Soc. Perkin Trans. I 2002, 2563–2585. [Google Scholar] [CrossRef]
- Plesniak, K.; Zarecki, A.; Wicha, J. The Smiles rearrangement and the Julia-Kocienski olefination reaction. Top. Curr. Chem. 2007, 275, 163–250. [Google Scholar] [CrossRef]
- Aïssa, C. Mechanistic manifold and new developments of the Julia-Kocienski reaction. Eur. J. Org. Chem. 2009, 1831–1844. [Google Scholar] [CrossRef]
- Blakemore, P.R. Olefination of carbonyl compounds by main-group element mediators. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Molander, G.A., Eds.; Elsevier Ltd: Oxford, UK, 2014; Volume 1, pp. 516–608. [Google Scholar]
- Zajc, B.; Kumar, R. Synthesis of fluoroolefins via Julia-Kocienski olefination. Synthesis 2010, 1822–1836. [Google Scholar] [CrossRef]
- Landelle, G.; Bergeron, M.; Turcotte-Savard, M.-O.; Paquin, J.-P. Synthetic approaches to monofluoroalkenes. Chem. Soc. Rev. 2011, 40, 2867–2908. [Google Scholar] [CrossRef]
- Yanai, H.; Taguchi, T. Synthetic methods for fluorinated olefins. Eur. J. Org. Chem. 2011, 5939–5954. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Zajc, B. High-yield synthesis of fluorinated benzothiazolyl sulfones: General synthons for fluoro-Julia olefinations. Org. Lett. 2006, 8, 1553–1556. [Google Scholar] [CrossRef]
- Zajc, B.; Kake, S. Exceptionally mild, high-yield synthesis of α-fluoro acrylates. Org. Lett. 2006, 8, 4457–4460. [Google Scholar] [CrossRef]
- He, M.; Ghosh, A.K.; Zajc, B. Julia olefination as a general route to phenyl (α-fluoro)vinyl sulfones. Synlett 2008, 999–1004. [Google Scholar]
- del Solar, M.; Ghosh, A.K.; Zajc, B. Fluoro-Julia olefination as a mild, high-yielding route to α-fluoro acrylonitriles. J. Org. Chem. 2008, 73, 8206–8211. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Banerjee, S.; Sinha, S.; Kang, S.B.; Zajc, B. α-Fluorovinyl Weinreb amides and α-fluoroenones from a common fluorinated building block. J. Org. Chem. 2009, 74, 3689–3697. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Zajc, B. Fluorinated 1-phenyl-1H-tetrazol-5-yl sulfonederivatives as general reagents for fluoroalkylidene synthesis. J. Org. Chem. 2009, 74, 8531–8540. [Google Scholar] [CrossRef]
- Kumar, R.; Pradhan, P.; Zajc, B. Facile synthesis of 4-vinyl- and 4-fluorovinyl-1,2,3-triazoles via bifunctional “click-olefination” reagents. Chem. Commun. 2011, 47, 3891–3893. [Google Scholar] [CrossRef]
- Mandal, S.K.; Ghosh, A.K.; Kumar, R.; Zajc, B. Expedient synthesis of α-substituted fluoroethenes. Org. Biomol. Chem. 2012, 10, 3164–3167. [Google Scholar] [CrossRef]
- Kumar, R.; Zajc, B. Stereoselective synthesis of conjugated fluoro enynes. J. Org. Chem. 2012, 77, 8417–8427. [Google Scholar] [CrossRef]
- Chevrie, D.; Lequeux, T.; Demoute, J.P.; Pazenok, S. A convenient one-step synthesis of fluoroethylidene derivatives. Tetrahedron Lett. 2003, 44, 8127–8130. [Google Scholar] [CrossRef]
- Pfund, E.; Lebargy, C.; Rouden, J.; Lequeux, T. Modified Julia fluoroolefination: Selective preparation of fluoroalkenoates. J. Org. Chem. 2007, 72, 7871–7877. [Google Scholar] [CrossRef]
- Alonso, D.A.; Fuensanta, M.; Gómez-Bengoa, E.; Nájera, C. Highly efficient and stereoselective Julia-Kocienski protocol for the synthesis of α-fluoro-α,β-unsaturated esters and Weinreb amides employing 3,5-bis(trifluoromethyl)phenyl (BTFP) sulfones. Adv. Synth. Catal. 2008, 350, 1823–1829. [Google Scholar] [CrossRef]
- Calata, C.; Catel, J.-M.; Pfund, E.; Lequeux, T. Scope and limitations of the Julia-Kocienski reaction with fluorinated sulfonylesters. Tetrahedron 2009, 65, 3967–3973. [Google Scholar] [CrossRef]
- Calata, C.; Pfund, E.; Lequeux, T. Toward the synthesis of benzothiazolyl fluoroaminosulfones. J. Org. Chem. 2009, 74, 9399–9405. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, W.; Zhu, L.; Hu, J. Difluoromethyl 2-pyridyl sulfone: A new gem-difluoroolefination reagent for aldehydes and ketones. Org. Lett. 2010, 12, 1444–1447. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Shakhmin, A.; Zibinsky, M.; Ledneczki, I.; Chacko, S.; Olah, G.A. Synthesis of monofluoroalkenes via Julia-Kocienski reaction. J. Fluorine Chem. 2010, 131, 1192–1197. [Google Scholar] [CrossRef]
- Allendörfer, N.; Es-Sayed, M.; Nieger, M.; Bräse, S. Novel aromatic fluoroolefins via fluoro-Julia-Kocienski olefination. Synthesis 2010, 3439–3448. [Google Scholar]
- Calata, C.; Pfund, E.; Lequeux, T. Convergent synthesis of functionalized fluoroallylamines by the Julia-Kocienski reaction. Tetrahedron 2011, 67, 1398–1405. [Google Scholar] [CrossRef]
- Jacobsen, C.B.; Nielsen, M.; Worgull, D.; Zweifel, T.; Fisker, E.; Jørgensen, K.A. Asymmetric organocatalytic monofluorovinylations. J. Am. Chem. Soc. 2011, 133, 7398–7404. [Google Scholar]
- Larnaud, F.; Malassis, J.; Pfund, E.; Linclau, B.; Lequeux, T. Ready synthetic access to enantiopure allylic α(F)-branched fluoroalkenes. Org. Lett. 2013, 15, 2450–2453. [Google Scholar] [CrossRef]
- Singh, G.; Kumar, R.; Swett, J.; Zajc, B. Modular Synthesis of N-Vinyl Benzotriazoles. Org. Lett. 2013, 15, 4086–4089. [Google Scholar] [CrossRef]
- Nahm, S.; Weinreb, S.M. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar] [CrossRef]
- Khlestkin, V.K.; Mazhukin, D.G. Recent advances in the application of N,O-dialkylhydroxylamines in organic chemistry. Curr. Org. Chem. 2003, 7, 967–993. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Aidhen, I.S. The growing synthetic utility of the Weinreb amide. Synthesis 2008, 3707–3738. [Google Scholar]
- For a recent example of the synthetic application of Weinreb amides please see: Pace, V.; Castoldi, L.; Holzer, W. Synthesis of α,β-unsaturated α’-haloketones through the chemoselective addition of halomethyllithiums to Weinreb amides. J. Org. Chem. 2013, 78, 7764–7770. [Google Scholar] [CrossRef]
- Benzothiazole-derived w-sulfonyl aldehydes, employed in intramolecular olefinations leading to αβ-unsaturated lactones, have been reported to be unstable under chromatographic conditions:Giesbrecht, H.E.; Knight, B.J.; Tanguileg, N.R.; Emerson, C.R.; Blakemore, P.R. Stereoselective synthesis of Z-configured αβ-unsaturated macrocyclic lactones and diolides by intramolecular Julia-Kocienski olefination. Synlett 2010, 374–378. [Google Scholar]
- Gaukroger, K.; Hadfield, J.A.; Hepworth, L.A.; Lawrence, N.J.; McGown, A.T. Novel syntheses of cis and trans isomers of combretastatin A-4. J. Org. Chem. 2001, 66, 8135–8138, and references therein. [Google Scholar]
- Manjunath, B.N.; Sane, N.P.; Aidhen, I.S. New reagent for convenient access to the α,β-unsaturated N-methoxy-N-methyl-amide functionality by a synthesis based on the Julia olefination protocol. Eur. J. Org. Chem. 2006, 2851–2855. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chowdhury, M.; Mandal, S.K.; Banerjee, S.; Zajc, B. Synthesis of Regiospecifically Fluorinated Conjugated Dienamides. Molecules 2014, 19, 4418-4432. https://doi.org/10.3390/molecules19044418
Chowdhury M, Mandal SK, Banerjee S, Zajc B. Synthesis of Regiospecifically Fluorinated Conjugated Dienamides. Molecules. 2014; 19(4):4418-4432. https://doi.org/10.3390/molecules19044418
Chicago/Turabian StyleChowdhury, Mohammad, Samir K. Mandal, Shaibal Banerjee, and Barbara Zajc. 2014. "Synthesis of Regiospecifically Fluorinated Conjugated Dienamides" Molecules 19, no. 4: 4418-4432. https://doi.org/10.3390/molecules19044418