2. Results and Discussion
Compound
1 was obtained as a white, amorphous powder. Positive mode HRESIMS indicated a molecular formula of C
29H
46O
5 ([M+Na]
+ m/
z 497.3235, calcd. 497.3237), implying seven degrees of unsaturation. The
1H-NMR spectrum of
1 (
Table 1) showed signals readily recognized for six tertiary methyl groups at
δ 1.32 (3H, Me-29), 1.06 (3H, Me-27), 1.03 (3H, Me-23), 0.82 (3H, Me-25), 0.81 (3H, Me-24) and 0.76 (3H, Me-26). In addition, proton signals for two oxymethines at
δ 3.77 (1H, H-2) and 3.09 (1H, H-3), and an olefinic proton at
δ 5.26 (1H, H-12) were also observed. The
13C-NMR and DEPT spectra (
Table 1) of
1 supported the above analysis, which showed 29 carbons including six methyls (
δC 29.2, 17.6, 16.7, 17.4, 25.9 and 25.6), nine methylenes, six methines [including one olefinic methine at
δ 122.5 (C-12), two oxy-methines at
δ 68.5 (C-2) and 83.7 (C-3)], and eight quaternary carbons [including an olefinic quaternary carbon at
δ 144.3 (C-13), a carboxyl carbon at
δ 179.9 (C-28), and an oxy-quaternary carbon at
δ 69.8 (C-20)]. These above findings accounted for two of the seven degrees of unsaturation, suggesting that
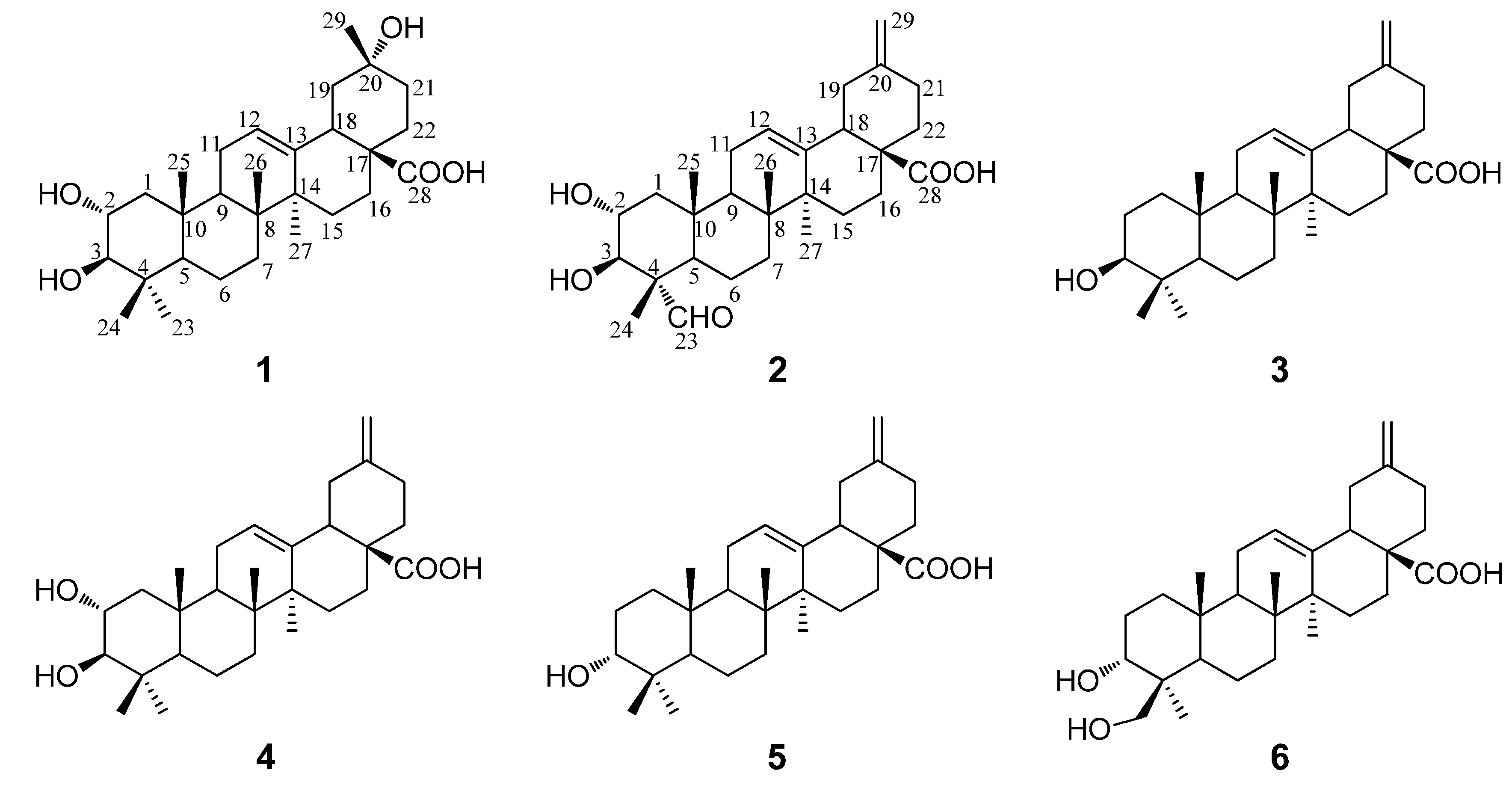
1 is a pentacyclic nortriterpenoid. Comparison of the NMR data of
1 with those of maslinic acid indicated that they were structurally closely related, with the major difference of a germinal methyl group attached at C-20 in maslinic acid being replaced by a hydroxyl group in
1 [
15]. This assignment was consistent with the molecular formula of
1 and in accord with the significant change of the chemical shift value for C-20 from
δC 30.8 in maslinic acid to
δC 69.7 in
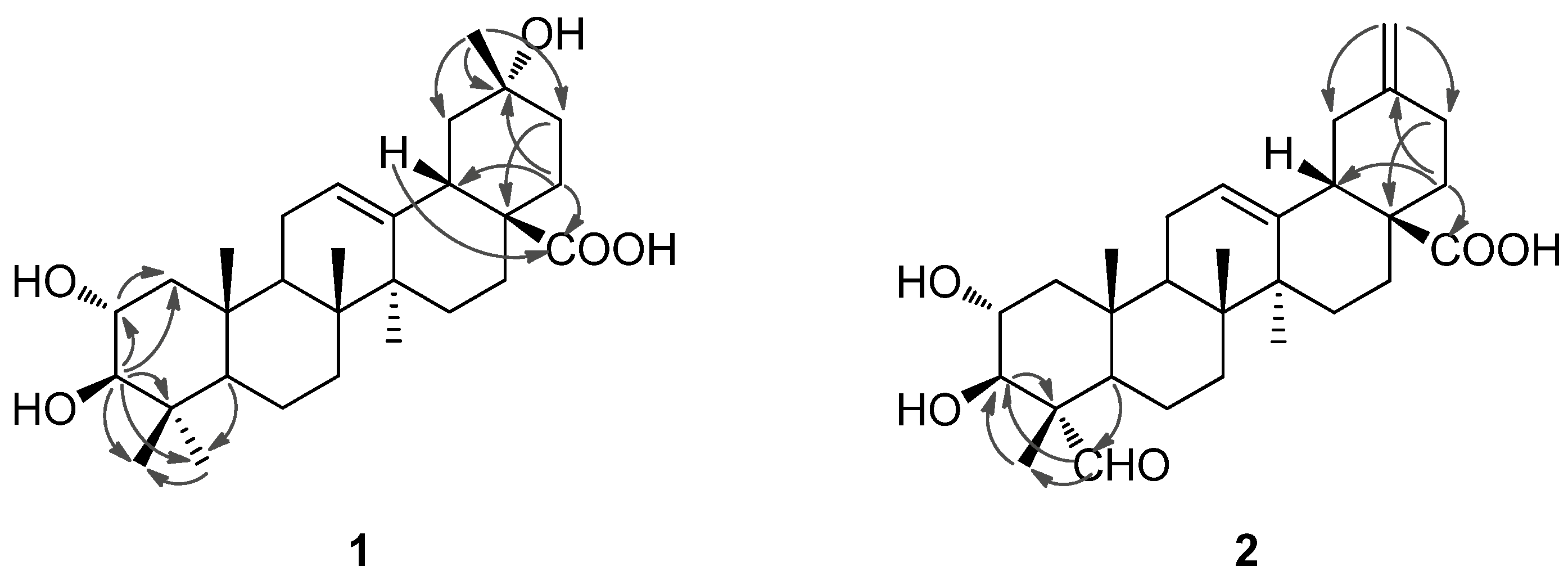
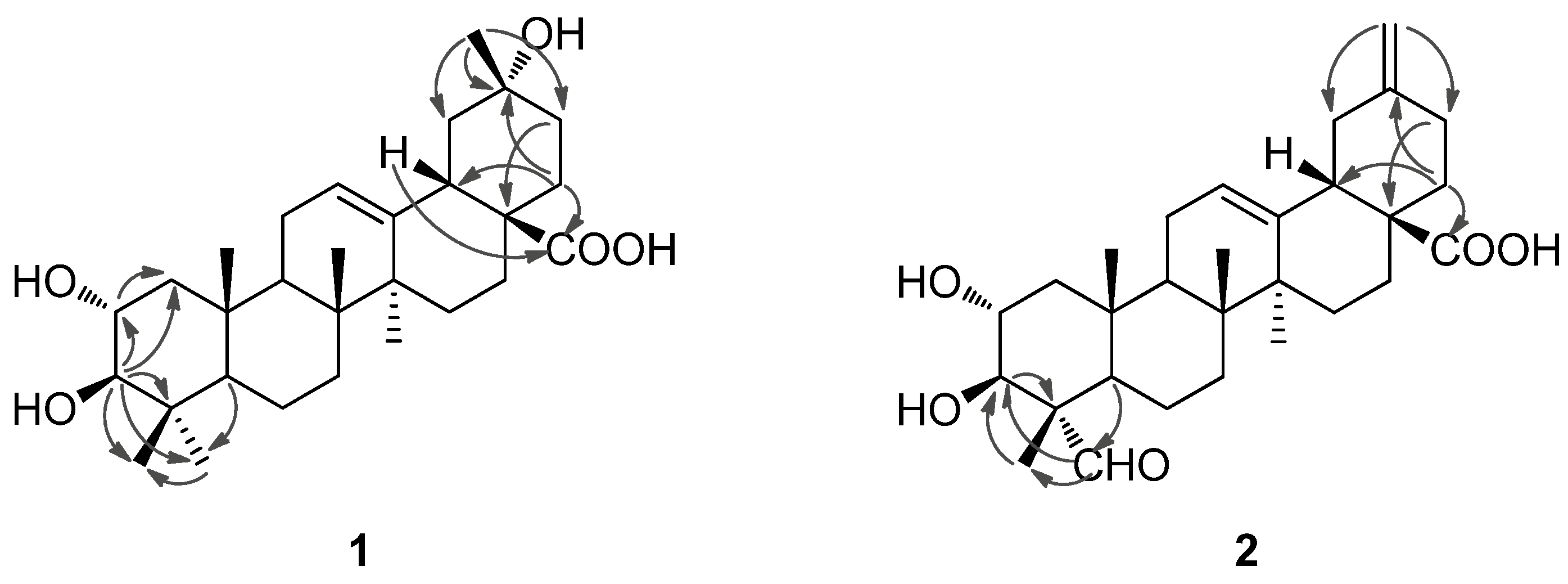
1. The location of the hydroxyl group at C-20 was furhter supported by HMBC correlations from
δH 1.32 (H
3-29) to
δC 47.6 (C-19), 69.8 (C-20), 36.1 (C-21) and from
δH 1.75 (H
2-22) to C-20. In the NOESY spectrum, the important NOE correlation between Me-29 (
δH 1.32) and H-18 (
δH 2.98) was observed, which supported the hydroxyl group at C-20 was
α-orientation (
Figure 2). In addition, the HMBC correlations (
Figure 3) from
δH 1.75 (H-22) and 2.98 (H-18) to
δC 179.9 (C-28) supported the location of the carboxyl group at C-17. The HMBC correlations from
δH 5.26 (H-12) to
δC 42.1 (C-14), 44.3 (C-18) and 47.9 (C-9) supported the location of double bond at C-12(13). The HMBC correlations from
δH 3.77 (H-2) to
δC 47.7 (C-1) and
δC 83.7 (C-3), and from
δH 3.09 (H-3) to
δC 47.7 (C-1), 68.5 (C-2), 39.7 (C-4), 29.2 (C-23) and 17.6 (C-24) supported that each of C-2 and C-3 was attached with a hydroxyl group.
Table 1.
1H-NMR and 13C-NMR data for compounds 1 and 2, δ in ppm and J in Hz.
Table 1.
1H-NMR and 13C-NMR data for compounds 1 and 2, δ in ppm and J in Hz.
| No. | δC (1) | δH (1) | δC (2) | δH (2) |
|---|
| 1 | 47.7 CH2 | 1.95 (dd, 11.8, 3.7), 0.97 (m) | 47.8 CH2 | 2.29 (dd, 12.0, 4.2), 1.41 (m) |
| 2 | 68.5 CH | 3.77 (td, 11.8, 9.5, 3.7) | 67.9 CH | 4.24 (td, 12.0, 9.3, 4.2) |
| 3 | 83.7 CH | 3.09 (d, 9.5) | 77.0 CH | 4.06 (d, 9.3) |
| 4 | 39.7 C | ― | 56.5 C | ― |
| 5 | 55.8 CH | 0.77 (m) | 47.9 CH | 1.64 (dd, 10.5, 1.8) |
| 6 | 18.7 CH2 | 1.41 (m), 1.23 (m) | 20.5 CH2 | 1.50 (m), 1.10 (m) |
| 7 | 33.1 CH2 | 1.34 (m), 1.13 (m) | 32.3 CH2 | 1.51 (m), 1.21 (m) |
| 8 | 39.7 C | ― | 39.7 C | ― |
| 9 | 47.9 CH | 1.53 (m) | 47.8 CH | 1.89 (m) |
| 10 | 38.4 C | ― | 38.2 C | ― |
| 11 | 23.8 CH2 | 2.03 (m), 1.75 (m) | 23.6 CH2 | 2.07 (d, 11.1), 1.97 (dd, 11,1, 3.4) |
| 12 | 122.5 CH | 5.26 (t, 3.2) | 122.5 CH | 5.48 (t, 3.4) |
| 13 | 144.3 C | ― | 144.1 C | ― |
| 14 | 42.1 C | ― | 42.0 C | ― |
| 15 | 28.2 CH2 | 1.86 (m), 0.98 (m) | 28.1 CH2 | 2.17 (m), 1.99 (m) |
| 16 | 23.7 CH2 | 2.03 (m), 1.76 (m) | 23.7 CH2 | 2.18 (m), 1.93 (m) |
| 17 | 46.6 C | ― | 46.9 C | ― |
| 18 | 44.3 CH | 2.98 (dd, 14.0, 3.7) | 47.4 CH | 3.23 (dd, 13.5, 4.6) |
| 19 | 47.6 CH2 | 2.10 (bt, 14.0), 1.57 (m) | 41.8 CH2 | 2.64 (bt, 13.5), 2.25 (m) |
| 20 | 69.8 C | ― | 148.9 C | ― |
| 21 | 36.1 CH2 | 1.73 (m), 1.57 (m) | 30.2 CH2 | 2.30 (m), 2.20 (m) |
| 22 | 35.0 CH2 | 1.75 (m, overlap) | 38.3 CH2 | 2.13 (m), 1.93 (m) |
| 23 | 29.2 CH3 | 1.03 (s) | 206.3 CH | 9.67 (s) |
| 24 | 17.6 CH3 | 0.81 (s) | 10.6 CH3 | 1.43 (s) |
| 25 | 16.7 CH3 | 0.82 (s) | 16.9 CH3 | 1.00 (s) |
| 26 | 17.4 CH3 | 0.76 (s) | 17.2 CH3 | 0.96 (s) |
| 27 | 25.9 CH3 | 1.06 (s) | 26.0 CH3 | 1.23 (s) |
| 28 | 179.9 C | ― | 179.4 C | ― |
| 29 | 25.6 CH3 | 1.32 (s) | 107.0 CH2 | 4.77 (s), 4.81 (s) |
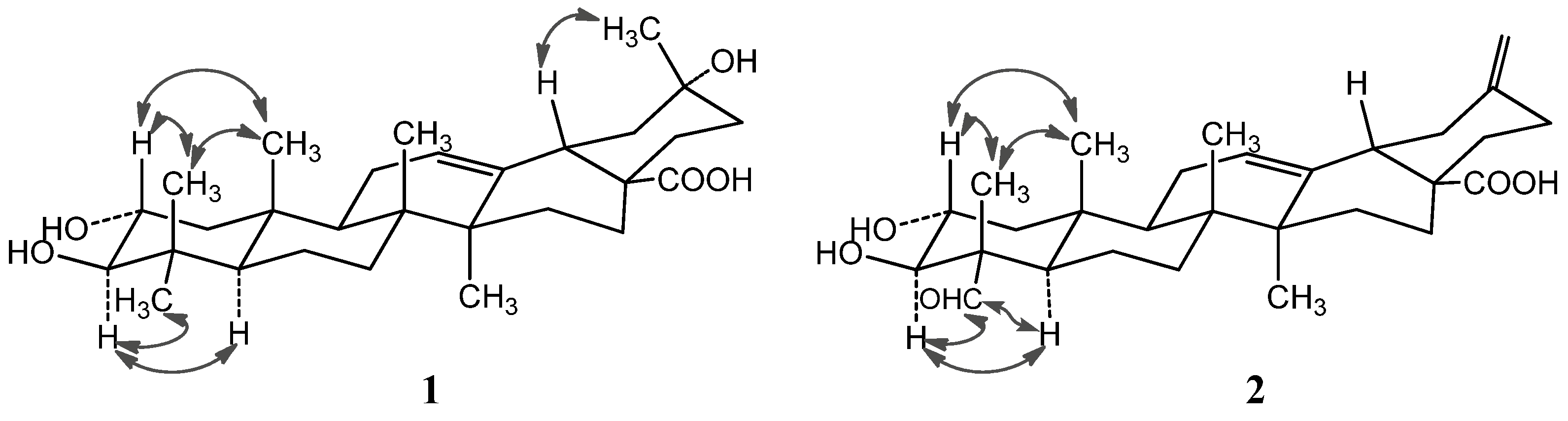
Figure 2.
Important NOESY correlations of compounds 1 and 2.
Figure 2.
Important NOESY correlations of compounds 1 and 2.
Figure 3.
Selected HMBC correlations of compounds 1 and 2.
Figure 3.
Selected HMBC correlations of compounds 1 and 2.
The NOE correlations of H-2 with Me-24 (
δH 0.81) and Me-25 (
δH 0.82), and the large proton spin-coupling constant of H-3 (
3JH-2,H-3 = 9.5 Hz) further supported that the hydroxyl groups at C-2 and C-3 were
α- and
β-orientation, respectively [
5]. Therefore,
1 was unambiguously identified as 2
α,3
β,20
α-trihydroxy-30-norolean-12-en-28-oic acid.
Compound
2 was obtained as a white amorphous powder with molecular formula C
29H
42O
5 as determined by HRESIMS. The
1H- and
13C-NMR data (see
Table 1) suggested that
2 is also a pentacyclic nortriterpenoid. Careful analysis of the
1H- and
13C-NMR spectra indicated that
2 closely resembled 2
α,3
β-dihydroxy-30-noroleana-12,20(29)-dien-28-oic acid [
5], a literature reported noroleanane triterpenoid which was also obtained in this study as compound
4. Except that the resonances for the methyl group at C-23 in
4 were replaced by signals [
δH 9.67 (1H, s, H-23);
δC 206.3 (C-23)] for an aldehyde group in
2. These findings led to establish the structure of
2 as shown in
Figure 1, which was well supported by the 2D NMR data. In the HMBC spectrum, the
1H−
13C long-range correlations from
δH 9.67 (1H, H-23) to
δC 10.6 (C-24), and from
δH 4.06 (1H, H-3) to
δC 206.3 (C-23) and 10.6 (C-24) were observed, which confirmed the attachment of the aldehyde group at C-4 (
Figure 3). The HMBC correlations from
δH 4.24 (1H, H-2) to
δC 76.9 (C-3), and from
δH 4.06 (1H, H-3) to
δC 67.9 (C-2), 56.5 (C-4), 206.3 (C-23) and 10.6 (C-24), and the large proton spin-coupling constant of H-3 (
3JH-2,H-3 = 9.3 Hz) supported the
β- and
α-orientation of the hydroxyls at C-3 and C-2, respectively [
5]. The HMBC correlations from H
2-29 (
δH 4.81, 4.77) to C-19 (
δC 41.8) and C-21 (
δC 30.2) confirmed the exocyclic double bond at C-20(29). The
α-orientation of the aldehyde group at C-4 was evidenced by significant NOE correlations in the NOESY spectrum of H-23 (
δH 9.67) with H-3 (
δH 4.06) and H-5 (
δH 1.64), and of H-2 with H
3-24 (
δH 1.43) and H
3-25 (
δH 1.00) (
Figure 2). Therefore, compound
2 was determined as 2
α,3
β-dihydroxy-23-oxo-30-norolean-12,20(29)-dien-28-oic acid.
The four known compounds were identified as 3
β-akebonoic acid (
3) [
16], 2
α,3
β-dihydroxy-30-noroleana-12,20(29)-dien-28-oic acid (
4) [
5], 3
α-akebonoic acid (
5) [
16] and quinatic acid (
6) [
17] by comparison of their spectral data (
1H and
13C-NMR and MS) to those reported in the literature. They were all reported here from the pericarps of
A. trifoliata for the first time.
The
in vitro antibacterial activity of the six isolated compounds against four Gram-positive bacteria [
St. aureus (CMCC26003),
B. cereus (CMCC63302),
B. subtilis (CMCC63501) and
MRSA] and three Gram-negative bacteria [
E. coli (CMCC44102),
Sa. typhimurium (CMCC44102) and
Sh. dysenteriae (CMCC51252)] were evaluated using a microdilution titre assay as described in the Experimental section. As shown in
Table 2, compound
3 displayed the best
in vitro bacteriostatic activity against all the Gram positive bacteria assayed with MICs from 12.5 to 25 μg/mL. Compounds
4,
5 and
6 showed bacteriostatic activity against
B. cereus CMCC63302 and
B. subtilis CMCC63501 with MICs of 50 and 25, of 25 and 50, and of 50 and 12.5 μg/mL, respectively. Neither compound showed antibacterial activity toward the three assayed Gram-negative bacteria strains (MICs > 200 μg/mL). It is noteworthy that compound
3 showed bacteriostatic activity against
MRSA with MIC 25 μg/mL, which was much stronger than reference compound of Kanamycin sulfate (MIC 125 μg/mL).
MRSA, firstly reported as a methicillin-resistant
Staphylococcus aureus in the U.K. in 1961, is now a multidrug-resistant strain responsible for a rapidly increasing number of serious infectious diseases throughout the world, which is lacking of effective antimicrobial agents for the control and therapy for its infection.
Table 2.
MIC values of compounds 1–6 in μg/mL against seven bacterial strains.
Table 2.
MIC values of compounds 1–6 in μg/mL against seven bacterial strains.
| Bacteria | 1 | 2 | 3 | 4 | 5 | 6 | K | C |
|---|
| S. aureus (CMCC26003) | >200 | >200 | 25 | 200 | >200 | >200 | 1.9 | 50 |
| MRSA | >200 | >200 | 25 | 200 | >200 | >200 | 125 | >200 |
| B. cereus (CMCC63302) | >200 | >200 | 25 | 50 | 25 | 50 | 3.9 | 200 |
| B. subtilis (CMCC63501) | >200 | >200 | 12.5 | 25 | 50 | 12.5 | 3.9 | 200 |
| E. coli (CMCC44102) | >200 | >200 | >200 | >200 | >200 | >200 | 3.9 | 12.5 |
| S. typhimurium (CMCC44102) | >200 | >200 | >200 | >200 | >200 | >200 | 3.9 | 12.5 |
| S. dysenteriae (CMCC51252) | >200 | >200 | >200 | >200 | >200 | >200 | 3.9 | 12.5 |
These compounds were also tested for their
in vitro cytotoxicity against three human tumor cell lines, A549 (human lung adenocarcinoma), HeLa (human cervical carcinoma) and HepG2 (human liver hepatocellular carcinoma), using the MTT method as described. The resulting IC
50 values are displayed in
Table 3. Compounds
4 and
5 showed interesting cytotoxicity against A549 and HeLa cell lines, with IC
50 values 8.8 and 5.6 μM, respectively. Compound
3 also showed cytotoxic activity (IC
50 49.48, 28.63 and 52.89 μM) against the three tested tumor cell lines, but it was weaker than
4 and
5. New compounds
1–
2 and compound
6 did not exhibit cytotoxic activity (IC
50 > 100 μM) in this bioassay. It could be deduced that the exocyclic double bond at C-20(29) might be an important active center for this type of nortriterpenoids to “maintain” their potential cytotoxicity, based on comparison of the structures and activities of
1 to
4. Comparison of the chemical structures of
3 and
4 versus 5 indicated that the
α-orientation of the hydroxyl group at C-3 could strengthen the cytotoxic activity of this type of nortriterpenoids, while the
α-hydroxyl group at C-2 seems not necessary. Moreover, a negative effect on the cytotoxicity was evident when Me-23 was oxidized (
i.e., replaced by an -CHO group), as supported by analyzing the structure-active relationship of
2 and
4.
Table 3.
Cytotoxicity of compounds 1–6 (IC50, µM).
Table 3.
Cytotoxicity of compounds 1–6 (IC50, µM).
| Compounds | A549 | HeLa | HepG2 |
|---|
| 1 | >100 | >100 | >100 |
| 2 | >100 | >100 | >100 |
| 3 | 49.48 ± 8.64 | 28.63 ± 7.41 | 52.89 ± 5.28 |
| 4 | 8.770 ± 0.59 | 16.33 ± 0.12 | 14.28 ± 0.49 |
| 5 | 10.59 ± 0.69 | 5.61 ± 0.00 | 10.39 ± 1.17 |
| 6 | >100 | >100 | >100 |
| Adriamycin | 0.69 ± 0.07 | 0.47 ± 0.06 | 1.22 ± 0.02 |
Compounds
1,
2,
5 and
6 were further tested for their
α-glucosidase inhibitory activity. The results are listed in
Table 4, with acarbose used as a reference compound. Compound
5 showed the best
α-glucosidase inhibitory activity with IC
50 value 0.035 mM, which was about twelve-fold stronger than acarbose (IC
50 0.409 mM). Compound
6 showed significant
α-glucosidase inhibitory activity with IC
50 value (0.10 mM) about four-fold stronger than the reference compound. Though the
α-glucosidase inhibitory activities of
1 and
2 (IC
50 0.367 and 0.220 mM, respectively) were inferior to
5 and
6, they were still more potent than acarbose. These results indicated that 30-noroleanane triterpenes in the pericarps of
A. trifoliata were effective
α-glucosidase inhibitors promising to be developed as effective and safe hypoglycemic agents for diabetes chemotherapy [
18].
Table 4.
α-Glucosidase inhibitory activity of compounds 1, 2, 5 and 6.
Table 4.
α-Glucosidase inhibitory activity of compounds 1, 2, 5 and 6.
| Compounds | IC50 (mM) |
|---|
| 1 | 0.367 ± 0.003 |
| 2 | 0.220 ± 0.004 |
| 5 | 0.035 ± 0.002 |
| 6 | 0.100 ± 0.001 |
| Acarbose | 0.409 ± 0.006 |
3. Experimental
3.1. General Information
Optical rotation were measured on a Perkin-Elmer 341 polarimeter (Perkin- Elmer, Waltham, MA, USA) with MeOH as solvent at the wavelength of 589 nm and 20 °C to gain their specific optical rotation [α] values after calculation. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Advance 600 NMR spectrometer (Bruker Biospin corporation, Billerica, MA, USA), Bruker advance 500M NMR spectrometer (Bruker) and a Bruker DRX-400 NMR spectrometer (Bruker Biospin, Rheistetten, Germany) with the solvent residual peaks of δH 7.22, 7.58, 8.74 and δC 123.4, 135.4, 149.7 for pyridine-d5, and δH 2.50 and δC 39.5 for DMSO-d6. HR-ESI-MS mass spectra were obtained on a Bruker maXis instrument (Bruker Daltonik GmbH, Bremen, Germany) in a positive ion mode after direct injection of the test solutions. ESI-MS data were obtained using a MDS SCIEX API 2000 LC/MS/MS system (Applied Biosystems, Foster City, CA, USA) in both positive and negative ion modes in the range of m/z 50–1000 after the test solutions were directly injected into the ESI source by a syringe pump. Preparative HPLC was carried out on a CXTH P3000 HPLC pump and a UV 3000 UV-Vis Detector with a Fuji-C18 column (10 um–100 A, ChuangXinTongHeng Science And Technology Co., Ltd, Beijing, China); the Performance of MPLC (medium pressure liquid chromatography) is a CXTH P3000 HPLC pump, a UV 3000 UV-Vis Detector and a C18 column (400 × 25 mM i.d, 50 μM, YMC Co. Ltd., Kyoto, Japan).
Column chromatography (CC) was performed with silica gel (80–100 and 200–300 mesh, Qingdao Haiyang Chemical Co., Qingdao, China), YMC ODS-A (50 μm, YMC Co. Ltd., Kyoto, Japan), Sephadex LH-20 (Pharmacia Fine Chemical Co. Ltd., Uppsala, Sweden), MCI gel CHP 20P (75–150 μM, Mitsubishi Chemical Corp., Tokyo, Japan). Analytical grade petroleum ether (b.p. 60–90 °C), methanol, ethyl acetate, chloroform, n-butanol, acetone were purchased from Tianjin Fuyu Fine Chemical Industry Co. (Tianjin, China); HPLC grade methanol was purchased from J&K Chemical Ltd. (Beijing, China); Fraction were monitored by precoated HSGF254 TLC (Yantai Jiangyou Silica Gel Co. Ltd, Yantai, China), and spot detection was performed under fluorescent light (λ = 254 nm), and then spraying 10% H2SO4 in ethanol, followed by heating. Kanamycin sulfate, resazurin, pyridine-d5, DMSO-d6, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and α-glucosidase were purchased from Sigma Chemical Co. (Sigma-Aldrich, St. Louis, MO, USA). Roswell Park Memorial Institute (RPMI)-1640 medium and fetal calf serum were purchased from Gibco BRL (Gaithersburg, MD, USA). Adriamycin was obtained from Pfizer Italia SRL (Roma, Italy). p-Nitrophenyl-α-d-glucopyranoside (PNPG) and acarbose were obtained from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan).
3.2. Plant Materials
The pericarps of Akebia trifoliata were collected in September 2009, at Liye of Longshan, Hunan Province, China, identified by Prof. Fu-Wu Xing at South China Botanical Garden, the Chinese Academy of Sciences (CAS). A voucher specimen (No. 20090920) was deposited at the Laboratory of Bioorganic Chemistry of the South China Botanical Garden, Chinese Academy of Sciences.
3.3. Extraction and Isolation
Air-dried pericarps of Akebia trifoliata (3 kg) were powdered and extracted three times (3 days each) with 95% EtOH (9 L × 3) at room temperature (25–32 °C). The concentration of the solution under vacuum gave a reddish solid. Then, the resulting residue was suspended in H2O (1.5 L) and successively partitioned with petroleum ether (1.5 L × 3) and ethyl acetate (1.5 L × 3) to afford petroleum ether-soluble (40.0 g) and EtOAc-soluble (180 g) fractions after condensation to dryness in vacuo. The petroleum ether-soluble fraction was subjected to silica gel column chromatography (1000 × 105 mM i.d.) using a gradient system of petroleum ether-acetone (100:0, 20:1, 10:1, 5:1, 2:1, 1:1, 0:100, v/v, each 1.5 L) to give nine fractions (E1-E9) after pooled according to their TLC profiles. E3 (0.45 g) was repeatedly chromatographied on Sephadex LH-20 column (1550 × 13.4 mM i.d.) eluted with acetone to yield compound 5 (6.5 mg). E4 (1.7 g) was purified on silica gel column chromatography (1000 × 105 mM i.d.) using petroleum ether-acetone (10:1, 9:1, 8:1, 7:1, 6:1, v/v, each 250 mL) as elution system, and then further purified by using MPLC eluted with MeOH-H2O (95:5, v/v) to yield compound 3 (2.2 mg). E7 (1.8 g) and E8 (2 g) were passed through a MCI gel column (200 × 40 mM i.d.) for depigmentation. The resultant methanolic eluate (0.9 g) of E7 was sequentially separated by MPLC eluted with a gradient of methanol in water (75:25, 80:20, 85:15, 90:10, 100:0, v/v, each 70 mL), and by Sephadex LH-20 column (1550 × 13.4 mM i.d.) chromatography eluted with MeOH, and silica gel CC using CHCl3-MeOH (98:2, v/v) elution to obtain compounds 2 (4 mg) and compound 4 (2.0 mg). The resultant methanolic eluate (1.1 g) of E8 was sequentially separated by MPLC eluted with a gradient of methanol in water (60:0, 70:30, 80:20, 90:10, 100:0, v/v, each 100 mL) to give eight fractions (E8-1-E8-8). The EtOAc-soluble fraction was subjected to silica gel CC (1000 × 105 mM i.d.) using a gradient of CHCl3-MeOH (97:3, 90:10, 85:15, 70:30, 60:40, 0:100, v/v, each 3 L) to give ten fractions (F1-F6). Fraction F3 (6.8 g), obtained by elution CHCl3-MeOH (85:15, v/v), was further subjected to silica gel CC (800 × 50 mM i.d.) and successively eluted with CHCl3-MeOH (98:2, 95:5, 90:10, v/v, each 0.5 L) to yield six sub-fractions (F5-1–F5-6), Sub-fraction F5-3 (1.6 g) was separated by MPLC using MeOH-H2O (60:40, 70:30, 80:20, 90:10, 100:0, v/v, each 350 mL) at a flow rate of 10 mL/min, and further purified by Sephadex LH-20 column (1550 × 13.4 mM i.d.) chromatography eluted with MeOH to obtain compound 6 (9 mg). Fraction F5 (20.5 g), obtained by elution CHCl3-MeOH (60:40, v/v), was further subjected to silica gel CC (1000 × 105 mM i.d.), and successively eluted with CHCl3-MeOH (90:10, 80:20, 70:30, 60:40, v/v, each 1.5 L) to obtain six sub-fractions (F5-1-F5-6). F5-3 (10 g) was separated by MPLC, and eluted with MeOH-H2O (70:30, v/v) to yield compound 1 (5 mg).
2α,3β,20α-Trihydroxy-30-norolean-12-en-28-oic acid (
1). White amorphous powder.
![Molecules 19 04301 i001]()
+106.4 (
c 0.70, MeOH). IR (KBr)
νmax 3428, 2939, 2873, 1699, 1459, 1380, 1286, 1450, 1116, 1047 cm
−1. ESI-MS (+)
m/
z: 497 [M+Na]+, 971 [2M+Na]+; ESI-MS (−)
m/
z: 473 [M−H]
–, 947 [2M−H]
–;. HR-ESI-MS
m/
z: 497.3235 [M+Na]+ (calcd for C29H46NaO5, 497.3237). For
1H-NMR (600 MHz, C
5D
5N) and
13C-NMR (150 MHz, C
5D
5N) data, see
Table 1.
2α,3β-Dihydroxy-23-oxo-30-norolean-12,20(29)-dien-28-oic acid (
2). White amorphous powder.
![Molecules 19 04301 i001]()
+190.0 (
c 0.17, MeOH); IR (KBr)
νmax 3413, 2935, 2861, 1720, 1689, 1456, 1384, 1292, 1220, 1054 cm
−1. ESI-MS (+)
m/
z: 493 [M+Na]
+, ESI-MS (−)
m/
z: 469 [M−H]
–. HR-ESI-MS
m/
z: 493.2922 [M+Na]
+ (calcd for C
29H
42NaO
5, 493.2924). For
1H-NMR (600 MHz, C
5D
5N) and
13C-NMR (150 MHz, C
5D
5N) data, see
Table 1.
3.4. Antibacterial Assay
The bacteriostatic activity of compounds
1–
6 were monitored according to the method of Rahman with slight modifications [
19]. Briefly, indicator solution (100 μL, resazurin in sterile water, 100 μg/mL) was first placed into each of the sterility control wells (11th column) on the 96 well plates, and indicator solution (about 7.5 mL, 100 μg/mL) was mixed with test organism (5 mL, 10
6 cfu/mL, OD
600 = 0.07) followed by transferring (100 μL, each) to growth control wells (12th column) and all test wells (1–10th column). Then, each of 100 μL of the test samples in beef extract peptone medium, the positive control solution were prepared by adding kanamycin sulfate and cefradine instead of the samples and the negative control solution (3% DMSO of beef extract peptone medium) were applied to the wells in the 1st column of the plates. Once all samples and controls were properly applied to the 1st column of wells on the plate, half of the homogenized content (100 μL) from these wells was then parallel transferred to the 2nd column of wells, and each subsequent well was treated similarly (doubling dilution) up to the 10th column, followed by discarding the last 100 μL aliquot. Finally, the plates were incubated at 37 °C for 5–6 h until the color of growth control change to pink. The lowest concentration for each test compound at which color change occurred was recorded as the MIC value of the test compound. The Gram-(+) bacteria,
Staphyloccocus aureus (CMCC26003),
Bacillus cereus (CMCC63302) and
Bacillus subtilis (CMCC63501), and Gram-(‒) bacteria
Escherichia coli (CMCC44102),
Salmonella typhimurium (CMCC44102) and
Shigella dysenteriae (CMCC51252) were obtained from the Guangdong Institute of Microbiology (Guangzhou, China). The multi-drug resistant
Staphyloccocus aureus (
MRSA) was kindly provided by Dr. Wei, X.Y. (South China Botanical Garden).
3.5. Cytotoxic Assay
Compounds
1−
6 were evaluated for their cytotoxicity against three human cancer cell lines, human lung adenocarcinoma (A549), human cervical carcinoma (HeLa) and human liver hepatocellular carcinoma (HepG2). The three tumor cell lines were generously provided by Kunming Institute of Zoology, Chinese Academy of Sciences. The cytotoxic activities of the tested compounds were assayed according to the MTT method by using 96 well plates [
20]. Briefly, the cells were cultured in RPMI-1640 medium, supplemented with 10% fetal bovine serum in a humidified atmosphere with 5% CO
2 at 37 °C. One hundred μL of adherent cells at the density of 5 × 10
4 cell/mL was seeded into each well of 96-well cell culture plates and incubated in 5% CO
2 at 37 °C for 24 h to form a monolayer on the flat bottoms. Then, removed the supernatant per well and subsequently added with 100 μL of fresh medium and 100 μL of medium containing a test compound. The plate was then incubated in 5% CO
2at 37 °C. After 72 h, 20 μL of 5 mg/mL MTT in DMSO was added into each well and incubated for 4 h. The supernatant per well was carefully removed and 150 μL of DMSO was added. The plate was then vortex shaken for 15 min to dissolve blue formazan crystals. The optical density (OD) of each well was measured on a Genois microplate reader (Tecan GENios, Männedorf, Switzerland) at the wavelength of 570 nm. All experiments were performed in triplicate and adriamycin was used as a positive control. In each experiment, each of the tumor cell lines was exposed to the test compound at concentrations of 50, 25, 12.5, 6.25, 3.125, 1.5625 µg/mL. The inhibitory rate of cell growth was calculated according to the following formula: Inhibition rate (%) = (OD
control− OD
treated)/OD
control× 100%. IC
50 values were calculated by SPSS 16.0 statistic software. The values were based on three individual experiments and expressed as means ± standard deviation (SD).
3.6. α-Glucosidase Inhibition Assay
The
a-glucosidase inhibitory activity of
1,
2,
5 and
6 were determined spectrophotometrically in a 96-well microtiter plate based on
p-nitrophenyl-
α-
d-glucopyranoside (PNPG) as a substrate following the method described in the literature with slight modifications [
21,
22]. In brief,
α-glucosidase (20 μL, 0.8 U/mL) and various concentrations (500, 250, 125, 62.5, 31.25, 15.625 µg/mL) of tested compounds (120 μL) in 67 mM phosphate buffer (pH 6.8) were mixed at room temperature for 10 min. Reactions were initiated by addition of 5.0 mM PNPG (20 μL). The reaction mixture was incubated for 15 min at 37 °C in a final volume of 160 μL. Then, 0.2 M Na
2CO
3 (80 μL) was added to the incubation solution to stop the reaction. The activities were detected in a 96-well plate, and the absorbance was determined at 405 nm (for
p-nitrophenol). The negative blank was set by adding phosphate buffer instead of the sample via the same way as the test. Acarbose was utilized as positive control. The blank was set by adding phosphate buffer instead of the
α-glucosidase using the same method. Inhibition rate (%) = [(OD
negative control− OD
blank) − (OD
test − OD
test blank)]/(OD
negative blank− OD
blank) × 100%. IC
50 values of the samples were calculated using the Microsoft Office Excel soft.



{kind=link}
{kind=link}
{kind=link}
 +106.4 (c 0.70, MeOH). IR (KBr) νmax 3428, 2939, 2873, 1699, 1459, 1380, 1286, 1450, 1116, 1047 cm−1. ESI-MS (+) m/z: 497 [M+Na]+, 971 [2M+Na]+; ESI-MS (−) m/z: 473 [M−H]–, 947 [2M−H]–;. HR-ESI-MS m/z: 497.3235 [M+Na]+ (calcd for C29H46NaO5, 497.3237). For 1H-NMR (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data, see Table 1.
+106.4 (c 0.70, MeOH). IR (KBr) νmax 3428, 2939, 2873, 1699, 1459, 1380, 1286, 1450, 1116, 1047 cm−1. ESI-MS (+) m/z: 497 [M+Na]+, 971 [2M+Na]+; ESI-MS (−) m/z: 473 [M−H]–, 947 [2M−H]–;. HR-ESI-MS m/z: 497.3235 [M+Na]+ (calcd for C29H46NaO5, 497.3237). For 1H-NMR (600 MHz, C5D5N) and 13C-NMR (150 MHz, C5D5N) data, see Table 1.