Synthesis, Spectroscopic and Theoretical Studies of New Quaternary N,N-Dimethyl-3-phthalimidopropylammonium Conjugates of Sterols and Bile Acids


Abstract
:1. Introduction
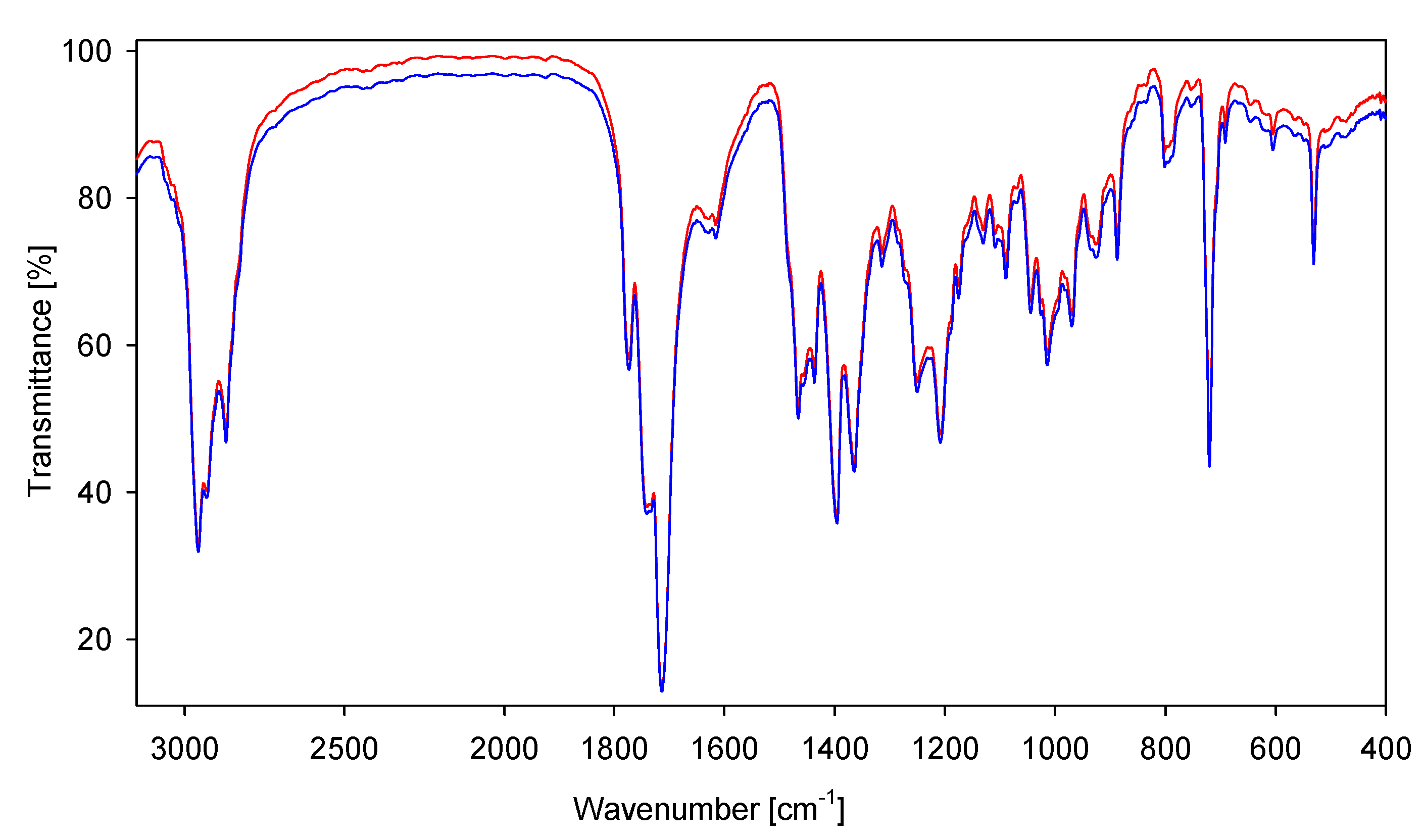
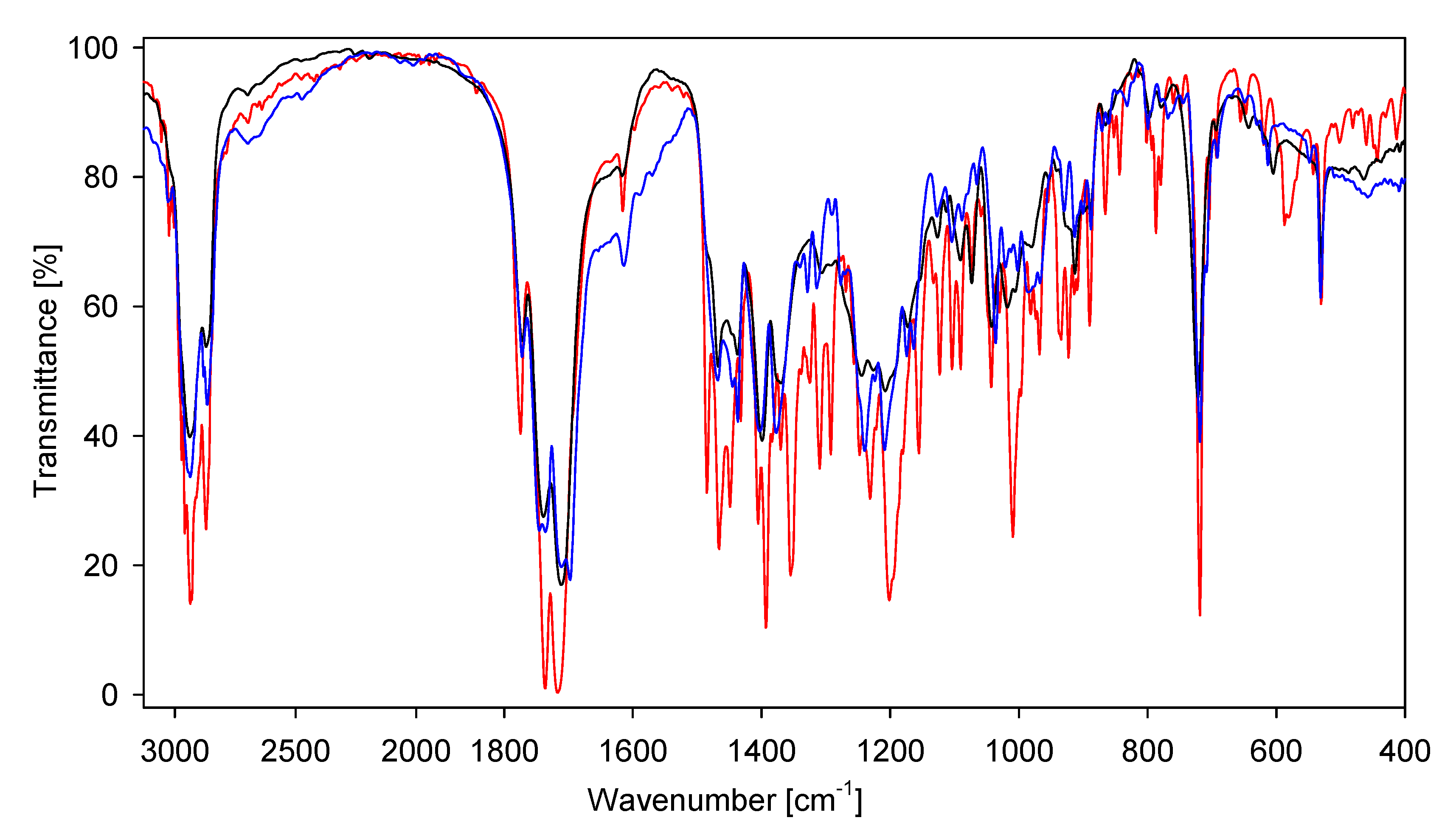
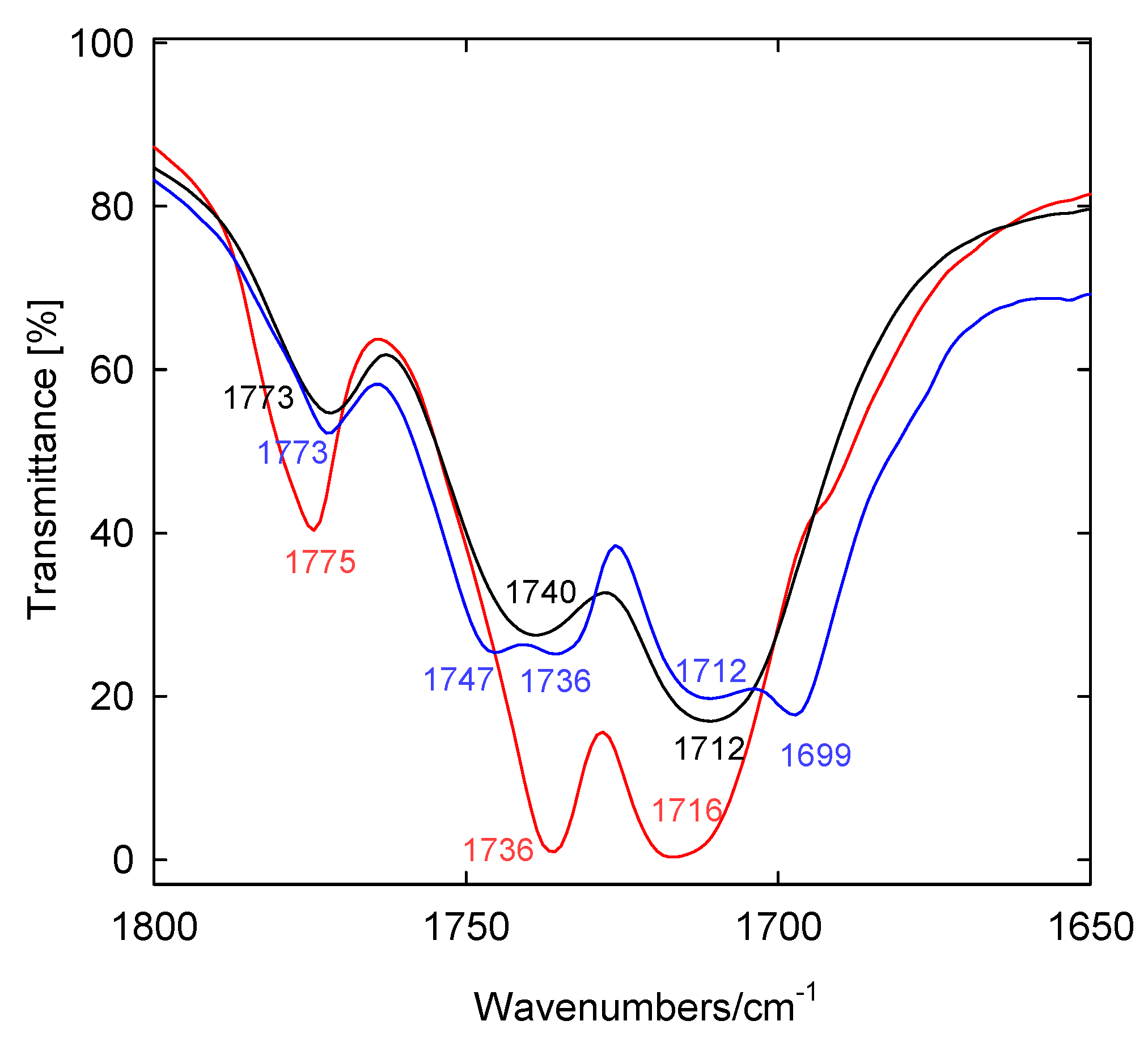
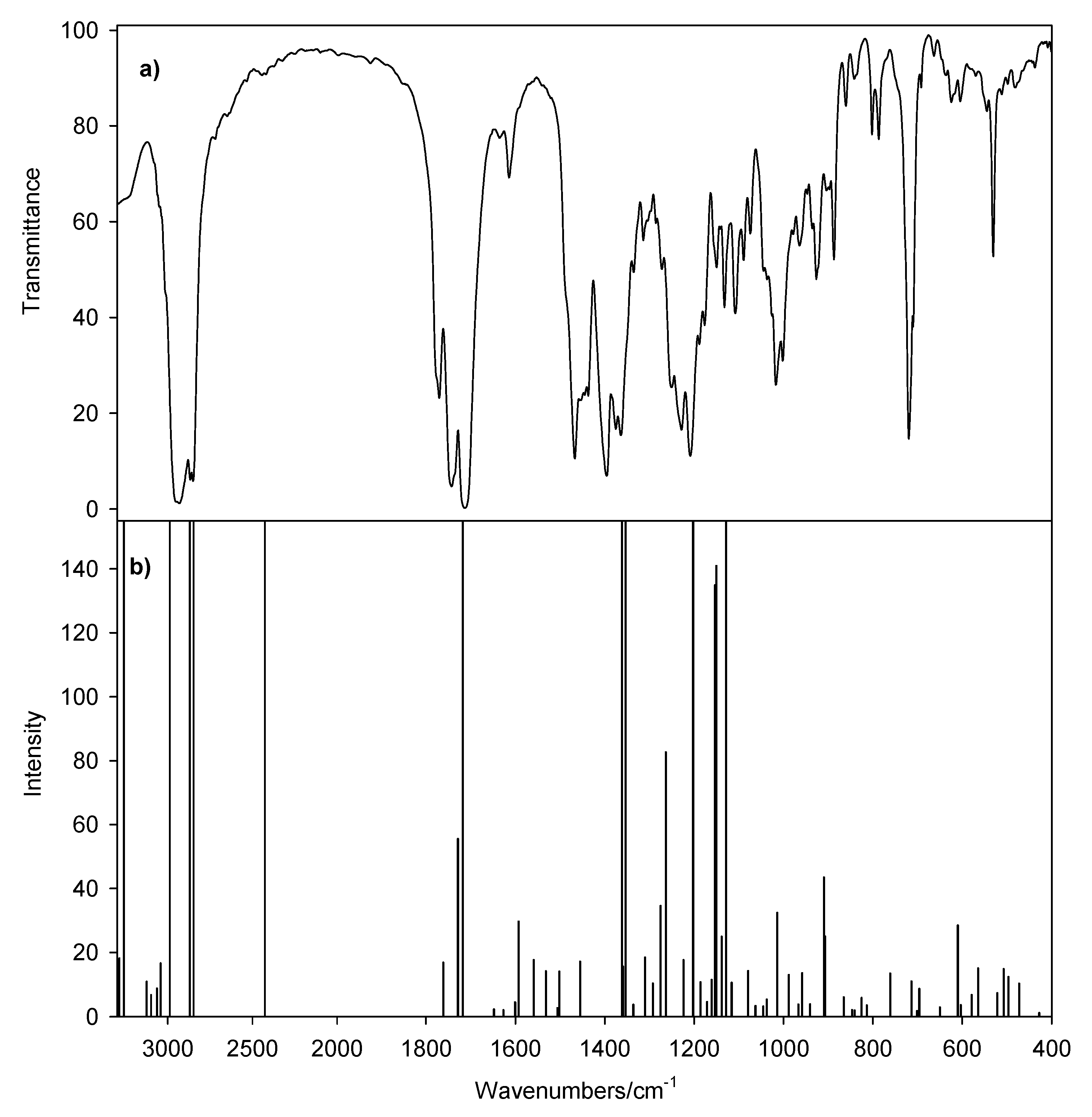
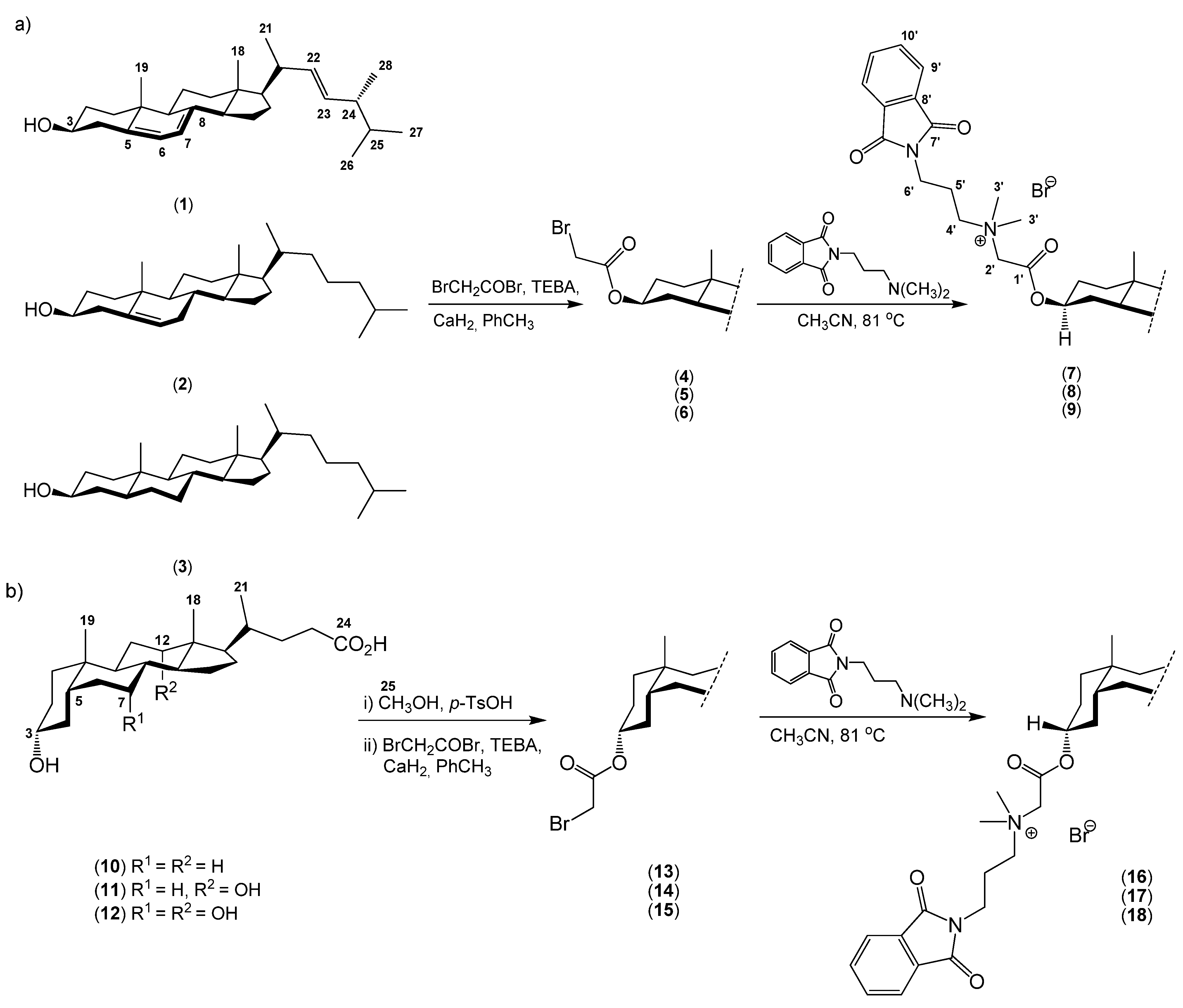
2. Results and Discussion


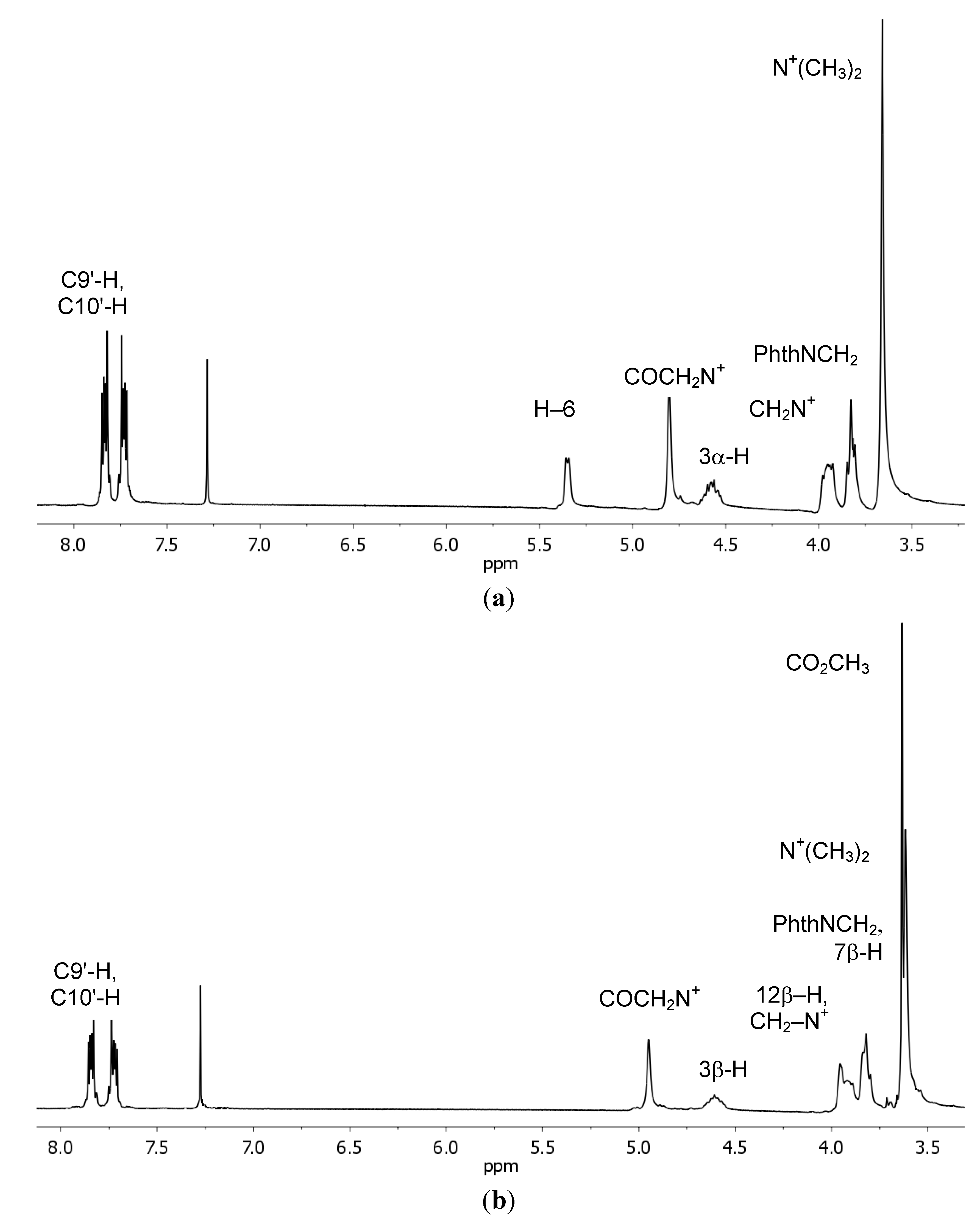{kind=link}
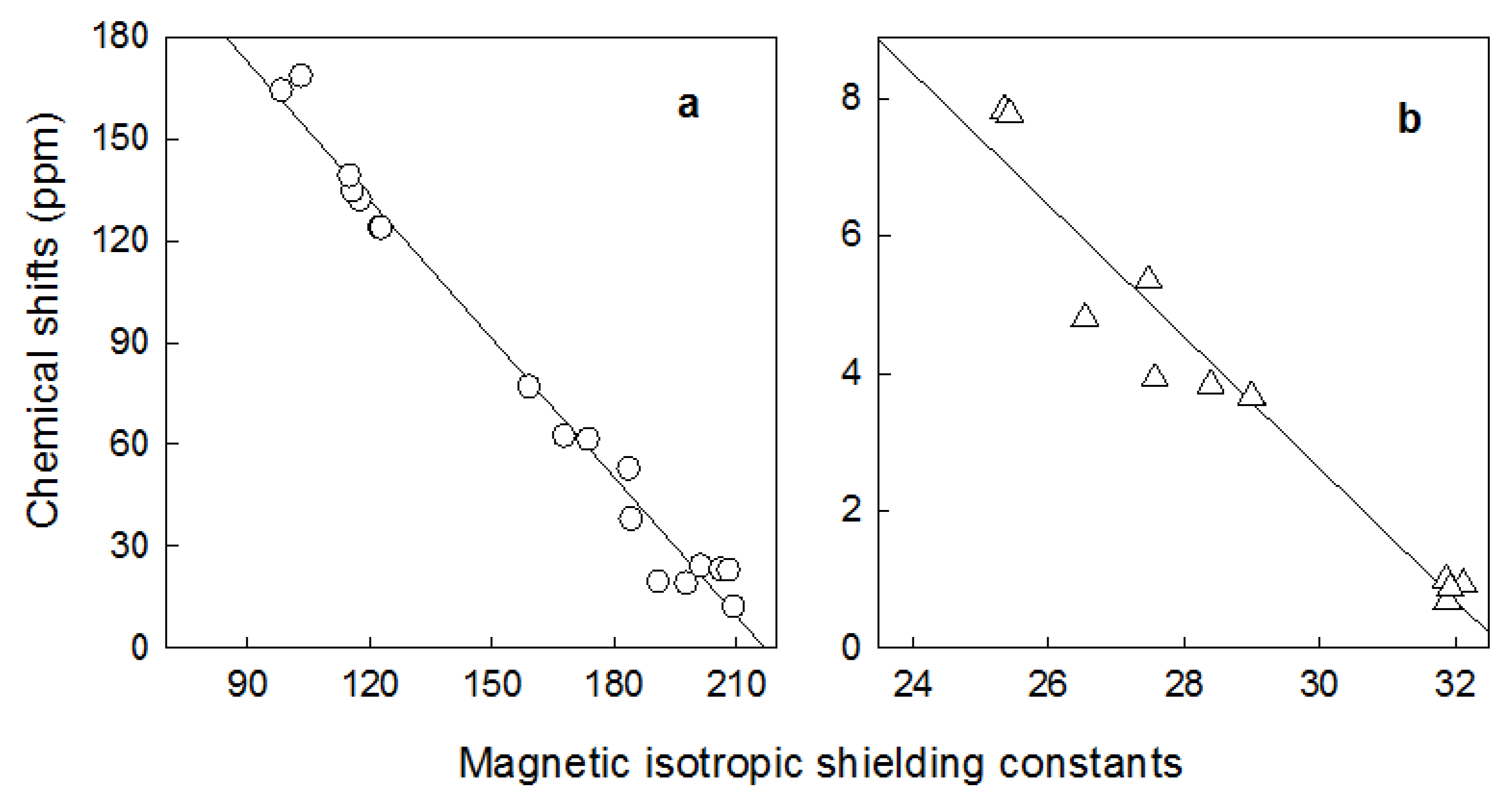{kind=link}
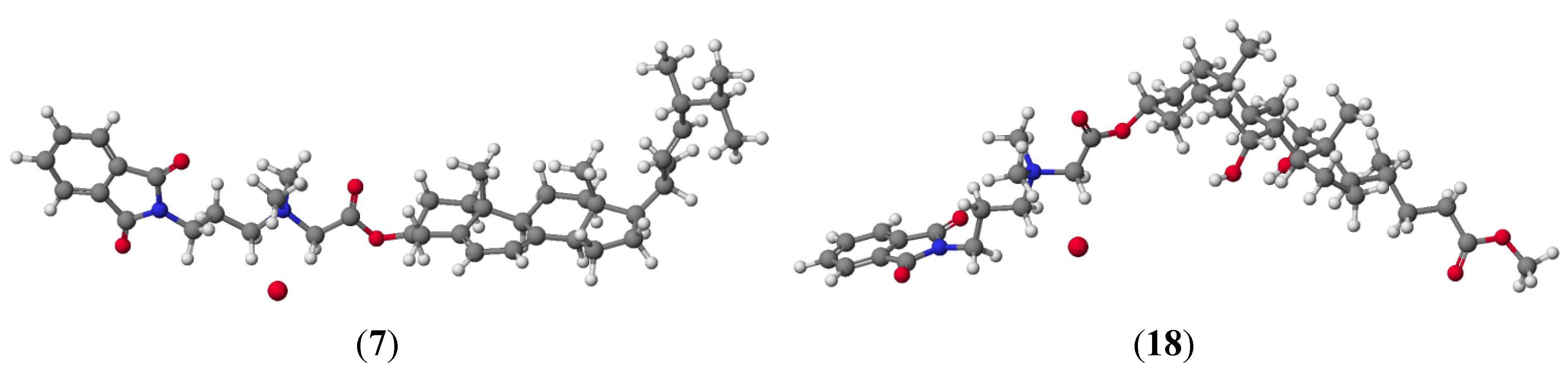{kind=link}
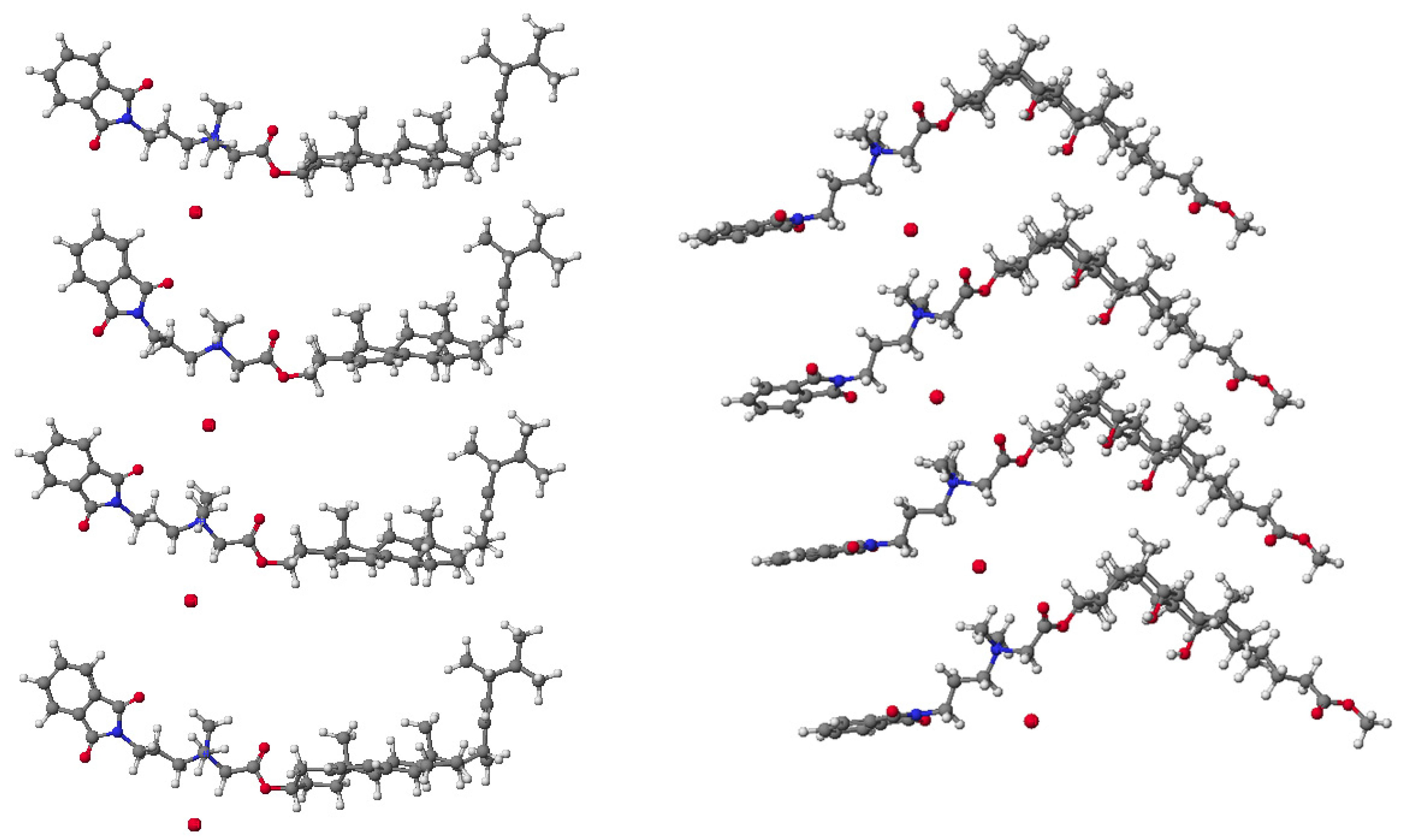{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Focal Predicted Activity (PA > 0.80) | Compounds | |||||
|---|---|---|---|---|---|---|
| 7 | 8 | 9 | 16 | 17 | 18 | |
| Glyceryl-ether mono-oxygenase inhibitor | 0.87 | 0.91 | 0.93 | 0.93 | 0.94 | 0.95 |
| Acylcarnitine hydrolase inhibitor | - | - | 0.81 | 0.83 | 0.91 | 0.94 |
| Alkylacetylglycerophosphatase inhibitor | - | - | - | 0.82 | 0.90 | 0.86 |
| Plasmanylethanolamine desaturase inhibitor | - | - | - | - | 0.71 | 0.78 |
| CYP3A4 substrate | - | - | - | - | 0.70 | 0.73 |
| N-(acyl)ethanolamine-deacylase inhibitor | - | - | - | - | 0.73 | 0.76 |
| Protein-disulfide reductase inhibitor | - | - | 0.72 | - | 0.75 | - |
| Alkenylglycerophosphocholine hydrolase inhibitor | - | - | - | - | 0.80 | - |
| Oxidoreductase inhibitor | 0.87 | 0.77 | - | - | - | - |
| Alcohol O-acetyl-transferase inhibitor | 0.88 | - | - | - | - | - |
| DELTA14-sterol reductase inhibitor | - | 0.73 | - | - | - | - |
| Alkylacetylglycerophosphatase inhibitor | - | - | 0.84 | - | - | - |
| Antieczematic | - | - | - | 0.72 | - | - |
| Glucan endo-1,3-β-D-glucosidase inhibitor | - | - | - | - | 0.73 | - |
| D-lactaldehyde dehydrogenase inhibitor | - | - | - | - | - | 0.74 |
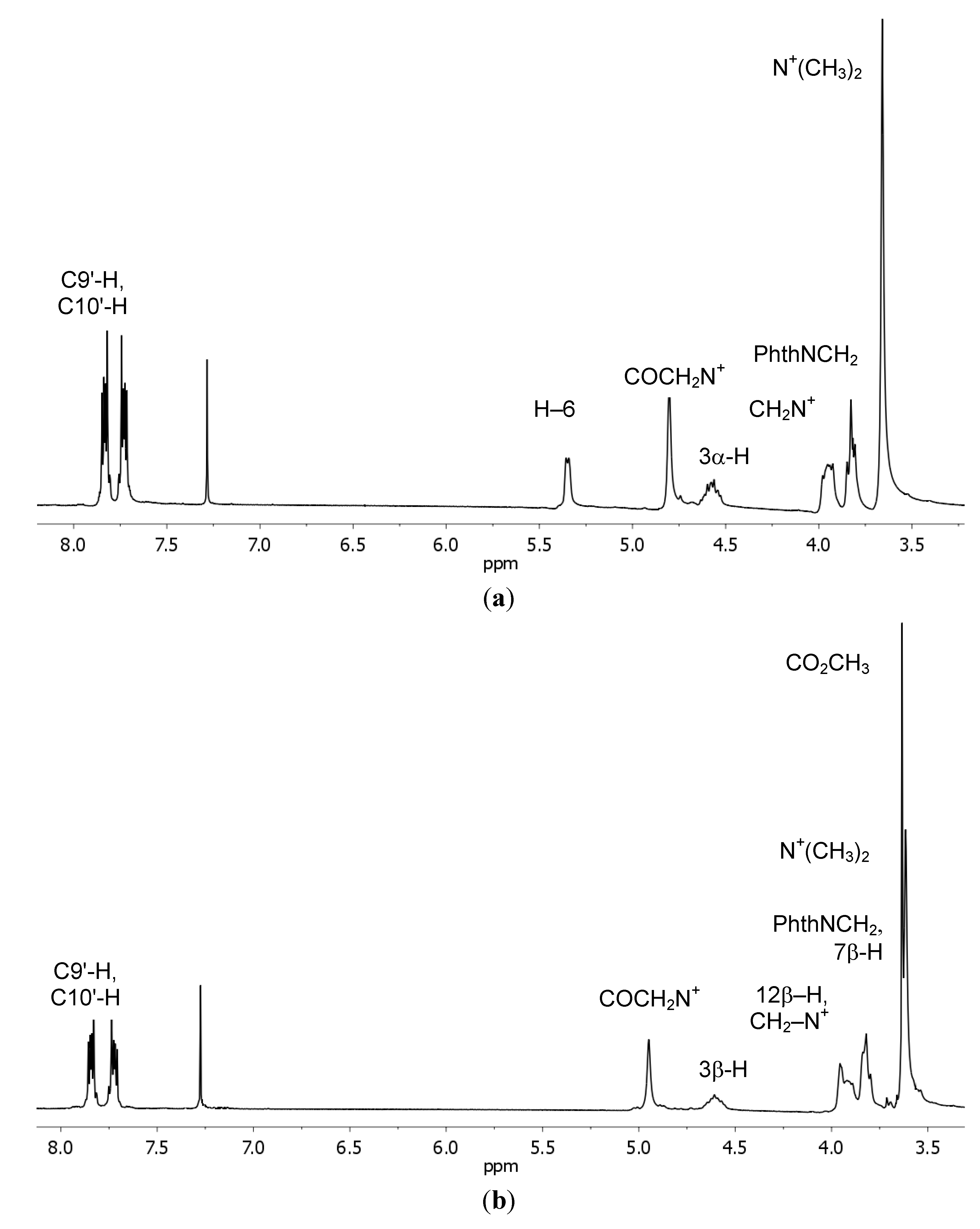
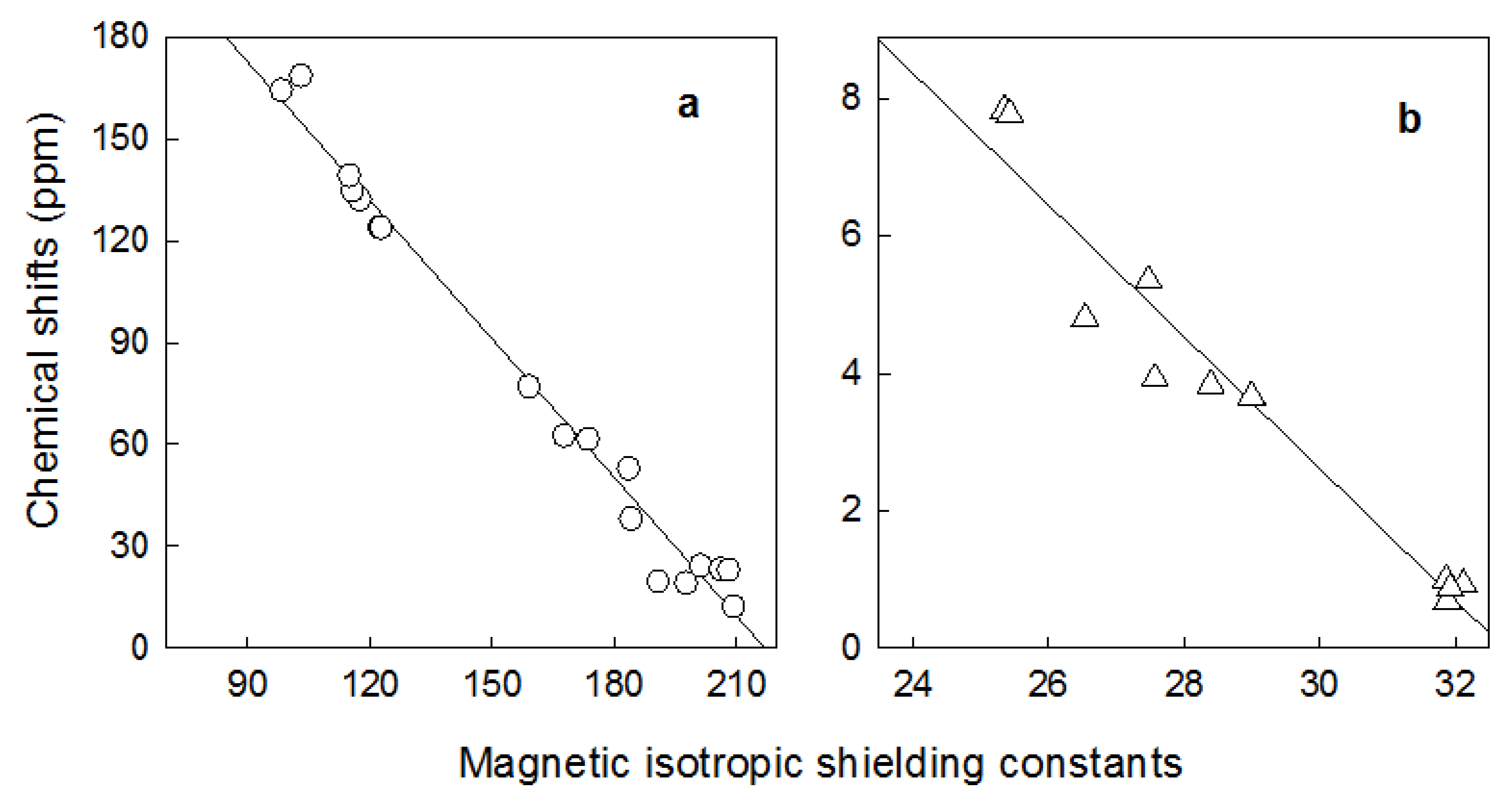
| δexp. | δcalc | σcalc | δexp. | δcalc | σcalc | ||
|---|---|---|---|---|---|---|---|
| C1' | 163.90 | 160.96 | 98.55 | H2' | 4.80 | 5.94 | 26.538 |
| C2' | 62.05 | 66.51 | 168.03 | H3' | 3.66 | 3.58 | 28.992 |
| C3' | 52.38 | 44.94 | 183.90 | H4' | 3.95 | 4.94 | 27.566 |
| C4' | 61.05 | 58.56 | 173.88 | H5' | - | - | 30.748 |
| C5' | 23.76 | 20.94 | 201.56 | H6' | 3.83 | 4.16 | 28.389 |
| C6' | 37.63 | 44.15 | 184.48 | H9' | 7.83 | 7.08 | 25.346 |
| C7' | 168.12 | 154.39 | 105.71 | H10' | 7.77 | 7.00 | 25.430 |
| C8' | 131.72 | 134.68 | 117.88 | H3 | 3.66 | 3.58 | 28.987 |
| C9' | 123.49 | 128.37 | 122.52 | H6 | 5.35 | 5.04 | 27.472 |
| C10' | 134.22 | 137.31 | 115.95 | H18 | 0.68 | 0.80 | 32.113 |
| C3 | 76.57 | 78.28 | 159.37 | H19 | 0.99 | 0.82 | 31.924 |
| C5 | 138.76 | 138.26 | 115.25 | H21 | 0.92 | 0.58 | 32.112 |
| C6 | 123.33 | 127.64 | 123.06 | H26,27 | 0.87 | 0.76 | 31.924 |
| C18 | 11.79 | 9.93 | 209.66 | - | - | - | - |
| C19 | 19.19 | 35.13 | 191.12 | - | - | - | - |
| C21 | 18.65 | 25.79 | 197.99 | - | - | - | - |
| C26 | 22.76 | 14.29 | 206.45 | - | - | - | - |
| C27 | 22.50 | 11.67 | 208.38 | - | - | - | - |
| a a | - | - | 294.9163 | - | - | 31.4274 | - |
| b b | - | - | -1.3593 | - | - | -0.9606 | - |
| r2 c | - | - | 0.9831 | - | - | 0.9443 | - |
| Compound | Heat of Formation | ΔHOF | Energy | ΔEnergy |
|---|---|---|---|---|
| [kcal/mol] | [kcal/mol] | [a.u.] | [a.u.] | |
| 1 | -97.1208 | - | -1113.175477 | - |
| 2 | -140.1058 | - | -1118.205536 | - |
| 3 | -162.7945 | - | -1119.450757 | - |
| 7 | -177.1668 | -80.0460 | -4569.562486 | -3456.387009 |
| 8 | -220.6382 | -80.5324 | -4570.807880 | -3452.602344 |
| 9 | -243.3231 | -80.5286 | -4605.917678 | -3486.466921 |
| 10 | -236.1585 | - | -1204.997432 | - |
| 11 | -278.4616 | - | -1263.040016 | - |
| 12 | -318.1685 | - | -1337.190617 | - |
| 16 | -306.6215 | -70.4630 | -4640.248926 | -3435.251494 |
| 17 | -348.9986 | -70.5370 | −4714.391510 | -3451.351494 |
| 18 | -388.2588 | −70.0903 | -4788.548913 | -3451.358296 |
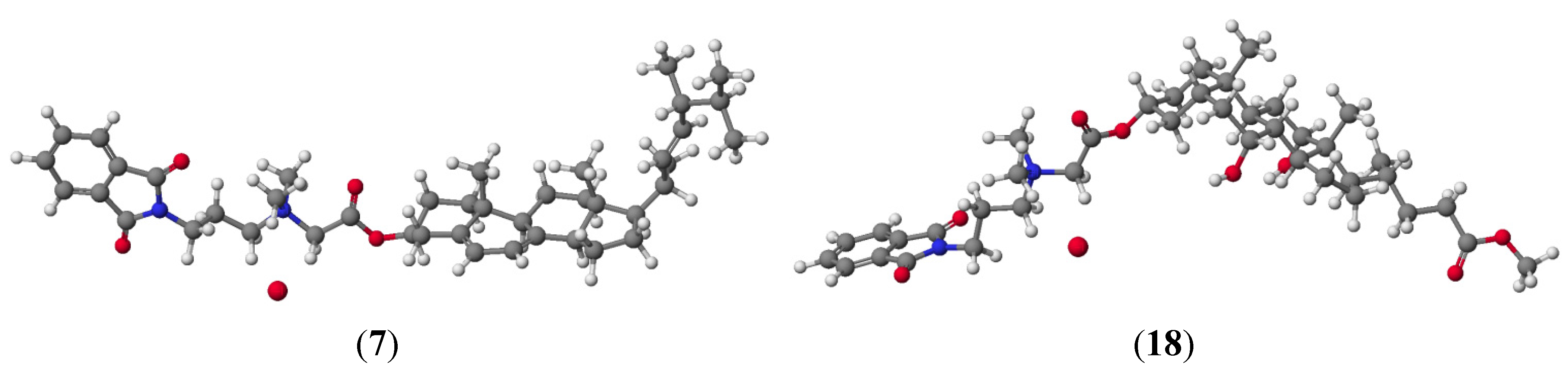
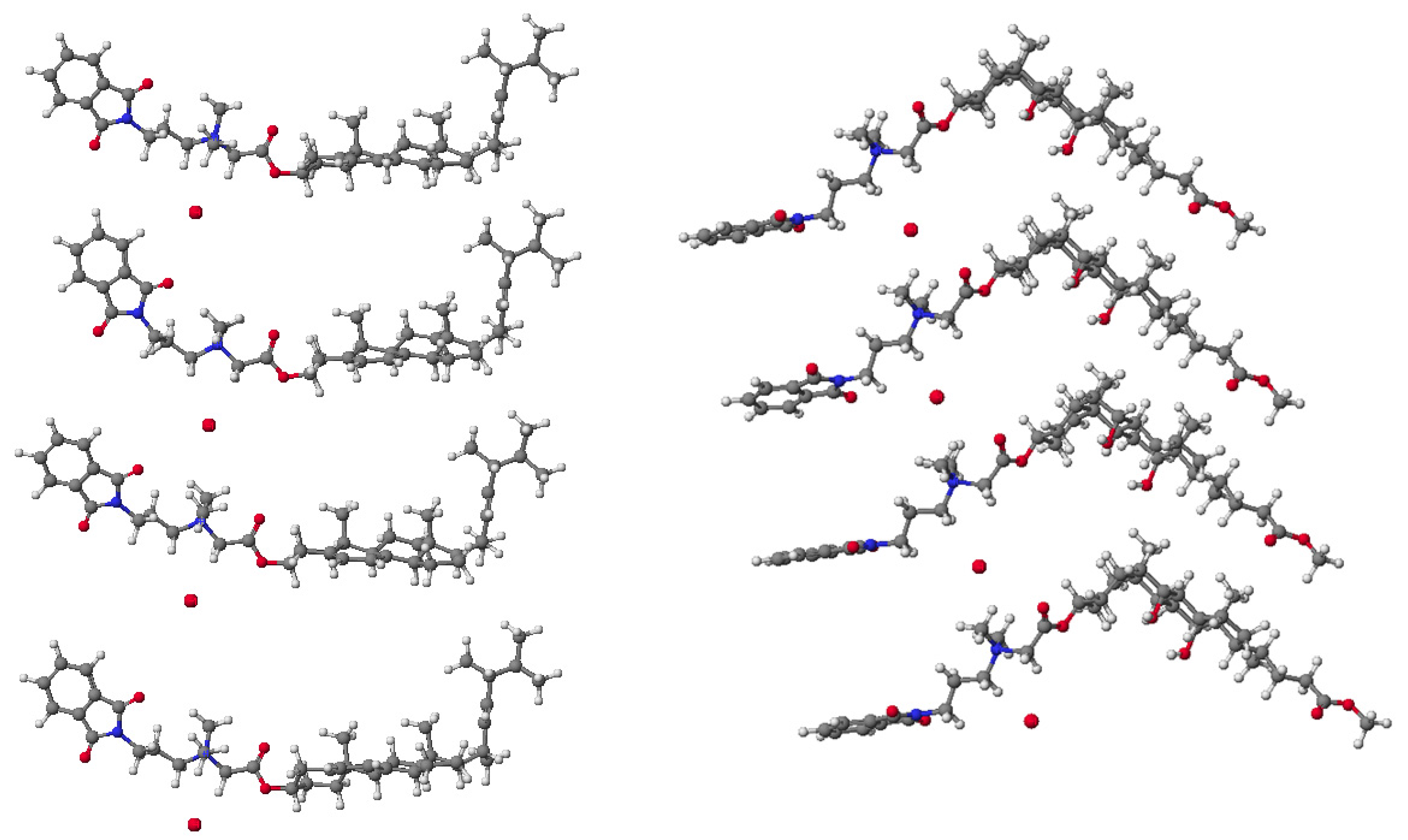
| Parameters | 7 | 8 | 9 | 16 | 17 | 18 |
|---|---|---|---|---|---|---|
| Dipole moments (Debye) | 9.0817 | 8.9725 | 8.8533 | 8.9578 | 8.4897 | 7.7646 |
| Bond lengths [Å] | ||||||
| N(1)-C(7') | 1.451 | 1.451 | 1.451 | 1.459 | 1.459 | 1.459 |
| 1.428 | 1.427 | 1.428 | 1.431 | 1.432 | 1.430 | |
| N(1)-C(3') | 1.495 | 1.494 | 1.494 | 1.492 | 1.492 | 1.451 |
| 1.471 | 1.471 | 1.471 | 1.469 | 1.469 | 1.469 | |
| C(1')-O(3) | 1.261 | 1.260 | 1.260 | 1.262 | 1.263 | 1.262 |
| 1.219 | 1.220 | 1.219 | 1.220 | 1.220 | 1.220 | |
| N(2)-C(4') | 1.573 | 1.570 | 1.573 | 1.572 | 1.572 | 1.571 |
| 1.546 | 1.546 | 1.546 | 1.543 | 1.544 | 1.543 | |
| N(2)-C(2') | 1.570 | 1.574 | 1.570 | 1.571 | 1.572 | 1.571 |
| 1.527 | 1.527 | 1.527 | 1.525 | 1.525 | 1.525 | |
| C(7')-O(1) | 1.260 | 1.261 | 1.260 | 1.255 | 1.255 | 1.255 |
| 1.210 | 1.210 | 1.210 | 1.208 | 1.208 | 1.208 | |
| Bond angles [°] | ||||||
| C(6')-C(5')-C(4') | 111.5 | 112.0 | 112.2 | 108.8 | 109.1 | 108.8 |
| 110.8 | 110.7 | 110.9 | 108.0 | 108.1 | 107.7 | |
| N(1)-C(6')-C(5') | 112.2 | 112.1 | 112.1 | 111.5 | 111.3 | 111.5 |
| 110.4 | 110.5 | 110.4 | 110.4 | 110.5 | 110.8 | |
| C(4')-N(2)-C(2') | 105.3 | 105.4 | 105.5 | 105.6 | 105.8 | 105.6 |
| 105.6 | 105.5 | 111.5 | 105.6 | 105.6 | 105.4 | |
| N(2)-C(2')-C(1') | 111.6 | 111.6 | 111.5 | 111.3 | 111.1 | 111.4 |
| 114.2 | 114.0 | 114.0 | 113.9 | 113.9 | 114.0 | |
| C(2')-C(1')-O(3) | 106.7 | 106.8 | 106.9 | 107.6 | 102.1 | 107.6 |
| 109.2 | 109.4 | 109.4 | 109.6 | 109.1 | 109.5 | |
| Torsion angles [°] | ||||||
| C(7')-N(1)-C(6')-C(5') | −58.1 | −60.0 | −60.8 | 58.3 | 57.3 | 58.5 |
| −82.2 | −80.3 | −79.4 | 86.8 | 85.5 | 77.6 | |
| C(8')-C(7')-N(1)-C(6') | −177.9 | −178.5 | −178.8 | 178.2 | 178.9 | 178.1 |
| 179.9 | −176.9 | −178.0 | −177.8 | −178.1 | 178.1 | |
| C(5')-C(4')-N(2)-C(2') | 170.0 | 171.4 | 172.1 | 178.3 | 178.0 | 178.2 |
| 172.9 | 172.1 | 172.4 | −169.2 | −168.5 | −170.6 | |
| C(3')-N(2)-C(2')-C(1') | 53.6 | 56.3 | 56.1 | 56.3 | 56.4 | 56.5 |
| 61.7 | 53.1 | 52.7 | 55.6 | 56.2 | 55.1 | |
| C(6')-C(5')-C(4')-N(2) | −99.3 | −98.4 | −98.0 | −179.8 | 179.1 | 179.8 |
| −104.8 | −103.8 | −103.2 | −167.7 | −167.3 | −172.0 | |
| Hydrogen bonds and short contacts lengths | ||||||
| Distances [Å] | ||||||
| C(4')-H···Br | 3.149 | 3.147 | 3.147 | 3.157 | 3.159 | 3.159 |
| 3.283 | 3.288 | 3.288 | 3.434 | 3.291 | 3.303 | |
| C(2')-H···Br | 3.077 | 3.076 | 3.075 | 3.067 | 3.064 | 3.068 |
| 3.075 | 3.080 | 3.075 | 3.051 | 3.045 | 3.303 | |
| N(2)···Br | 3.370 | 3.372 | 3.373 | 3.369 | 3.368 | 3.370 |
| 3.459 | 3.464 | 3.463 | 3.458 | 3.463 | 3.459 | |
| Angles [deg] | ||||||
| C(4')-H···Br | 153.0 | 153.6 | 153.9 | 153.5 | 153.3 | 153.4 |
| 144.1 | 143.8 | 143.9 | 139.1 | 138.2 | 139.4 | |
| C(2')-H···Br | 157.7 | 158.0 | 158.0 | 158.4 | 158.5 | 158.4 |
| 149.6 | 150.5 | 150.1 | 152.0 | 153.1 | 152.5 |

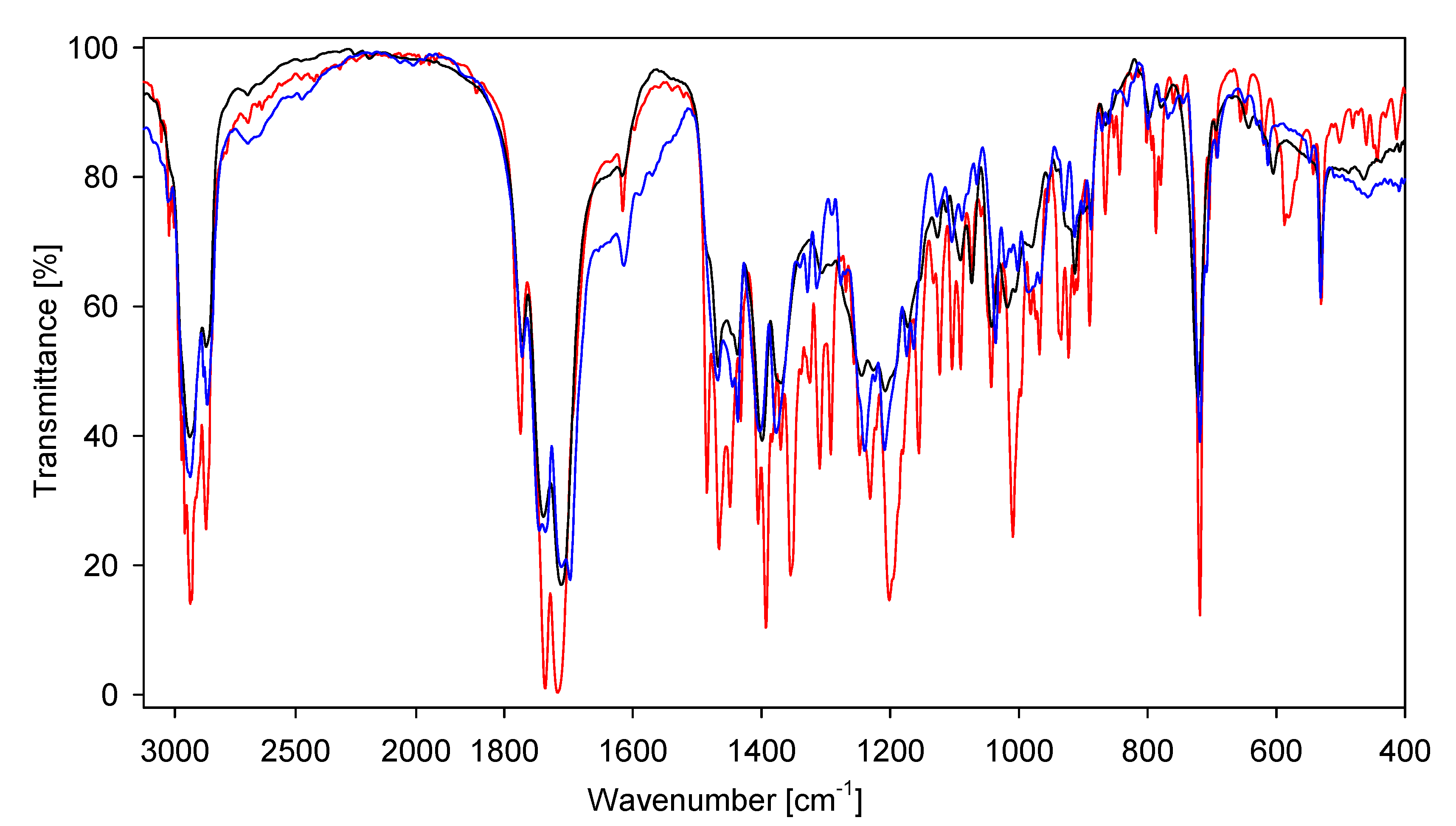

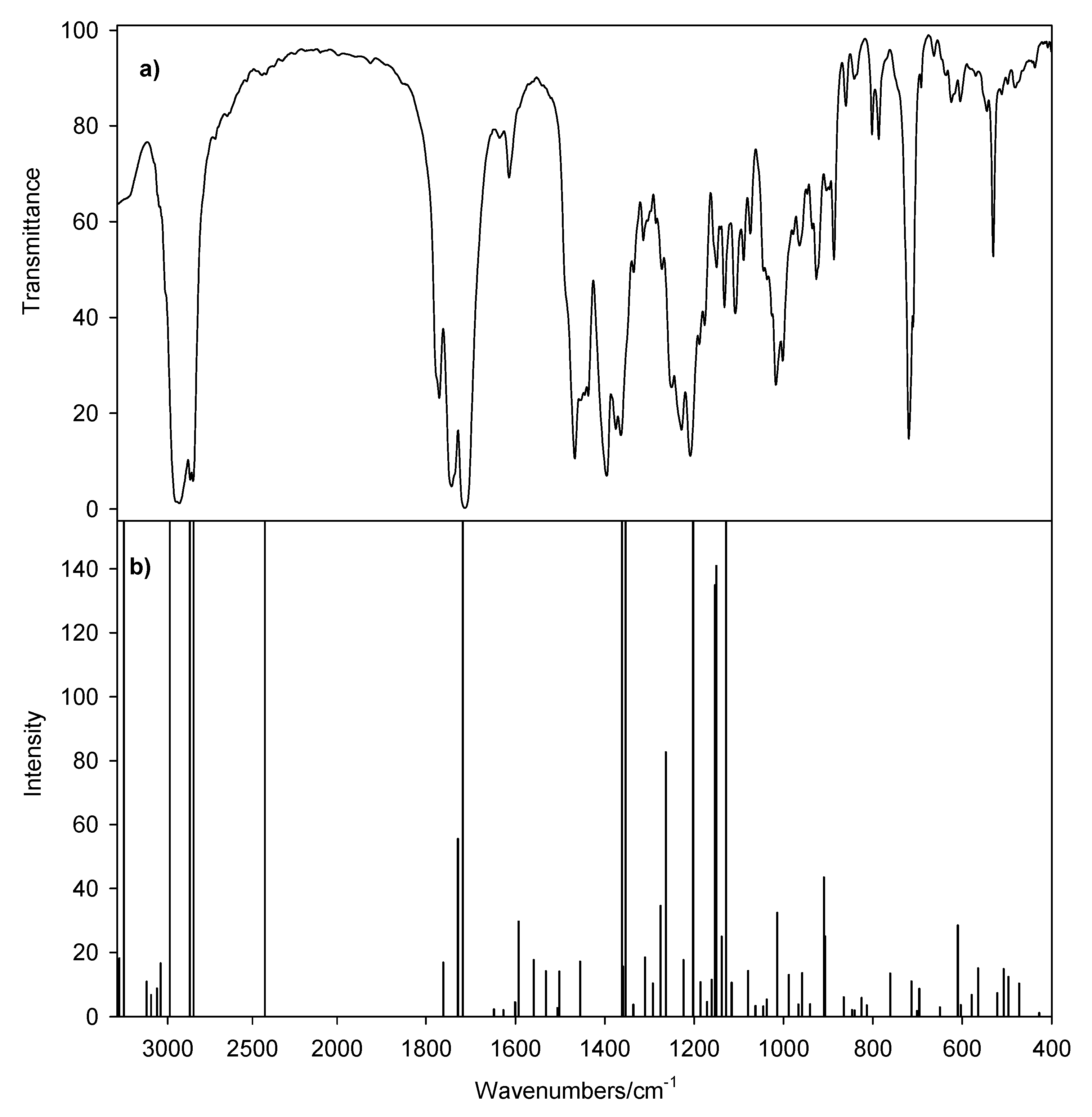
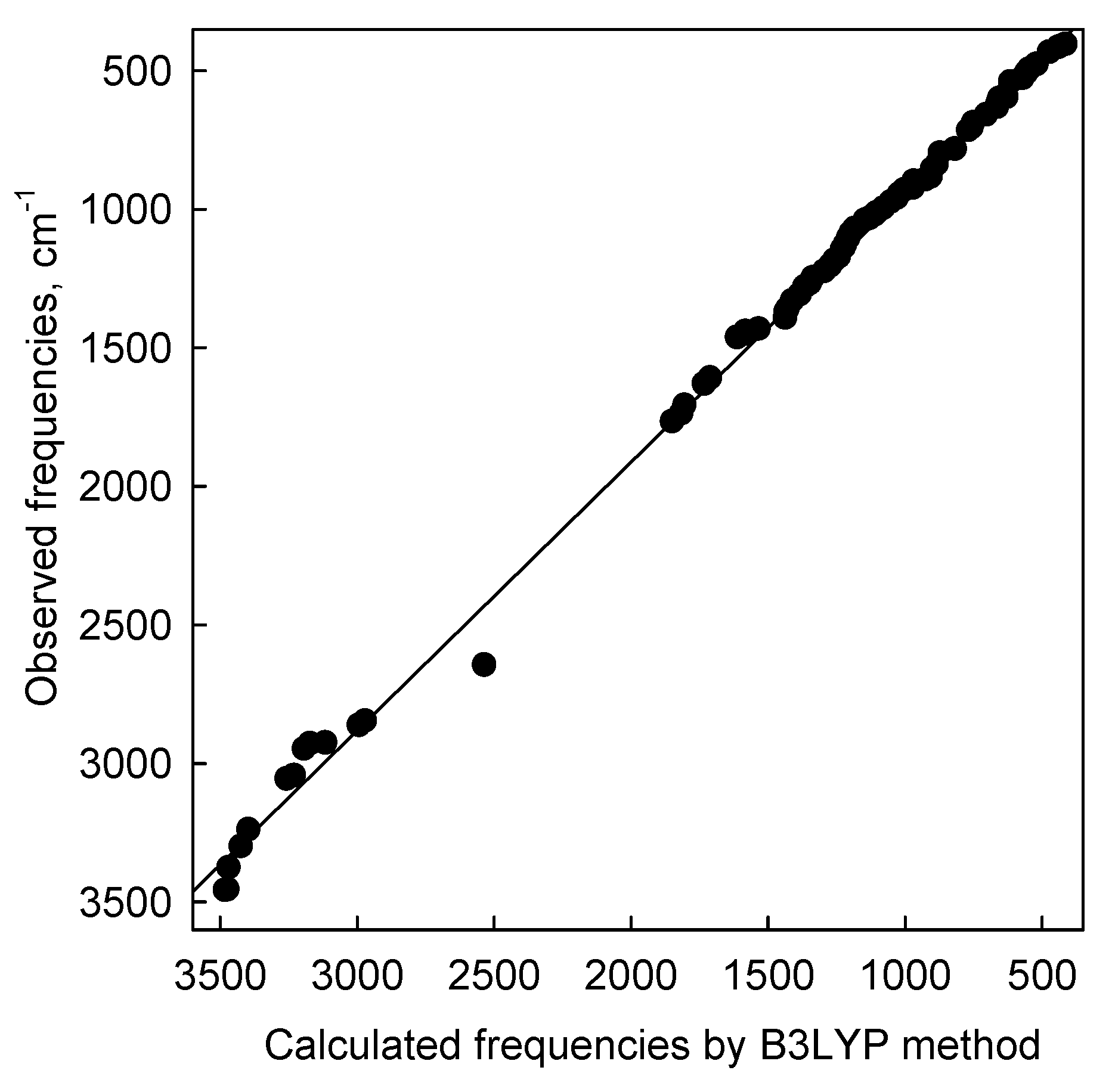
| IR | IRcalc. | IRcalc.scaled | INT | Proposed Assignment | |
|---|---|---|---|---|---|
| 3,462w | 3,477 | 3,344 | 3.34 | nCH | |
| 3,459w | 3,467 | 3,333 | 17.8 | nCH | |
| 3,380w | 3,464 | 3,330 | 5.26 | nCH | |
| 3,304w | 3,419 | 3,286 | 18.2 | nCH2 | |
| 3,243w | 3,391 | 3,259 | 381 | nCH2 | |
| 3,060w | 3,252 | 3,125 | 10.9 | nCH2 | |
| 3,048s | 3,225 | 3,098 | 6.74 | nCH2 | |
| 2,952s | 3,189 | 3,063 | 8.81 | nCH3 | |
| 2,933s | 3,167 | 3,042 | 16.7 | nCH3 | |
| 2,930s | 3,111 | 2,988 | 485 | nCH2 | |
| 2,867s | 2,988 | 2,869 | 515 | nCH2 | |
| 2,851s | 2,966 | 2,847 | 994 | nCH2 | |
| 2,719w | - | - | - | nCH∙∙∙Br | |
| 2,649w | 2,531 | 2,486 | 1982 | nCH∙∙∙Br | |
| 2,534w | - | - | - | nCH∙∙∙Br | |
| 1,770m | 1,845 | 1,761 | 16.9 | nasCO | |
| 1,742s | 1,811 | 1,728 | 55.6 | nCOO | |
| 1,712s | 1,800 | 1,718 | 190 | nsCO | |
| 1,635vw | 1,728 | 1,650 | 2.38 | nCC | |
| 1,614w | 1,706 | 1,627 | 2.13 | nCC | |
| - | 1,679 | 1,600 | 4.59 | nCC | |
| - | 1,671 | 1,593 | 29.7 | nCC | |
| - | 1,636 | 1,558 | 17.7 | nCC | |
| 1,467s | 1,608 | 1,532 | 14.2 | nCC | |
| 1,454m | 1,581 | 1,505 | 2.76 | nCC, βCH2 | |
| 1,445 m | 1,577 | 1,502 | 14.1 | βasCH3 | |
| 1,437m | 1,529 | 1,455 | 17.2 | βCH2 | |
| 1,396s | 1,432 | 1,361 | 175 | βOH | |
| 1,375 m | 1,430 | 1,359 | 15.7 | βsCH3 | |
| 1,364 m | 1,424 | 1,353 | 178 | nCO | |
| 1,335 w | 1,406 | 1,336 | 3.82 | nCC | |
| 1,313 w | 1,379 | 1,310 | 18.5 | nCN | |
| 1,285 w | 1,361 | 1,292 | 10.4 | nCC | |
| 1,272 w | 1,343 | 1,275 | 34.6 | nCC, βCH2 | |
| 1,251 m | 1,331 | 1,263 | 82.7 | nCO | |
| 1228 m | 1,290 | 1,224 | 17.7 | βCH2 | |
| 1,209 s | 1,268 | 1,202 | 232 | nCC | |
| 1,188 m | 1,251 | 1,186 | 10.8 | nCC | |
| 1,176 m | 1,236 | 1,172 | 4.70 | nCC | |
| 1,150 w | 1,225 | 1,161 | 11.5 | nCN | |
| 1,142 w | 1,217 | 1,153 | 135 | nCN | |
| 1,132 w | 1,214 | 1,150 | 141 | βCH | |
| 1,108 w | 1,202 | 1,138 | 25.0 | nCN | |
| 1,089 w | 1,192 | 1,128 | 169 | nCN | |
| 1,074 w | 1,179 | - | 10.6 | γCH2 | |
| 1,044 w | 1,141 | 1,116 | 14.3 | γCH2 | |
| 1,037 w | 1,124 | 1,079 | 3.42 | γCH2 | |
| 1,026 w | 1,106 | 1,063 | 3.28 | δCH2 | |
| 1,017 m | 1,098 | 1,045 | 5.41 | βCH2 | |
| 1,001 m | 1,074 | 1,037 | 32.5 | γCH2 | |
| 978 w | 1,047 | 1,014 | 13.0 | βCCC | |
| 964 w | 1,025 | 988 | 3.87 | βCO | |
| 950 w | 1,016 | 967 | 13.6 | γCH2 | |
| 935 w | 998 | 958 | 3.96 | βCH2 | |
| 927 w | 966 | 941 | 43.5 | γCH2 | |
| 904 w | 963 | 909 | 25.0 | γCH2 | |
| 897 w | 920 | 907 | 6.08 | βCCC | |
| 887 w | 901 | 907 | 2.12 | βCCC | |
| 860 vw | 895 | 865 | 2.04 | tring | |
| 842 vw | 879 | 847 | 5.89 | nCC | |
| 802 vw | 867 | 825 | 3.62 | βCCC | |
| 787 vw | 813 | 814 | 13.5 | βCCC | |
| 720 m | 764 | 761 | 11.0 | βCCC | |
| 710 vw | 751 | 714 | 1.86 | βCNC | |
| 692 vw | 746 | 701 | 8.70 | βring | |
| 663 vw | 698 | 694 | 3.00 | γCH | |
| 636 vw | 659 | 612 | 0.13 | γCH | |
| 625 vw | 657 | 610 | 28.5 | γCH | |
| 605 vw | 650 | 603 | 3.71 | βring | |
| 600 vw | 625 | 579 | 6.76 | βNCC | |
| 545 vw | 610 | 565 | 15.1 | γCC | |
| 531 w | 566 | 522 | 7.31 | βCCC | |
| 512 vw | 551 | 508 | 14.9 | tring | |
| 498 vw | 540 | 497 | 12.4 | tring | |
| 482 vw | 515 | 472 | 10.3 | γCCC | |
| 438 vw | 469 | 428 | 1.23 | Lattice mode | |
| 419 vw | 434 | 394 | 11.4 | Lattice mode | |
| 409 vw | 409 | 370 | 9.39 | Lattice mode | |
| - | 386 | 348 | 5.04 | Lattice mode | |
| - | 345 | 308 | 22.0 | Lattice mode | |
| - | 329 | 292 | 57.4 | Lattice mode | |
| - | 282 | 247 | 3.62 | Lattice mode | |
| - | 257 | 223 | 10.3 | Lattice mode | |
| - | 205 | 172 | 5.27 | Lattice mode | |
| - | 177 | 145 | 1.08 | Lattice mode | |
| - | 102 | 72 | 1.63 | Lattice mode | |
| - | 56 | 28 | 2.59 | Lattice mode |
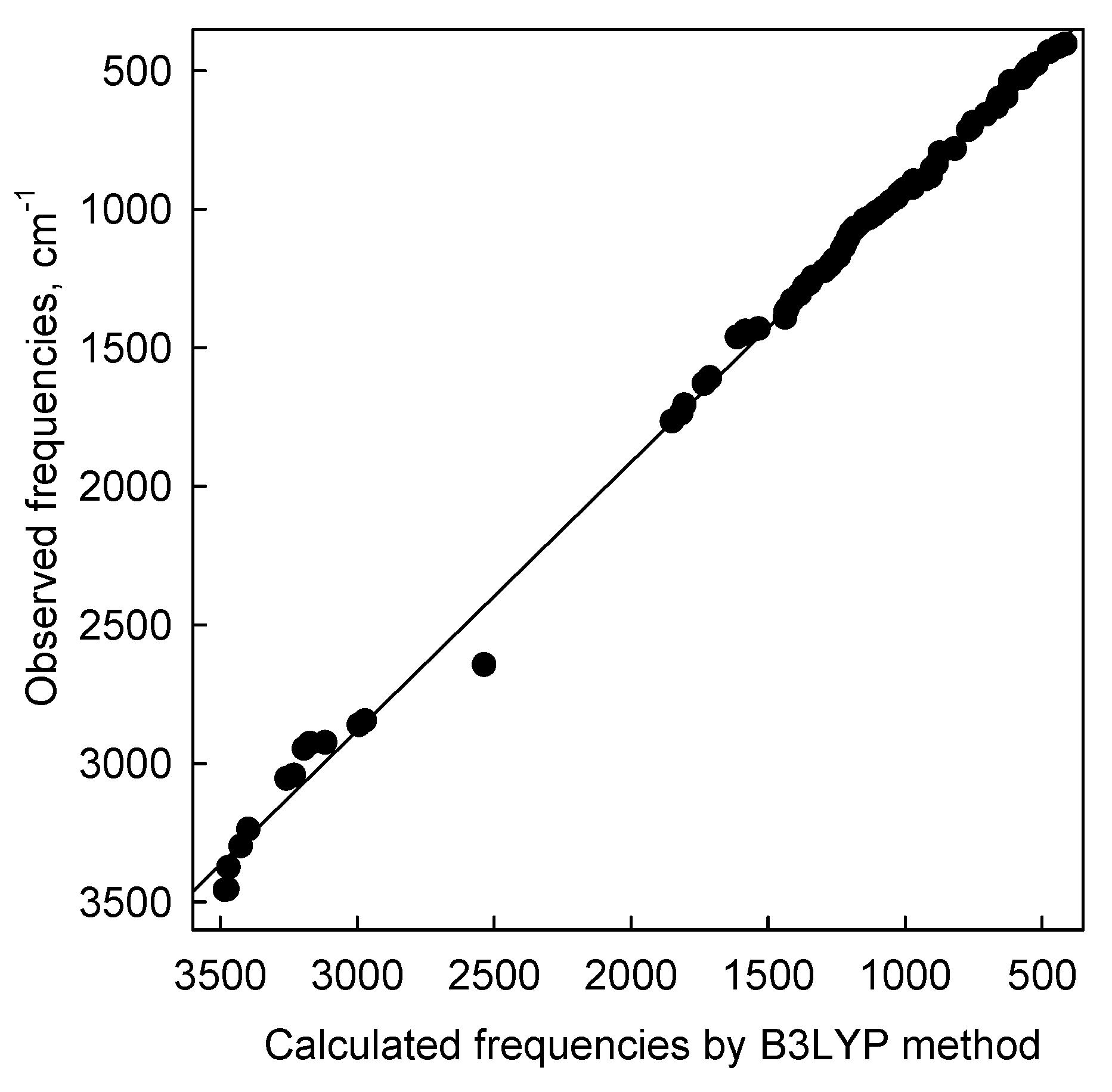
3. Experimental
3.1. General
3.2. Synthesis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vance, D.E.; van den Bosch, H. Cholesterol in the year 2000. Biochim. Biophys. Acta 2000, 1529, 1–8. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Montagnon, T. Steroids and the Pill. In Molecules that Changed the World; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 79–90. [Google Scholar]
- Dewick, P.M. Steroids. In Medicinal Natural Products A Biosynthetic Approach, 3rd ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2009; pp. 275–277. [Google Scholar]
- Bloch, K. Sterol molecule: Structure, biosynthesis, and function. Steroids 1997, 57, 378–383. [Google Scholar] [CrossRef]
- Bloch, K. 50 Years ago, the structure of cholesterol and of the bile acids. Trends Biochem. Sci. 1982, 7, 334–336. [Google Scholar] [CrossRef]
- Murry, K.R.; Granner, D.K.; Mayes, P.A.; Rodwell, V.W. Cholesterol Synthesis, Transport & Excretion. In Harper’s Biochemistry, 26th ed.; McGraw-Hill: New York, NY, USA; Volume 1996, pp. 219–230.
- Risley, J.M. Cholesterol biosynthesis: Lanosterol to cholesterol. J. Chem. Educ. 2002, 79, 377–384. [Google Scholar] [CrossRef]
- Hanukoglu, I. Steroidogenic enzymes: Structure, function, and role in regulation of steroid hormone biosynthesis. J. Steroid Biochem. Mol. Biol. 1992, 43, 779–804. [Google Scholar] [CrossRef]
- Nagrady, T.; Weaver, D.F. Steroid Hormones: Cholesterol as a Biosynthetic Precursor. In Medicinal Chemistry A Molecular and Biochemical Approach, 3rd ed.; Oxford University Press: New York, NY, USA, 2005; pp. 316–320. [Google Scholar]
- Koskinen, A.M.P. Terpens. In Asymmetric Synthesis of Natural Products; John Wiley & Sons, Ltd.: Chichester, UK, 2012; pp. 235–244. [Google Scholar]
- Ikan, R. Isoprenoids. In Natural Products a Laboratory Guide, 2nd ed.; Academic Press: London, UK, 1991; pp. 154–159. [Google Scholar]
- Rajakumar, K.; Greenspan, S.L.; Thomas, S.B.; Holick, M.F. SOLAR ultraviolet radiation and vitamin D: A historical perspective. Am. J. Public Health 2007, 97, 1746–1754. [Google Scholar] [CrossRef]
- Parish, E.J.; Nes, W.D. Biochemistry and Function of Sterols; CRC-Press: Boca Raton, FL, USA, 1997. [Google Scholar]
- Schaller, H. The role of sterols in plant growth and development. Prog. Lipid Res. 2003, 42, 163–175. [Google Scholar] [CrossRef]
- Fieser, L.F.; Fieser, M. Structures of the Bile Acids and of Cholesterol and Sterols. In Steroids; Reinhold Publishing Corporation: New York, NY, USA, 1959; pp. 53–90, 341–364. [Google Scholar]
- Templeton, W. The Sterols, Bile Acids and Related Compounds. In An Introduction to the Chemistry of Terpenoids and Steroids; Butterworths: London, UK, 1969; pp. 158–190. [Google Scholar]
- Lednicer, D. Steroid Chemistry at a Glance; John Wiley & Sons, Ltd.: Chichester, UK, 2011. [Google Scholar]
- Pikuleva, I.A. Cytochrome P450s and cholesterol homeostasis. Pharmacol. Ther. 2006, 112, 761–773. [Google Scholar] [CrossRef]
- Zollner, G.; Marschall, H.U.; Wagner, M.; Trauner, M. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: Pathogenetic and therapeutic considerations. Mol. Pharm. 2006, 3, 231–251. [Google Scholar] [CrossRef]
- Valkonen, A.; Sievänen, E.; Ikonen, S.; Lukashev, N.V.; Dones, P.A.; Averin, A.D.; Lathinen, M.; Kolehmainen, E. Novel lithocholaphanes: Syntheses, NMR, MS, and molecular modeling studies. J. Mol. Struct. 2007, 846, 65–73. [Google Scholar] [CrossRef]
- Kritchevsky, D.; Nair, P.P. Chemistry of the Bile Acids. In The Bile Acids: Chemistry, Physiology, and Metabolism. Volume 1 Chemistry; Plenum: New York, NY, USA, 1971; pp. 3–9. [Google Scholar]
- Nonappa; Maitra, U. Unlocking the potentials of bile acids in synthesis, Supramolecular/materials chemistry and nanoscience. Org. Biomol. Chem. 2008, 6, 657–669. [Google Scholar] [CrossRef]
- Salunke, D.B.; Hazra, B.G.; Pore, V.S. Steroidal conjugates and their pharmacological applications. Curr. Med. Chem. 2006, 13, 813–847. [Google Scholar] [CrossRef]
- Karigiannis, G.; Papaioannou, D. Structure, biological activity and synthesis of polyamine analogues and conjugates. Eur. J. Org. Chem. 2000, 10, 1841–1863. [Google Scholar] [CrossRef]
- Wallace, H.M.; Fraser, A.V.; Hughes, A. A perspective of polyamine metabolism. Biochem. J. 2003, 376, 1–14. [Google Scholar] [CrossRef]
- Majtan, V.; Majtanova, L. Effect of quaternary ammonium salts and amine oxides on the surface hydrophobicity of Enterobacter cloacae. Chem. Papers 2000, 54, 49–52. [Google Scholar]
- Devinsky, F.; Masarova, L.; Lacko, L.; Mlynarcik, D. Synthesis, IR spectra, and antimicrobial activity of some bis-ammonium salts of N,N'-bis(2-dimethylaminoethyl)methylamine. Coll. Czech. Chem. Commun. 1984, 49, 2819–2827. [Google Scholar] [CrossRef]
- Jones, R.A. Quaternary Ammonium Salts: Their Use in Phase-Transfer Catalysis; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Baregama, L.K.; Ahmed, M.; Dak, G.; Sharma, K.; Talesara, G.I. Evaluation of the antimicrobial activity of some novel alpha substituted hydroxylamine derivatives. Indian J. Pharmacol. 2004, 36, 312–313. [Google Scholar]
- Matijevic-Sosa, J.; Cvetic, Z. Antimicrobial activity of N-phthaloylamino acid Hydroxamates. Acta Pharm. 2005, 55, 387–399. [Google Scholar]
- Pluempanupat, W.; Adisakwattana, S.; Yibchok-Anun, S.; Chavasiri, W. Synthesis of N-phenylphthalimide derivatives as alpha-glucosidase inhibitors. Arch. Pharm. Res. 2007, 30, 1501–1506. [Google Scholar] [CrossRef]
- Manivannan, G. Disinfection and Decontamination: Principles, Applications and Related Issues; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2008. [Google Scholar]
- Owens, R.C.; Lautenbach, E. Antimicrobial Resistance: Problem Pathogens and Clinical Countermeasures; Informa Healthcare: New York, NY, USA, 2008. [Google Scholar]
- Paulus, W. Directory of Microbiocides for the Protection of Materials: A. Handbook; Springer: Dordrecht, The Netherlands, 2005. [Google Scholar]
- Brycki, B.; Werner, J.; Kowalczyk, I. Synthesis and spectroscopic properties of N,N-bis-(3-aminopropyl)-N,N-dialkylammonium salts. Ann. Pol. Chem. 2005, 1, 192–195. [Google Scholar]
- Brycki, B.; Kowalczyk, I.; Krysman, M. Synthesis and spectra properties of quaternary aminoalkylammonium salts. Ann. Pol. Chem. Soc. 2006, 1, 47–52. [Google Scholar]
- Brycki, B.; Koenig, H.; Kowalczyk, I.; Pospieszny, T. Synthesis, spectroscopic and semiempirical studies of new quaternary alkylammonium conjugates of sterols. Molecules 2013, 18, 14961–14976. [Google Scholar] [CrossRef]
- Bhat, S.; Maitra, Y. Low molecular mass cationic gelators derived from deoxycholic acid: Remarkable gelation of aqueous solvents. Tetrahedron 2007, 63, 7309–7320. [Google Scholar] [CrossRef]
- Vida, N.; Svobodova, H.; Rarova, L.; Drašar, P.; Šaman, D.; Cvačka, J.; Wimmer, Z. Polyamine conjugates of stigmasterol. Steroids 2012, 77, 1212–1218. [Google Scholar] [CrossRef]
- Avinash, B.; Sandhya, B.; Somanath, K.; Ashima, S.; Manish, S. Deciphering the role of charge, hydration, and hydrophobicity for cytotoxic activities and membrane interactions of bile acid based facial amphiphiles. BBA–Biomembranes 2013, 8, 1926–1937. [Google Scholar]
- Aher, N.G.; Pore, V.S.; Patil, S.P. Design, synthesis, and micellar properties of bile acid dimers and oligomers linked with a 1,2,3-triazole ring. Tetrahedron 2007, 63, 12927–12934. [Google Scholar] [CrossRef]
- Kowalczyk, I. Study of N,N-dimethyl(carboethoxymethyl)-3-phthalimidopropylammonium chloride dihydrate by DFT calculations, NMR and FTIR spectroscopy. J. Mol. Struct. 2009, 928, 12–17. [Google Scholar] [CrossRef]
- PharmaExpert Predictive Services. Available online: http://www.pharmaexpert.ru/PASSOnline/ (accessed on 1 November 2013).
- Poroikov, V.V.; Filimonov, D.A.; Borodina, Y.V.; Lagunin, A.A.; Kos, A. Robustness of biological activity spectra predicting by computer program PASS for noncongeneric sets of chemical compounds. J. Chem. Inf. Comput. Sci. 2000, 40, 1349–1355. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A. How to acquire new biological activities in old compounds by computer prediction. J. Comput. Aided Mol. Des. 2002, 16, 819–824. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A. Predictive Toxicology; Helma, C., Ed.; Taylor and Francis: Boca Raton, FL, USA, 2005; pp. 459–478. [Google Scholar]
- Stepanchikova, A.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. Prediction of biological activity spectra for substances: Evaluation on the diverse sets of drug-like structures. Curr. Med. Chem. 2003, 10, 225–233. [Google Scholar] [CrossRef]
- Gulis, I.M.; Komyak, A.I.; Saechnikov, K.A.; Tsvirko, V.A. Stepwise intramolecular vibrational-energy redistribution in the S1 state of 3-amino and 3-amino-n-methylphthalimide molecules. J. Appl. Spectrosc. 1993, 58, 36–39. [Google Scholar] [CrossRef]
- Poyor, B.A.; Palmer, P.M.; Andrews, P.M.; Berger, M.B.; Troxler, T.; Topp, M.R. Spectroscopy of jet-cooled polar complexes of aminophthalimides. Chem. Phys. Lett. 1997, 271, 19–26. [Google Scholar] [CrossRef]
- Brycki, B.; Kowalczyk, I.; Werner, J.; Borowiak, T.; Wolska, I. Polyamines. I. Spectroscopic properties of N,N-bis-(phthalimidopropyl)-N-propylamine and supramolecular interactions in ist crystals. J. Mol. Struct. 2006, 791, 137–143. [Google Scholar] [CrossRef]
- Brycki, B.; Kowalczyk, I.; Zieliński, A.; Borowiak, T.; Wolska, I. Spectroscopic properties of N-n-hexyltetrachlorophthalimide and supramolecular interactions in its crystals. J. Mol. Struct. 2008, 874, 145–150. [Google Scholar] [CrossRef]
- Borowiak, T.; Wolska, I.; Jensz, P.; Kowalczyk, I.; Brycki, B.; Sztul, A. Polyamines. Part II: Spectroscopic properties of N,N-dimethyl-3-phthalimidopropylammonium acetate and hydrochloride and supramolecular interactions in their crystals. J. Mol. Struct. 2008, 891, 205–213. [Google Scholar] [CrossRef]
- Palafox, M.A. Scaling factors for the prediction of vibrational spectra. I. Benzene molecule. Int. J. Quant. Chem. 2000, 77, 661–684. [Google Scholar] [CrossRef]
- Palafox, M.A.; Iza, N. Tautomerism of the natural thymidine nucleoside and the antiviral analogue D4T. Structure and influence of an aqueous environment using MP2 and DFT methods. Phys. Chem. Chem. Phys. 2010, 12, 881–893. [Google Scholar] [CrossRef]
- Dennington, R., II; Keith, T.; Millam, J. Gauss View, Version 4.1.2. Semichem. Inc.: Shawnee Mission, KS, USA, 2007. [Google Scholar]
- CAChe, version 5.04. UserGuide. Fujitsu: Chiba, Japan, 2003.
- Stewart, J.J.P. Optimization of parameters for semiempirical methods. III Extension of PM3 to Be, Mg, Zn, Ga, Ge, As, Se, Cd, In, Sn, Sb, Te, Hg, Tl, Pb, and Bi. J. Comput. Chem. 1991, 12, 320–341. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods. I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision C.01. Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange correlation functional. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, G.R. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, B37, 785–789. [Google Scholar]
- Hehre, W.J.; Random, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Dichfield, R. Self-consistent perturbation theory of diamagnetism: I. A gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 4–9 and 13–18 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Brycki, B.; Koenig, H.; Kowalczyk, I.; Pospieszny, T. Synthesis, Spectroscopic and Theoretical Studies of New Quaternary N,N-Dimethyl-3-phthalimidopropylammonium Conjugates of Sterols and Bile Acids. Molecules 2014, 19, 4212-4233. https://doi.org/10.3390/molecules19044212
Brycki B, Koenig H, Kowalczyk I, Pospieszny T. Synthesis, Spectroscopic and Theoretical Studies of New Quaternary N,N-Dimethyl-3-phthalimidopropylammonium Conjugates of Sterols and Bile Acids. Molecules. 2014; 19(4):4212-4233. https://doi.org/10.3390/molecules19044212
Chicago/Turabian StyleBrycki, Bogumil, Hanna Koenig, Iwona Kowalczyk, and Tomasz Pospieszny. 2014. "Synthesis, Spectroscopic and Theoretical Studies of New Quaternary N,N-Dimethyl-3-phthalimidopropylammonium Conjugates of Sterols and Bile Acids" Molecules 19, no. 4: 4212-4233. https://doi.org/10.3390/molecules19044212
APA StyleBrycki, B., Koenig, H., Kowalczyk, I., & Pospieszny, T. (2014). Synthesis, Spectroscopic and Theoretical Studies of New Quaternary N,N-Dimethyl-3-phthalimidopropylammonium Conjugates of Sterols and Bile Acids. Molecules, 19(4), 4212-4233. https://doi.org/10.3390/molecules19044212