Synthesis of PNA Oligoether Conjugates

Abstract
:1. Introduction
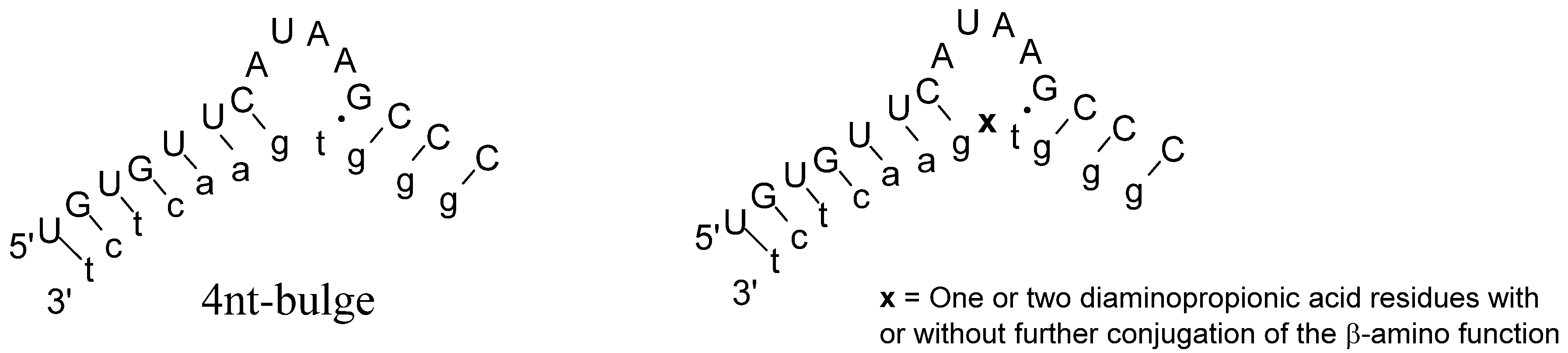
2. Results and Discussion
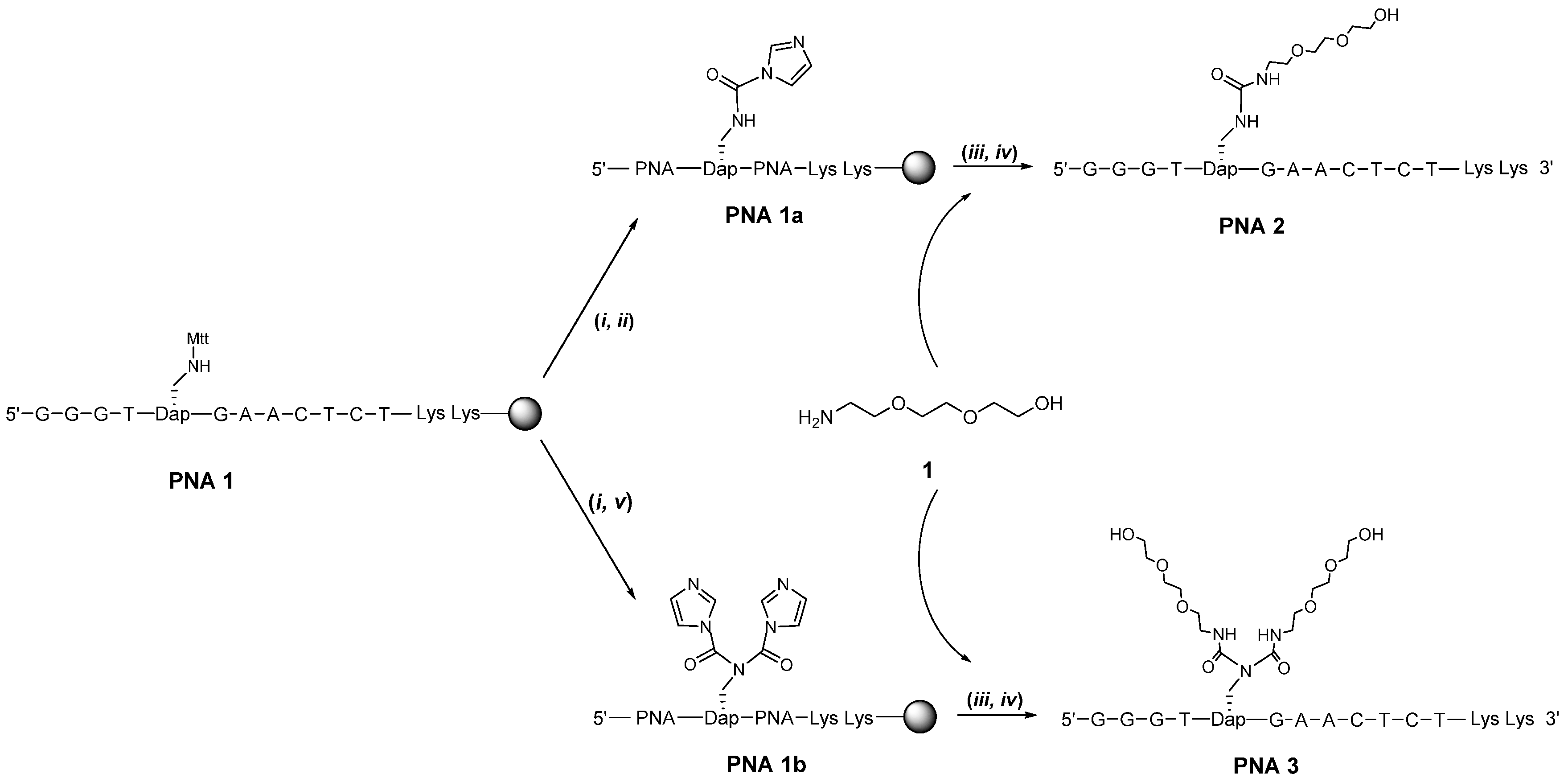

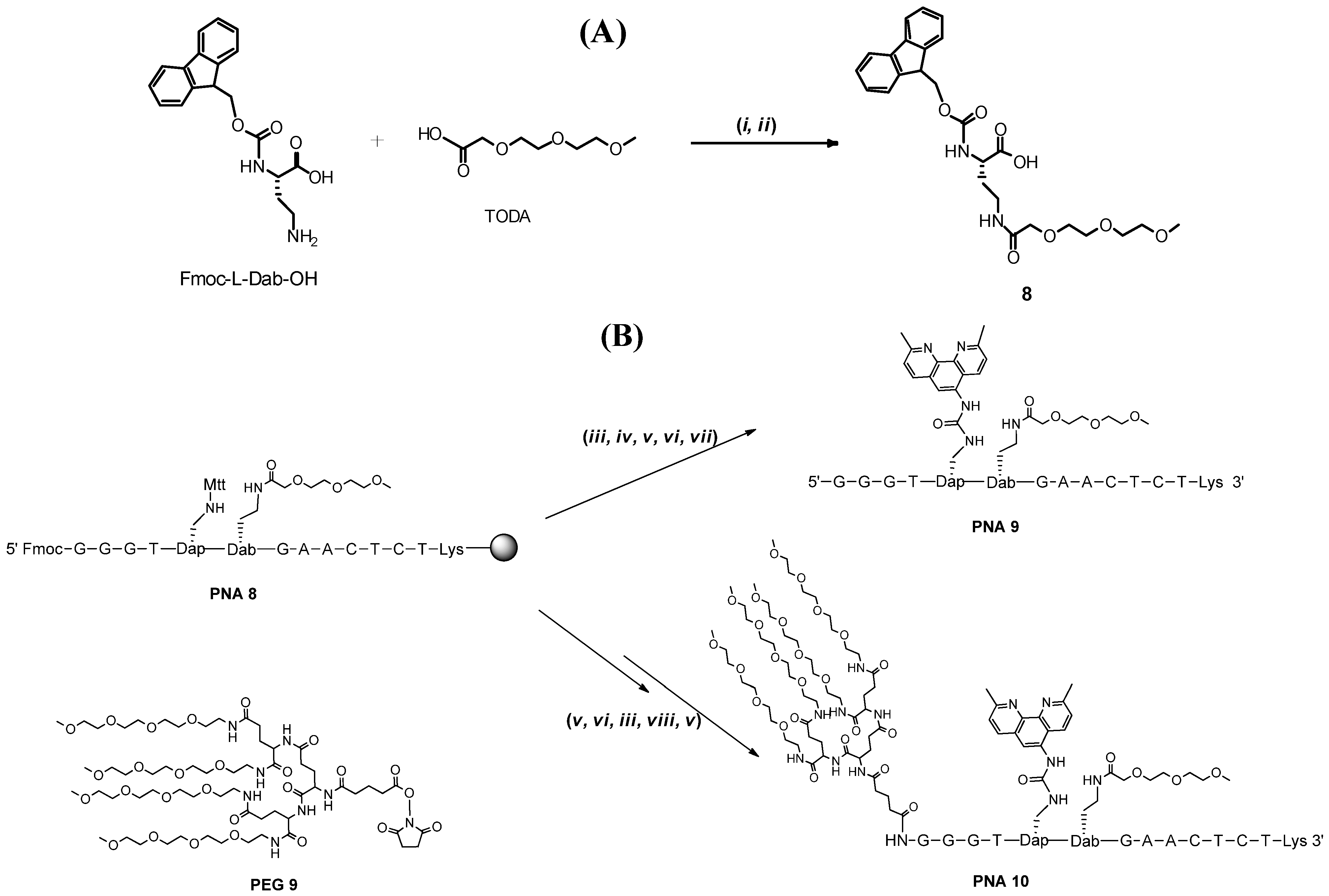

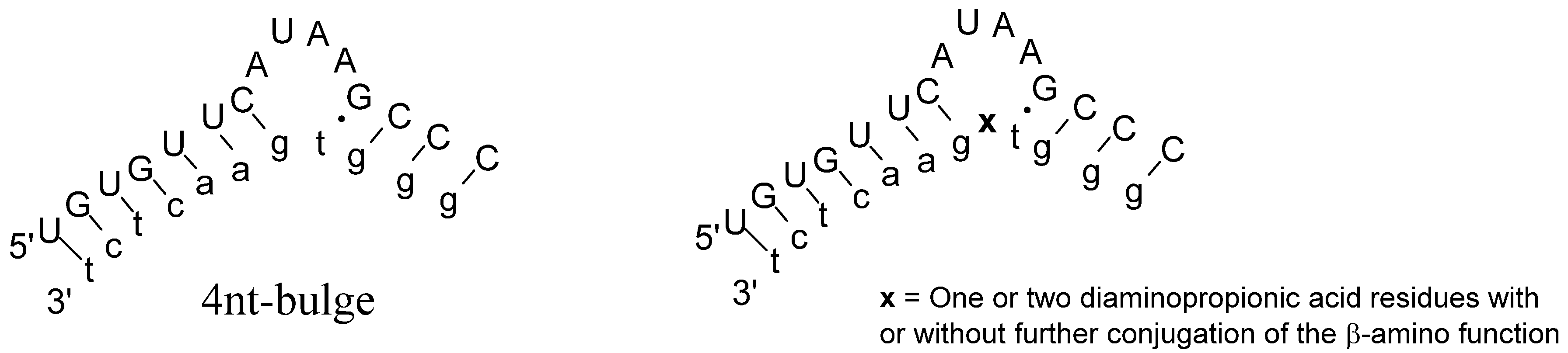{kind=link}
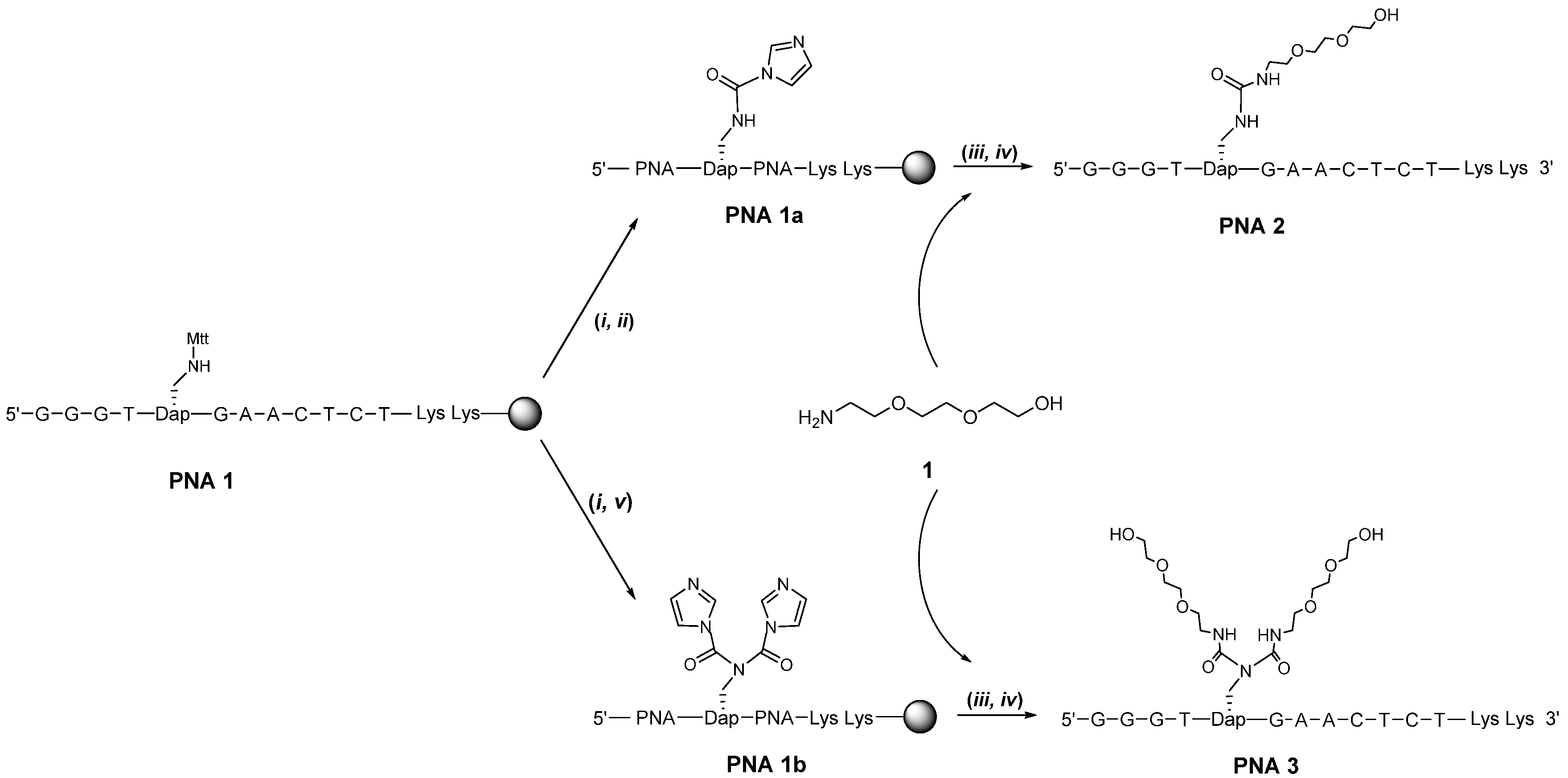{kind=link}
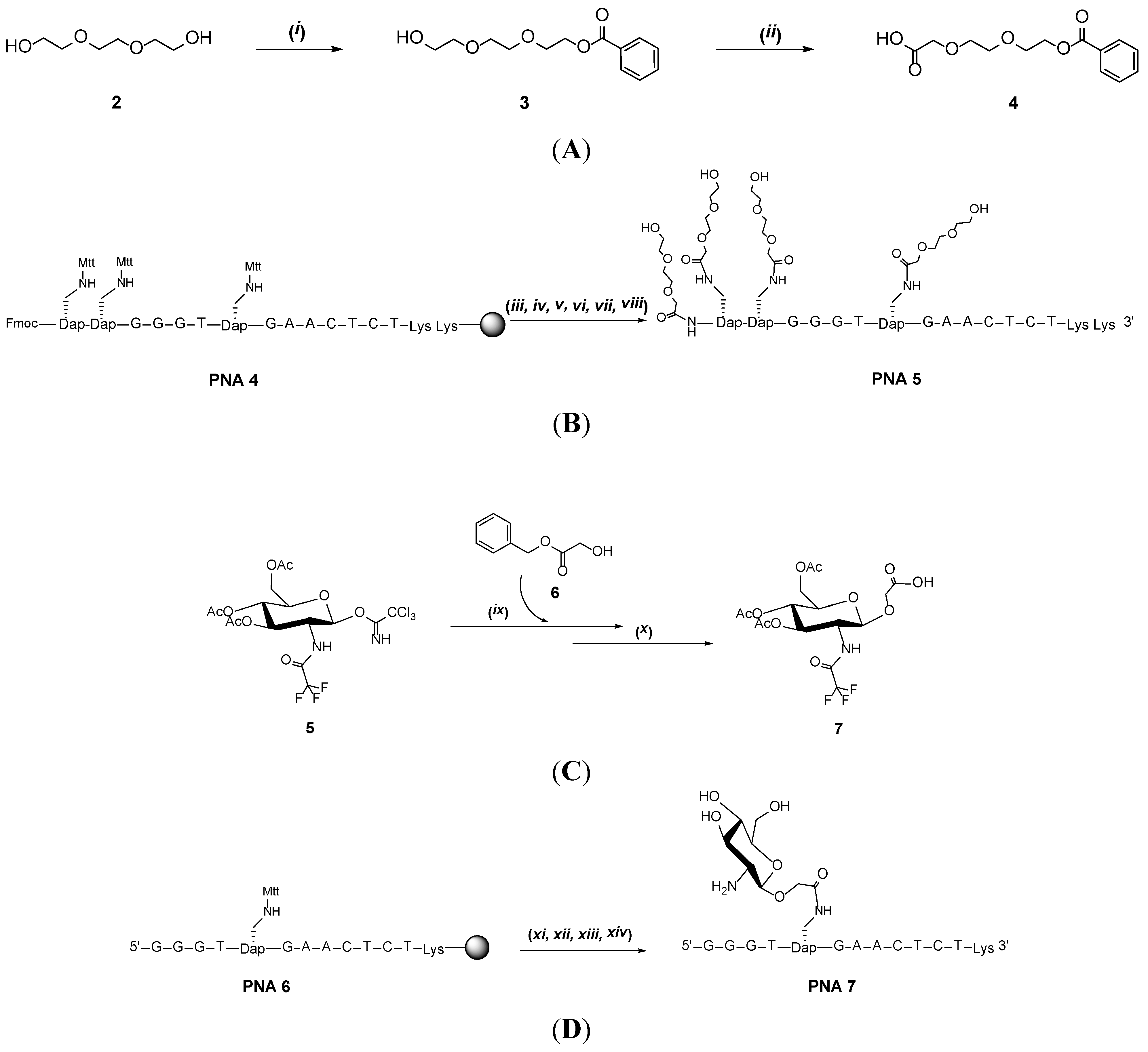{kind=link}
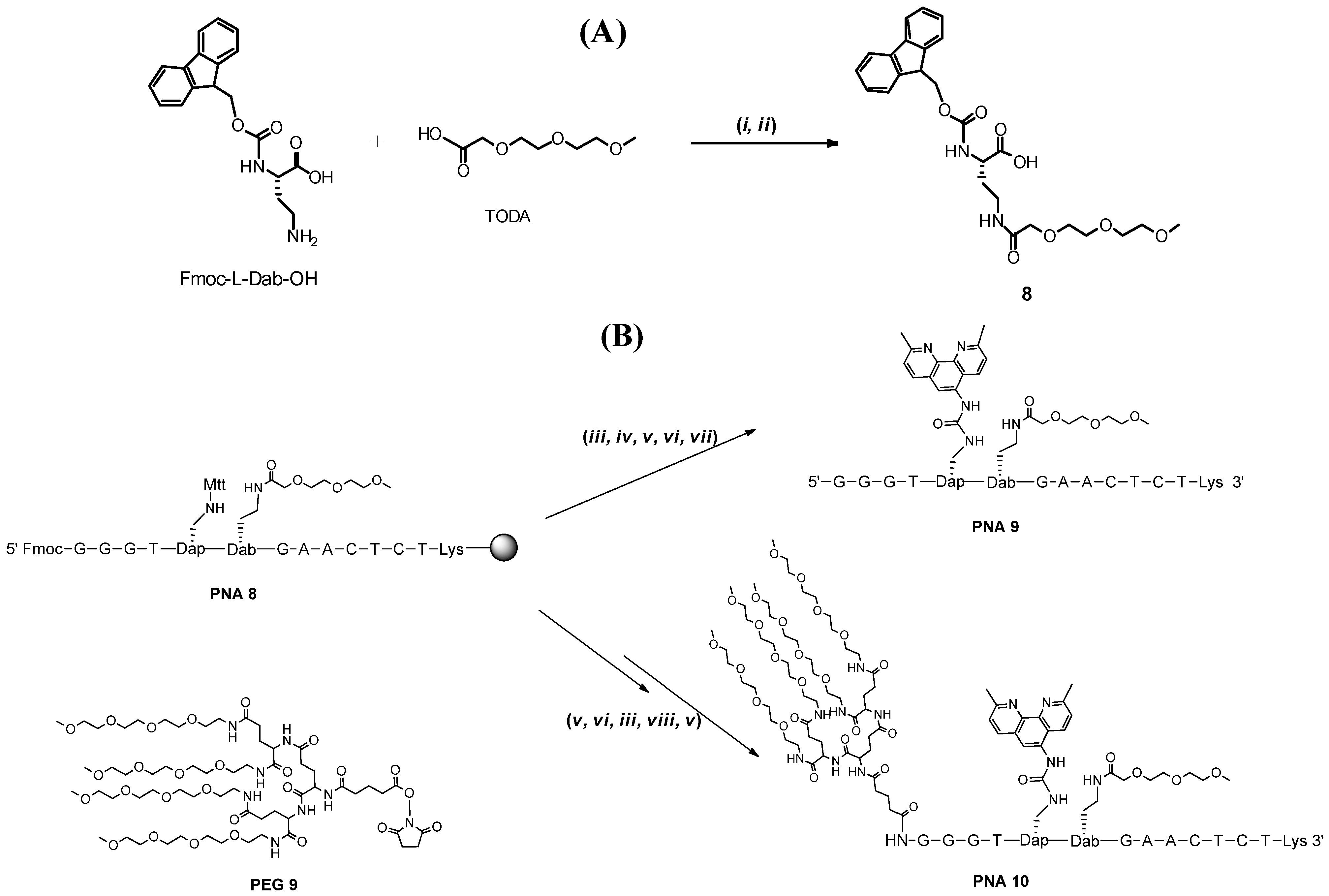{kind=link}
| Entry | PNA Construct | RP-18 HPLC Ret. Time (min) a | Type and Number of Conjugated Entities |
|---|---|---|---|
| 1 | PNA-Dapa-PNA 1 | 13.7 | 1 Dapa |
| 2 | PNA 2 | 15.7 | 1 hydroxyoligoether |
| 3 | PNA 3 | 21.7 | 2 hydroxyoligoethers |
| 4 | PNA 5 | 21.1 | 1 central and 3 terminal hydroxyoligoethers |
| 5 | PNA-Dapa-PNA 6 | 17.7 | 1 Dapa |
| 6 | PNA-Gly-PNA | 17.6 | 1 glycine |
| 7 | PNA 7 | 15.8 | 1 aminosugar |
| 8 | PNA-GlyNeo-PNA | 22.1 | 1 glycine and 1 neocuproine |
| 9 | PNA 9 | 25.5 | 1 methoxyoligoether and 1 neocuproine |
| 10 | PNA 10 | 27.9 b | 1 methoxyoligoether and 1 neocuproine and 4 terminal methoxyoligoethers |
3. Experimental
3.1. Materials and Methods
3.2. Synthesis of PNAs
3.3. Post-Conjugation of Oligoethers to PNA
3.4. Introduction of a New Amino Acid Building Block in the PNA Sequence
4. Conclusions
Supplementary Materials
Acknowledgments
Conflictts of Interest
References
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar]
- Nielsen, P.E. Peptide nucleic acid. A molecule with two identities. Acc. Chem. Res. 1999, 32, 624–630. [Google Scholar] [CrossRef]
- Ganesh, K.N.; Nielsen, P.E. Peptide nucleic acids: Analogs and derivatives. Curr. Org. Chem. 2000, 4, 931–943. [Google Scholar] [CrossRef]
- Rozners, E. Recent advances in chemical modification of peptide nucleic acids. J. Nucleic Acids 2012. [Google Scholar] [CrossRef]
- Lundin, K.E.; Good, L.; Strömberg, R.; Gräslund, A.; Smith, C.I.E. Biological activity and biotechnological aspects of peptide nucleic acid. Adv. Genet. 2006, 56, 1–51. [Google Scholar] [CrossRef]
- Nielsen, P.E. Peptide nucleic acids (PNA) in chemical biology and drug discovery. Chem. Biodivers. 2010, 7, 786–804. [Google Scholar] [CrossRef]
- Greenwald, R.B.; Choe, Y.H.; McGuire, J.; Conover, C.D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliver. Rev. 2003, 55, 217–250. [Google Scholar] [CrossRef]
- Ryan, S.M.; Mantovani, G.; Wang, X.; Haddleton, D.M.; Brayden, D.J. Advances in PEGylation of important biotech molecules: Delivery aspects. Expert Opin. Drug Del. 2008, 5, 371–383. [Google Scholar] [CrossRef]
- Dettin, M.; Silvestri, D.; Danesin, R.; Cretaio, E.; Picariello, G.; Casarin, E.; Sonato, A.; Romanato, F.; Morpurgo, M. Synthesis and chromatography-free purification of PNA-PEO conjugates for the functionalisation of gold sensors. Molecules 2012, 17, 11026–11045. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Y.; Jarreau, C.; Welch, M.J.; Taylor, J.-S.A. Nucleic acid-directed self-assembly of multifunctional gold nanoparticle imaging agents. Biomater. Sci. 2013, 1, 1055–1064. [Google Scholar] [CrossRef]
- Millili, P.G.; Yin, D.H.; Fan, H.; Naik, U.P.; O’Sullivan, M. Formulation of a peptide nucleic acid based nucleic acid delivery construct. Bioconjugate Chem. 2010, 21, 445–455. [Google Scholar]
- Brown, P.N.; Yin, H. PNA-based microRNA inhibitors elicit anti-inflammatory effects in microglia cells. Chem. Commun. 2013, 49, 4415–4417. [Google Scholar] [CrossRef]
- Gaglione, M.; Milano, G.; Chambery, A.; Moggio, L.; Romanelli, A.; Messere, A. PNA-based artificial nucleases as antisense and anti-miRNA oligonucleotide agents. Mol. Biosystem. 2011, 7, 2490–2499. [Google Scholar] [CrossRef]
- Sahu, B.; Sacui, I.; Rapireddy, S.; Zanotti, K.J.; Bahal, R.; Armitage, B.A.; Ly, D.H. Synthesis and characterization of conformationally preorganized, (r)-diethylene glycol-containing γ-peptide nucleic acids with superior hybridization properties and water solubility. J. Org. Chem. 2011, 76, 5614–5627. [Google Scholar] [CrossRef]
- Nelson, J.W.; Martin, F.H.; Tinoco, I., Jr. DNA and RNA oligomer thermodynamics: The effect of mismatched bases on double-helix stability. Biopolymers 1981, 20, 2509–2531. [Google Scholar] [CrossRef]
- Madder, A.; Ehrl, R.; Strömberg, R. Stabilization of RNA bulges by oligonucleotide complements containing an adenosine analogue. ChemBioChem 2003, 4, 1194–1200. [Google Scholar] [CrossRef]
- Sandbrink, J.; Ossipov, D.; Åström, H.; Strömberg, R. Investigation of potential RNA bulge stabilizing elements. J. Mol. Recognit. 2005, 18, 318–326. [Google Scholar]
- Åström, H.; Williams, N.H.; Strömberg, R. Oligonucleotide based artificial nuclease (OBAN) systems. Bulge size dependence and positioning of catalytic group in cleavage of RNA-bulges. Org. Biomol. Chem. 2003, 1, 1461–1465. [Google Scholar] [CrossRef]
- Åström, H.; Strömberg, R. Synthesis of new OBAN's and further studies on positioning of the catalytic group. Org. Biomol. Chem. 2004, 2, 1901–1907. [Google Scholar] [CrossRef]
- Murtola, M.; Strömberg, R. 2'-O-Methyloligoribonucleotide based artificial nucleases (2'-O-MeOBANs) cleaving a model of the leukemia related m-bcr/abl m-RNA. ARKIVOC 2008, 3, 84–94. [Google Scholar] [CrossRef]
- Murtola, M.; Strömberg, R. PNA based artificial nucleases displaying catalysis with turnover in the cleavage of a leukemia related RNA model. Org. Biomol. Chem. 2008, 6, 3837–3842. [Google Scholar] [CrossRef]
- Murtola, M.; Wenska, M.; Strömberg, R. PNAzymes that are artificial RNA restriction enzymes. J. Am. Chem. Soc. 2010, 132, 8984–8990. [Google Scholar] [CrossRef]
- Bourel, L.; Carion, O.; Gras-Masse, H.; Melnyk, O. The deprotection of Lys (Mtt) Revisited. J. Pept. Sci. 2000, 6, 264–270. [Google Scholar]
- Kaneno, J.; Kubo, T.; Fujii, M. Synthesis of DNA-peptide conjugates by solid phase fragment condensation. J. Pept. Sci. 2002, 38, 339–342. [Google Scholar]
- Sandbrink, S.; Murtola, M.; Strömberg, R. Solid support post-conjugation of amino acids and a phenanthroline derivative to a central position in peptide nucleic acids. Nucleos. Nucleot. Nucl. 2007, 26, 1485–1489. [Google Scholar] [CrossRef]
- Aerts, J.M.; Franciscus, G.; Overkleeft, H.S. Preparation of steroidal aminodeoxy glycosides and disaccharides as anti-inflammatory prodrugs. PCT/NL2008/050379, 18 December 2008. [Google Scholar]
- Zall, A.; Kieser, D.; Hoettecke, N.; Naumann, E.C.; Thomaszewski, B.; Schneider, K.; Steinbacher, D.T.; Schubenel, R.; Masur, S.; Baumann, K.; et al. NSAID-Derived γ-Secretase modulation requires an acidic moiety on the carbazole scaffold. Bioorg. Med. Chem. 2011, 19, 4903–4909. [Google Scholar] [CrossRef]
- Makino, A.; Kurosaki, K.; Ohmae, M.; Kobayashi, S. Chitinase-Catalyzed Synthesis of Alternatingly N-Deacetylated Chitin: A chitin-chitosan hybrid polysaccharide. Biomacromolecules 2006, 7, 950–957. [Google Scholar] [CrossRef]
- Huang, Y.; Dey, S.; Zhang, X.; Soennichsen, F.; Garner, P. The Alpha-Helical peptide nucleic acid concept: Merger of peptide secondary structure and codified nucleic acid recognition. J. Am. Chem. Soc. 2004, 126, 4626–4640. [Google Scholar] [CrossRef]
- Mehta, S.; Meldal, M.; Duus, J.O.; Bock, K. Internally quenched fluorogenic α-Helical dimeric peptides and glycopeptides for the evaluation of the effect of glycosylation on the conformation of peptides. J. Chem. Soc. Perkin Trans. 1997, 1365–1374. [Google Scholar]
- Puglisi, J.D.; Tinoco, I., Jr. Absorbance melting curves of RNA. Meth. Enzymol. 1989, 180, 304–325. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds PNA 2, PNA 3, PNA 5, PNA 7, PNA 9, PNA 10 and compound 4, 7 and 8 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ghidini, A.; Steunenberg, P.; Murtola, M.; Strömberg, R. Synthesis of PNA Oligoether Conjugates. Molecules 2014, 19, 3135-3148. https://doi.org/10.3390/molecules19033135
Ghidini A, Steunenberg P, Murtola M, Strömberg R. Synthesis of PNA Oligoether Conjugates. Molecules. 2014; 19(3):3135-3148. https://doi.org/10.3390/molecules19033135
Chicago/Turabian StyleGhidini, Alice, Peter Steunenberg, Merita Murtola, and Roger Strömberg. 2014. "Synthesis of PNA Oligoether Conjugates" Molecules 19, no. 3: 3135-3148. https://doi.org/10.3390/molecules19033135