Pharmacokinetic Study in Mice of Galphimine-A, an Anxiolytic Compound from Galphimia glauca


 ,
,
Abstract
:1. Introduction
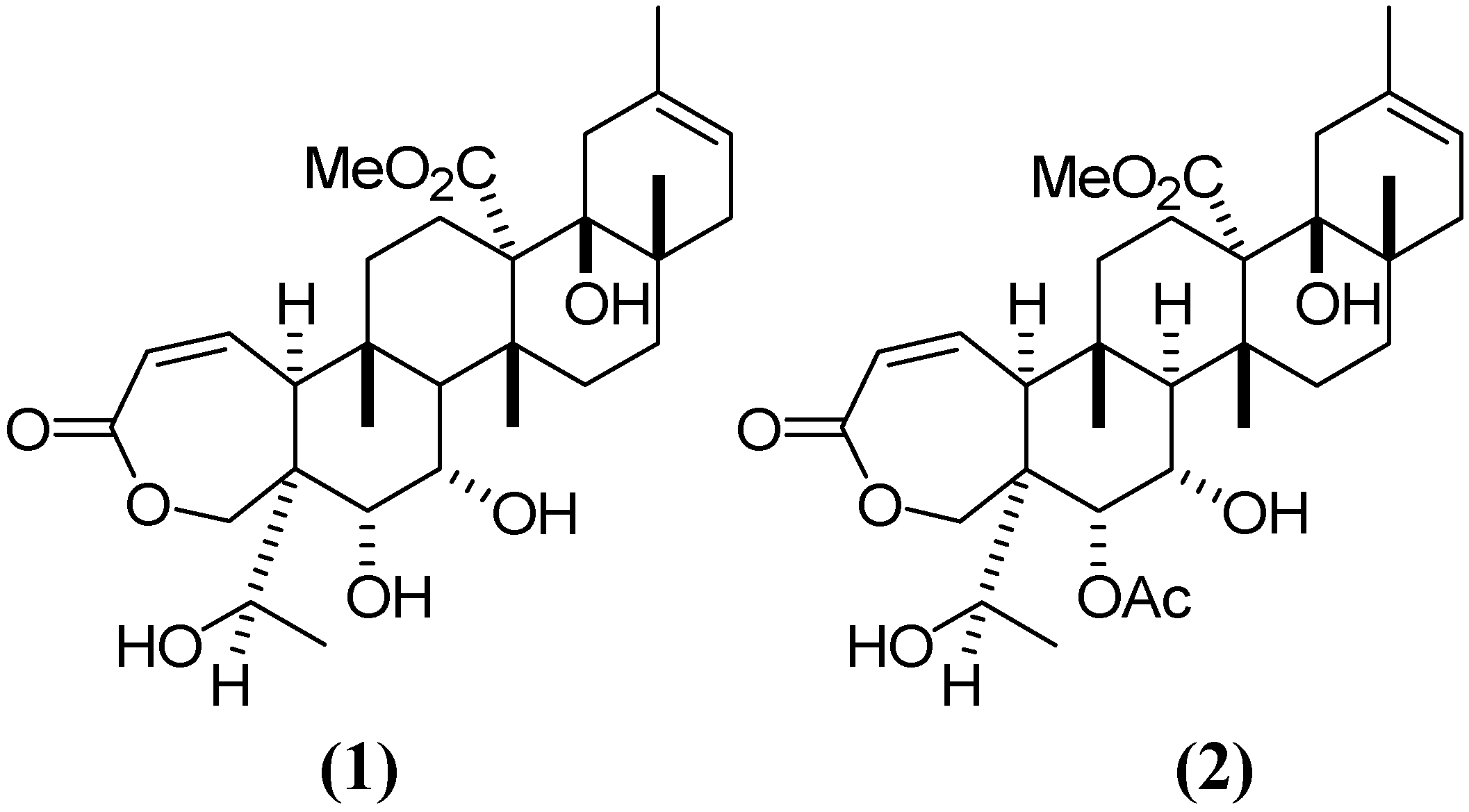
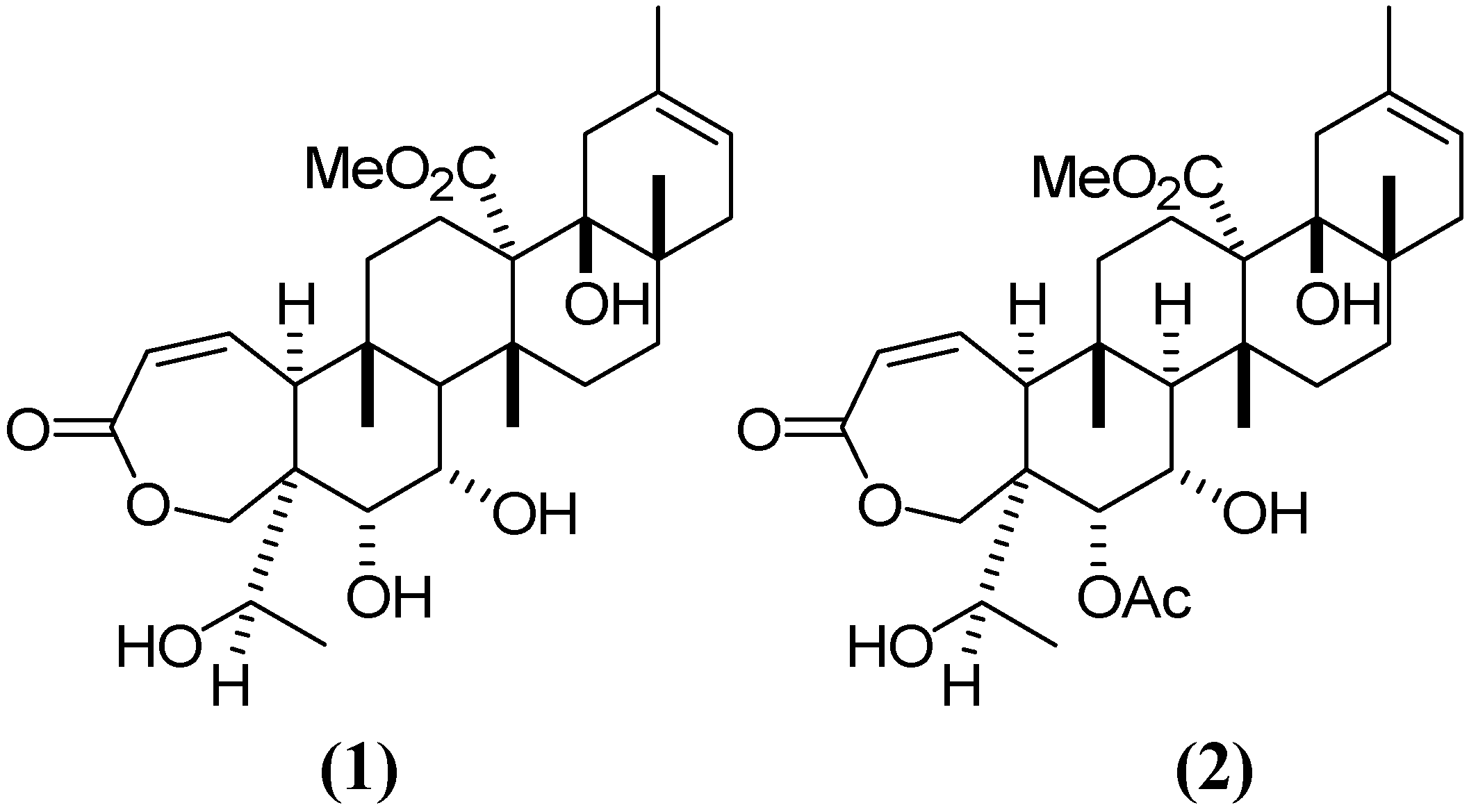
2. Results and Discussion
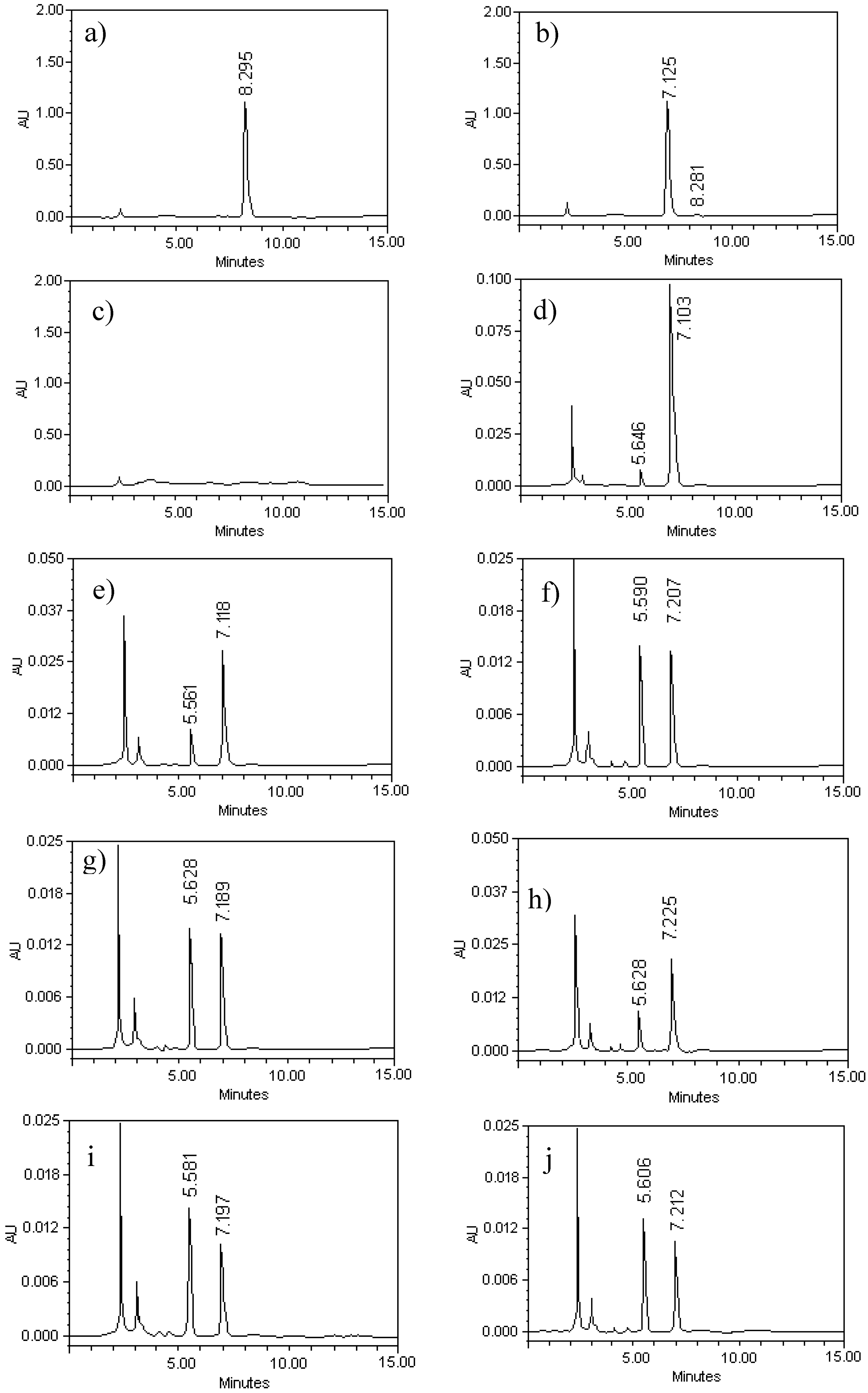
2.1. Chromatographic Analysis of G–A and G–E
2.2. Method Validation
Standardization of the Chromatographic Process
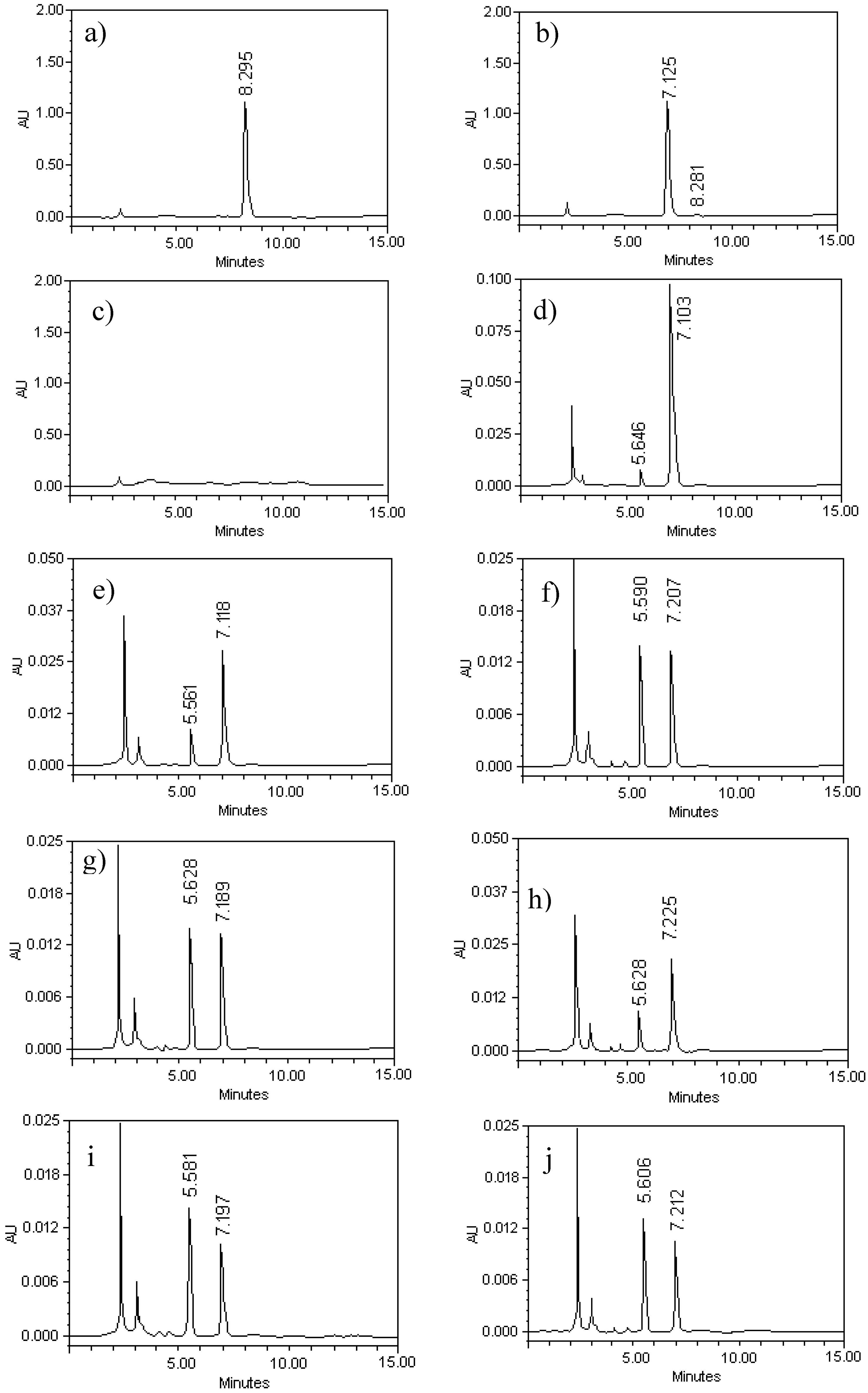

{kind=link}
{kind=link}
{kind=link}
| Matrix Sample | Nominal Concentration (μg/mL) | Observed Concentration (μg/mL) ± S.D. | Accuracy (% Bias) | RSD (%) |
|---|---|---|---|---|
| Plasma Intra-day | 0.80 | 0.77 ± 0.04 | −3 | 5.2 |
| 4.00 | 3.96 ± 0.09 | −4 | 2.3 | |
| 10.00 | 9.98 ± 0.97 | −2 | 9.7 | |
| Plasma Inter-day | 0.80 | 0.75 ± 0.07 | −5 | 9.3 |
| 4.00 | 3.94 ± 0.10 | −6 | 2.5 | |
| 10.00 | 10.01 ± 1.04 | 1 | 10.4 | |
| Brain tissue Intra-day | 0.80 | 0.78 ± 0.06 | −2 | 7.7 |
| 4.00 | 3.94 ± 0.12 | −6 | 3.0 | |
| 10.00 | 10.01 ± 0.73 | 1 | 7.3 | |
| Brain tissue Inter-day | 0.80 | 0.77 ± 0.08 | −3 | 10.4 |
| 4.00 | 3.96 ± 0.11 | −4 | 2.8 | |
| 10.00 | 10.02 ± 0.17 | 2 | 1.7 |
| Matrix Sample | Spiked Concentration (μg/mL) | Recovery Index ± S.D. | RSD (%) |
|---|---|---|---|
| Plasma | 0.80 | 0.854 ± 0.11 | 13.2 |
| 4.00 | 0.874 ± 0.09 | 10.3 | |
| 10.00 | 0.978 ± 0.14 | 14.3 | |
| Tissue brain | 0.80 | 0.951 ± 0.09 | 8.5 |
| 4.00 | 0.968 ± 0.08 | 8.3 | |
| 10.00 | 0.943 ± 0.13 | 13.8 |
| Matrix Sample | Handling Conditions | Nominal Concentration μg/mL) | Observed Concentration (μg/mL) ± S.D. | AccuracyBias (%) | RSD (%) |
|---|---|---|---|---|---|
| Plasma | 3 Freeze/thaw cycles (−70 °C) | 0.80 | 0.79 ± 0.08 | −1.00 | 10.1 |
| 4.00 | 4.01 ± 0.14 | 0.25 | 3.5 | ||
| 10.0 | 9.93 ± 0.67 | −0.70 | 6.7 | ||
| Autosampler stability (4 °C; 8 h) | 0.80 | 0.83 ± 0.05 | 3.75 | 6.0 | |
| 4.00 | 3.93 ± 0.43 | −1.75 | 10.9 | ||
| 10.0 | 10.10 ± 0.85 | 10.00 | 8.4 | ||
| Long–term stability (−70 °C; 1 month) | 0.80 | 0.77 ± 0.06 | −3.00 | 7.8 | |
| 4.00 | 4.26 ± 0.31 | 6.50 | 7.2 | ||
| 10.0 | 9.43 ± 0.83 | −5.70 | 8.8 | ||
| Brain tissue | 3 Freeze/thaw cycles (−70 °C) | 0.80 | 0.83 ± 0.08 | 3.75 | 9.6 |
| 4.00 | 4.36 ± 0.34 | 9.00 | 7.8 | ||
| 10.0 | 10.36 ± 0.52 | 3.60 | 5.0 | ||
| Autosampler stability (4 °C; 8 h) | 0.80 | 0.87 ± 0.11 | 8.75 | 12.6 | |
| 4.00 | 3.60 ± 0.33 | −10.0 | 9.1 | ||
| 10.0 | 9.83 ± 0.43 | −1.70 | 4.3 | ||
| Long–term stability (−70 °C; 1 month) | 0.80 | 0.72 ± 0.10 | −10.00 | 13.9 | |
| 4.00 | 3.73 ± 0.41 | −6.75 | 10.9 | ||
| 10.0 | 10.43 ± 0.98 | 4.30 | 9.4 |
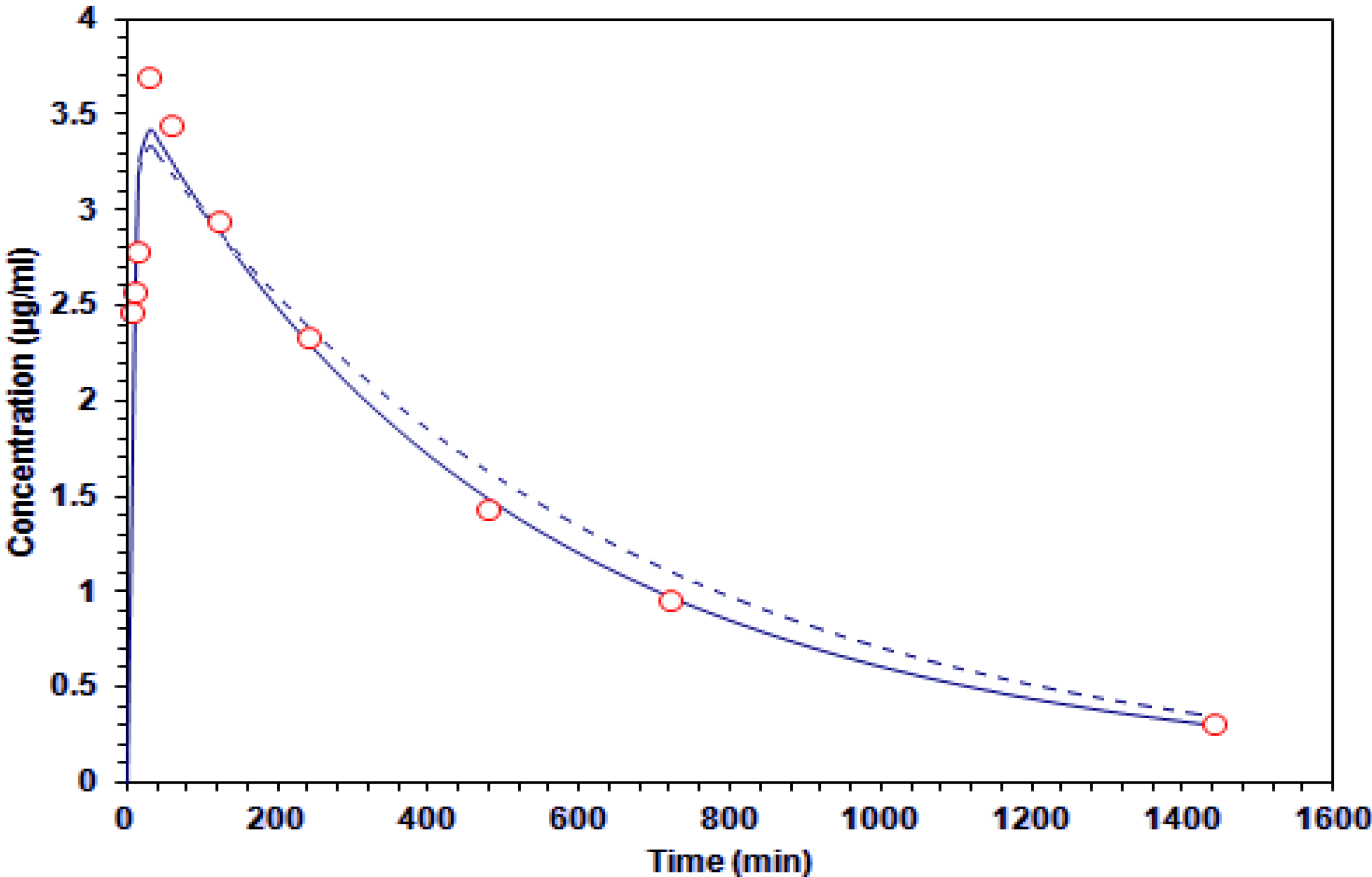
2.3. G–A Plasma Levels and Brain
| G–A Concentration (μg/mL) | |||
|---|---|---|---|
| Time (min) | Plasma | Brain | |
| 5 | 2.47 ± 0.43 | 0.00 | |
| 10 | 2.57 ± 0.52 | 0.88 ± 0.33 * | |
| 15 | 2.79 ± 0.79 | 1.48 ± 0.58 * | |
| 30 | 3.70 ± 0.70 | 3.44 ± 0.50 | |
| 60 | 3.44 ± 0.35 | 3.01 ± 0.59 | |
| 120 | 2.95 ± 0.43 | 2.50 ± 0.58 | |
| 240 | 2.33 ± 0.42 | 2.18 ± 0.48 | |
| 480 | 1.44 ± 0.37 | 1.78 ± 0.37 | |
| 720 | 0.96 ± 0.21 | 0.55 ± 0.18 * | |
| 1440 | 0.31 ± 0.02 | 0.32 ± 0.14 | |
| Parameter | One-Compartament Value | Two-Compartament Value | Units |
|---|---|---|---|
| A | 3.50 | 1.83 | μg/mL |
| B | --- | 1.82 | μg/mL |
| ka | 0.182 | 0.164 | 1/min |
| k10 | 0.0016 | 0.0017 | 1/min |
| k12 | --- | 0.00017 | 1/min |
| k21 | --- | 0.00195 | 1/min |
| t1/2ka | 3.79 | 4.22 | 1/min |
| α | --- | 0.0025 | 1/min |
| β | --- | 0.0014 | 1/min |
| V/F | 57.59 | 55.49 | (mg/kg)/(μg/mL) |
| CL/F | 0.092 | 0.098 | (mg/kg)/(μg/mL)/min |
| Tmax | 26.16 | 27.35 | min |
| Cmax | 3.33 | 3.42 | μg/mL |
| AUC0→∞ | 2170.15 | 2030.23 | μg/mL·min |
| AUC0-1440 min | 1951.58 | 1824.95 | μg/mL·min |
| MRT | 630.40 | 620.45 | min |
| Diagnostics | |||
| SS | 0.7217 | 0.6331 | |
| R2 | 0.9885 | 0.9899 | |
| AIC | −6.1575 | −4.6565 | |
| SC | −5.2498 | −3.1436 | |
| Parameter | Value | Unit |
|---|---|---|
| A | 2.98 | μg/mL |
| ka | 0.051 | 1/min |
| k10 | 0.0008 | 1/min |
| t1/2 ka | 13.50 | 1/min |
| V/F | 68.16 | (mg/kg)/(μg/mL) |
| CL/F | 0.057 | (mg/kg)/(μg/mL)/min |
| Tmax | 81.59 | min |
| Cmax | 2.74 | μg/mL |
| AUC0→∞ | 3519.23 | μg/mL·min |
| AUC0-1440 min | 1912.49 | μg/mL·min |
| MRT | 1218.86 | min |
 ) and two-compartment (-----) models.
) and two-compartment (-----) models.
) and two-compartment (-----) models.
) and two-compartment (-----) models.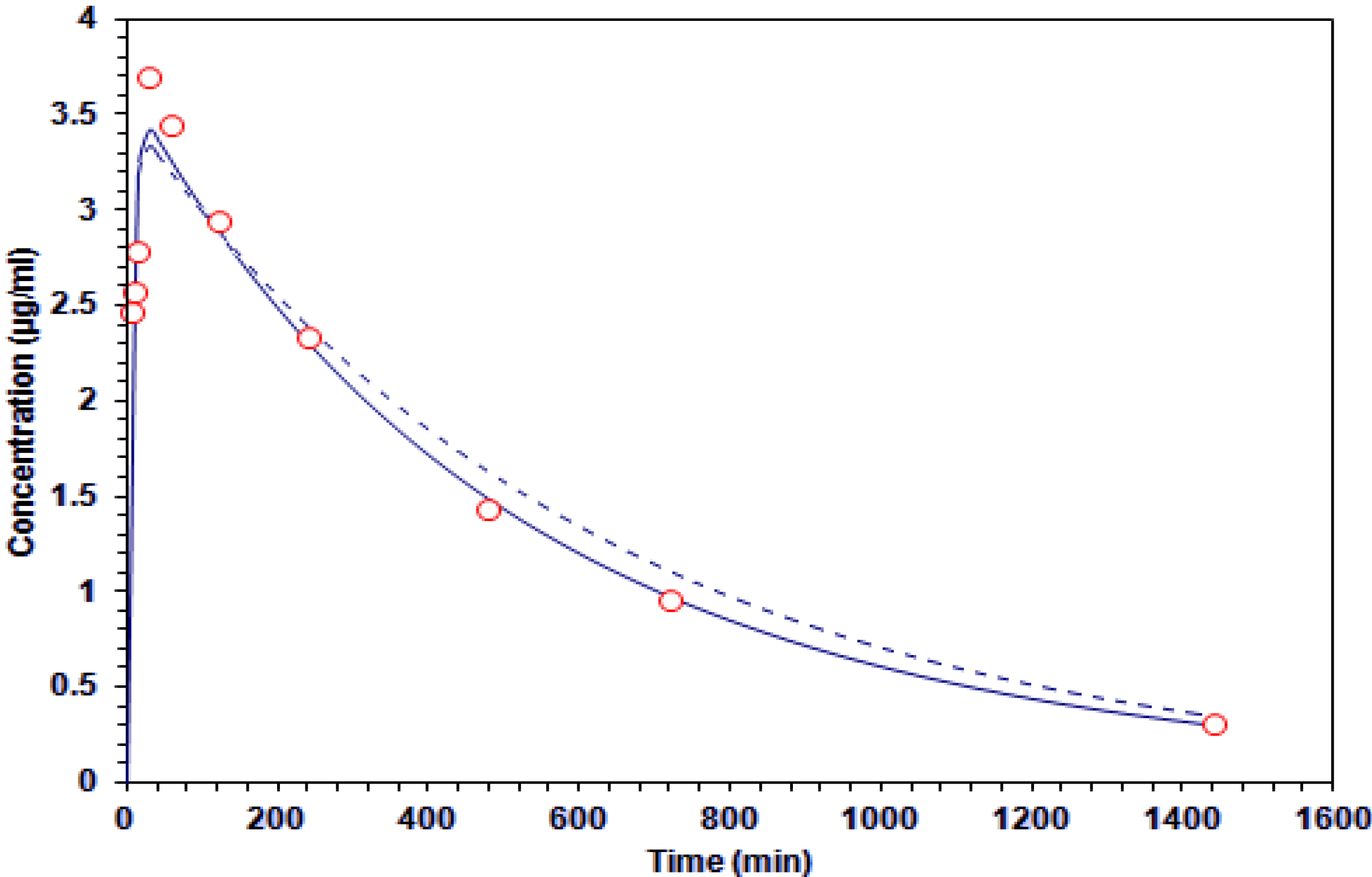
3. Experimental
3.1. Chemicals
3.2. Plants Material
3.3. Extraction and G–A Purification
3.4. Animals
3.5. Preparation of Standard and Internal Standard Stock Solution
3.6. HPLC Calibration Curve of G–A
3.7. Experimental Design of G–A Administration and Blood and Brain Sample Collection
3.8. Quantification of G–A in Plasma and Brain Samples
3.9. HPLC Calibration Curve of G–A
3.10. Method Validation
3.11. Linearity and Sensitivity
3.12. Specificity
3.13. Accuracy and Precision
3.14. Recovery (Extraction Efficiency)
3.15. Stability Study
3.16. Pharmacokinetic Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflictts of Interest
References
- Estrada, E. Jardín Botánico de Plantas Medicinales “Maximino Martínez”; (in Spanish). Universidad Autónoma de Chapingo: Chapingo Edo. Méx, México, 1985; p. 15. [Google Scholar]
- Tortoriello, J.; Lozoya, X. Effect of Galphimia glauca methanolic extract on neuropharmacological test. Planta Med. 1992, 58, 234–236. [Google Scholar] [CrossRef]
- Herrera-Ruiz, M.; Jiménez-Ferrer, J.E.; de Lima, T.C.M.; Avilés-Montes, D.; Pérez-García, D.; González-Cortazar, M.; Tortoriello, J. Anxiolytic and antidepressant-like activity of a standardized extract from Galphimia glauca. Phytomedicine 2005, 13, 23–28. [Google Scholar]
- Herrera-Ruiz, M.; González-Cortazar, M.; Jiménez-Ferrer, E.; Zamilpa, A.; Alvarez, L.; Ramírez, G.; Tortoriello, J. Anxiolytic effect of natural galphimines from Galphimia glauca and their chemical derivatives. J. Nat. Prod. 2006, 69, 59–61. [Google Scholar] [CrossRef]
- Herrera-Arellano, A.; Jiménez-Ferrer, E.; Zamilpa, A.; Morales-Valdéz, M.; García-Valencia, C.E.; Tortoriello, J. Efficacy and tolerability of a standardized herbal product from Galphimia glauca on generalized anxiety disorder. A randomized, double-blind clinical trial controlled with lorazepam. Planta Med. 2007, 73, 713–717. [Google Scholar] [CrossRef]
- González-Cortazar, M.; Tortoriello, J.; Alvarez, L. Norsecofridelanes as spasmolytics, advances of structure-activity relationships. Planta Med. 2005, 71, 711–716. [Google Scholar] [CrossRef]
- United States Health and Human Services. Guidance for industry: Bioanalytical method validation. 2001. Available online: http://www.fda.gov./downloads/Drugs/GuidancemplianceRegulatoryInformation/Guidance/UCM070107.pdf (accessed on 23 January 2014). [Google Scholar]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Makino, T.; Ohtake, N.; Watanabe, A.; Tsuchiya, N.; Imamura, S.; Iizuka, S.; Inoue, M.; Mizukami, H. Down-regulation of a hepatic transporter multidrug resistance-associated protein 2 is involved in alteration of pharmacokinetics of glycyrrhizin and its metabolites in a rat model of chronic liver injury. Drug Metab. Dispos. 2008, 36, 1438–1443. [Google Scholar] [CrossRef]
- Gao, Q.T.; Chen, X.H.; Bi, K.S. Comparative pharmacokinetic behavior of glycyrrhetic acid after oral administration of glycyrrhizic acid and gancao-fuzi-tang. Biol. Pharm. Bull. 2004, 27, 226–228. [Google Scholar] [CrossRef]
- Zhao, W.J.; Wang, B.J.; Wei, C.M.; Yuan, G.Y.; Bu, F.L.; Guo, R.C. Determination of glycyrrhetic acid in human plasma by HPLC-MS method and investigation of its pharmacokinetics. J. Clin. Pharm. Ther. 2008, 33, 289–294. [Google Scholar] [CrossRef]
- Ludden, T.M.; Beal, S.L.; Sheiner, L.B. Comparison of the Akaike Information Criterion, the Schwarz criterion and the F test as guide to model selection. J. Pharmacokinet. Biopharm. 1994, 22, 431–445. [Google Scholar] [CrossRef]
- Sample Availability: Sample of the compound galphimine-A (G–A) is available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vargas, R.A.; Zamilpa, A.; Aguilar, F.A.; Herrera-Ruiz, M.; Tortoriello, J.; Jiménez-Ferrer, E. Pharmacokinetic Study in Mice of Galphimine-A, an Anxiolytic Compound from Galphimia glauca. Molecules 2014, 19, 3120-3134. https://doi.org/10.3390/molecules19033120
Vargas RA, Zamilpa A, Aguilar FA, Herrera-Ruiz M, Tortoriello J, Jiménez-Ferrer E. Pharmacokinetic Study in Mice of Galphimine-A, an Anxiolytic Compound from Galphimia glauca. Molecules. 2014; 19(3):3120-3134. https://doi.org/10.3390/molecules19033120
Chicago/Turabian StyleVargas, Rodolfo Abarca, Alejandro Zamilpa, Francisco Alarcón Aguilar, Maribel Herrera-Ruiz, Jaime Tortoriello, and Enrique Jiménez-Ferrer. 2014. "Pharmacokinetic Study in Mice of Galphimine-A, an Anxiolytic Compound from Galphimia glauca" Molecules 19, no. 3: 3120-3134. https://doi.org/10.3390/molecules19033120
APA StyleVargas, R. A., Zamilpa, A., Aguilar, F. A., Herrera-Ruiz, M., Tortoriello, J., & Jiménez-Ferrer, E. (2014). Pharmacokinetic Study in Mice of Galphimine-A, an Anxiolytic Compound from Galphimia glauca. Molecules, 19(3), 3120-3134. https://doi.org/10.3390/molecules19033120