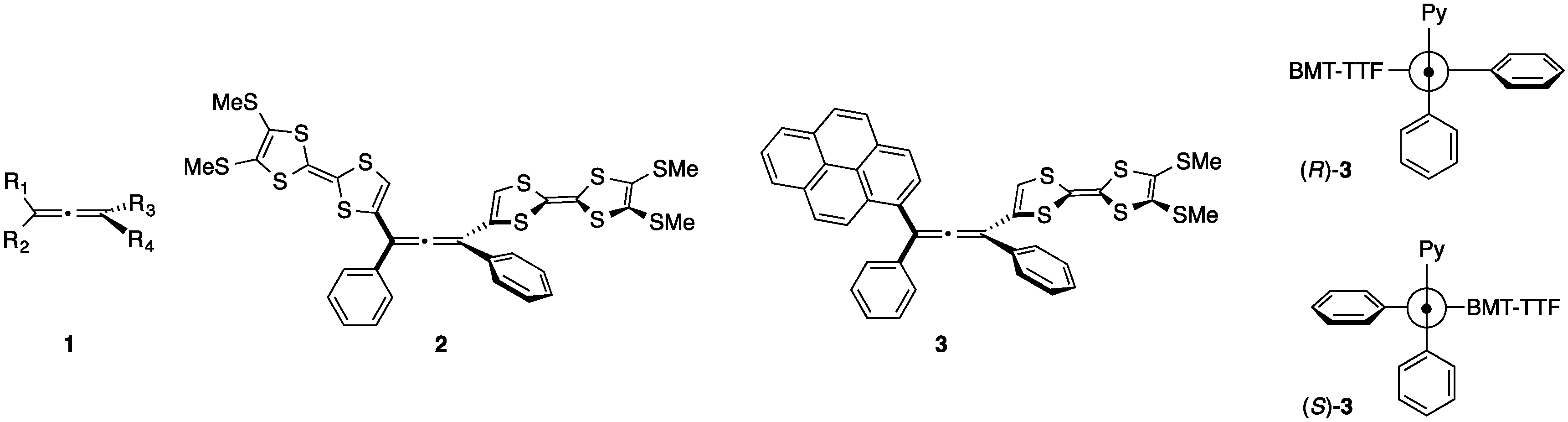
Chiroptical Properties and the Racemization of Pyrene and Tetrathiafulvalene-Substituted Allene: Substitution and Solvent Effects on Racemization in Tetrathiafulvalenylallene

Abstract
:
1. Introduction
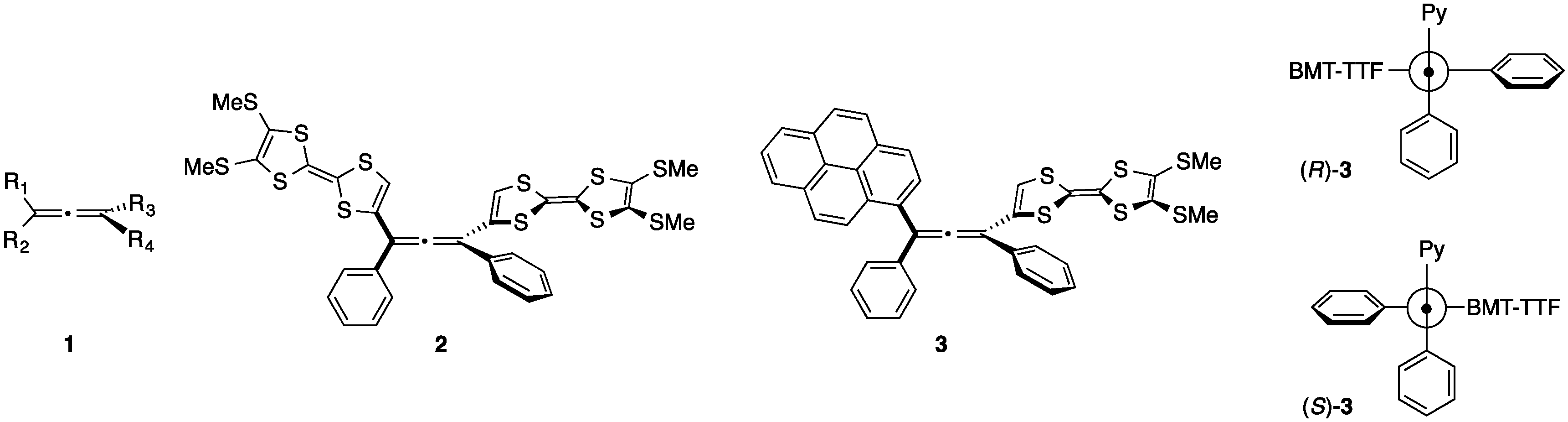
2. Results and Discussion
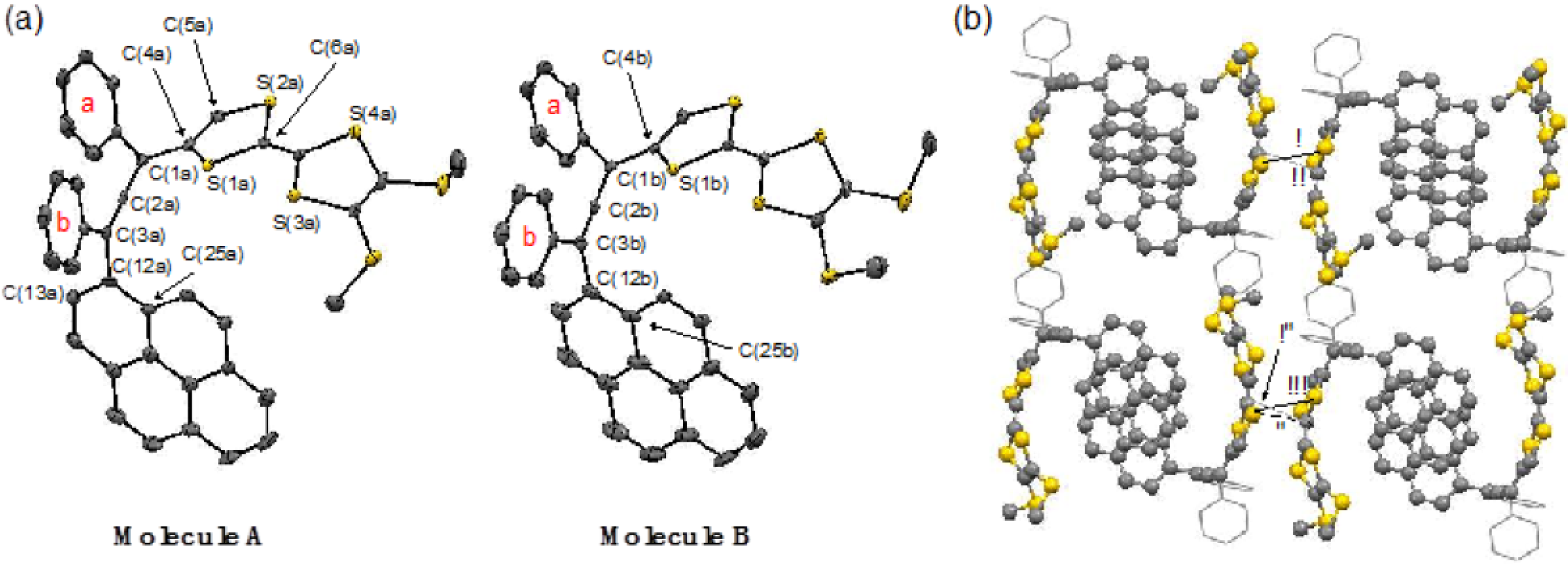
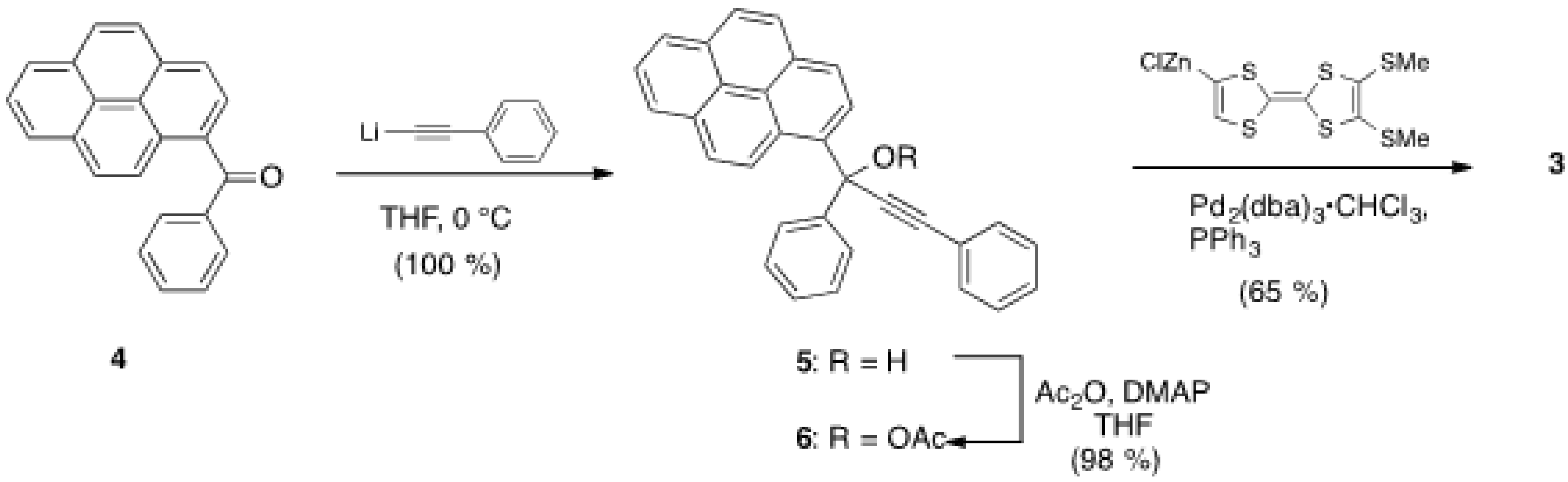
2.1. Synthesis of Pyrene and Tetrathiafulvalene-substituted Allene 3 and X-ray Determination
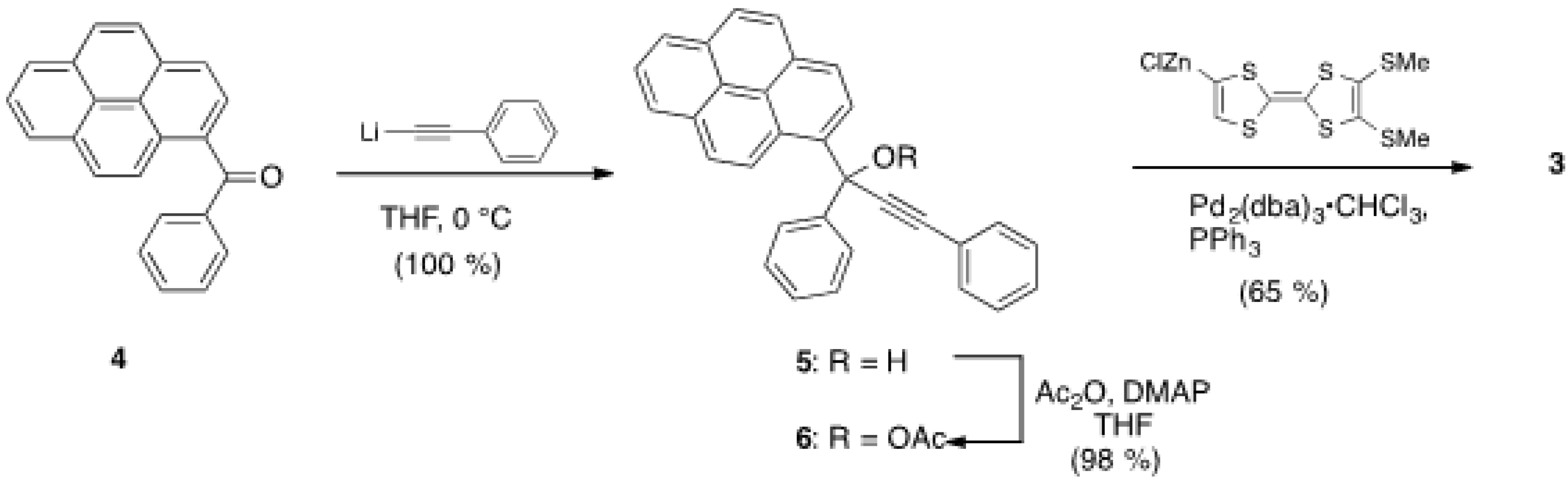
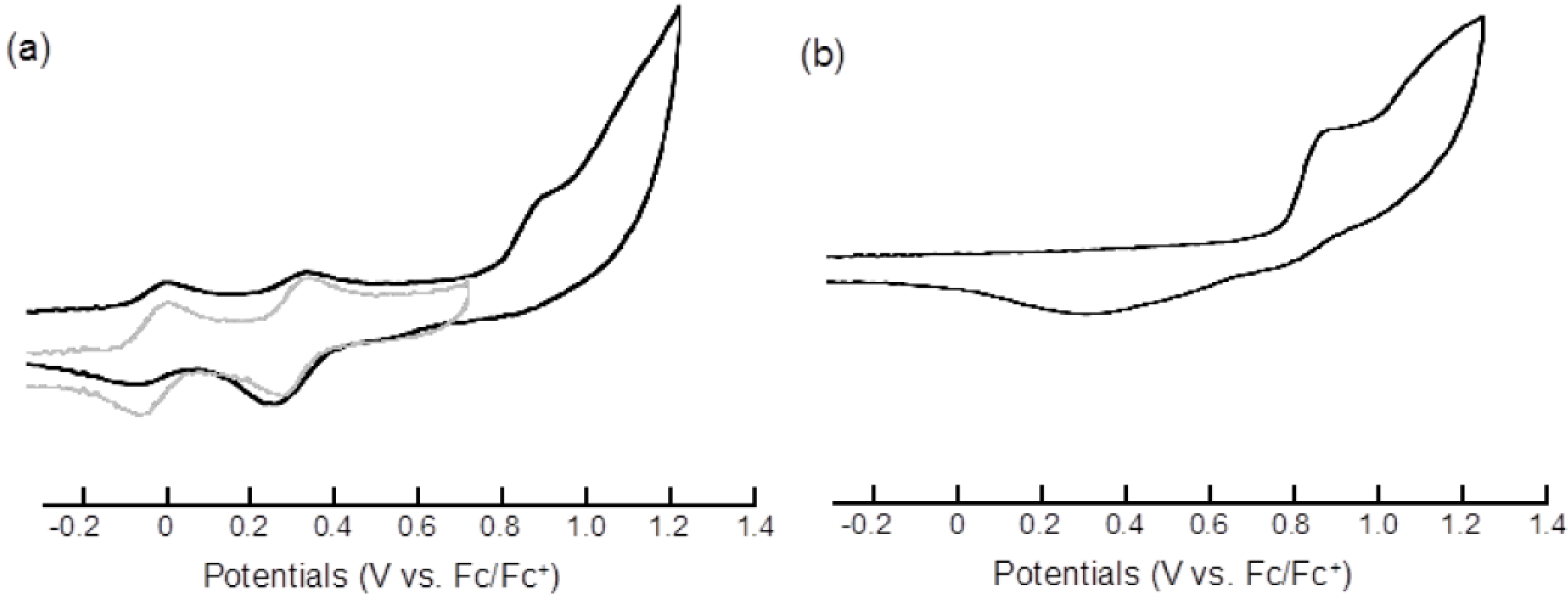
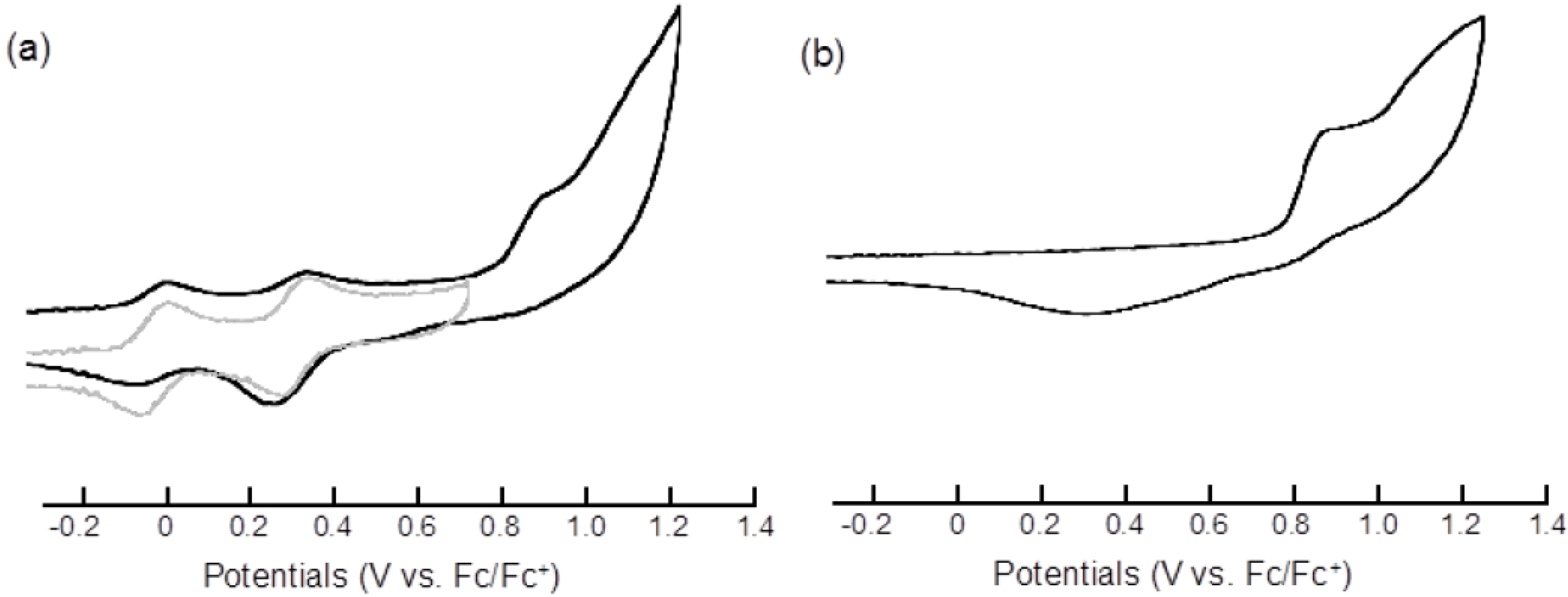
2.2. Electrochemical Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | E11/2 | E21/2 | Epa3 | Epc3 |
|---|---|---|---|---|
| 3 | 0.00 | 0.33 | 0.92 b | 0.31 b,c |
| 7 | −0.02 | 0.34 | ||
| pyrene | 0.92 b | 0.33 b,c |
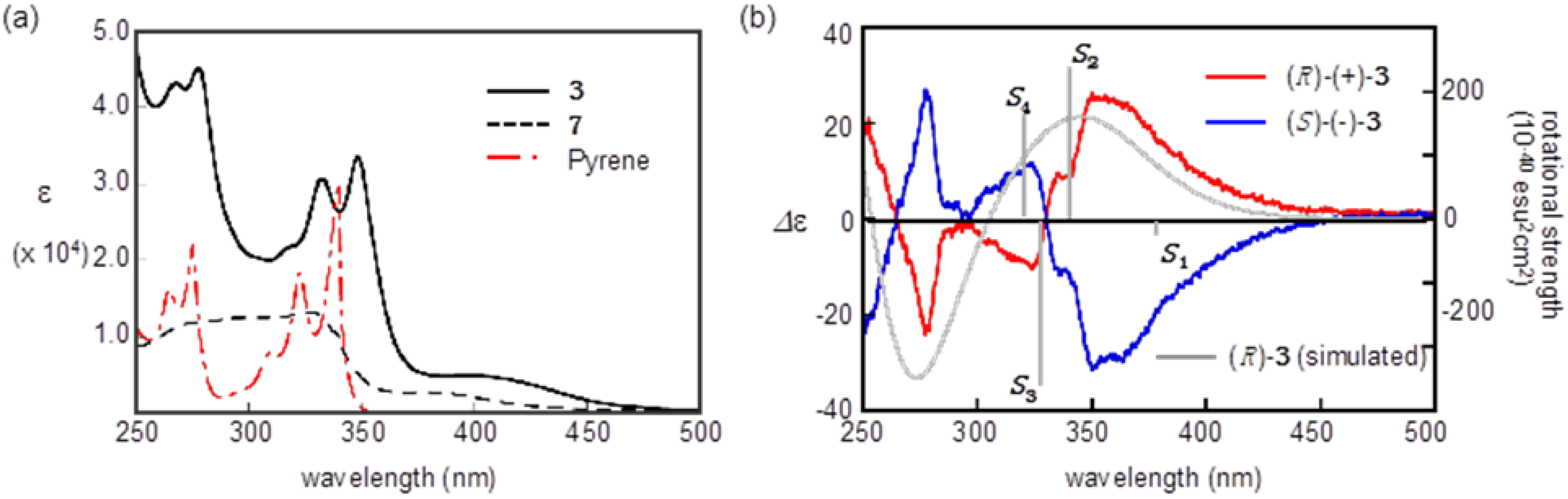
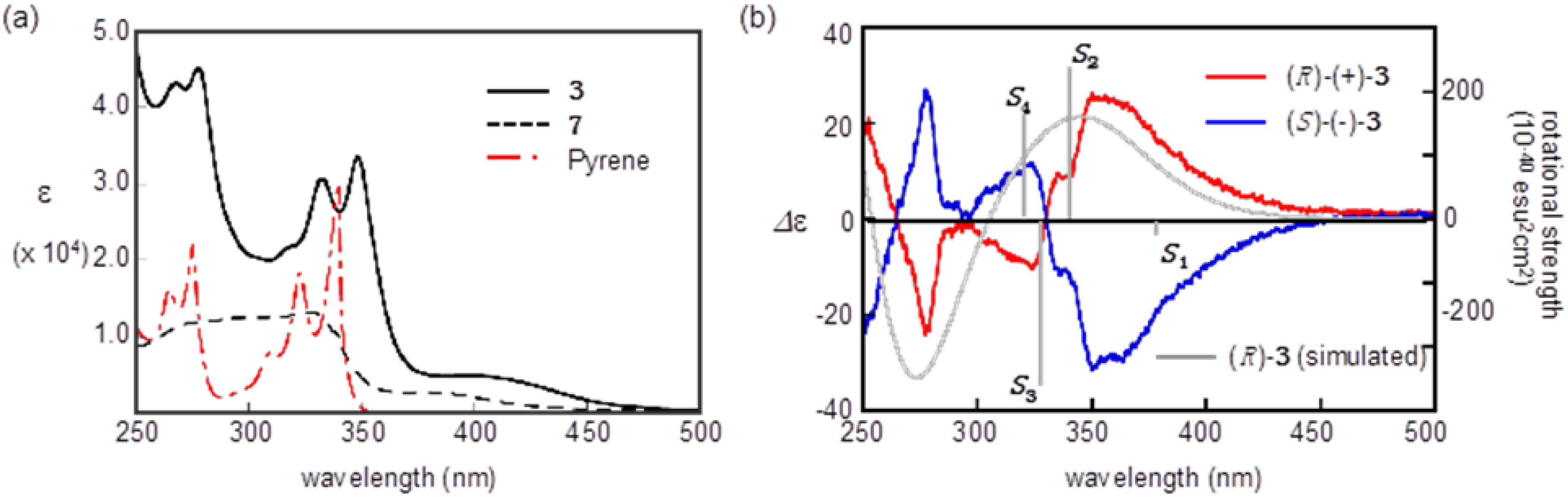
2.3. Optical Resolution and ECD Spectra

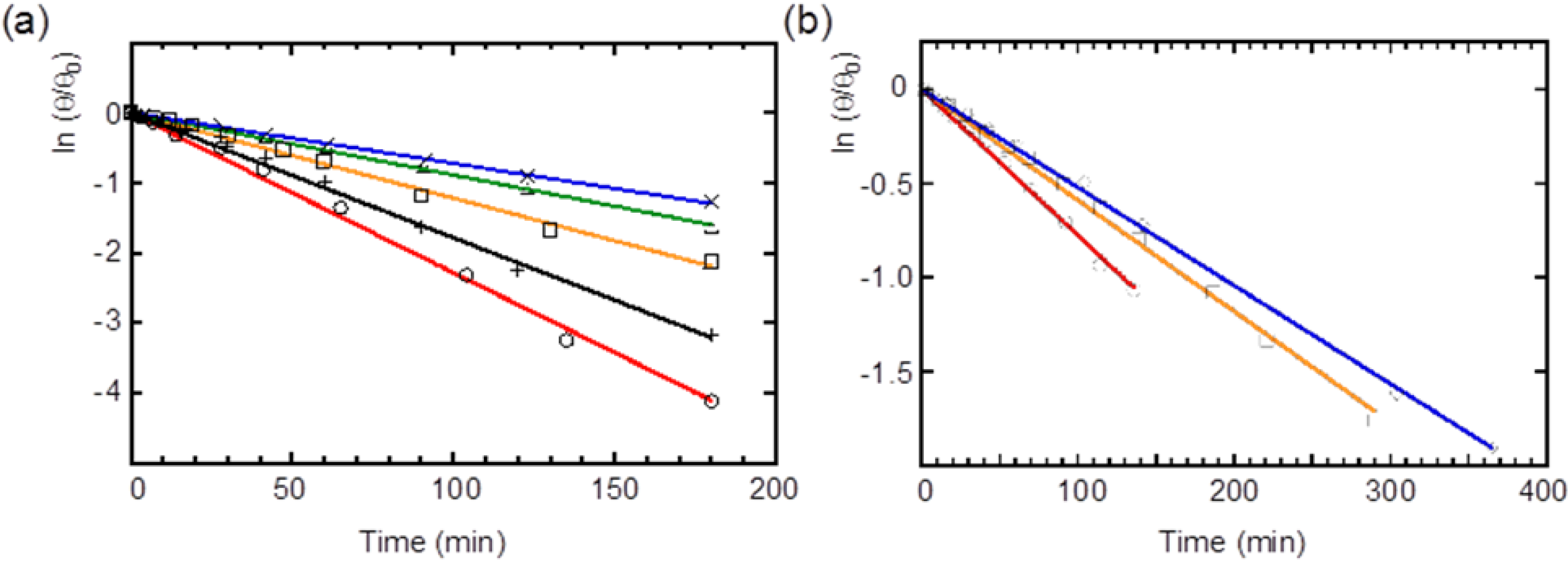
2.4. Racemization of 2 and 3 in Various Solvents
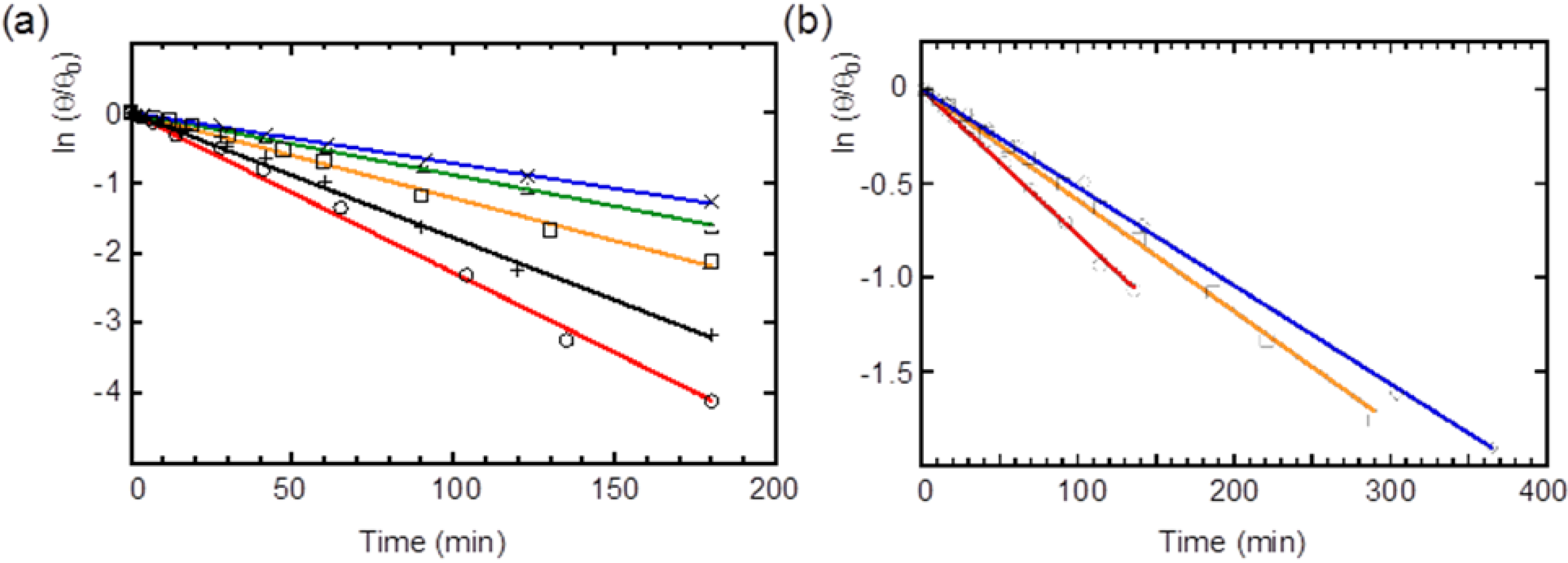
| Compound | Solvent | Rate (×10−3·min−1) | Half-life (min) |
|---|---|---|---|
| 2 | Benzene | 22.8 ± 0.37 | 30 |
| MCH | 17.8 ± 0.26 | 39 | |
| CH2Cl2 | 12.1 ± 0.24 | 57 | |
| CH2Cl2–MeCN (v/v = 4:1) | 8.87 ± 0.08 | 78 | |
| CH2Cl2–MeCN (v/v = 1:1) | 7.18 ± 0.08 | 96 | |
| 3 | Benzene | 7.78 ± 0.01 | 89 |
| CH2Cl2 | 5.91 ± 0.06 | 117 | |
| CH2Cl2–MeCN (v/v = 1:1) | 5.22 ± 0.04 | 133 |
3. Experimental
3.1. General Information
3.2. Synthesis of 1,3-Diphenyl-2-propyne-1-pyrenyl-1-ol 5
3.3. Synthesis of 1,3-Diphenyl-2-propyne-1-pyrenylacetate (6)
3.4. Synthesis of the Allene 3
3.5. X-ray Crystallographic Study of 3
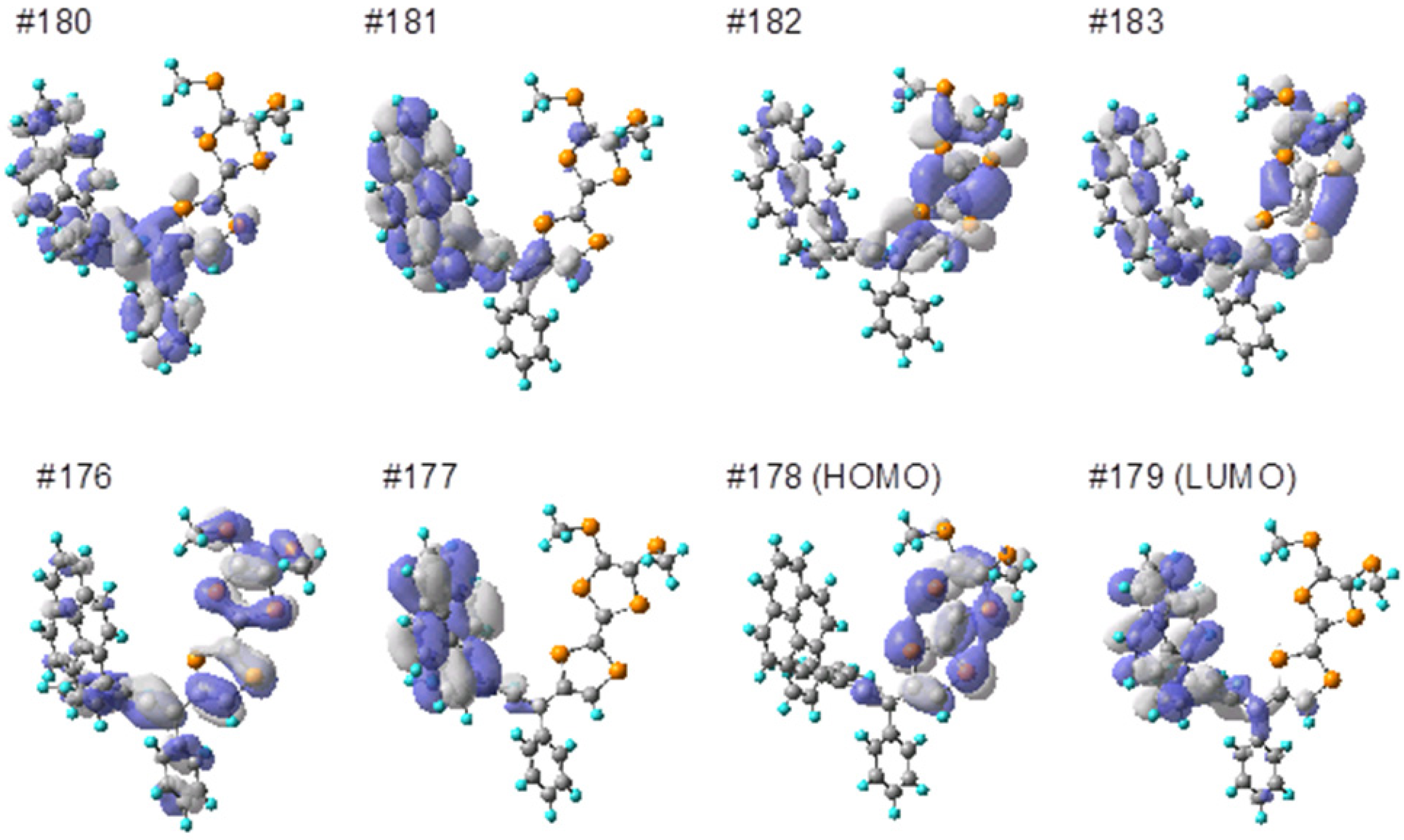
3.6. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflictts of Interest
References and Notes
- Krause, N.; Hashimi, A.S.K. Modern Allene Chemistry; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Hopf, H. Allenea and cumulenes. In Classics in Hyderocarbon Chemistry; Wiley-VCH: Weinheim, Germany, 2000; pp. 171–196. [Google Scholar]
- Yu, S.; Ma, S. How easy are the synthesis of allenes? Chem. Commun. 2011, 5384–5418. [Google Scholar]
- Canary, J.W. Redox-triggered chiroptical molecular switches. Chem. Soc. Rev. 2009, 38, 747–756. [Google Scholar] [CrossRef]
- Rivera-Fuentes, P.; Diederich, F. Allenes in molecular materials. Angew. Chem. Int. Ed. 2012, 51, 2818–2828. [Google Scholar] [CrossRef]
- Campolo, D.; Gastaldi, S.; Roussel, C.; Bertrand, M.P.; Nechab, M. Axial-to-central chirality transfer in cyclization processes. Chem. Soc. Rev. 2013, 42, 8434–8466. [Google Scholar]
- Wolf, C. Racemization, enantiomerization, and diastereomerization. In Dynamic Stereochemsitry of Chiral Compounds, Principles and Applications; RSC Publishing: Cambridge, UK, 2008; pp. 29–135. [Google Scholar]
- Roth, W.R.; Ruf, G.; Ford, P.W. Rotationsbarriere in 1,2-Dienen; Resonanzenergie des Allyl-Radikals. Chem. Ber. 1974, 107, 48–52. [Google Scholar]
- Roth, W.R.; Bastigkeit, T. Die Allen-Racemisierung, eine zweistufige Reaktion. Liebigs Ann. 1996, 1996, 2171–2183. [Google Scholar] [CrossRef]
- Schurig, V.; Keller, F.; Reich, S.; Fluck, M. Dynamic phenomena involving chiral dimethyl-2,3-pentadienedioate in enantioselective gas chromatography and NMR spectroscopy. Tetrahedron: Asymm. 1997, 8, 3475–3480. [Google Scholar] [CrossRef]
- Trapp, V.; Scurig, V. Novel direct access to enantimerization barriers from peak profiles in enantioselective dynamic chromatography: Enantiomerization of dialkyl-1,3-allenedicarboxylates. Chirality 2002, 14, 465–470. [Google Scholar] [CrossRef]
- Rodriguez, O.; Morrisonm, H. Photosensitized racemization of an optically active allene. J. Chem. Soc. D Chem. Commun. 1971, 679. [Google Scholar] [CrossRef]
- Stierman, T.; Johnson, R.P. Cumulene photochemistry: Singlet and triplet photorearrangements of 1,2-cyclononadiene. J. Am. Chem. Soc. 1985, 107, 3971–3980. [Google Scholar] [CrossRef]
- Horváth, K.; Bäckvall, J.-E. Mild and efficient palladium(II)-catalyzed racemization of allenes. Chem. Commun. 2004, 964–965. [Google Scholar] [CrossRef]
- Claesson, A.; Olsson, L.-I. Chiral allenes are racemised by organocuprates. J. Chem. Soc. Chem. Commun. 1979, 524–525. [Google Scholar] [CrossRef]
- Sherry, B.D.; Toste, F.D. Gold(I)-Catalyzed Propargyl Claisen Rearrangement. J. Am. Chem. Soc. 2004, 126, 15978–15979. [Google Scholar] [CrossRef]
- Alonso-Gómez, J.L.; Schanen, P.; Rivera-Fuentes, P.; Seiler, P.; Diederich, F. 1,3-Diethynylallenes (DEAs): Enantioselective synthesis, absolute configuration, and chiral induction in 1,1,4,4-tetracyanobuta-1,3-dienes (TCBDs). Chem. Eur. J. 2008, 14, 10564–10568. [Google Scholar] [CrossRef]
- Odermatt, S.; Alonso-Gómez, L.; Seiler, P.; Cid, M.M.; Diederich, F. Shape-persistent chiral alleno-acetylenic macrocycles and cyclophanes by acetylenic scaffolding with 1,3-diethynylallenes. Angew. Chem. Int. Ed. 2005, 44, 5074–5078. [Google Scholar] [CrossRef]
- Hasegawa, M.; Sone, Y.; Iwata, S.; Matsuzawa, H.; Mazaki, Y. Tetrathiafulvalenylallene: A new class of donor molecules having strong chiroptical properties in neutral and doped states. Org. Lett. 2011, 13, 4688–4691. [Google Scholar] [CrossRef]
- Deretey, E.; Shapiro, M.; Brumer, P. Chiral molecules with achiral excited states: A computational study of 1,3-dimethylallene. J. Phys. Chem. A 2001, 105, 9509–9517. [Google Scholar] [CrossRef]
- Jarowski, P.D.; Diederich, F.; Houk, K.N. Butatrienes as extended alkenes: Barriers to internal rotation and substitution effects on the stabilities of the ground states and transition states. J. Phys. Chem. A 2006, 110, 7237–7246. [Google Scholar] [CrossRef]
- Drucker, C.S.; Toscano, V.G.; Weiss, R.G. A general method for the determination of steric effects during collisional energy transfer. Partial photoresolution of penta-2,3-diene. J. Am. Chem. Soc. 1973, 95, 6482–6484. [Google Scholar] [CrossRef]
- Löhr, S.; Averbeck, J.; Schurmann, M.; Krause, N. Synthesis and complexation properties of allenic bipyridines, a new class of axially chiral ligands for transition metal catalysis. Eur. J. Inorg. Chem. 2008, 2008, 552–556. [Google Scholar]
- Zelder, C.; Krause, N. Enantioselective synthesis and circular dichroism of endocyclic allenes. Eur. J. Org. Chem. 2004, 2004, 3968–3971. [Google Scholar] [CrossRef]
- Hasegawa, M.; Daigoku, K.; Hashimoto, K.; Nishikawa, H.; Iyoda, M. Face-to-face dimeric tetrathiafulvalenes and their cation radical and dication species as models of mixed valence and π-dimer states. Bull. Chem. Soc. Jpn. 2012, 85, 51–60. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C. 01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- See Supporting Information.
- Waldeck, D.H. Photoisomerization dynamics of stilbenes. Chem. Rev. 1991, 91, 415–438. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hasegawa, M.; Iwata, S.; Sone, Y.; Endo, J.; Matsuzawa, H.; Mazaki, Y. Chiroptical Properties and the Racemization of Pyrene and Tetrathiafulvalene-Substituted Allene: Substitution and Solvent Effects on Racemization in Tetrathiafulvalenylallene. Molecules 2014, 19, 2829-2841. https://doi.org/10.3390/molecules19032829
Hasegawa M, Iwata S, Sone Y, Endo J, Matsuzawa H, Mazaki Y. Chiroptical Properties and the Racemization of Pyrene and Tetrathiafulvalene-Substituted Allene: Substitution and Solvent Effects on Racemization in Tetrathiafulvalenylallene. Molecules. 2014; 19(3):2829-2841. https://doi.org/10.3390/molecules19032829
Chicago/Turabian StyleHasegawa, Masashi, Seiya Iwata, Yasuto Sone, Junta Endo, Hideyo Matsuzawa, and Yasuhiro Mazaki. 2014. "Chiroptical Properties and the Racemization of Pyrene and Tetrathiafulvalene-Substituted Allene: Substitution and Solvent Effects on Racemization in Tetrathiafulvalenylallene" Molecules 19, no. 3: 2829-2841. https://doi.org/10.3390/molecules19032829
APA StyleHasegawa, M., Iwata, S., Sone, Y., Endo, J., Matsuzawa, H., & Mazaki, Y. (2014). Chiroptical Properties and the Racemization of Pyrene and Tetrathiafulvalene-Substituted Allene: Substitution and Solvent Effects on Racemization in Tetrathiafulvalenylallene. Molecules, 19(3), 2829-2841. https://doi.org/10.3390/molecules19032829