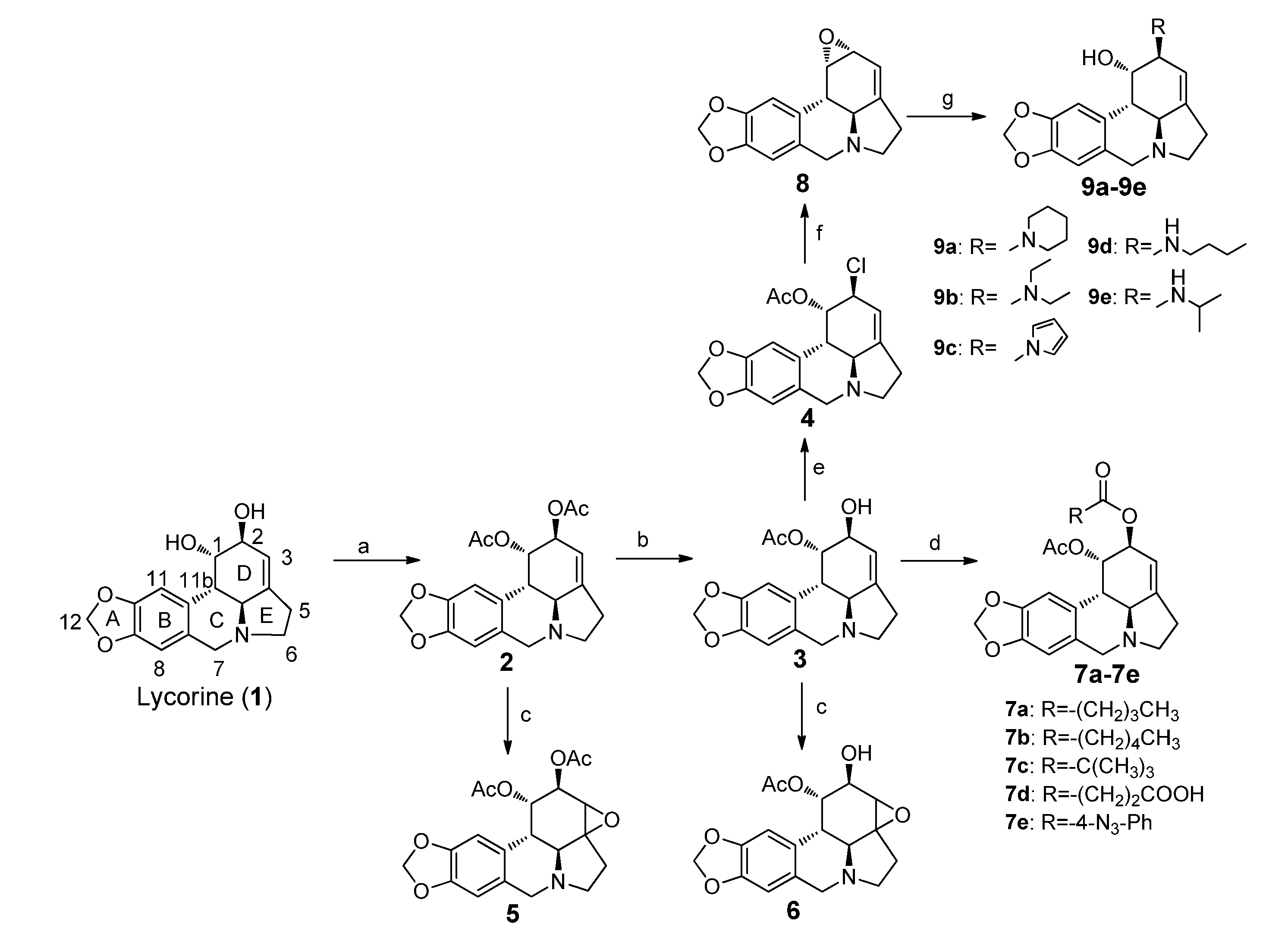
3.4. Chemistry
(1S,2S,3a1S,12bS)-2,3a1,4,5,7,12b-Hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridine-1,2-diyl diacetate (1,2-diacetyllycorine, 2). To a solution of lycorine (3.17 g, 11 mmol) in pyridine (8.0 mL) was added acetic anhydride (9.5 mL) and the solution was stirred at 50 °C for 12 h. Subsequently methanol (25 mL) was added. The mixture was stirred at r.t. for 3 h. The solvent was removed under reduced pressure followed by the addition of DCM (50 mL) and water (40 mL). The organic layer was washed with aqueous NaHCO3 solution and brine, respectively, and dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated and the crude residue was purified using a silica gel chromatography and was eluted with DCM/EtOAc/CH3OH (10:10:1) to give 2 as a white solid (3.78 g, 93.3%). 1H-NMR (400 MHz, CDCl3) δ: 6.75 (s, 1H), 6.57 (s, 1H), 5.91 (s, 2H), 5.74 (s, 1H), 5.53 (s, 1H), 5.26 (s, 1H), 4.16 (d, J = 14.0 Hz, 1H), 3.53 (d, J = 14.0 Hz, 1H), 3.37 (m, 1H), 2.86 (d, J = 10.0 Hz, 1H), 2.77 (d, J = 10.4 Hz, 1H), 2.65 (m, 2H), 2.40 (m, 1H), 2.08 (s, 3H), 1.95 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 170.0, 169.8, 146.4, 146.2, 129.4, 126.5, 113.8, 107.3, 105.0, 101.0, 70.9, 69.2, 61.2, 56.9, 53.6, 40.5, 28.7, 21.2, 21.0; ESI-MS: m/z 372 [M + H]+; HRMS: m/z calcd. for C20H22NO6 372.1442 [M + H]+; found: 372.1445.
(1S,2S,3a1S,12bS)-2-Hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]-phenanthridin-1-yl acetate (1-acetyllycorine, 3). To a solution of 2 (0.96 g, 2.58 mmol) in methanol (100 mL) was added conc. HCl (20 mL), and the solution was stirred at 55 °C for 1 h. The mixture was made alkaline (pH = 8) with aqueous NaHCO3 solution, followed by the addition of DCM (25 mL). The organic layer was washed with brine (25 mL), dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated under reduced pressure and the crude residue was purified using silica gel chromatography and was eluted with EtOAc/CH3OH (10:1) to give 3 as a white solid (0.50 g, 58.9%). 1H-NMR (400 MHz, CDCl3) δ: 6.66 (s, 1H), 6.57 (s, 1H), 5.92 (s, 2H), 5.61 (s, 1H), 5.54 (s, 1H), 4.18 (s, 1H), 4.25 (d, J = 14.4 Hz, 1H), 3.50 (d, J = 14.0 Hz, 1H), 3.35 (m, 1H), 2.85 (d, J = 10.4 Hz, 1H), 2.74 (d, J = 10.4 Hz, 1H), 2.63 (m, 2H), 2.37 (m, 1H), 1.94 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 170.8, 146.4, 146.2, 143.3, 129.1, 127.0, 117.6, 107.3, 104.9, 101.0, 72.7, 69.2, 61.6, 56.8, 53.7, 39.0, 28.5, 21.1; ESI-MS: m/z 330 [M + H]+; HRMS: m/z calcd. for C18H20NO5 330.1341 [M + H]+; found: 330.1344.
(1S,2S,3a1S,12bS)-2-Chloro-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]-phenan-thridin-1-yl acetate (1-acetyl-2-chlorollycorine, 4). To a solution of NaCl (5 mg, 0.1 mmol) in phosphorus oxychloride (2 mL) was added 3 (156 mg, 0.47 mmol), and the mixture was stirred at 30 °C for 1 h, followed by the addition of conc. HCl solution (0.1 mL). After stirred at 30 °C for another 2 h, the mixture was poured into ice water, made alkaline (pH = 8) with aqueous ammonia solution and was extracted with DCM (25 mL). The organic layer was dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated under reduced pressure and the crude residue was purified using silica gel chromatography and eluted with EtOAc/CH3OH (10:1) to give 4 as a white solid (132 mg, 80.9%). 1H-NMR (400 MHz, CDCl3) δ: 6.72 (s, 1H), 6.58 (s, 1H), 5.92 (s, 2H), 5.88 (s, 1H), 5.55 (s, 1H), 4.06 (s, 1H), 4.15 (d, J = 14.4 Hz, 1H), 3.55 (d, J = 14.0 Hz, 1H), 3.37 (m, 1H), 3.18 (d, J = 10.4 Hz, 1H), 2.87 (d, J = 10.0 Hz, 1H), 2.66 (m, 2H), 2.42 (m, 1H), 1.96 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 170.2, 146.5, 146.4, 144.0, 129.4, 126.5, 116.2, 107.4, 104.8, 101.0, 71.8, 61.3, 56.9, 56.7, 53.6, 38.4, 28.7, 20.9; ESI-MS: m/z 348 [M + H]+; HRMS: m/z calcd. for C18H19ClNO4 348.0997 [M + H]+; found: 348.0990.
(1a1S,10bS,11S,12R)-1a1,2,3,5,10b,11,12,12a-Octahydro-[1,3]dioxolo[4,5-j]oxireno[2,3-c]pyrrolo-[3,2,1-de]phenanthridine-11,12-diyl diacetate (1,2-diacetyl-3,4-epoxyllycorine, 5). To a solution of 2 (371 mg, 1 mmol) in 5 mL DCM was added m-CPBA (260 mg, 1.5 mmol), and the mixture was stirred at r.t. for 1 h. After solvent evaporation, the crude residue was purified using a silica gel chromatography eluted with DCM/CH3OH (3:1) to give 5 as a white solid (324 mg, 83.7%). 1H-NMR (400 MHz, CD3OD) δ: 6.76 (s, 1H), 6.59 (s, 1H), 5.86 (s, 2H), 5.82 (s, 1H), 5.67 (s, 1H), 5.20 (s, 1H), 4.74 (d, J = 15.2 Hz, 1H), 4.48 (d, J = 14.8 Hz, 1H), 4.13 (d, J = 11.6 Hz, 1H), 3.83 (m, 1H), 3.49 (d, J = 11.6 Hz, 1H), 2.96 (m, 1H), 2.79 (m, 1H), 1.99 (s, 3H), 1.83 (s, 3H); 13C-NMR (100 MHz, CD3OD) δ: 171.3, 171.2, 149.0, 148.8, 141.3, 125.9, 125.7, 119.0, 108.6, 106.0, 103.0, 72.5, 70.7, 70.0, 69.0, 68.6, 35.7, 27.6, 20.9, 20.6; ESI-MS: m/z 388 [M + H]+; HRMS: m/z calcd. for C20H22NO7 388.1391 [M + H]+; found: 388.1386.
(1a1S,10bS,11S,12R)-12-Hydroxy-1a1,2,3,5,10b,11,12,12a-octahydro-[1,3]dioxolo[4,5-j]oxireno[2,3-c]pyrrolo[3,2,1-de]phenanthridin-11-yl acetate (1-acetyl-3,4-epoxyllycorine, 6). To a solution of 3 (200 mg, 0.61 mmol) in DCM (4 mL) was added m-CPBA (210 mg, 1.22 mmol), and the mixture was stirred at r.t. for 1 h. After solvent evaporation, the crude residue was purified using silica gel chromatography and eluted with DCM/CH3OH (3:1) to give 6 as a white solid (153 mg, 72.7%). 1H-NMR (400 MHz, CD3OD) δ: 6.78 (s, 1H), 6.63 (s, 1H), 5.90 (s, 2H), 5.78 (s, 1H), 5.73 (s, 1H), 4.77 (d, J = 15.2 Hz, 1H), 4.52 (d, J = 15.2 Hz, 1H), 4.13 (s, 1H), 4.12 (d, J = 10.4 Hz, 1H), 3.84 (m, 2H), 3.52 (d, J = 11.2 Hz, 1H), 2.98 (m, 1H), 2.82 (m, 1H), 1.86 (s, 3H); 13C-NMR (100 MHz, CD3OD) δ: 171.7, 148.9, 148.6, 137.9, 126.7, 125.6, 122.7, 108.5, 105.6, 102.9, 72.8, 72.6, 69.3, 69.0, 68.6, 34.4, 27.37, 20.7; ESI-MS: m/z 346 [M + H]+; HRMS: m/z calcd. for C18H20NO6 346.1258 [M + H]+; found: 346.1257.
3.4.1. General Procedure for 7
To a solution of 3 (1 equiv.) in anhydrous pyridine (2 mL) was added acyl chloride or anhydride (1.2 equiv.) in anhydrous DCM (5 mL) over 15 min at 0 °C, and the solution was stirred at 0 °C for another 3 h. Subsequently, DCM and water were added, and the organic layer was washed using aqueous NaHCO3 solution and brine, dried over anhydrous Na2SO4 and filtered. The filtrate was concentrated under reduced pressure and the crude residue was purified using a silica gel chromatography to give 7a–e.
(1S,2S,3a1S,12bS)-1-Acetoxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]-phenanthridin-2-yl pentanoate (1-acetyl-2-valeryllycorine, 7a). Following the previously described procedure, 95 mg (0.28 mmol) of 3 gave 7a as a pale solid (163 mg, 64.7%). 1H-NMR (400 MHz, CDCl3) δ: 6.75 (s, 1H), 6.57 (s, 1H), 5.92 (s, 2H), 5.73 (s, 1H), 5.53 (s, 1H), 5.26 (s, 1H), 4.16 (d, J = 14.1 Hz, 1H), 3.55 (d, J = 14.0 Hz, 1H), 3.38 (dt, J = 9.1, 4.7 Hz, 1H), 2.89 (d, J = 10.5 Hz, 1H), 2.81 (d, J = 10.5 Hz, 1H), 2.66 (s, 2H), 2.43 (dd, J = 17.5, 8.7 Hz, 1H), 2.33 (td, J = 7.4, 3.3 Hz, 2H), 1.95 (s, 3H), 1.61 (dd, J = 15.2, 7.6 Hz, 2H), 1.36 (dd, J = 15.0, 7.4 Hz, 3H), 0.91 (t, J = 7.3 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ: 172.5, 170.0, 146.5, 146.3, 145.8, 129.3, 126.6, 114.0, 107.3, 105.1, 101.0, 70.6, 69.2, 61.2, 56.7, 53.6, 40.4, 34.1, 28.7, 26.9, 22.2, 20.9, 13.7; ESI-MS: m/z 414 [M + H]+; HRMS: m/z calcd. for C23H28NO6 414.1917 [M + H]+; found: 414.1905.
(1S,2S,3a1S,12bS)-1-Acetoxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]-phenanthridin-2-yl hexanoate (1-acetyl-2-hexanoyllycorine, 7b). Following the previously described procedure, 95 mg (0.28 mmol) of 3 gave 7b as a white solid (99 mg, 75.0%). 1H-NMR (400 MHz, CDCl3) δ: 6.71 (s, 1H), 6.54 (s, 1H), 5.88 (s, 2H), 5.70 (s, 1H), 5.49 (s, 1H), 5.23 (s, 1H), 4.13 (d, J = 14.0 Hz, 1H), 3.50 (d, J = 14.0 Hz, 1H), 3.34 (m, 1H), 2.85 (d, J = 10.0 Hz, 1H), 2.75 (d, J = 10.4 Hz, 1H), 2.62 (m, 2H), 2.37 (m, 1H), 2.29 (m, 2H), 1.92 (s, 3H), 1.61 (m, 2H), 1.28 (m, 4H), 0.86 (m, 3H); 13C-NMR (100 MHz, CDCl3) δ: 172.5, 170.0, 146.4, 146.3, 145.9, 129.4, 126.5, 114.0, 107.3, 105.0, 101.0, 70.6, 69.2, 61.2, 56.9, 53.6, 40.5, 34.3, 31.2, 28.7, 24.6, 22.3, 21.0, 14.0; ESI-MS: m/z 428 [M + H]+; HRMS: m/z calcd. for C24H30NO6 428.2073 [M + H]+; found: 428.2067.
(1S,2S,3a1S,12bS)-1-Acetoxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]-phenanthridin-2-yl pivalate (1-acetyl-2-pivaloyllycorine, 7c). Following the previously described procedure, 95 mg (0.28 mmol) of 3 gave 7c as a pale solid (64mg, 56%). 1H-NMR (400 MHz, CDCl3) δ: 6.77 (s, 1H), 6.59 (s, 1H), 5.93 (s, 2H), 5.72 (s, 1H), 5.52 (s, 1H), 5.24 (s, 1H), 4.19 (d, J = 14.2 Hz, 1H), 3.56 (d, J = 14.1 Hz, 1H), 3.41 (dt, J = 9.0, 4.7 Hz, 1H), 2.90 (d, J = 10.4 Hz, 1H), 2.81 (d, J = 10.6 Hz, 1H), 2.68 (s, 2H), 2.44 (m, 1H), 1.97 (s, 3H), 1.22 (s, 9H); 13C-NMR (100 MHz, CDCl3) δ: 177.2, 169.9, 146.5, 146.3, 145.8, 129.3, 126.6, 113.9, 107.3, 105.1, 101.0, 70.6, 69.1, 61.3, 56.8, 53.6, 40.6, 38.7, 28.6, 27.1, 20.9; ESI-MS: m/z 414 [M + H]+; HRMS: m/z calcd. for C23H28NO6 414.1917 [M + H]+; found: 414.1912.
4-(((1S,2S,3a1S,12bS)-1-Acetoxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]-phenanthridin-2-yl)oxy)-4-oxobutanoic acid (1-acetyl-2-(4-carboxyl-propionate)lycorine, 7d). Following the previously described procedure, 150 mg (0.46 mmol) of 3 gave 7d as a white solid (178mg, 90.5%). 1H-NMR (400 MHz, CDCl3) δ: 6.72 (s, 1H), 6.62 (s, 1H), 5.92 (s, 2H), 5.76 (s, 1H), 5.58 (s, 1H), 5.27 (s, 1H), 4.19 (d, J = 14.0 Hz, 1H), 3.81 (d, J = 14.0 Hz, 1H), 3.50 (brs, 1H), 3.21 (d, J = 10.8 Hz, 1H), 3.02 (d, J = 10.4 Hz, 1H), 2.80 (m, 1H), 2.74 (m, 2H), 2.54–2.60 (m, 4H), 1.96 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 175.7, 171.3, 169.8, 147.1, 146.7, 143.7, 127.4, 126.4, 115.2, 107.6, 105.0, 101.2, 70.0, 68.4, 60.7, 55.0, 53.2, 38.5, 29.9, 29.8, 28.7, 20.9; ESI-MS: m/z 428 [M − H]−; HRMS: m/z calcd. for C22H24NO8 430.1502 [M + H]+; found: 430.1489.
(1S,2S,3a1S,12bS)-1-Acetoxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-2-yl 4-azidobenzoate (1-acetyl-2-(4-azidobenzoyl)lycorine, 7e). Following the previously described procedure described above, 115 mg (0.35 mmol) of 3 gave 7e as a white solid (120 mg, 72.0%). 1H-NMR (400 MHz, CDCl3) δ: 8.02 (d, J = 6.8, 2H), 7.02 (d, J = 6.8, 2H), 6.77 (s, 1H), 6.63 (s, 1H), 5.92 (s, 1H), 5.91 (s, 2H), 5.68 (s, 1H), 5.53 (s, 1H), 4.23 (d, J = 14.0, 1H), 3.72 (d, J = 14.0, 1H), 3.47 (m, 1H), 3.14 (d, J = 10.8, 1H), 3.09 (d, J = 10.8, 1H), 2.74 (m, 2H), 2.68 (m, 1H), 2.00 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 170.0, 164.4, 146.8, 146.6, 145.4, 145.0, 144.3, 131.7, 128.5, 126.5, 126.3, 118.8, 118.6, 114.5, 107.5, 105.0, 101.1, 70.9, 68.9, 61.0, 56.0, 53.5, 39.7, 28.8, 21.0; ESI-MS: m/z 475 [M + H]+; HRMS: m/z calcd. for C25H23N4O6 475.1612 [M + H]+; found: 475.1610.
(1aR,2a1S,11bS,11cS)-2a1,3,4,6,11b,11c-Hexahydro-1aH-[1,3]dioxolo[4,5-j]oxireno[2,3-a]pyrrolo-[3,2,1-de]phenanthridine (1,2-α-epoxylycorine, 8). To a solution of 4 (500 mg, 1.44 mmol) in 100 mL of CH3OH was added 1.0 g of CH3ONa, and the mixture was stirred in ice bath for 20 min. DCM and water was added, and the organic layer was dried over anhydrous Na2SO4. After filtration, the filtrate was concentrated under reduced pressure and the crude residue was purified using a silica gel chromatography eluted with EtOAc to give 8 as a white solid (166 mg, 42.9%). 1H-NMR (400 MHz, CDCl3) δ: 7.04 (s, 1H), 6.60 (s, 1H), 5.94 (d, J = 4.4 Hz, 2H), 5.76 (d, J = 1.6 Hz, 1H), 4.08 (d, J = 14.0 Hz, 1H), 3.96 (d, J = 4.0 Hz, 1H), 3.58 (d, J = 14.0 Hz, 1H), 3.51 (t, J = 4.0 Hz, 1H), 3.21 (m, 1H), 2.80 (t, J = 13.6 Hz, 2H), 2.61 (m, 1H), 2.43 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ: 147.9, 146.4, 146.2, 129.5, 129.2, 112.6, 107.5, 105.4, 101.0, 63.1, 57.0, 54.5, 54.0, 49.5, 40.8, 29.3; ESI-MS: m/z 270 [M + H]+; HRMS: m/z calcd. for C16H16NO3 270.1125 [M + H]+; found: 270.1130.
3.4.2. General Procedure for the Preparation of 9
Compound 8 was dissolved in amine (5 mL, piperidine, diethylamine, pyrrole, n-butylamine, isopropylamine for 9a–e, respectively), and the solution was heated to reflux overnight. The amine was removed under reduced pressure and the crude residue was purified using silica gel chromatography to give the products.
(1S,2S,3a1S,12bS)-2-(Piperidin-1-yl)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-ol (2-piperidinelycorine, 9a). Following the previously described procedure, 175 mg (0.65 mmol) of 8 gave 9a as a white solid (102 mg, 44.0%). 1H-NMR (400 MHz, CDCl 3) δ: 6.89 (s, 1H), 6.61 (s, 1H), 5.93 (d, J = 4.8 Hz, 2H), 5.47 (m, 1H), 4.73 (d, J = 2.0 Hz, 1H), 4.13 (d, J = 14.0 Hz, 1H), 3.52 (d, J = 14.0 Hz, 1H), 3.32–3.36 (m, 2H), 2.70–2.74 (m, 2H), 2.62–2.65 (m, 3H), 2.47–2.50 (m, 3H), 2.37 (m, 2H), 1.55–1.59 (m,4H), 1.45 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ: 146.6, 146.3, 142.3, 130.4, 127.5, 117.8, 107.9, 104.8, 101.0, 69.1, 65.2, 60.7, 56.9, 53.7, 50.6, 44.4, 28.7, 26.6, 24.5; ESI-MS: m/z 355 [M + H]+; HRMS: m/z calcd. for C21H27N2O3 355.2016 [M + H]+; found: 355.2015.
(1S,2S,3a1S,12bS)-2-(Diethylamino)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-ol (2-diethylaminelycorine, 9b). Following the previously described procedure, 200 mg (0.74 mmol) of 8 gave 9b as a gray solid (143 mg, 56.5%). 1H-NMR (400 MHz, CDCl3) δ: 6.88 (s, 1H), 6.59 (s, 1H), 5.93 (d, J = 11.2 Hz, 2H), 5.45 (s, 1H), 4.83 (s, 1H), 4.13 (d, J = 14.0 Hz, 1H), 3.91 (s, 1H), 3.58 (d, J = 14.0 Hz, 1H), 3.36 (m, 1H), 2.80 (m, 5H), 2.67 (brs, 2H), 2.55–2.38 (m, 2H), 1.25 (t, J = 7.1 Hz, 6H); 13C-NMR (100 MHz, CDCl3) δ: 146.6, 146.3, 145.8, 129.7, 127.1, 113.0, 107.5, 105.5, 101.0, 66.8, 65.1, 60.6, 56.5, 53.4, 45.0, 43.9, 29.0, 12.3; ESI-MS: m/z 343 [M + H]+; HRMS: m/z calcd. for C20H27N2O3 343.2016 [M + H]+; found:343.2016.
(1S,2S,3a1S,12bS)-2-(1H-Pyrrol-1-yl)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-ol (2-pyrrolelycorine, 9c). Following the previously described procedure, 200 mg (0.74 mmol) of 8 gave 9c as a gray solid (161 mg, 65.3%). 1H-NMR (400 MHz, CDCl3) δ: 6.62 (s, 1H), 6.58 (s, 1H), 6.56 (s, 1H), 6.10 (s, 1H), 5.98 (s, 1H), 5.88 (d, J = 5.2 Hz, 2H), 5.50 (s, 1H), 4.49 (s, 1H), 4.15 (d, J = 14.9 Hz, 1H), 3.77 (s, 1H), 3.51 (d, J = 15.2 Hz, 1H), 3.35–3.27 (m, 1H), 2.88 (d, J = 10.6 Hz, 1H), 2.77 (d, J = 10.6 Hz, 1H), 2.67 (m, 2H), 2.44–2.32 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ: 146.6, 146.3, 139.8, 132.5, 127.8, 117.2, 116.6, 108.3, 107.6, 106.0, 104.8, 101.0, 71.8, 61.1, 57.0, 53.9, 45.3, 41.0, 28.5; ESI-MS: m/z 337 [M + H]+; HRMS: m/z calcd. for C20H21N2O3 337.1547 [M + H]+; found:337.1543.
(1S,2S,3a1S,12bS)-2-(Butylamino)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-ol (2-butylaminelycorine, 9d). Following the previously described procedure described above, 143 mg (0.36 mmol) of 8 gave 9d as a pale yellow solid (132 mg, 72.8%). 1H-NMR (400 MHz, CDCl3) δ: 6.85 (s, 1H), 6.58 (s, 1H), 5.92 (d, J = 5.6 Hz, 2H), 5.48 (s, 1H), 4.50 (s, 1H), 4.13 (d, J = 14.4 Hz, 1H), 3.48 (d, J = 14.0 Hz, 1H), 3.33 (m, 1H), 3.24 (s, 1H), 2.74 (m, 3H), 2.60 (m, 3H), 2.32 (m, 1H), 1.44 (m, 2H), 1.34 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ: 146.5, 146.2, 141.5, 130.4, 128.0, 117.6, 107.7, 104.7, 100.9, 69.8, 61.6, 61.2, 57.2, 53.9, 48.3, 41.7, 32.6, 28.6, 20.4, 14.0; ESI-MS: m/z 343 [M + H]+; HRMS: m/z calcd. for C20H27N2O3 343.2016 [M + H]+; found: 343.2022.
(1S,2S,3a1S,12bS)-2-(Isopropylamino)-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo-[3,2,1-de]phenanthridin-1-ol (2-isopropylaminelycorine, 9e). Following the previously described procedure, 160 mg (0.59 mmol) of 8 gave 9e as a pale yellow solid (175 mg, 90.4%). 1H-NMR (400 MHz, CDCl3) δ: 6.85 (s, 1H), 6.57 (s, 1H), 5.91 (d, J = 5.2 Hz, 2H), 5.45 (s, 1H), 4.45 (s, 1H), 4.12 (d, J = 14.4 Hz, 1H), 3.48 (d, J = 14.0 Hz, 1H), 3.31 (m, 1H), 3.29 (s, 1H), 3.01 (m, 1H), 2.72 (d, J = 10.8 Hz, 1H), 2.60 (d, J = 10.8 Hz, 1H), 2.58 (m, 2H), 2.31 (m, 1H), 1.08 (s, 3H), 1.06 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ: 145.5, 145.1, 139.9, 129.3, 127.1, 116.8, 106.6, 103.7, 99.9, 69.8, 60.1, 57.7, 56.2, 52.9, 46.4, 40.4, 27.6, 22.5, 22.2; ESI-MS: m/z 329 [M + H]+; HRMS: m/z calcd. for C19H25N2O3 329.1860 [M + H]+; found: 329.1855.



{kind=link}
{kind=link}