Abstract
A series of 1-aryl-3-(2H-chromen-5-yl)urea and 1-aryl-3-(chroman-5-yl)urea derivatives were designed, synthesized and evaluated for their inhibitory activities towards TNF-α production in lipopolysaccharide-stimulated THP-1 cells. The most active compound, 40g, inhibited TNF-α release with an IC50 value of 0.033 μM, which is equipotent to that of BIRB796 (IC50 = 0.032 μM).
1. Introduction
The p38 mitogen-activated protein kinase (p38MAPK) plays a key role in inflammatory responses through the production of cytokines and inflammatory mediators such as TNF-α and IL-1β [1]. At least four distinct homologues, standardized in the nomenclature as p38α, β, γ and δ, have been identified. The inhibition of p38MAPKα is considered to be a promising therapeutic strategy for chronic inflammatory diseases such as rheumatoid arthritis [2], psoriasis, inflammatory bowel disease [2], and chronic obstructive pulmonary disease [3]. Recent studies have also revealed that p38MAPKα inhibitors may have therapeutic potential in the treatment of cancer [4,5], neuropathic pain [6] and periodontal diseases [7]. Consequently, considerable effort has been directed toward the development of p38MAPKα inhibitors as potential anti-inflammatory and anticancer drug.
p38MAPKα inhibitors, like many other kinase inhibitors, can be classified into two types based on their mode of action: ATP-competitive inhibitors, which bind to an ATP-binding site, and non-ATP-competitive or allosteric inhibitors. Allosteric inhibitors utilize the ATP binding cleft and a hydrophobic allosteric pocket created when the activation loop adopts the inactive “Asp-Phe-Gly (DFG)-out” conformation. Because allosteric inhibitors do not compete directly with ATP or substrate, they can offer a significant kinetic advantage over ATP competitive inhibitors. In addition, because the allosteric pocket is less conserved than the ATP binding region, allosteric inhibitors usually have better kinase selectivity profiles than ATP competitive inhibitors [8]. BIRB796 is a typical allosteric p38MAPKα inhibitor with an N-pyrazole-N'-naphthyl urea scaffold. The crystal structure of the p38MAPKα/BIRB796 complex shows that BIRB796 fits well into the DFG-out conformation by forming several tight interactions. (Figure 1 graphically depicts a two-dimensional p38MAPKα/BIRB796 interaction map.) By taking advantage of these interactions, BIRB796 achieves potent p38MAPKα inhibition with a Kd value of 0.1 nM [9]. The major drawback of BIRB796 is its hepatotoxicity [10], which may be caused by one of its metabolic intermediates, naphthalene epoxide [11]. We therefore performed scaffold modification and structure–activity relationship (SAR) investigations of BIRB796 and its analogues to discover novel p38MAPKα inhibitors with drug-like properties.
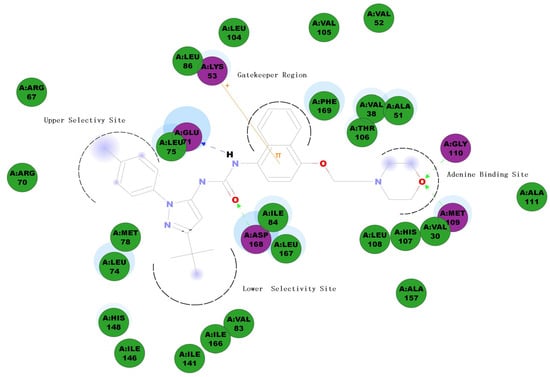
Figure 1.
Structure of the compound BIRB796 and two-dimensional p38 MAPKα/BIRB796 interaction map.
According to the crystal structure of the p38MAPKα/BIRB796 complex (PDB 1VK2) [9], the carbonyl from the urea linkage of BIRB796 accepts hydrogen bonds from the backbone NH of Asp168, while the NH from the urea linkage forms hydrogen bonds to the Glu71 side chain. Such a hydrogen bonding network is essential to maintain the p38MAPKα inhibition activity, so we preserved this urea linkage in the compounds we designed. The naphthyl ring of BIRB796 pushes very deep into the hydrophobic gatekeeper pocket. It is known that such a hydrophobic interaction is essential to obtain high activity and selectivity for a p38MAPKα inhibitor [12]. A pi-cation interaction has also been observed between the naphthyl ring and the cationic amino groups in the side chains of Lys53. To preserve the activity and selectivity, and to eliminate the hepatotoxicity caused by the naphthyl ring, we attempted to replace the naphthalene ring with two other aromatic hydrophobic scaffolds: 2H-chromene and chromane. In BIRB796, the morpholinoethoxy group occupies the adenine binding site, and forms hydrogen bond interactions with the residue of Met109 and Gly110. According to SAR information of kinase inhibitors, a variety of functional groups could be well tolerated by this adenine binding site. We therefore attempted to either replace this morpholinoethoxy moiety with a 2-morpholino-2-oxoethoxy moiety, or to replace the morpholine ring with a larger aliphatic group (2,6-dimethylmorpholine) or aromatic hydrogen bond-accepting moieties (triazole, pyridine or imidazole) with the aim of increasing the binding affinity. BIRB796 also utilizes a unique allosteric pocket created when the activation loop adopts the “DFG-out” conformation. This allosteric pocket can be divided into two selectivity sites: the lower selectivity site, occupied by the t-butyl group, and the upper selectivity site, occupied by the p-tolyl group. It has been reported that the t-butyl group fits well with the highly conserved lower selectivity site, while the upper selectivity site is less conserved and offers a unique position to enable p38MAPKα inhibitory activity and kinase selectivity [13]. On the basis of this knowledge, the t-butyl group was preserved in our compounds, while we attempted to substitute the 4-tolyl group with another substituted phenyl to investigate the SAR around the phenyl ring unit. It was also of interest to us that whether substituting the pyrazole ring with its isosteres, such as oxazole and imidazole, would increase the inhibitory activity for p38MAPKα. On the basis of the above information, a series of N-aryl-N'-chromenyl urea and N-aryl-N'-chromanyl urea derivatives was designed, synthesized and evaluated for their inhibitory activity against TNF-α release.
2. Results and Discussion
To obtain our target compounds, two series of aromatic amines [one series of 5-alkoxy-2H-chromen-8-amines (Scheme 1) and 5-alkoxychroman-8-amines (Scheme 2) and another series containing pyrazol-5-amines (Scheme 3) and their isosteres (Scheme 4)] were first prepared, and then the target urea derivatives obtained by coupling these two series of aromatic amines. The key to the success of this synthetic strategy was to prepare the 5-alkoxychroman-8-amines and 5-alkoxy-2H-chromen-8-amines efficiently. A simple procedure for the synthesis of 5-morpholinoethoxy-2H-chromen-8-amines, starting from commercially available 2-nitro-5-fluorophenol, had been developed by our group [13]. In this paper, an improved and more diverse procedure, using 5-hydroxy-8-nitro-2H-chromene as a key intermediate, was developed and executed to prepare the desired 5-alkoxy-2H-chromen-8-amines and 5-alkoxychroman-8-amines.
The general synthetic approach to the 5-alkoxy-2H-chromen-8-amines 10a–c is outlined in Scheme 1. 2-nitro-5-fluorophenol (1) reacted with propargyl bromide in dimethylformamide in the presence of potassium carbonate to produce compound 2 [14]. Compound 4 was obtained through the reaction of 2 with sodium 4-methoxyphenylethoxylate (3). Claisen thermal rearrangement of 4 in N,N-diethyl aniline at 180–200 °C led to the production of 5-(4-methoxybenzyloxy)-8-nitro-2H-chromene (5) [15]. Compound 5 was debenzylated with trifluoroacetic acid in dichloromethane to produce 5-hydroxyl-8-nitro-2H-chromene (6). 5-(2-Bromoethoxy)-8-nitro-2H-chromene (7) was obtained by reacting 6 with 1,2-dibromoethane in CH3CN under reflux for 24 h. Compound 7 reacted with morpholine (8a), cis-2,6-dimethylmorpholine (8b), and triazole (8c) in dimethylformamide (DMF) at 80 °C in the presence of K2CO3 to yield the corresponding compounds 9a–c, which were then reduced to the 5-alkoxy-2H-chromen-8-amines 10a–c with SnCl2 in refluxing EtOH.
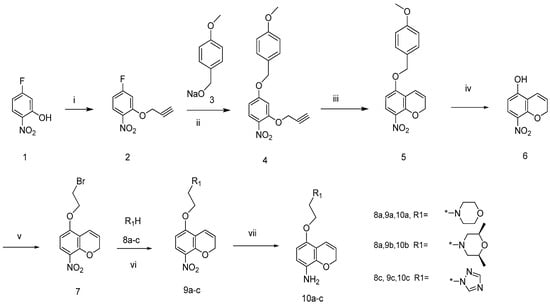
Scheme 1.
Synthesis of 5-alkoxy-2H-chromen-8-amines.
The synthetic approach to the chroman-8-amine derivatives 17a–f is outlined in Scheme 2. 5-hydroxy-8-nitro-2H-chromene (7), prepared as described in Scheme 1, was reduced with H2NNH2/H2O in the presence of Pd/C in ethanol to give 5-hydroxy-8-aminochromane (11). The amino group of 11 was protected with di-tert-butyldicarbonate to produce N-Boc-5-hydroxy-8-amino-chromane (12). Compound 12 reacted with 1,2-dibromoethane (13) through an intramolecular SN2 reaction to give 5-(2-bromoethoxy)-8-aminochromane (14), which was then substituted with morpholine (15a), cis-2,6-dimethylmorpholine (15b), imidazole (15c) and triazole (15d) to yield the corresponding compounds 16a–d. Deprotection of 16a–d by treatment with trifluoroacetic acid in dichloromethane produced the 8-aminochromane derivatives 17a–f. The preparation of 5-(2-(pyridin-4-yl)ethoxy)chroman-8-amine (17e) was accomplished by a Mitsunobu reaction of N-boc-5-hydroxy-8-aminochromane (12) and 2-(pyridin-4-yl)ethanol (18) with triphenylphosphine (Ph3P) and diisopropyl azodicarboxylate (DIAD) in CH2Cl2 [15] and subsequent cleavage of the Boc group by employing trifluoroacetic acid in CH2Cl2. Another 8-aminochroman derivative (compound 17f) was obtained by oxyalkylation of compound 12 with 4-(2-chloroacetyl)morpholine (20), which was prepared by the reaction of the morpholine (15a) and chloroacetyl chloride (19) in the presence of triethylamine, followed by cleavage of the Boc group.
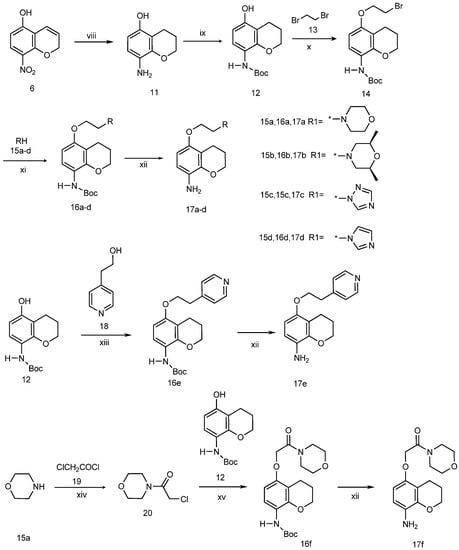
Scheme 2.
Synthesis of 5-alkoxychroman-8-amines.
The preparation of 1-aryl-3-tert-butyl-1H-pyrazol-5-amines 25a–g, 25l was conducted according to the procedures of Regan et al. (Scheme 3) [16] Diazotization of arylamine 21 with NaNO2, followed by reduction with SnCl2 in HCl at 0 °C, led to the corresponding arylhydrazine hydrochlorides 23a–j [17], which were condensed with pivaloylacetonitrile (24) in ethanolic solution of HCl at reflux to give 25a–g and 25l [18]. The other four pyrazol-5-amines 25k–n are commercially available. The structures of 25a–l are listed in Table 1.
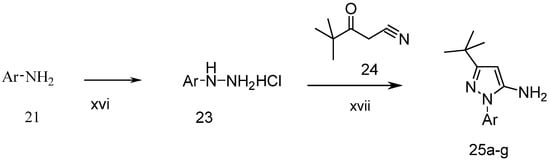
Scheme 3.
Synthesis of 1-aryl-3-tert-butyl-1H-pyrazol-5-amines.
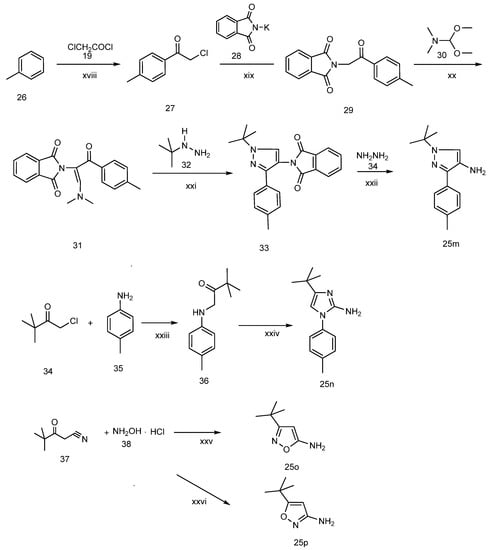
Scheme 4.
The synthesis of arylamines 25m, 25n, 25o, 25p.

Table 1.
1-Aryl-3-tert-butyl-1H-pyrazol-5-amines 25a–l. 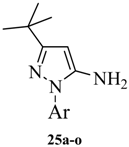
| Compound | Ar | Compound | Ar | Compound | Ar | Compound | Ar |
|---|---|---|---|---|---|---|---|
| 25a |  | 25b |  | 25c | 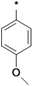 | 25d | 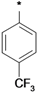 |
| 25e | 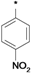 | 25f | 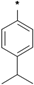 | 25g |  | 25h | 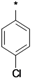 |
| 25i | 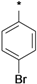 | 25j | 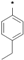 | 25k | 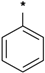 | 25l |  |
The other four arylamines (compounds 25m, 25n, 25o, 25p) were prepared as shown in Scheme 4. 1-tert-Butyl-3-p-tolyl-1H-pyrazol-4-amine (25m) was prepared by referring to similar reaction conditions [19]. 2-Chloro-1-p-tolylethanone (27) was obtained by Friedel-Crafts acylation of toluene with chloroacetyl chloride. Condensation of 27 with phthalimide potassium salt (28) in DMF gave a good yield of 2-(1,3-dioxoisoindolin-2-yl)-1-p-tolylethanone (29). Reaction of 29 with dimethyl-formamide dimethyl acetal (30) at reflux gave the enamine 31 in quantitative yield. Reaction of 31 with hydrazine in ethanol at room temperature for one hour gave 4-aminopyrazole (25m 78%. 4-tert-butyl-1-p-tolyl-1H-imidazol-2-amine (25n) was obtained referring the method described in literature [20]. N-(pivaloylmethyl)-4-methylaniline (36), which was synthesized from 4-methylaniline (34) and α-chloropinacolone (35) in DMF using sodium bicarbonate as a deacidifying reagent at 75 °C for 48 h, condensed with cyanamide (37) upon heating in ethanol at reflux for 12 h to give 25n at a yield of 77%. 3-tert-Butylisoxazol-5-amine (25o) was prepared in good yield (88%) by condensation of pivaloylacetonitrile (37) and hydroxylamine hydrochloride (38) in alkaline aqueous sodium hydroxide solution at 50 °C [21]. 5-tert-Butylisoxazol-3-amine (25o), the positional isomer of 25p, was prepared following the method described in [16]. Thus, pivaloylacetonitrile (24) reacted with hydroxylamine hydrochloride (35) at pH 10–11 and 50 °C for 10 h and then at pH 4–5 and 50 °C for 3 h to produce 25p at a yield of 77%.
Finally, the target N-aryl-N'-chromenylurea and N-aryl-N'-chromanylurea derivatives were prepared as shown in Scheme 5. The 5-alkoxy-2H-chromen-8-amines 10a–c or the 5-alkoxychroman-8-amines 17a–f reacted with triphosgene in dichloromethane at −15 °C to form the corresponding isocyanate compounds [22], which then reacted with aryl amines 25a–p in the presence of triethylamine at room temperature for 12–24 h to yield the target compounds 39a–j and 40a–i. The structures of 39a–j and 40a–i are listed in Table 1.
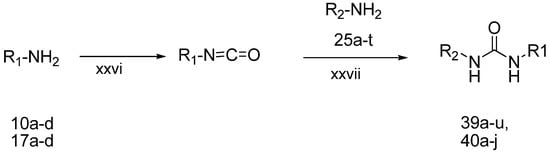
Scheme 5.
The synthesis of N-aryl-N'-chromenylurea and N-aryl-N'-chromanylurea derivatives.
The potential inhibitors 39a–j and 40a–r were profiled for their ability to inhibit TNF-α release in lipopolysaccharide (LPS)-stimulated THP-1 cells [18], with BIRB796 as positive control. The results are shown in Table 2 and Table 3. Among our potential inhibitors, the 2H-chromen-5-ylurea compounds 40a, 40k, 40l and 40m exhibited approximately an order of magnitude more potent anti-TNF-α activity than their corresponding chroman-5-ylurea analogues 39a, 39c, 39d and 39e. Compounds 39a and 40a were identical to BIRB796, except the 2H-chromen-5-ylurea ring in BIRB796 was changed to a 2H-chromen and chroman moiety, respectively. The TNF-α inhibitory activities decreased in sequence of BIRB796 (IC50 = 0.032 μM), 40a (IC50 = 0.050 μM) and 39a (IC50 = 0.31 μM), which is consistent with the downward tendency of the length of the conjugated system of the naphthyl, 2H-chromen and chroman rings. We presume that the conjugated system length of the naphthyl ring, 2H-chromen and chroman is related to the strength of the π-cation interaction between the conjugated system and the cationic amino groups in the side chains of Lys53, which in turn affects the anti-TNF-α activity of the corresponding derivatives. The activity of 2H-chromen-5-yl urea compounds was very close to that of the the corresponding naphthyl urea analogues, which indicates that 3-(2H-chromen-5-yl)ureas may serve as a novel chemotype for the development of p38MAPKα inhibitors.
When the left side of our molecules utilized the 1-aryl-3-tert-butyl-1H-pyrazol-5-amine group from BIRB796 (compounds 39f–j and compounds 40q–r), an obvious decrease in TNF-α inhibition activity was seen when the morpholine group was replaced with 1,2,4-triazole (39h, 40r) or pyridine (39j). In contrast, a relatively small decrease in activity was observed with the replacement of the morpholine group with 2,6-dimethylmorpholine (39g, 40q) and 1,3-imidazole (39i). These results, taken together, indicate that the TNF-α inhibitory activity of our compounds was very sensitive to substitutions in this region. Aromatic hydrogen bond accepting moieties such as 1,2,4-triazole, pyridine and 1,3-imidazole are inferior to the aliphatic morpholine. Introducing two methyl groups to the position ortho to the oxygen atom of morpholine is also not conducive to TNF-α inhibitory activity. In our molecules, we also attempted to modify the ethoxy linker of the morpholinoethoxy group with a 2-oxoethoxy. Unfortunately, the resultant compound 39f exhibited little activity against TNF-α release, which may indicate that a hydrophobic linkage is more suitable than a hydrophilic one for this region.
Table 2.
Structure and TNF-α inhibitory activity of 1-aryl-3-(chroman-5-yl)urea compounds 40a–i. 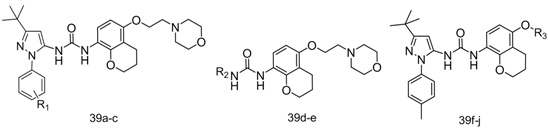
| Compound | R1 | R2 | R3 | IC50 (μM) |
|---|---|---|---|---|
| 39a | 4-Methyl | 0.31 | ||
| 39b | 4-H | 1.17 | ||
| 39c | 3-Methyl | 0.895 | ||
| 39d | 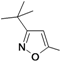 | 2.0 | ||
| 39e | 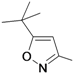 | 2.5 | ||
| 39f | 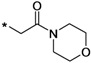 | >10 | ||
| 39g | 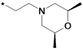 | 0.50 | ||
| 39h | 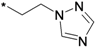 | >10 | ||
| 39i | 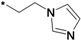 | 0.48 | ||
| 39j |  | >10 | ||
| BIRB796 | 0.032 | |||
When the right side of our molecules utilized the 2-morpholinoethoxy group from BIRB796 (compounds 39a–e and 40a–p), changing the 1-(p-tolyl)-3-tert-butyl-1H-pyrazol-5-amine group (compounds 40a, 39a) on the left side to 5-tert-butylisoxazol-3-amine (compounds 40l, 39g) or 3-tert-butylisoxazol-5-amine (compounds 40m, 39h) led to an apparent decrease in potency. Likewise, in the 2H-chromenylurea series, when the 1-(p-tolyl)-3-tert-butyl-1H-pyrazol-5-amine group of 40a was replaced with its positional isomer 4-tert-butyl-1-(p-tolyl)-1H-imidazol-2-amine (40n) or 1-tert-butyl-3-(p-tolyl)-1H-pyrazol-4-amine (40p), a dramatic loss of activity was observed. These results demonstrate that the p-tolyl group is important to maintain TNF-α inhibitory activity, and the electronic configuration model of the pyrazole ring also significantly affects the activity. To further probe the SAR in this particular region, we also attempted to replace the methyl group of the p-tolyl group with other substitution. As seen in Table 3, replacing the 4-methyl group with a larger substituent, such as 4-chloro (40c), 4-bromo (40d), 4-methoxy (40e), 4-trifluoromethyl (40f), 4-ethyl (40h), 4-isopropyl (40i), or 3-chloro-4-fluoro (40j) resulted in almost complete disappearance of the TNF-α inhibitory activity (IC50 < 10 μM). Changing the p-tolyl group for a naphthyl group also abolished the activity (IC50 < 10 μM). In contrast, changing the 4-methyl group to a small polar substituent such as 4-fluoro (40b) was relatively tolerated and resulted in 4-fold loss of activity, while changing the 4-methyl group to a powererfully polar nitro group provided a potent p38MAPKα inhibitor (40g) with an IC50 value of 0.033 μM, which is comparable to that of BIRB796 (0.032 μM) and higher than that of compound 40a (0.050 μM).
Table 3.
Structure and TNF-α inhibitory activity of 1-aryl-3-(2H-chromen-5-yl) urea compounds 39a–r. 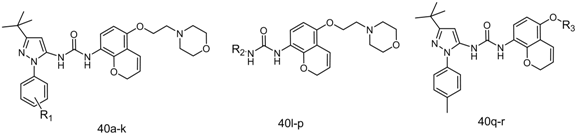
| Compound | R1 | R2 | R3 | IC50 (μM) |
|---|---|---|---|---|
| 40a | 4-Methyl | 0.050 | ||
| 40b | 4-Fluoro | 0.223 | ||
| 40c | 4-Chloro | >10 | ||
| 40d | 4-Bromo | >10 | ||
| 40e | 4-Methoxy | >10 | ||
| 40f | 4-Trifluoromethyl | >10 | ||
| 40g | 4-Nitro | 0.033 | ||
| 40h | 4-Ethyl | >10 | ||
| 40i | 4-Isopropyl | >10 | ||
| 40j | 3-Chloro-4-fluoro | >10 | ||
| 40k | 3-Methyl | 0.058 | ||
| 40l |  | 0.25 | ||
| 40m | 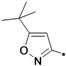 | 0.42 | ||
| 40n | 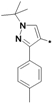 | >10 | ||
| 40o | 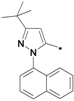 | >10 | ||
| 40p | 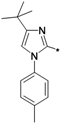 | >10 | ||
| 40q | 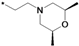 | 0.16 | ||
| 40r | 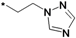 | >10 | ||
| BIRB796 | 0.032 | |||
These results indicate that the TNF-α inhibitory activity of our compounds was also sensitive to the substitutions of the phenyl ring, and 4-nitrophenyl was at least as effective as the 4-methylphenyl group.
3. Experimental
3.1. General Information
All the reagents were commercially available and used without further purification. 1H-NMR spectra were measured using a Bruker-400 (Bruker Company, karlsruhe, Germany) or YS-300 instrument. Mass spectra were obtained from VG300, ZAD-2F or API3000 instruments.
3.2. Chemistry
4-Fluoro-1-nitro-2-(prop-2-ynyloxy) benzene (2). To a stirred solution of 5-fluoro-2-nitrophenol (30 g, 191 mmol) in DMF (300 mL) anhydrous K2CO3 (52.83 g, 382 mmol) and 3-bromopropyne (27.27 g, 229 mmol) were successively added The mixture was stirred at room temperature for 5 h and then poured into ice water (1,500 mL). A buff precipitate separated out upon standing overnight. The solid was collected by filtration and air-dried to give compound 2 as a buff solid (33.91 g, yield: 85%), mp: 50–52 °C, 1H-NMR (CDCl3, 400 MHz), δ: 7.98 (m, 1H), 6.98 (m, 1H), 6.80 (m, 1H), 4.86 (s, 2H), 2.66 (s, 1H). FAB-MS (m/z): 196 [M+H]+.
4-(4-Methoxybenzyloxy)-1-nitro-2-(prop-2-ynyloxy)benzene (4). To a stirred solution of (4-methoxyphenyl)methanol (22.63 g, 163.8 mmol) in anhydrous DMF (200 mL), 60% sodium hydride (8.74 g, 218.4 mmol) was added in portions. The mixture was stirred at ambient temperature until no further gas was released, then cooled to −35 °C. To this solution, compound 2 (21.84 g, 109.2 mmol) was added in one batch. The reaction mixture was stirred for a further 6 h at −35 °C under nitrogen, and then was poured into ice water (1,000 mL). The precipitate was collected, washed with water and air-dried to give compound 4 as a buff solid (27.82 g, yield: 93%), mp: 53–54 °C, 1H-NMR (CDCl3, 400 MHz), δ: 8.03 (d, J = 9.24 Hz, 1H), 7.36 (2H, d, J = 8.44 Hz), 6.94 (2H, d, J = 8.44 Hz), 6.79 (1H, d, J = 2.52 Hz), 6.66 (dd, J = 9.24 Hz, 2.52 Hz, 1H), 5.08 (2H, s), 4.82 (2H, d, J = 2.24 Hz), 384 (3H, s), 2.59 (1H, t, J = 2.52 Hz).
5-(4-Methoxybenzyloxy)-8-nitro-2H-chromene (5). Compound 4 (22 g, 71.8 mmol) was dissolved in N,N-diethylaniline (330 mL). The reaction mixture was heated to 195 °C and kept at this temperature for 1 h. After cooling to room temperature, the solvent was distilled off under reduced pressure. The residue was purified by column chromatography on silica gel with ethyl acetate/petroleum ether (3:2) as eluent to give compound 5 as a yellow solid (6.47 g, yield: 29%). 1H-NMR (CDCl3, 400 MHz), δ: 7.85 (1H, d, J = 9.52 Hz), 7.32 (2H, d, J = 5.64 Hz), 6.94 (2H, d, J = 5.64 Hz), 6.79 (1H, dt, J = 10.36 Hz, 1.68 Hz), 6.55 (1H, d, J = 9.52 Hz), 5.83 (1H, dt, J = 10.36 Hz, 3.64 Hz), 5.08 (2H, s), 4.94 (2H, dd, J = 3.64 Hz), 3.81(3H, s). ESI-MS (+Q, m/z): 314 [M+H]+, 336 [M+Na]+.
8-Nitro-2H-chromen-5-ol (6). To a solution of compound 5 (18.2 g, 58 mmol) in dichloromethane (200 mL), trifluoroethylacetic acid (10.0 mL) was added dropwise with stirring at −10 °C. The reaction mixture was stirred at this temperature for 8 h, then quenched by addition of ice water (5 mL). The aqueous solution was then adjusted to pH = 10 with 1 N sodium hydroxide and the two phases were separated. The water phase was extracted twice with dichloromethane. The organic phases were combined and washed with water and brine, dried with Na2SO4 and the solvent removed under vacuum to yield the crude product, which was purified by column chromatography (dichloromethane/methanol 100:2) to give compound 6 as a yellow solid (7.5 g, yield: 67%). 1H-NMR (CDCl3, 400 MHz), δ: 11.11 (1H, s) 7.73 (d, 1H, J = 9.24 Hz), 6.68 (dt, 1H, J = 10.08 Hz, 1.96 Hz), 6.53 (1H, d, J = 9.24 Hz), 5.92 (1H, dt, J = 10.08 Hz, 3.64 Hz), 4.87 (2H, dd, J = 3.64 Hz, 1.96 Hz).
5-(2-Bromoethoxy)-8-nitro-2H-chromene (7). To a stirred solution of compound 6 (5.97 g, 31 mmol) in acetonitrile (150 mL) potassium carbonate (5.13 g, 37 mmol) and 1,2-dibromoethane (23.23 g, 124 mmol) were continuously added. The resulting mixture was heated to reflux for 2.5 h and then concentrated. The residue was partitioned between water (50 mL) and ethyl acetate (50 mL). The organic layer was separated, and the aqueous phase extracted with several additional portions of ethyl acetate. The combined organic phase was washed with brine, dried (MgSO4) and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (1/1) as eluent to give compound 7 as a yellow solid (4.69 g, yield: 34%). 1H-NMR (CDCl3, 400 MHz), δ: 7.84 (d, 1H, J = 9.2 Hz), 6.82 (dd, 1H, J = 10.8 Hz, 1.7 Hz), 6.41 (1H, d, J = 9.52 Hz), 6.80 (1H, dt, J = 10.36 Hz, 1.68 Hz), 6.44 (1H, d, J = 9.52 Hz), 5.88 (1H, dt, J = 10.08 Hz, 3.68 Hz). ESI-MS (+Q, m/z): 300 [M+H]+, 302 [M+H]+, 322 [M+Na]+, 324 [M+Na]+.
4-(2-(8-Nitro-2H-chromen-5-yloxy)ethyl)morpholine (9a). To a stirred solution of compound 7 (2 g, 6.66 mmol) in DMF (60 mL) potassium carbonate (1.4 g, 10.1 mmol) and morpholine (908 mg, 10.42 mmol) were continuously added. The resulting mixture was heated to 80 °C for 2 h. and then poured into cold water and extracted with ethyl acetate. The organic phase was washed with brine, dried over Na2SO4, and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (2/1) as eluent to give compound 9a as a white solid (1.06 g, yield: 52%). mp: 92–94 °C, 1H-NMR (CDCl3, 400), δ: 7.86 (d, J = 9.2Hz, 1H), 6.73 (dt, J = 10.0, 2.0 Hz, 1H), 6.47 (d, J = 9.2 Hz, 1H), 5.86 (dt, J = 10, 3.6 Hz, 1H), 4.95 (m, 2H), 4.22 (m, 2H), 3.75 (m, 4H), 2.86 (m, 2H), 2.61 (m, 4H). FAB-MS(m/z): 307 [M+H]+.
cis-2,6-Dimethyl-4-(2-(8-nitro-2H-chromen-5-yloxy)ethyl) morpholine (9b). Compound 9b was prepared following the method described for compound 9a, except cis-2,6-dimetylmopholine was used instead of morpholine as a reactant. Treating compound 7 (2 g, 6.63 mmol) in this manner to afford the title compound resulted in 1.80 g (81% yield) of compound 9b as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.85 (1H, d, J = 9.24 Hz), 6.74 (1H, dt, J = 10.12 Hz, 1.96 Hz), 6.47 (1H, d, J = 9.24 Hz), 6.74 (1H, dt, J = 10.12 Hz, 3.64 Hz), 4.94 (2H, dd, J = 3.64 Hz, 1.96 Hz), 4.19 (2H, m), 3.68 (2H, m), 2.81 (4H, m), 1.92 (2H, m), 1.17 (3H, s), 1.16 (3H, s). ESI-MS (+Q, m/z): 335 [M+H]+, 142.
1-(2-(8-Nitro-2H-chromen-5-yloxy)ethyl)-1H-1,2,4-triazole (9c). Compound 9c was prepared following the method described for compound 9a, except 1H-1,2,4-triazole was used instead of morpholine as a reactant. Treating compound 7 (2 g, 6.66 mmol) by this method resulted in 1.11 g (58% yield) of compound 9c as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 8.21 (1H, s), 8.00 (1H, s), 7.81 (1H, d, J = 9.24 Hz), 6.56 (1H, dt, J = 10.08 Hz, 1.96 Hz), 6.42 (1H, d, J = 9.52 Hz), 5.87 (1H, dt, J = 10.08 Hz, 3.64 Hz), 4.94 (2H, dd, J = 3.64 Hz, 1.96 Hz), 4.64 (2H, m), 4.54 (2H, m). ESI-MS (+Q, m/z): 289 [M+H]+, 311 [M+Na]+.
5-Hydroxy-8-aminochromane (11). To a solution of 5-hydroxy-8-nitro-2H-chromene (7, 5.0 g, 25.9 mmol) in anhydrous ethanol (100 mL) two drops of hydrochloric acid and palladium/carbon (2.0 g, 10%) were added. The mixture was heated to 60 °C, then hydrazine hydrate (10.0 mL, 85%) was added dropwise. After refluxing for 6 h, the mixture was cooled and filtered. The filtrate was dried and concentrated to give 2.12 g (50% yield) of compound 11 as an oily product which was sufficiently pure for use in the next step without further purification. 1H-NMR (CDCl3, 400 MHz), δ: 6.44 (d, 1H, J = 8.0 Hz), 6.19 (d, 1H, J = 8.4 Hz), 4.18 (t, 2H, J = 5.2 Hz), 3.54 (s, 2H), 2.65 (t, 2H, J = 6.8 Hz), 2.00 (p, 2H).
N-Boc-5-hydroxy-8-aminochromane (12). To a stirred solution of crude 11 (2.12 g, 12.8 mmol) and triethylamine (3.0 mL) in dichloromethane (100 mL), a solution of (Boc)2O (4.0 g, 18.3 mmol) in dichloromethane (20 mL) was added at 0 °C. The mixture was stirred at room temperature overnight, poured into water, extracted with dichloromethane, dried, and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (2/1) as eluent to give 2.69 g (79% yield) of compound 12 as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.61 (d, 1H), 6.76 (s, 1H), 6.30 (d, 1H, J = 12 Hz), 5.30 (s, 1H), 4.17 (t, 2H, J = 6 Hz), 2.65 (t, 2H, J = 8 Hz), 1.98 (p, 2H), 1.52 (s, 9H).
N-Boc-5-(2-bromoethoxy)-8-aminochromane (14). A mixture of compound 12 (2.20 g, 8.3 mmol), anhydrous potassium carbonate (1.38 g, 10 mmol), and 1,2-dibromopropane (6.23 g, 33 mmol) in acetonitrile (50 mL) was heated under reflux for 72 h. The resulting mixture was cooled to room temperature and water was added to the mixture, which was then extracted three times with ethyl acetate. The combined ethyl acetate layers were washed with water, brine, and then dried over Na2SO4, filtered and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (1/1) as eluent to give 0.83 g (27% yield) of compound 14 as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.78 (d, 1H), 6.85 (s, 1H), 6.35 (d, 1H, J = 8 Hz), 4.27 (t, 2H, J = 6 Hz), 4.20 (t, 2H, J = 8 Hz), 3.65 (t, 2H, J = 6 Hz), 2.73 (t, 2H, J = 6 Hz), 2.00 (p, 2H), 1.53 (s, 9H).
N-Boc-5-(2-morpholinoethoxy)-8-aminochromane (16a). A mixture of compound 14 (1.86 g, 5 mmol), anhydrous potassium carbonate (0.83 g, 6.01 mmol) and morpholine (6 mmol) in DMF (20 mL) was heated to 80 °C for 2 h. The resulting mixture was cooled to room temperature and poured onto cold water, and extracted three times with ethyl acetate. The combined ethyl acetate layers were washed with water, brine, and then dried over Na2SO4, filtered and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (2/1) as eluent to give 1.56 g (82.4% yield) of compound 16a as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.77 (d, 1H), 6.81 (s, 1H), 6.35 (d, 1H, J = 9.2 Hz), 4.17 (t, 2H, J = 5.6 Hz), 4.08 (t, 2H), 3.74 (t, 2H, J = 4.4 Hz), 2.80 (t, 2H), 2.60–2.65 (m, 6H), 1.97 (p, 2H), 1.50 (s, 9H).
N-Boc-5-(2-cis-2,6-dimethyl)-morpholinoethoxy)-8-aminochromane (16b). Compound 16b was prepared following the method described for compound 16a, except cis-1,6-dimetylmophiline was used instead of morpholine as a reactant. Treating compound 14 (2 g, 5.37 mmol) in this manner produced 1.71 g (90% yield) of compound 16b as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.76 (d, 1H), 6.81 (s, 1H), 6.35 (d, 1H, J = 8.8 Hz), 4.17 (t, 2H, J = 4.8 Hz), 4.07 (t, 2H), 3.70 (m, 2H), 2.77–2.84 (m, 4H), 2.65 (t, 2H, J = 6.8 Hz), 1.91–1.98 (m, 4H), 1.50 (s, 9H), 1.17 (d, 3H), 1.15 (d, 3H).
N-Boc-5-(2-(1H-1,2,4-triazol-1-yl)-ethoxy)-8-aminochromane (16c). Compound 16c was prepared following the method described for compound 16a, except 1,2,4-triazole was used instead of morpholine as a reactant. Treating compound 14 (2 g, 5.37 mmol) in this manner produced 1.28 g (66% yield) of compound 16c as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 8.23 (s, 1H), 7.97 (s, 1H), 7.77 (d, 1H), 6.81 (s, 1H), 6.31 (d, 1H, J = 8.8 Hz), 4.57 (t, 2H, J = 5.2 Hz), 4.29 (t, 2H, J = 5.2 Hz), 4.15 (t, 2H, J = 5.2 Hz), 2.51 (t, 2H, J = 6.4 Hz), 1.94 (p, 2H), 1.50 (s, 9H).
N-Boc-5-(2-(1H-imidazol-1-yl)-ethoxy)-8-aminochromane (16d). Compound 16d was prepared following the method described for compound 16a, except imidazole was used instead of morpholine as a reactant. Treating compound 14 (2 g, 5.37 mmol) in this manner produced 1.29 g (67% yield) of compound 16d as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.76 (d, 1H), 7.66 (s, 1H), 7.08 (s, 1H), 7.02 (s, 1H), 6.82 (s, 1H), 6.30 (d, 1H, J = 8.8 Hz), 4.34 (t, 2H), 4.17 (t, 2H), 2.58 (t, 2H, J = 6.6 Hz), 1.96 (p, 2H), 1.50 (s, 9H).
N-Boc-5-(2-(pyridin-4-yl)ethoxy)-8-aminochromane (16e). A solution of diethyl azodicarboxylate (DEAD, 1.05g, 5.0 mmol) in THF (25 mL) was slowly added to a solution of triphenylphosphine (1.57 g, 5.0 mmol), compound 12 (1.59 g, 6 mmol), and 2-(pyridin-4-yl)ethanol (616 mg, 5.0 mmol) in CH2Cl2 (25 mL), and the resulting cloudy mixture was stirred at room temperature for 3 h. After filtration of the mixture, the filtrate was concentrated in vacuo. Flash chromatography of the crude product (ethyl acetate/petroleum ether(1/1) afforded 1.76 g (95% yield) of compound 16e as a colourless oil that gradually crystallized upon standing at room temperature. 1H-NMR (CDCl3, 400 MHz), δ: 8.45 (d, 2H, J = 6.0 Hz), 7.80 (d, 1H), 6.87 (d, 2H, J = 5.2 Hz), 6.86 (s, 1H), 6.39 (d, 2H, J = 8.8 Hz), 4.36 (t, 2H), 4.30 (t, 2H), 4.17 (t, 2H), 2.62 (t, 2H, J = 6.4 Hz), 1.94 (p, 2H), 1.51 (s, 9H).
3.2.1. N-Boc-5-(2-morpholino-2-oxoethoxy)-8-aminochromane (16f)
Step 1: preparation of 4-(2-chloroacetyl)morpholine (20). To a stirred solution of morpholine (17.4 g, 200 mmol) and triethylamine (24.24 g, 220 mmol) in CH2Cl2 (200 mL), acetyl chloride (24.00 g, 210 mmol) in CH2Cl2 (volume) was added dropwise. The resulting reaction mixture was stirred at this temperature for a further 4 h, then poured into water, and the aqueous layer extracted twice with methylene chloride. The organic chloride phase was combined, washed with dilute hydrochloric acid and water, and dried over Na2SO4, and evaporated to dryness to give compound 20 (22.66 g, 69.2% yield) as a pale yellow oil.
Step 2: preparation of N-boc-5-(2-morpholino-2-oxoethoxy)-8-aminochromane (16f). A solution of compound 12 (1.86 g, 5.0 mmol), anhydrous potassium carbonate (0.83 g, 6.0 mmol) and 4-(2-chloroacetyl)-morpholine (20, 982 mg, 6.0 mmol) in DMF (20 mL) was heated to 80 °C for 2 h. The resulting mixture was cooled to room temperature, poured onto cold water and extracted three times with ethyl acetate. The combined ethyl acetate layers were washed with water, brine, and then dried over Na2SO4, filtrated and concentrated to dryness. The residue was separated by column chromatography on silica gel with ethyl acetate/petroleum ether (1/1) as eluent to give 1.35 g (69% yield) of compound 16f as a white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.77 (d, 1H), 6.89 (s, 1H), 6.35 (d, 1H, J = 8.8 Hz), 4.63 (s, 2H), 4.19 (t, 2H), 3.60–3.68 (m, 8H), 2.68 (t, 2H, J = 6.0 Hz), 1.99 (p, 2H), 1.50 (s, 9H).
Step 3: preparation of 5-(morpholinoethoxy)-8-aminochromane (17a). To a solution of compound 16a (1.48 g, 4.0 mmol) in CH2Cl2 (40 mL), precooled trifluoroacetic acid (4.0 mL) was added at 0–4 °C, and the reaction was stirred at this temperature for 5 h. After evaporation, water was added to the residue, and the pH of the mixture was adjusted to 10 by addition of 1 M aqueous NaOH solution. The aqueous layer was extracted with ethyl acetate, washed with water, dried over anhydrous sodium sulfate, filtered, and concentrated to give 930 mg (86% yield) of compound 17a as a grey solid. This product is unstable and was therefore used without delay for the next step.
5-(2-(2,6-Dimethyl)-morpholinoethoxy)-8-aminochromane (17b). Compound 17b was prepared following the method described for compound 17a employing 16b (1.64 g, 4.0 mmol) as a reactant, producing compound 17b as a pale white solid (906 mg, 74% yield). This product is unstable and was therefore used without delay for the next step.
5-(2-(1,2,4-Triazole)-ethoxy)-8-aminochromane (17c). Compound 17c was prepared following the method described for the compound 17a employing 16c (1.23 g, 4.0 mmol) as reactant, to produce compound 17c as a white solid (729 mg, 70% yield). This product is unstable and was therefore used without delay for the next step.
5-(2-Imidazolylethoxy)-8-aminochromane (17d). Compound 17d was prepared following the method described for the compound 17a employing 16d (1.23 g, 4.0 mmol) as reactant, to produce compound 17d as a white solid (778 mg, 75% yield). This product is unstable and was therefore used without delay for the next step.
5-(2-(Pyridin-4-yl)ethoxy)-8-aminechromane (17e). Compound 17e was prepared following the method described for compound 17a employing 16e (1.48 g, 4 mmol) as reactant, to produce compound 17e as a white solid (789 mg, 73% yield). This product is unstable and was therefore used without delay for the next step.
5-(2-Morpholino-2-oxoethoxy)-8-aminochroman (17f). Compound 17f was prepared following the method described for compound 17a employing 16f (1.17 g, 4 mmol) as reactant, to afford the compound 17f as a pale white solid (795 mg, 68% yield). This product is unstable and was therefore used without delay for the next step.
3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-amine (25a). A solution of 4-tolyllhydrazine hydrochloride (5.20 g, 33 mmol) and pentylacyl acetonitrile (3.75 g, 30 mmol) in 0.4 M ethanolic solution of HCl (100 mL) was heated under reflux during 48 h. After cooling to room temperature, 1M NaOH was added to the mixture until the pH reached 10–11. The mixture was partitioned between water and ethyl acetate. The water phase was extracted twice with dichloromethane. The organic phases were combined and washed with water and brine, then dried with Na2SO4. Evaporation of the solvent in vacuo afforded the crude product, which was subjected to recrystallization from ethyl acetate and petroleum ether to produce compound 25a as a white solid (5.88 g, 86% yield). 1H-NMR (CDCl3, 400 MHz), δ: 7.40 (d, 2H, J = 8.4 Hz), 7.25 (d, 2H, J = 3.08 Hz), 5.50 (s, 1H), 3.72 (brs, 2H, NH2), 2.37 (s, 3H), 1.32 (s, 9H).
3-tert-Butyl-1-(4-fluorophenyl)-1H-pyrazol-5-amine (25b). The title compound was prepared according to the method used for 25a except (4-fluorophenyl)hydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 79%. 1H-NMR (CDCl3, 400 MHz), δ: 7.59 (d, 2H), 7.10 (d, 2H), 5.58 (s, 1 H), 3.62 (brs, 2H, NH2), 1.32 (s, 9H).
3-tert-Butyl-1-(4-methoxyphenyl)-1H-pyrazol-5-amine (25c). The title compound was prepared according to the method used for 25a, except (4-methoxyphenyl)hydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 87%. 1H-NMR (CDCl3, 400 MHz): 7.41 (d, 2H), 6.97 (d, 2H), 5.43 is, 1H), 3.83 (s, 3H), 1.35 (s, 9H).
3-tert-Butyl-1-(4-trifluoromethylphenyl)-1H-pyrazol-5-amine (25d). The title compound was prepared according to the method used for 25a, except (4-trifluoromethylphenyl)hydrazine hydrochloride was used instead of 4-tolyllhydrazine hydrochloride. Yield: 79%. 1H-NMR (CDCl3, 400 MHz), δ: 7.77 (d, 2H, J = 8.4 Hz), 7.69 (d, 2H, J = 8.4 Hz), 5.56 (s, 1H), 3.76 (s, 2H), 1.31 (s, 9H).
3-tert-Butyl-1-(4-nitrophenyl)-1H-pyrazol-5-amine (25e). The title compound was prepared according to the method used for 25a, except 4-nitrohydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 83%. 1H-NMR (DMSO-d6, 400 MHz), δ: 8.28 (d, J = 6.9 Hz, 2H), 7.93 (d, J = 6.9 Hz, 2H), 5.55 (s, 2H), 5.46 (s, 1H), 1.20 (s, 9H).
3-tert-Butyl-1-(4-isopropylphenyl)-1H-pyrazol-5-amine (25f). The title compound was prepared according to the method used for 25a, except (4-isopropylphenyl) hydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 81%. 1H-NMR (CDCl3, 400 MHz), δ: 7.44 (d, 2H, J = 8.4 Hz), 7.29 (d, 2H, J = 8.4 Hz), 5.51 (s, 1H), 3.72 (s, 2H), 2.93 (m, 1H), 1.31 (s, 9H), 1.25 (d, 3H), 1.24 (d, 3H).
3-tert-Butyl-1-m-tolyl-1H-pyrazol-5-amine (25g). The title compound was prepared according to the method used for 25a, except m-tolylhydrazine hydrochloride was used instead of p-tolylhydrazine hydrochloride. Yield: 79%. 1H-NMR (CDCl3, 400 MHz), δ: 7.38 (s, 1H), 7.31–7.33 (m, 2H), 7.13 (m, 1H), 5.51 (s, 1H), 3.73 (bs, 2H), 2.39 (s, 3H), 1.31 (s, 9H).
3-tert-Butyl-1-(4-chlorophenyl)-1H-pyrazol-5-amine (25h), 3-tert-butyl-1-(4-bromophenyl)-1H-pyrazol-5-amine (25i), 3-tert-butyl-1-(4-ethylphenyl)-1H-pyrazol-5-amine (25j) and 3-tert-butyl-1-phenyl-1H-pyrazol-5-amine (25n) are all commercially available.
3-tert-Butyl-1-(naphthalen-1-yl)-1H-pyrazol-5-amine (25o). Compound 25o was prepared according to the method used for 1, except (naphthalen-1-yl)hydrazine hydrochloride was used instead of 4-tolylhydrazine hydrochloride. Yield: 82%. 1H-NMR (CDCl3, 400 MHz), δ: 7.89–7.91 (m, 2H), 7.51–7.55 (m, 5H), 5.59 (s, 1H), 3.50 (s, 2H), 1.34 (s, 9H).
3.2.2. 1-tert-Butyl-3-(p-tolyl)-4-amine-1H-pyrazole (25p)
Step 1: preparation of 2-chloro-1-p-tolylethanone (27). To a suspension of anhydrous aluminum chloride (5.60 g, 40 mmol) in anhydrous toluene (40 mL), chloroacetyl chloride (4.52 g, 40 mmol) was slowly added dropwise. After the aluminum chloride has dissolved, the mixture was heated slowly to 80 °C and held at that temperature for 2 h, then cooled and poured into crushed ice (100 g) containing concentrated hydrochloric acid (10 mL). The aqueous layer was extracted three times with toluene. The combined toluene layers were washed successively with 10% aqueous sodium hydroxide, water and brine, dried over anhydrous sodium sulfate and evaporated under reduced pressure. The residue was purified by crystallization from diethyl ether to give the pure compound 27(5.6 g, yield: 86%). 1H-NMR (CDCl3, 400 MHz), δ: 7.46 (d, 2H, J = 7.5 Hz), 7.01 (d, 2H, J = 7.5 Hz), 4.63 (s, 2H), 2.35 (s, 3H).
Step 2: preparation of 2-(1,3-dioxoisoindolin-2-yl)-1-p-tolylethanone (29). To a solution of compound 27 (5.06 g, 30 mol) in DMF (30 mL), phthalimide potassium salt (5.56 g, 30 mol) was added, and the resulting mixture was stirred at 70 °C for 2 h, cooled, and then poured into ice water (300 mL). The precipitated white solid was filtered and dried to give compound 29 (7.37 g, yield: 88%). 1H-NMR (400 MHz, CDCl3), δ: 7.88–7.83 (m, 4H), 7.73–7.69 (m, 2H), 7.27 (d, J = 7.9 Hz, 2H), 5.08 (s, 2H), 2.40 (s, 3H).
Step 3: preparation of N,N-dimethyl-3-(isoindoline-1,3-dione-2yl)-2-p-tolylprop-1-en-1-amine (31). To a solution of compound 29 (5.58 g, 20 mmol) in DMF (40 mL), N,N-dimethylformamide dimethyl acetal (5.68 g, 24 mmol) was added dropwise. The mixture was heated to 100 °C and kept at this temperature for 24 h, then cooled, poured into ice water (200 mL) and extracted with ethyl acetate. The combined organic phase was dried over anhydrous sodium sulfate, and evaporated in vacuo to provide a residue which was purified by silica gel chromatography using ethyl acetate and petroleum ether (1/3) as eluent to obtain compound 31 as a white solid (4.0 g, yield: 62%).
Step 4: preparation of 1-tert-butyl-3-(p-tolyl)-4-phthalimido-pyrazole (33). A solution of compound 31 (4.0 g, 14.3 mmol) and tert-butyl hydrazine (1.39 g, 15.7 mmol) in 90% ethanol (200 mL) was heated under reflux for 12 h. After cooling to room temperature, the resulting white crystalline solid was filtered, washed with anhydrous ether and dried to give compound 33 as a white solid (3.73 g, yield: 73%).
Step 5: preparation of 1-tert-Butyl-3-(p-tolyl)-4-amine-1H-pyrazole (25p). To a solution of compound 33 (3.59 g, 10 mol) in ethanol (100 mL), 85% hydrazine hydrate solution (2.36 g, 40 mmol) was added dropwise. The mixture was heated to reflux for 2 h, cooled to room temperature, and concentrated, and ether (20 mL) was added. The precipitated white solid was filtered, washed with anhydrous ether, purified by silica gel chromatography using ethyl acetate and petroleum ether (1/1) as eluent to give compound 25p as a white solid (1.89 g, yield 83%). 1H-NMR (CDCl3, 400 MHz) δ: 7.21–7.26 (m, 5H), 2.68 (s, 2H), 2.41 (s, 3H), 1.41 (s, 9H).
3.2.3. 1-(4-Methylphenyl)-2-amino-4-tert-butyl-imidazole (25q)
Step 1: preparation of N-(pivaloylmethyl)-4-methylaniline (36). To a solution of tert-butylchloromethyl ketone (3.23 g, 0.024 mol) and p-toluidine (2.14 g, 0.020 mol) in DMF (20 mL), sodium bicarbonate (2.52 g, 0.030 mol) was added. The mixture was stirred at 75 °C for 48 h, then cooled, and poured into ice water (200 mL). The precipitated white solid was filtered and dried to give compound 36 as a white solid (4.08 g, yield: 99%).
Step 2: preparation of 1-(4-methylphenyl)-2-amino-4-tert-butyl-imidazole (25q). A solution of compound 36 (5.0 g, 20 mmol) and 50% aqueous cyanamide (16.92 g, 20 mmol) in ethanol (200 mL) was refluxed for 12 h and concentrated, water was added to the residue. the mixture was extracted three times with ethyl acetate, then the combined organic layers washed with dilute sodium hydroxide solution, water and saturated brine, and dried. The residue was purified by silica gel chromatography using dichloromethane and methanol (100/1) to obtain a white solid product (2.62 g, yield: 57%). 1H-NMR (CDCl3, 400 MHz), δ: 7.26 (m, 4H), 6.35 (s, 1H), 4.18 (s, 2H), 2.39 (s, 3H), 1.27 (s, 9H).
3-tert-Butyl-5-aminoisoxazole (25r). To an aqueous solution of sodium hydroxide solution (0.84 g, 21 mmol) in water (10 mL), pivaloylacetonitrile (1.25 g, 10 mmol) and hydroxylamine hydrochloride (0.76 g, 11 mmol) were added. The resulting solution was stirred at 50° C for 3 h. The reaction mixture was cooled and the resultant white crystalline solid was filtered, washed with water and dried to provide compound 25r as a white crystalline solid (1.23 g, yield 88%). 1H-NMR (CDCl3, 400 MHz), δ: 5.03 (s, 1H), 4.32 (bs, 2H), 1.27 (s, 9H).
5-(tert-Butyl)-3-aminoisoxazole (25s). To a solution of pivaloylacetonitrile (3 g, 23.97 mmol) in water (20 mL), NaOH (1.06 g, 26.4 mmol) and hydroxylamine hydrochloride (1.83 g, 26.4 mmol) were added continuously with stirring. The resulting solution was stirred for approximately 30 min at room temperature, and the pH adjusted to 10–11 with 1 M NaOH. After stirring for 10 h or more at 50 °C, the mixture was cooled and washed two to three times with carbon tetrachloride. The aqueous layer was acidified with concentrated HC1 until the pH = 4–5, and then further stirred for approximately 3 h at 50 °C. The reaction mixture was cooled to room temperature, and adjusted to pH 12 by adding an aqueous solution of 1 N NaOH. The resulting solid was filtered, washed with distilled water, and dried in air to obtain compound 25s as a white solid (2.0 g, yield: 70%). 1H-NMR (DMSO-d6, 300 MHz), δ: 5.49 (s, 1H), 5.40 (s, 2H), 1.21 (s, 9H).
3.2.4. General Procedure for the Preparation of Chromanylureas (39a–j) and 2H-Chromenylureas (40a–r)
A solution of compounds 10a–d or compounds 17a–d (1.0 mmol) in dichloromethane (10 mL) was slowly added to a stirred solution of triphosgene (109 mg, 0.36 mmol) in dichloromethane (50 mL) over a period of 30 min using a syringe. After stirring for a further 30 min, a solution of compound 25a–r (0.6 mmol) and triethylamine (0.4 mL, 2.77 mmol) in dichloromethane (10 mL) was added in one portion. The reaction mixture was stirred for 2 h at room temperature. After completion of the reaction, the reaction was poured into water (50 mL) and extracted three times with dichloromethane. The organic layer was washed with water (5 mL), sat. NaCl solution (5 mL), and dried over Na2SO4. After evaporation of solvent under vacuum, the residue was purified by silica gel chromatography to give the desired chromanylurea or 2H-chromenylurea compounds.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39a). Yield: 19%, 1H-NMR (CDCl3, 400 MHz), δ: 7.69 (d, 1H, J = 8.4 Hz), 7.29–7.31 (m, 3H), 7.16 (m, 2H), 6.84 (s, 1H), 6.31–6.34 (m, 2H), 4.04–4.07 (m, 4H), 3.72 (t, 4H), 2.80 (t, 2H, J = 5.2 Hz), 2.60 (t, 2H), 2.59 (t, 4H), 2.31 (s, 3H), 1.91 (p, 2H), 1.34 (s, 9H). FAB-MS (m/z): 534.2 [M+H]+.
1-(3-tert-Butyl-1-phenyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39b). White solid, yield: 26%. 1H-NMR (CDCl3, 400 MHz), δ: 7.67 (d, 1H, J = 8.4 Hz), 7.32–7.45 (m, 2H), 7.34 (m, 3H), 7.28 (m, 1H), 7.03 (s, 1H), 6.35 (s, 1H), 6.31 (d, 1H, J = 10.8 Hz), 4.04–4.06 (m, 4H), 3.70 (t, 4H, J = 4.8 Hz), 2.77 (t, 2H, J = 5.6 Hz), 2.56–2.61 (m, 6H), 1.90 (p, 2H), 1.34 (s, 9H). FAB-MS (m/z): 520.2 [M+H]+.
1-(3-tert-Butyl-1-m-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39c). White solid, yield: 18%, 1H-NMR (CDCl3, 400 MHz), δ:7.68 (d, 1H, J = 8.4 Hz), 7.21–7.26 (m, 4H), 7.11 (d, 1H), 6.83 (s, 1H), 6.36 (s, 1H), 6.32 (d, 1H, J = 8.8 Hz), 4.05–4.08 (m, 4H), 3.73 (t, 4H, J = 4.4 Hz), 2.80 (t, 2H, J = 5.6 Hz), 2.57–2.62 (m, 6H), 2.32 (s, 3H), 1.91 (p, 2H), 1.35 (s, 9H). FAB-MS (m/z): 534.2 [M+H]+.
1-(3-tert-Butylisoxazol-5-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39d). White solid, yield: 19%, 1H-NMR (CDCl3, 400 MHz), δ: 8.59 (s, 1H), 7.67 (d, 1H, J = 8.0 Hz), 6.36 (d, 1H, J = 9.2 Hz), 6.12 (s, 1H), 4.08–4.10 (m, 4H), 3.75 (t, 4H, J = 4.4 Hz), 2.83 (t, 2H, J = 5.6 Hz), 2.60–2.64 (m, 6H), 1.92 (p, 2H), 1.30 (s, 9H). FAB-MS (m/z): 445.1 [M+H]+.
1-(5-tert-Butylisoxazol-3-yl)-3-(5-(2-morpholinoethoxy)-chroman-8-yl)urea (39e). White solid, yield: 31%. 1H-NMR (CDCl3, 400 MHz), δ: 8.75 (s, 2H), 7.86 (d, 1H, J = 8.8 Hz), 6.37 (d, 1H, J = 8.8 Hz), 6.07 (s, 1H), 4.23 (t, 2H, J = 4.8 Hz), 4.10 (t. 2H), 3.74 (t, 4H, J = 4.4 Hz), 2.82 (t, 2H, J = 5.2 Hz), 2.61–2.67 (m, 6H), 1.99 (p, 2H), 1.34 (s, 9H). FAB-MS (m/z): 445.1 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholino-2-oxoethoxy)chroman-8-yl)urea (39f). White solid, yield: 16%, 1H-NMR (CDCl3, 400 MHz), δ: 7.75 (d, 1H, J = 9.2 Hz), 7.34 (d, 2H, J = 8.4 Hz), 7.25 (d, 2H), 6.38 (s, 1H), 6.34 (d, 1H, J = 9.2 Hz), 4.64 (s, 2H), 4.11 (t, 2H, J = 3.6 Hz), 3.63–3.67 (m, 8H), 2.67 (t, 2H, J = 6.4 Hz), 2.37 (s, 3H), 1.95 (p, 2H), 1.36 (s, 9H). FAB-MS (m/z): 548.1 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-(sn-2,6-dimethylmorpholino)ethoxy)chroman-8-yl)-urea (39g). White solid, yield: 21%, 1H-NMR (CDCl3, 400 MHz), δ: 7.71 (d, 1H, J = 9.2 Hz), 7.31 (d, 2H, J = 8.4 Hz), 7.21 (d, 2H), 7.17 (s, 1H), 6.31–6.36 (m, 3H), 4.08–4.10 (m, 4H), 3.71 (m, 2H), 2.80–2.86 (m, 4H), 2.63 (t, 2H, J = 6.4 Hz), 2.36 (s, 3H), 1.92–1.95 (m, 4H), 1.35 (s, 9H), 1.17 (d, 3H), 1.15 (d, 3H). FAB-MS (m/z): 562.2 [M+H]+.
3-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-1-(5-(2-(1H-1,2,4-triazol-1-yl)ethoxy)chroman-8-yl)urea (39h). White solid, yield: 15%, 1H-NMR (CDCl3, 400 MHz), δ: 8.08 (s 1H), 7.82–7.84 (m, 2H), 7.45 (s, 1H), 7.32 (d, 2H, J = 8.4 Hz), 7.17 (d, 2H, J = 8.4 Hz), 6.40 (s, 1H), 6.27 (d, 1H, J = 9.2 Hz), 4.56 (t, 2H, J = 4.8 Hz), 4.23 (t, 2H, J = 4.8 Hz), 3.77 (t, 2H), 2.33–2.36 (m, 5H), 1.69 (p, 2H), 1.35 (s, 9H). FAB-MS (m/z): 516.3 [M+H]+.
3-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-1-(5-(2-(1H-imidazol-1-yl)ethoxy)chroman-8-yl)urea (39i). White solid, yield: 21%, 1H-NMR (CDCl3, 400 MHz), δ: 7.88 (d, 1H, J = 8.8 Hz), 7.73 (s, 1H), 7.53 (s, 1H), 7.32 (d, 2H, J = 8.0 Hz), 7.15 (d, 2H, J = 8.0 Hz), 6.93 (s, 1H), 6.89 (s, 1H), 6.41 (s, 1H), 6.22 (d, 1H, J = 8.8 Hz), 4.32 (t, 2H, J = 4.8 Hz), 4.12 (t, 2H, J = 5.6 Hz), 3.62 (t, 2H), 2.35 (t, 2H), 2.32 (s, 3H), 1.61 (p, 2H), 1.35 (s, 9H). FAB-MS (m/z): 515.2 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-(pyridin-4-yl)ethoxy)chroman-8-yl)urea (39j). White solid, yield: 17%, 1H-NMR (CDCl3, 400 MHz), δ: 7.71 (d, 1H), 7.50–7.53 (m, 2H), 7.31 (d, 2H), 7.21 (d, 2H, J = 8.0 Hz), 6.37 (s, 1H), 6.30 (d, 1H, J = 8.4 Hz), 6.26 (t, 1H), 4.52 (t, 2H, J = 5.2 Hz), 4.28 (t, 2H, J = 5.2 Hz), 4.07 (t, 2H, J = 5.6 Hz), 2.54 (t, 2H, J = 6.4 Hz), 2.35 (s, 3H), 1.91 (p, 2H), 1.36 (s, 9H). FAB-MS (m/z): 515.1 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40a). Yield: 27%, white solid. 1H-NMR (CDCl3, 400 MHz), δ: 7.74 (d, 1H, J = 8.8 Hz), 7.33 (d, 2H, J = 8.4 Hz), 7.21 (d, 2H, J = 8.0 Hz), 7.13 (s, 1H), 6.72–6.74 (dd, 1H, J = 8.4 Hz, 1.6 Hz), 6.39 (d, 1H, J = 9.2 Hz), 6.35 (s, 1H), 6.34 (s, 1H), 5.72 (m, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.11 (t, 2H), 3.74 (t, 4H), 2.82 (t, 2H), 2.60 (t, 4H), 2.36 (s, 3H), 1.35 (s, 9H). FAB-MS (m/z): 532.2 [M+H]+.
1-(3-tert-Butyl-1-(4-fluorophenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40b). Yield: 22%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.67 (d, 1H, J = 8.4 Hz), 7.43 (m, 2H), 7.07-–7.11 (m, 3H), 6.73 (d, 1H, J = 10.4 Hz), 6.58 (s, 1H), 6.38 (d, 1H, J = 9.2 Hz), 6.35 (s, 1H), 5.73 (m, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.11 (t, 2H, J = 5.2 Hz), 3.75 (t, 4H), 2.83 (t, 2H), 2.61 (t, 4H), 1.35 (s, 9H). FAB-MS (m/z): 536.2 [M+H]+.
1-(3-tert-Butyl-1-(4-chlorophenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40c). Yield: 29%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.65 (d, 1H, J = 8.8 Hz), 7.43 (d, 2H), 7.35 (d, 2H, J = 8.8 Hz), 7.10 (s, 1H), 6.74 (d, 1H, J = 10.0 Hz), 6.68 (s, 1H), 6.36–6.38 (m, 2H), 5.72 (m, 1H), 4.69 (t, 2H, J = 2.0 Hz), 4.10 (t, 2H, J = 5.2 Hz), 3.74 (t, 4H, J = 4.4 Hz), 2.81 (t, 2H, J = 4.8 Hz), 2.59 (t, 4H), 1.35 (s, 9H). FAB-MS (m/z): 552.2 [M+H]+.
1-(1-(4-Bromophenyl)-3-tert-butyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40d). Yield: 26%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.65 (d, 1H), 7.50 (d, 2H, J = 8.8 Hz), 7.36 (d, 2H, J = 8.8 Hz), 7.12 (s, 1H), 6.77 (s, 1H), 6.73 (d, 1H, J = 10.4 Hz), 6.38 (d, 1H, J = 7.2 Hz), 6.36 (s, 1H), 5.72 (m, 1H), 4.69 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H, J = 1.6 Hz), 3.74 (t, 4H, J = 4.4 Hz), 2.82 (t, 2H, J = 4.8 Hz), 2.60 (t, 4H), 1.34 (s, 9H). FAB-MS (m/z): 598.0 [M+H]+.
1-(3-tert-Butyl-1-(4-methoxyphenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40e). Yield: 26%, white solid, 1H-NMR (CDCl3, 400 MHz), δ:7.72 (d, 1H, J = 8.8 Hz), 7.33 (d, 2H, J = 9.2 Hz), 7.25 (s, 1H), 6.88 (d, 2H, J = 9.2 Hz), 6.75 (d, 1H), 6.73 (s, 1H), 6.36 (d, 1H), 6.33 (s, 1H), 5.70 (m, 1H), 4.67 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H), 3.77 (s, 3H), 3.74 (t, 4H, J = 4.4 Hz), 2.80 (t, 2H, J = 5.6 Hz), 2.59 (t, 4H), 1.35 (s, 9H). FAB-MS (m/z): 548.1 [M+H]+.
1-(3-tert-Butyl-1-(4-(trifluoromethyl)phenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40f). Yield: 17%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.65 (m, 5H), 7.07 (s, 1H), 6.76 (s, 1H), 6.73 (d, 1H, J = 9.6 Hz), 6.40 (s, 1H), 6.38 (d, 1H, J = 8.8 Hz), 5.70 (m, 1H), 4.70 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H, J = 5.2 Hz), 3.74 (t, 4H, J = 4.0 Hz), 2.82 (t, 2H), 2.60 (t, 4H), 1.36 (s, 9H). FAB-MS (m/z): 586.1 [M+H]+.
1-(3-tert-Butyl-1-(4-nitrophenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40g). Yield: 37%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 8.18 (d, 2H, J = 9.2 Hz), 7.74 (d, 2H, J = 8.8 Hz), 7.55 (d, 1H), 7.30 (s, 1H), 6.71 (d, 1H, J = 10.0 Hz), 6.39 (s, 1H), 6.34 (d, 1H, J = 9.2 Hz), 5.70 (m, 1H), 4.68 (t, 2H, J = 1.6 Hz), 4.09 (t, 2H, J = 6.0 Hz), 3.74 (t, 4H, J = 4.4 Hz), 2.81 (t, 2H, J = 6.0 Hz), 2.60 (t, 4H), 1.34 (s, 9H). FAB-MS (m/z): 563.0 [M+H]+.
1-(3-tert-Butyl-1-(4-ethylphenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40h). Yield: 27%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.74 (d, 1H, J = 8.8 Hz), 7.36 (d, 2H, J = 8.4 Hz), 7.25 (d, 2H), 7.14 (s, 1H), 6.73 (d, 1H, J = 10.0 Hz), 6.35–6.40 (m, 3H), 5.72 (m, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.12 (t, 2H), 3.75 (t, 4H), 2.84 (t, 2H), 2.62–2.68 (m, 6H), 1.36 (s, 9H), 1.22 (t, 3H). FAB-MS (m/z): 546.0 [M+H]+.
1-(3-tert-Butyl-1-(4-isopropylphenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40i). Yield: 19%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.74 (d, 1H, J = 9.2 Hz), 7.34 (d, 2H), 7.26 (d, 2H), 7.15 (s, 1H), 6.73 (d, 1H, J = 10.0 Hz), 6.43 (s, 1H), 6.39 (d, 1H, J = 9.2 Hz), 6.35 (s, 1H), 5.72 (m, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.09 (t, 2H, J = 5.2 Hz), 3.73 (t, 4H), 2.81 (t, 2H), 2.59 (t, 4H), 1.36 (s, 9H), 1.24 (d, 3H), 1.22 (d, 3H). FAB-MS (m/z): 560.1 [M+H]+.
1-(3-tert-Butyl-1-(3-chloro-4-fluorophenyl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40j). Yield: 27%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.60 (m, 2H), 7.38 (m, 1H), 7.08–7.14 (m, 3H), 6.72 (d, 1H, J = 9.6 Hz), 6.38 (d, 1H, J = 8.8 Hz), 6.35 (s, 1H), 5.72 (m, 1H), 4.70 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H, J = 5.2 Hz), 3.74 (t, 4H, J = 4.4 Hz), 2.83 (t, 2H), 2.61(t, 4H), 1.34 (s, 9H). FAB-MS (m/z): 570.0 [M+H]+.
1-(3-tert-Butyl-1-m-tolyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40k). Yield: 21%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.22 (d, 1H, J = 8.8 Hz), 7.26–7.29 (m, 3H), 7.15 (d, 2H), 6.73 (d, 1H, J = 10.0 Hz), 6.58 (s, 1H), 6.38 (d, 1H, J = 9.6 Hz), 6.36 (s, 1H), 5.72 (m, 1H), 4.71 (t, 2H, J = 1.6Hz), 4.12 (t, 2H), 3.76 (t, 4H), 2.84 (t, 2H), 2.63 (t, 4H), 2.36 (s, 3H), 1.36 (s, 9H). FAB-MS (m/z): 532.1 [M+H]+.
1-(3-tert-Butylisoxazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40l). Yield: 39%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 9.27 (s, 1H), 7.74 (d, 1H, J = 8.8 Hz), 7.40 (s, 1H), 6.67 (d, 1H, J = 10.0 Hz), 6.37 (d, 1H, J = 9.2 Hz), 6.13 (s, 1H), 5.63 (m, 1H), 4.62 (t, 2H, J = 1.6 Hz), 4.10 (t, 2H, J = 5.6 Hz), 3.77 (t, 4H, J = 4.4 Hz), 2.83 (t, 2H, J = 5.2 Hz), 2.69 (t, 4H), 1.30 (s, 9H). FAB-MS (m/z): 443.0 [M+H]+.
1-(5-tert-Butylisoxazol-3-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40m). Yield: 33%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 8.77 (s, 1H), 8.27 (s, 1H), 7.86 (d, 1H, J = 8.8 Hz), 6.75 (d, 1H, J = 10.0 Hz), 6.42 (d, 1H, J = 9.6 Hz), 6.02 (s, 1H), 5.76 (m, 1H), 4.84 (t, 2H, J = 2.0 Hz), 4.12 (t, 2H, J = 6.8 Hz), 3.76 (t, 4H), 2.84 (t, 2H), 2.62 (t, 4H), 1.36 (s, 9H). FAB-MS (m/z): 443.0 [M+H]+.
1-(1-tert-Butyl-3-p-tolyl-1H-pyrazol-4-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40n). Yield: 21%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.67–7.70 (m, 2H), 7.18–7.20 (m, 4H), 6.73 (d, 1H, J = 10.0 Hz), 6.68 (s, 1H), 6.36 (d, 1H, J = 9.2 Hz), 5.72 (m, 1H), 5.49 (s, 1H), 4.71 (t, 2H, J = 2.0 Hz), 4.11 (t, 2H), 3.76 (t, 4H), 2.83 (t, 2H), 2.62 (t, 4H), 2.39 (s, 3H), 1.46 (s, 9H). FAB-MS (m/z): 532.1 [M+H]+.
1-(3-tert-Butyl-1-(naphthalen-1-yl)-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)-urea (40o). Yield: 22%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.88–7.90 (m, 2H), 7.48–7.50 (m, 5H), 7.38 (d, 1H), 6.95 (s, 1H), 6.68 (d, 1H, J = 10.0 Hz), 6.48 (s, 1H), 6.24–6.27 (m, 2H), 5.67 (m, 1H), 4.63 (t, 2H, J = 1.6 Hz), 4.07 (t, 2H, J = 5.2 Hz), 3.75 (t, 4H, J = 4.4 Hz), 2.82 (t, 2H), 2.61 (t, 4H), 1.40 (s, 9H). FAB-MS (m/z): 568.1 [M+H]+.
1-(4-tert-Butyl-1-p-tolyl-1H-imidazol-2-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl)urea (40p). Yield: 27%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 8.03 (d, 1H, J = 9.2 Hz), 7.26–7.30 (m, 5H), 6.78 (d, 1H, J = 10.0 Hz), 6.44 (s, 1H), 6.41 (d, 1H, J = 9.2 Hz), 5.77 (m, 1H), 4.88 (t, 2H), 4.13 (t, 2H, J = 6.8 Hz), 3.76 (t, 4H), 2.84 (t, 2H), 2.62 (t, 4H), 2.42 (s, 3H), 1.34 (s, 9H). FAB-MS (m/z): 532.4 [M+H]+.
1-(3-tert-Butyl-1-p-tolyl-1H-pyrazol-5-yl)-3-(5-(2-(cis-2,6-dimethylmorpholino)ethoxy)-2H-chromen-8-yl)urea (40q). Yield: 28%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 7.74 (d, 1H, J = 9.2 Hz), 7.33 (d, 2H, J = 8.0 Hz), 7.22 (d, 2H), 7.14 (s, 1H), 6.74 (d, 1H, J = 10.0 Hz), 6.45 (s, 1H), 6.39 (d, 1H, J = 9.2 Hz), 6.35 (s, 1H), 5.72 (m, 1H), 4.71 (t, 2H, J = 1.6 Hz), 4.12 (t, 2H, J = 4.0 Hz), 3.74 (m, 2H), 2.83 (m, 4H), 2.36 (s, 3H), 1.94 (m, 2H), 1.36 (s, 9H), 1.17 (d, 3H), 1.16 (d, 3H). FAB-MS (m/z): 532.4 [M+H]+.
1-(5-(2-(1H-1,2,4-Triazol-1-yl)ethoxy)-2H-chromen-8-yl)-3-(3-tert-butyl-1-p-tolyl-1H-pyrazol-5-yl)-urea (40r). Yield: 16%, white solid, 1H-NMR (CDCl3, 400 MHz), δ: 8.10 (s, 1H), 7.86 (s, 1H), 7.83 (d, 1H, J = 8.0 Hz), 7.38 (s, 1H), 7.32 (d, 2H, J = 8.4 Hz), 7.18 (d, 2H), 6.39 (s, 1H), 6.36 (d, 1H), 6.29 (d, 1H, J = 9.2 Hz), 5.47 (m, 1H), 4.55 (t, 2H, J = 4.8 Hz), 4.44 (t, 2H), 4.28 (t, 2H, J = 4.8 Hz), 2.33 (s, 3H), 1.35 (s, 9H). FAB-MS (m/z): 514.2 [M+H]+.
3.3. Pharmacology
THP-1 cells from a human monocytic cell line (obtained from the National Platform of Experimental Cell Resource for Sci-Tech of China, Beijing, China) were suspended in culture medium [RPM1 (Gibco-BRL, Gaithersburg, MD, USA) containing 15% fetal bovine serum and 0.02 mM 2-mercaptoethanol], at a concentration of 2.5 × 106 cells/mL and then plated in a 96-well plate (0.2 mL aliquots in each well). Test compounds were dissolved in DMSO then diluted with the culture medium such that the final DMSO concentration was 0.5%. Aliquots (20 μL) of test solution or medium with DMSO (control) were added to each well. The cells were incubated at 37 °C for 30 min. LPS (Sigma, St. Louis, MO, USA) was added to the wells at a final concentration of 0.5 μg/mL, and cells were incubated for an additional 2 h. At the end of the incubation period, culture supernatants were collected and the amount of TNF-α present was determined using an ELISA assay (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. The data were evaluated by nonlinear regression analysis (Origin software, version 7.5; OriginLab Corp., Northampton, MA, USA) using a sigmoidal model, and the IC50 value for each compound was calculated from the sigmoidal curve.
4. Conclusions
A series of 3-(2H-chromen-5-yl)urea and 3-(chroman-5-yl)urea derivatives was designed as potential p38MAPKα inhibitors based on knowledge of the crystallographic structure of the p38MAPKα/BIRB796 complex. To obtain these target compounds, a simple and efficient synthetic method was developed to construct 5-alkoxy-2H-chromen-8-amine and 5-alkoxychroman-8-amine skeletons. The synthesized compounds were evaluated for their inhibitory activity against TNF-α release in LPS-stimulated THP-1 cells. Among our compounds, the 3-(2H-chromen-5-yl)urea derivatives exhibited relatively good anti-TNFα activity, with compound 40g blocking TNF-α release with an IC50 value of 0.033 μM, which is equipotent to that of BIRB796 (IC50 = 0.032 μM). These results indicate that 3-(2H-chromen-5-yl)urea compounds may serve as a novel chemotype for the development of p38MAPKα inhibitors.
Acknowledgments
We gratefully acknowledge financial support from the National Nature Science Foundation of China (Grant No. 81072529).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ashwell, J.D. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat. Rev. Immunol. 2006, 6, 532–540. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. P38 MAPK: Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef]
- Singh, D.; Smyth, L.; Borrill, Z.; Sweeney, L.; Singer, R.T. A randomized, placebo-controlled study of the effects of the p38 MAPK inhibitor SB-681323 on blood biomarkers of inflammation in COPD patients. J. Clin. Pharmacol. 2010, 50, 94–100. [Google Scholar] [CrossRef]
- Erwin, F.W.; Ángel, R.N. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- James, W.A.; Melyssa, R.B.; Lori, M.G. Pharmacology and anti-tumor activity of RWJ67657, a novel inhibitor of p38 mitogen activated protein kinase. Am. J. Cancer Res. 2012, 2, 446–458. [Google Scholar]
- Anand, P.; Shenoy, R.; Palmer, J.E.; Amanda, J.B.; Lai, R.Y.K.; Robertson, J.; Bird, N.; Ostenfeld, T.; Chizh, B.A. Clinical trial of the p38 MAP kinase inhibitor dilmapimod in neuropathic pain following nerve injury. Eur. J. Pain 2011, 15, 1040–1048. [Google Scholar]
- Keith, L.K.; Carlos, R.J. The potential of p38 MAPK inhibitors to modulate periodontal infections. Curr. Drug Metab. 2009, 10, 55–67. [Google Scholar]
- Barun, O.; Advait, N.; Francisco, J.A. A general strategy for creating “inactive-conformation’’ abl inhibitors. Chem. Biol. 2006, 13, 779–786. [Google Scholar] [CrossRef]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar] [CrossRef]
- Toru, A.; Hitoshi, Y.; Chiyoshi, K. Identification, synthesis, and biological evaluation of 6-[(6R)-2-(4-fluorophenyl)-6-(hydroxymethyl)-4,5,6,7-tetrahydropyrazolo [1,5-a]pyrimidin-3-yl]-2-(2-methylphenyl)pyridazin-3(2H)-one (AS1940477), a potent p38 MAP kinase inhibitor. J. Med. Chem. 2012, 55, 7772–7785. [Google Scholar]
- Shunsuke, I.; Yoshiji, A.; Hideo, A. A possible mechanism for hepatotoxicity induced by BIRB-796, an orally active p38 mitogen-activated protein kinase inhibitor. J. Appl. Toxicol. 2011, 31, 671–677. [Google Scholar]
- Justin, D.; Christopher, H.; Laurence, H.H. The design, synthesis, and evaluation of 8 hybrid DFG-out allostertic kinase inhibitors: A structural analysis of the binding interactiona of Gleevec, Nexavar, and BIRB-796. Bioorg. Med. Chem. 2010, 18, 5738–5748. [Google Scholar] [CrossRef]
- Li, X.Z.; Zhou, X.M.; Zheng, Z.B.; Zhong, W.; Xiao, J.H.; Li, S. Short synthesis of 1-(3-tert-butyl-1-phenyl-1H-pyrazol-5-yl)-3-(5-(2-morpholinoethoxy)-2H-chromen-8-yl) urea derivatives. Synth. Commun. 2009, 39, 1–11. [Google Scholar]
- Usha, R.; Kalpattu, K.B. Claisen rearrangement of meta-substituted aryl propargyl ethers in poly(ethylene glycol). Heterocycles 1984, 22, 1351–1357. [Google Scholar] [CrossRef]
- Tetsuto, T.; Yoskiko, Y.; Itô, S. 1,1'-(Azodicarbonyl)dipiperidine-tributylphosphine, a new reagent system for Mitsunobu reaction. Tetrahedron Letts. 1993, 34, 1639–1642. [Google Scholar] [CrossRef]
- Bae, I.H.; Son, J.B.; Han, S.M. Thieno[3,2-d]pyrimidine Derivatives Having Inhibitory Activity for Protein Kinases. WO Patent WO2013/100632, 4 July 2013. [Google Scholar]
- Tetsuo, M.; Edward, M.K. An improved synthesis of the antibiotic. Hexahydrospinamysin. J. Med. Chem. 1972, 15, 339–340. [Google Scholar]
- John, R.; Steffen, B.; Pier, C.; Thomas, G.; Anne, G.G.; Eugene, H.; Bernhard, K.; Jeffrey, M.; Monica, M.; Neil, M.; et al. Pyrazole urea-based inhibitors of p38 MAP kinase: From lead compound to clinical candidate. J. Med. Chem. 2002, 45, 2994–3008. [Google Scholar] [CrossRef]
- Chen, C.; Keith, W.; James, R.M. A convenient one-pot synthesis of 4-amino-3-arylpyrazoles from α-phthaloylaminoacetophenones. Tetrahedron Letts. 1998, 39, 8229–8232. [Google Scholar] [CrossRef]
- Peter, D.H.; Anne, C.; Derek, N.E.; Helen, F.; Paul, W.S.; Steven, L.S.; Neil, T.; Alan, J.W. 4-Acetylamino-3-(imidazol-1-yl)-benzoic acids as novel inhibitors of influenza sialidase. Eur. J. Med. Chem. 1999, 34, 225–234. [Google Scholar] [CrossRef]
- Akira, T.; Akira, M.; Shinzaburo, S.; Shiro, U.; Yasuo, M. Practical synthesis of 3-amino-5-tert-butylisoxazole from pivaloylacetonitrile with hydroxylamine. Heterocycles 1991, 32, 1153–1158. [Google Scholar] [CrossRef]
- Pavel, M.; Ramnarayan, S.R. A safe and efficient method for preparation of N,N'-unsymmetrically disubstituted ureas utilizing triphosgene. J. Org. Chem. 1994, 59, 1937–1938. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).