Docking Studies in Target Proteins Involved in Antibacterial Action Mechanisms: Extending the Knowledge on Standard Antibiotics to Antimicrobial Mushroom Compounds





Abstract
:
1. Introduction
2. Results and Discussion
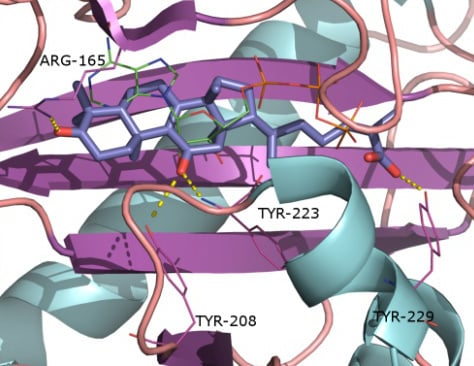
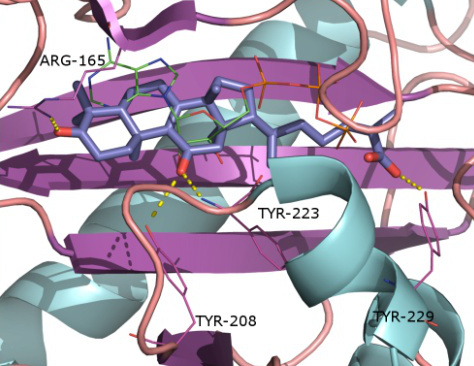
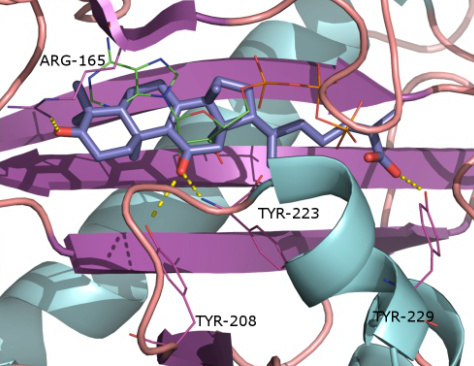
2.1. Protein Targets
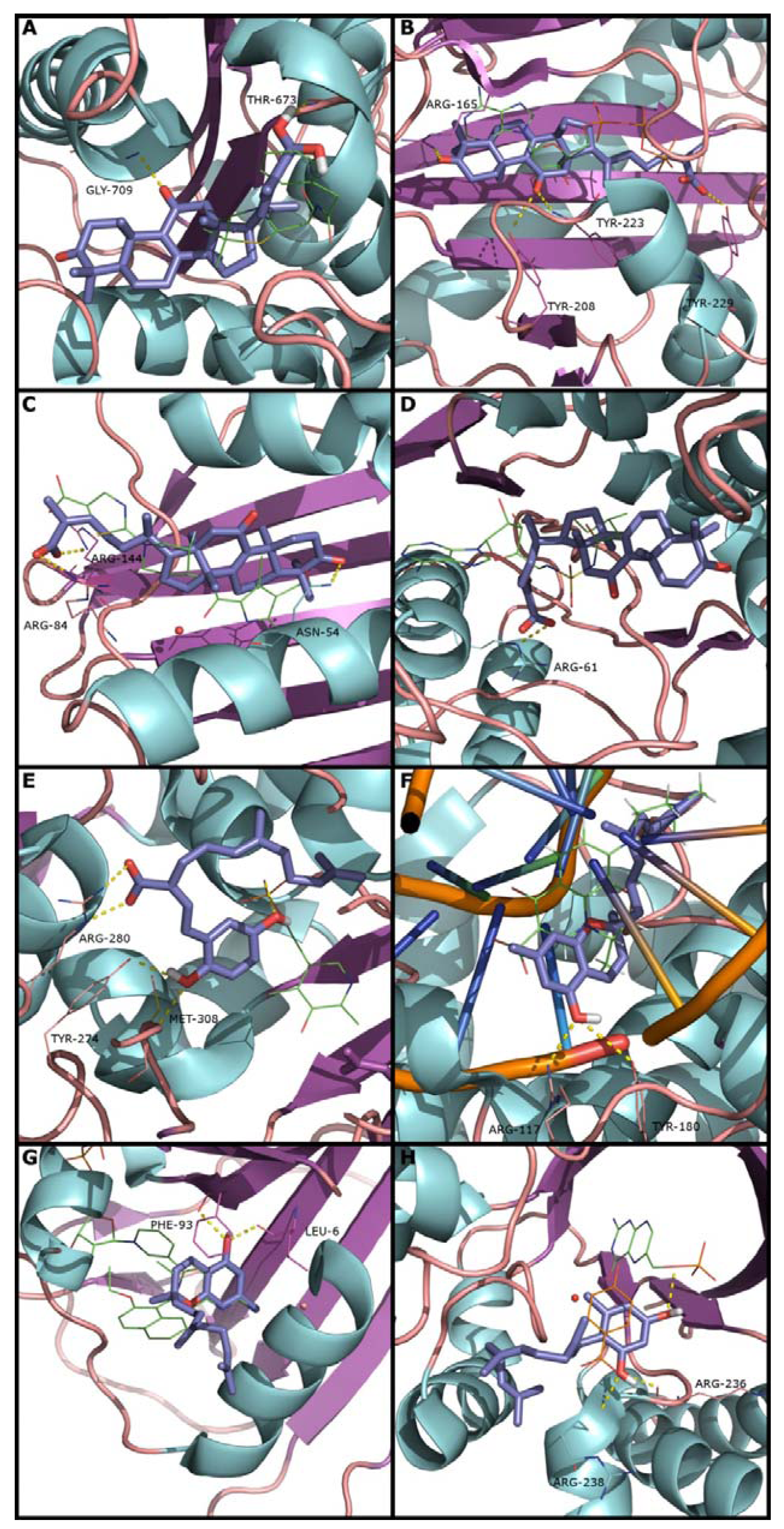
2.2. Docking and Scoring Validation

{kind=link}
{kind=link}
{kind=link}
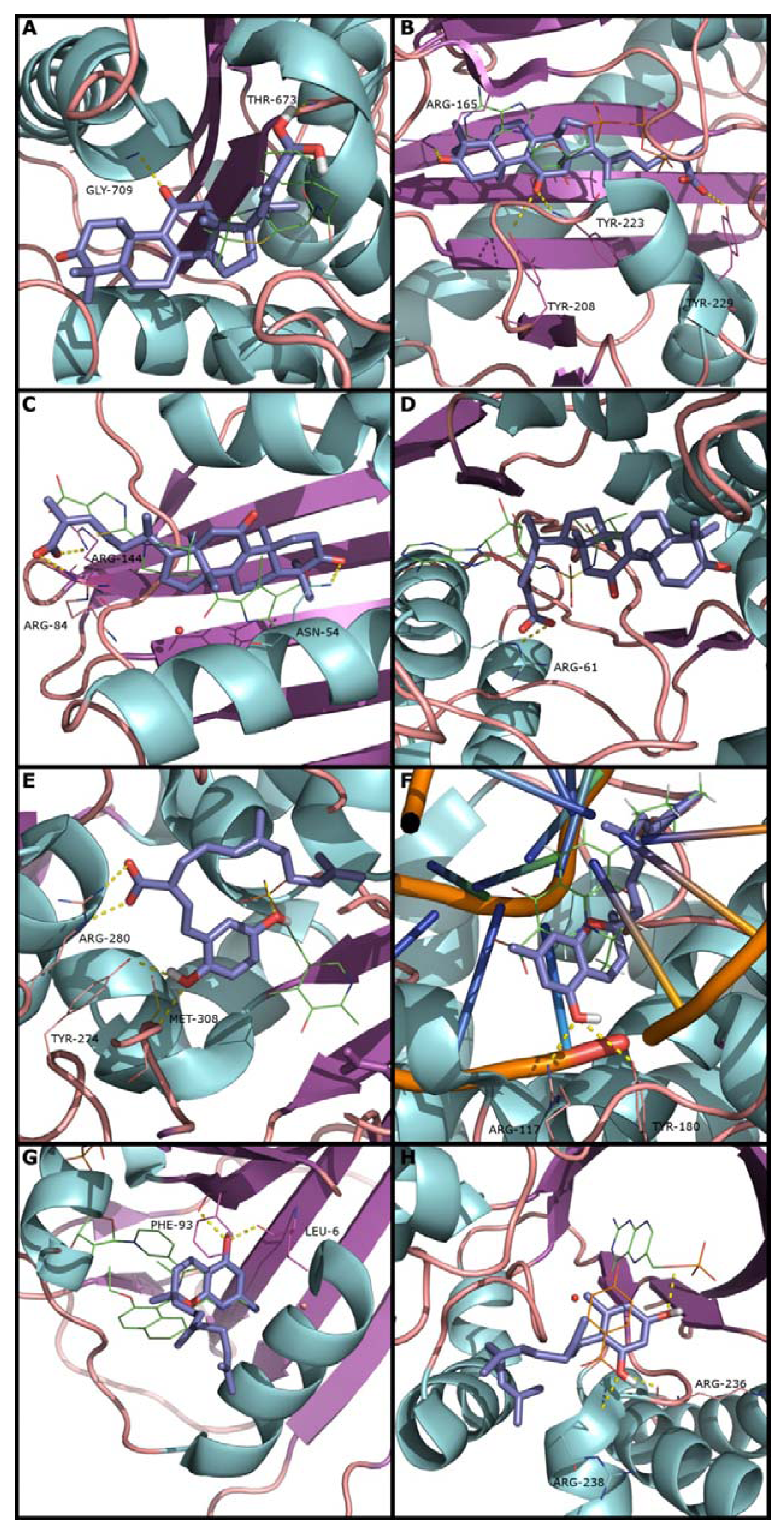{kind=link}
| Protein | Class | Ligand | PDB (ID) | RMSD (Å) | Predicted AutoDock4 Ki (µM) | Predicted Xscore Ki (µM) | Experimental Ki (µM) |
|---|---|---|---|---|---|---|---|
| PBP1a | A | PNM b | 3UDI | 1.33 | 5.49 | 1.862 | - |
| Ddl | A | ATP a | 2ZDQ | 1.49 | 0.006 | 0.478 | - |
| IARS | B | ILA b | 1JZQ | 1.13 | 0.203 | 0.707 | 0.006 |
| DNA Gyrase | C | 07N b | 3TTZ | 0.23 | 0.419 | 0.457 | 0.004 |
| TopoIV | C | LFX b | 3RAE | 1.66 | 0.139 | 0.245 | - |
| DHPS | D | PMM a | 2VEG | 0.57 | 4.382 | 7.413 | 33 |
| DHFR | D | Q2 b | 3SRW | 1.85 | 0.050 | 0.089 | 0.00003 |
| r | 0.90 | 0.99 | style="border-top: solid thin" | ||||
| ρ | 0.80 | 1 |
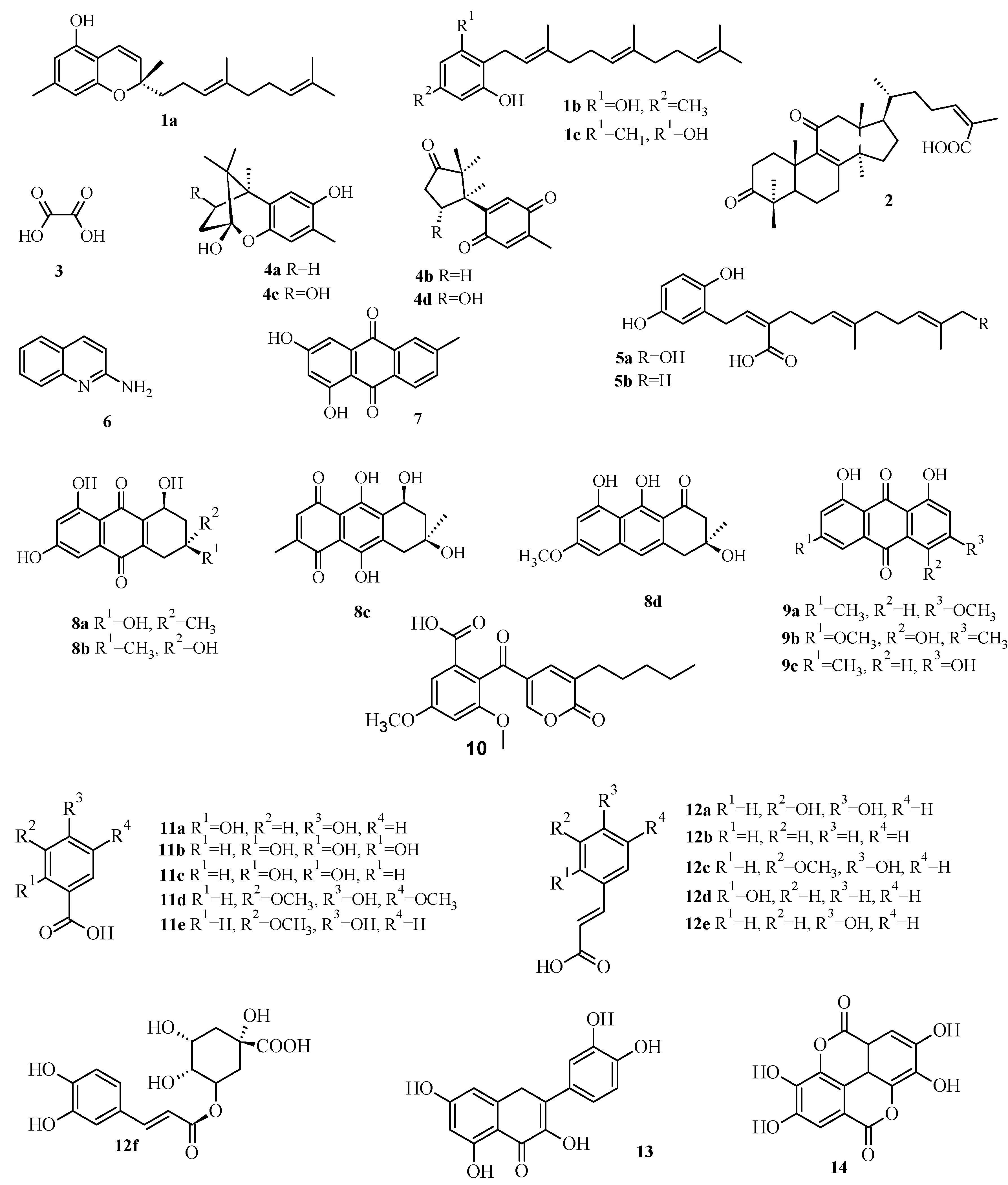
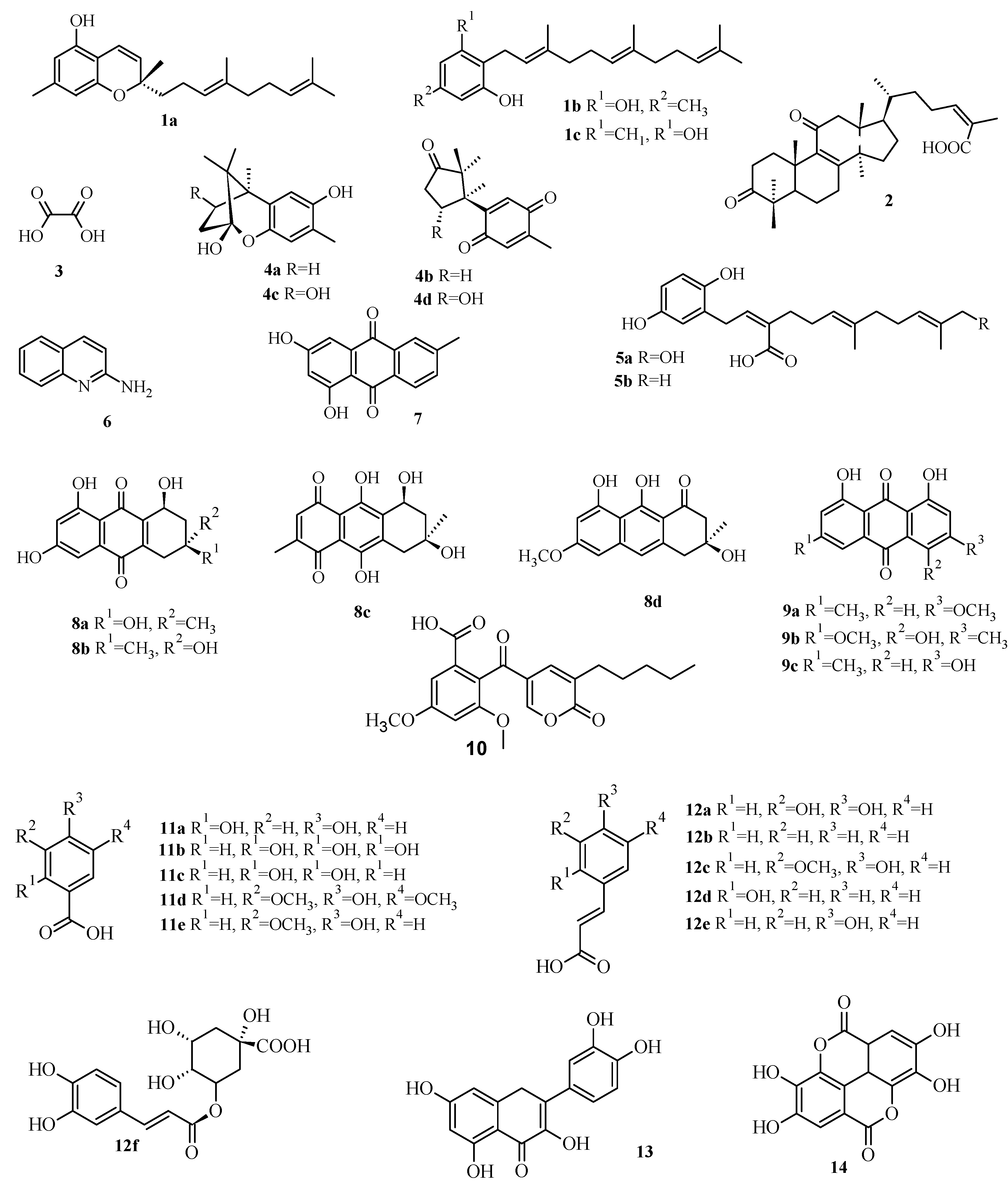
2.3. Virtual Screening of Antimicrobial Mushroom Compounds
| Compound | Code | PBP1a | Alr | Ddl | IARS | DNA Gyrase | TopoIV | DHPS | DHFR |
|---|---|---|---|---|---|---|---|---|---|
| Confluentin | 1a | 10.72 | 0.17 | 1.45 | 162.18 | 8.56 | 0.26 | 6.46 | 0.44 |
| Grifolin | 1b | 1.91 | 1.35 | 0.1 | 251.19 | 28.18 | 0.83 | 4.47 | 1.75 |
| Neogrifolin | 1c | 0.91 | 1.12 | 0.09 | 45.71 | 37.15 | 1.07 | 1.51 | 2.33 |
| 3,11-Dioxolanosta-8,24(Z)-diene-26-oic acid | 2 | 0.07 | 14.45 | 0.01 | 3.16 | 6.31 | 0.23 | 4.27 | 0.15 |
| Oxalic acid | 3 | 112.2 | 134.9 | 87.1 | 354.81 | 89.13 | 77.62 | 158.49 | 124.9 |
| Enokipodins A | 4a | 1.23 | 0.36 | 0.15 | 50.12 | 9.55 | 2.75 | 63.1 | 3.55 |
| Enokipodins B | 4b | 1.62 | 0.51 | 4.57 | 23.44 | 21.38 | 7.76 | 131.8 | 5.62 |
| Enokipodins C | 4c | 1.02 | 0.25 | 0.19 | 60.26 | 10.23 | 6.03 | 70.79 | 4.04 |
| Enokipodins D | 4d | 1.17 | 0.52 | 0.34 | 218.78 | 32.36 | 7.59 | 2.4 | 4.68 |
| Ganomycin A | 5a | 1.78 | 0.49 | 0.33 | 457.09 | 69.18 | 1.62 | 14.45 | 0.86 |
| Ganomycin B | 5b | 1.66 | 0.15 | 0.3 | 40.74 | 48.98 | 0.66 | 5.37 | 0.46 |
| 2-Aminoquinoline | 6 | 12.59 | 5.37 | 2.4 | 4.47 | 54.95 | 25.12 | 25.12 | 5.71 |
| 6-Methylxanthopurpurin-3- O-methyl | 7 | 2.95 | 0.66 | 0.38 | 28.84 | 30.9 | 0.93 | 3.09 | 1.25 |
| Austrocortilutein A | 8a | 3.24 | 0.3 | 0.4 | 12.88 | 14.13 | 11.75 | 2.14 | 5.37 |
| Austrocortilutein B | 8b | 2.51 | 0.3 | 0.35 | 13.18 | 19.5 | 0.54 | 2.14 | 5.5 |
| Austrocortirubin | 8c | 2.88 | 0.4 | 0.32 | 41.69 | 36.31 | 22.39 | 21.38 | 25.51 |
| Torosachrysone | 8d | 2.04 | 0.26 | 0.32 | 58.88 | 16.98 | 10.23 | 2.95 | 4.33 |
| Physcion | 9a | 1.91 | 0.6 | 0.3 | 30.2 | 7.08 | 0.72 | 3.39 | 1.62 |
| Erythroglaucin | 9b | 2.69 | 0.55 | 0.19 | 26.3 | 28.84 | 0.62 | 2.63 | 2.15 |
| Emodin | 9c | 2.57 | 0.41 | 0.32 | 40.74 | 11.75 | 0.72 | 2.57 | 1.12 |
| Coloratin A | 10 | 1.12 | 2 | 0.25 | 61.66 | 16.98 | 0.54 | 1.58 | 0.91 |
| 2,4-Dihydroxybenzoic acid | 11a | 16.22 | 7.94 | 9.55 | 7.76 | 2.34 | 9.55 | 16.98 | 15.37 |
| Gallic acid | 11b | 19.05 | 8.32 | 17.38 | 6.76 | 35.48 | 8.91 | 87.1 | 19.65 |
| Protocatechuic acid | 11c | 18.2 | 6.17 | 15.85 | 8.13 | 33.88 | 10.96 | 17.78 | 17.65 |
| Syringic acid | 11d | 18.2 | 10 | 14.13 | 316.23 | 74.13 | 9.55 | 15.14 | 25.31 |
| Vanillic acid | 11e | 19.05 | 9.77 | 13.8 | 7.94 | 34.67 | 10.47 | 18.62 | 22.91 |
| Caffeic acid | 12a | 7.94 | 3.8 | 5.37 | 114.82 | 46.77 | 5.89 | 195 | 33.11 |
| Cinammic | 12b | 10.23 | 4.27 | 5.01 | 3.63 | 46.77 | 8.71 | 12.02 | 18.48 |
| Ferulic acid | 12c | 12.88 | 4.37 | 5.89 | 269.15 | 20.89 | 6.03 | 9.55 | 36.59 |
| o-Coumaric acid | 12d | 12.3 | 4.17 | 4.07 | 204.17 | 22.91 | 6.46 | 30.2 | 11.66 |
| p-Coumaric acid | 12e | 10.72 | 4.9 | 6.03 | 154.88 | 16.98 | 6.46 | 40.74 | 19.8 |
| Chlorogenic acid | 12f | 1.55 | 38.02 | 0.21 | 316.23 | 23.99 | 7.59 | 42.66 | 3.6 |
| Quercetin | 13 | 1.74 | 0.52 | 0.35 | 97.72 | 19.5 | 0.52 | 13.8 | 2.53 |
| Ellagic acid | 14 | 4.27 | 0.56 | 0.58 | 58.88 | 46.77 | 4.68 | 2.63 | 8.45 |
3. Experimental
3.1. Proteins and Natural Compounds Structure Preparation
3.2. Molecular Docking
3.3. Docking and Score Validation
3.4. Virtual Screening
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tenover, F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Med. 2006, 119, S3–S10. [Google Scholar] [CrossRef]
- Murray, P.R.; Rosenthal, K.S.; Pfaller, M.A. Microbiologia Médica, 5th ed.; Mosby, Elsevier: Rio de Janeiro, Brasil, 2006. [Google Scholar]
- Calvo, J.; Martínez-Martínez, L. Mecanismos de acción de los antimicrobianos. Enferm. Infec. Microbiol. Clin. 2009, 27, 44–52. [Google Scholar] [CrossRef]
- Scholar, E.M.; Pratt, W.B. The inhibitors of cell wall synthesis, I. Mechanism of Action of the Penicillins, Cephalosporins, Vancomycin, and Other Inhibitors of Cell Wall Synthesis. In The Antimicrobial Drugs; Scholar, E.M., Pratt, W.B., Eds.; Oxford University Press: Oxford, UK, 2000; pp. 51–80. [Google Scholar]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Kourelis, T.G.; Mamos, P.; Magoulas, G.E.; Kalpaxis, D.L. Insights into the chloramphenicol inhibition effect on peptidyl transferase. Activity, using two new analogs of the drug. Open Enz. Inhib. J. 2009, 4, 1–10. [Google Scholar]
- Golan, D.E. Princípios de Farmacologia: A Base Fisiopatológica da Farmacoterapia, 2nd ed.; Guanabara Koogan: Rio de Janeiro, Brasil, 2009; pp. 547–561. [Google Scholar]
- Brugueras, M.; García, M.; Díaz, R. Actualidad de las quinolonas. Rev. Cubana Farm. 2005, 39, 1–15. [Google Scholar]
- Huang, L.; Crothers, K.; Atzori, C.; Benfield, T.; Miller, R.; Rabodonirina, M.; Helweg-Larsen, J. Dihydropteroate synthase gene mutations in pneumocystis and sulfa resistance. Emerg. Infect. Dis. 2004, 10, 1721–1728. [Google Scholar] [CrossRef]
- Radulović, N.S.; Blagojević, P.D.; Stojanović-Radić, Z.Z.; Stojanović, N.M. Antimicrobial plant metabolites: Structural diversity and mechanism of action. Curr. Med. Chem. 2013, 20, 932–952. [Google Scholar]
- Alves, M.J.; Ferreira, I.C.F.R.; Dias, J.; Teixeira, V.; Martins, A; Pintado, M. A review on antimicrobial activity of mushroom (Basidiomycetes) extracts and isolated compounds. Planta Med. 2012, 78, 1707–1718. [Google Scholar] [CrossRef]
- Alves, M.J.; Ferreira, I.C.F.R.; Froufe, H.C.; Abreu, R.M.V.; Martins, A.; Pintado, M. Antimicrobial activity of phenolic compounds identified in wild mushrooms, SAR analysis and docking studies. J. Appl. Microbiol. 2013, 115, 346–357. [Google Scholar] [CrossRef]
- Yun, M.-K.; Wu, Y.; Li, Z.; Zhao, Y.; Waddell, M.B.; Ferreira, A.M.; Lee, R.E.; Bashford, D.; White, S.W. Catalysis and sulfa drug resistance in dihydropteroate synthase. Science 2012, 335, 1110–1114. [Google Scholar] [CrossRef]
- Berglez, J.; Pilling, P.; Macreadie, I.; Fernley, R.T. Purification, properties, and crystallization of Saccharomyces cerevisiae dihydropterin pyrophosphokinase-dihydropteroate synthase. Prot. Express Purif. 2005, 41, 355–362. [Google Scholar] [CrossRef]
- Han, S.; Caspers, N.; Zaniewski, R.P.; Lacey, B.M.; Tomaras, A.P.; Feng, X.; Geoghegan, K.F.; Shanmugasundaram, V. Distinctive attributes of β-lactam target proteins in Acinetobacter baumannii relevant to development of new antibiotics. J. Am. Chem. Soc. 2011, 133, 20536–20545. [Google Scholar] [CrossRef]
- Wu, D.; Hu, T.; Zhang, L.; Chen, J.; Du, J.; Ding, J.; Jiang, H.; Shen, X. Residues Asp164 and Glu165 at the substrate entryway function potently in substrate orientation of alanine racemase from E. coli: Enzymatic characterization with crystal structure analysis. Protein Sci. 2008, 17, 1066–1076. [Google Scholar] [CrossRef]
- Nakama, T.; Nureki, O.; Yokoyama, S. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. J. Biol. Chem. 2001, 276, 47387–47393. [Google Scholar] [CrossRef]
- Sherer, B.A.; Hull, K.; Green, O.; Basarab, G.; Hauck, S.; Hill, P.; Loch, J.T., 3rd; Mullen, G.; Bist, S.; Bryant, J.; et al. Pyrrolamide DNA gyrase inhibitors: optimization of antibacterial activity and efficacy. Bioorg. Med. Chem. Lett. 2011, 21, 7416–7420. [Google Scholar] [CrossRef]
- Levy, C.; Minnis, D.; Derrick, J.P. Dihydropteroate synthase from Streptococcus pneumoniae: structure, ligand recognition and mechanism of sulfonamide resistance. Biochem. J. 2008, 412, 379–388. [Google Scholar]
- Li, X.; Hilgers, M.; Cunningham, M.; Chen, Z.; Trzoss, M.; Zhang, J.; Kohnen, L.; Lam, T.; Creighton, C.G.C.K.; Nelson, K.; et al. Structure-based design of new DHFR-based antibacterial agents: 7-aryl-2,4-diaminoquinazolines. Bioorg. Med. Chem. Lett. 2011, 21, 5171–5176. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–33. [Google Scholar] [CrossRef]
- Abreu, R.M.V.; Froufe, H.J.C.; Queiroz, M.J.R.P.; Ferreira, I.C.F.R. MOLA: A bootable, self-configuring system for virtual screening using AutoDock4/Vina on computer clusters. J. Cheminform. 2010, 2, 1–10. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System. Version 1.3, Schrödinger, LLC.
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alves, M.J.; Froufe, H.J.C.; Costa, A.F.T.; Santos, A.F.; Oliveira, L.G.; Osório, S.R.M.; Abreu, R.M.V.; Pintado, M.; Ferreira, I.C.F.R. Docking Studies in Target Proteins Involved in Antibacterial Action Mechanisms: Extending the Knowledge on Standard Antibiotics to Antimicrobial Mushroom Compounds. Molecules 2014, 19, 1672-1684. https://doi.org/10.3390/molecules19021672
Alves MJ, Froufe HJC, Costa AFT, Santos AF, Oliveira LG, Osório SRM, Abreu RMV, Pintado M, Ferreira ICFR. Docking Studies in Target Proteins Involved in Antibacterial Action Mechanisms: Extending the Knowledge on Standard Antibiotics to Antimicrobial Mushroom Compounds. Molecules. 2014; 19(2):1672-1684. https://doi.org/10.3390/molecules19021672
Chicago/Turabian StyleAlves, Maria José, Hugo J. C. Froufe, Ana F. T. Costa, Anabela F. Santos, Liliana G. Oliveira, Sara R. M. Osório, Rui M. V. Abreu, Manuela Pintado, and Isabel C. F. R. Ferreira. 2014. "Docking Studies in Target Proteins Involved in Antibacterial Action Mechanisms: Extending the Knowledge on Standard Antibiotics to Antimicrobial Mushroom Compounds" Molecules 19, no. 2: 1672-1684. https://doi.org/10.3390/molecules19021672
APA StyleAlves, M. J., Froufe, H. J. C., Costa, A. F. T., Santos, A. F., Oliveira, L. G., Osório, S. R. M., Abreu, R. M. V., Pintado, M., & Ferreira, I. C. F. R. (2014). Docking Studies in Target Proteins Involved in Antibacterial Action Mechanisms: Extending the Knowledge on Standard Antibiotics to Antimicrobial Mushroom Compounds. Molecules, 19(2), 1672-1684. https://doi.org/10.3390/molecules19021672