Biopharmaceutical Profiling of New Antitumor Pyrazole Derivatives


Abstract
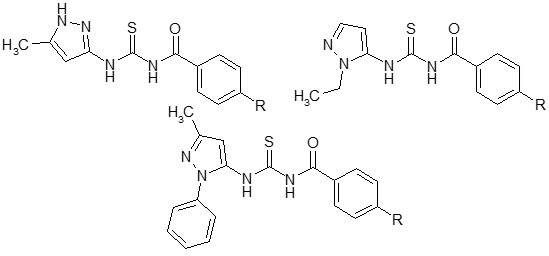
:
1. Introduction

2. Results and Discussion
2.1. Preliminary Screening
2.1.1. Molecular Descriptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | MW | pKa(a) | nHDon | nHAcc | nBM | RBN | nAT | TPSA | MlogP | AlogP | AMR | Ui | Hy | Mv | ARR | Mp |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BC2 | 294.8 | 9.85 | 3 | 4 | 13 | 2 | 30 | 101.9 | 2.59 | 3.09 | 79.2 | 3.81 | 1.25 | 0.69 | 0.55 | 0.73 |
| BC3 | 308.8 | 8.5 | 2 | 4 | 13 | 3 | 33 | 91.0 | 2.45 | 3.08 | 82.9 | 3.81 | 0.46 | 0.67 | 0.52 | 0.72 |
| BC5 | 370.9 | 8.84 | 2 | 4 | 19 | 3 | 40 | 91.0 | 3.7 | 4.59 | 102.6 | 4.32 | 0.31 | 0.7 | 0.63 | 0.74 |
| BM2 | 274.4 | 8.91 | 2 | 4 | 13 | 3 | 33 | 91.0 | 1.91 | 2.42 | 78.1 | 3.81 | 0.45 | 0.65 | 0.55 | 0.69 |
| BM3 | 288.4 | 8.05 | 2 | 4 | 13 | 3 | 36 | 91.0 | 2.18 | 2.9 | 83.2 | 3.81 | 0.41 | 0.64 | 0.52 | 0.69 |
| BM5 | 350.5 | 8.25 | 2 | 4 | 19 | 3 | 43 | 91.0 | 3.44 | 4.41 | 102.8 | 4.32 | 0.27 | 0.67 | 0.63 | 0.71 |
| BT2 | 260.4 | 8.8 | 3 | 4 | 13 | 2 | 30 | 101.9 | 2.05 | 2.42 | 74.4 | 3.81 | 1.25 | 0.67 | 0.58 | 0.71 |
| BT3 | 274.4 | 7.91 | 3 | 4 | 13 | 2 | 33 | 101.9 | 2.32 | 2.91 | 79.4 | 3.81 | 1.2 | 0.65 | 0.55 | 0.69 |
| BT5 | 336.5 | 8.1 | 2 | 4 | 19 | 3 | 40 | 91.0 | 3.20 | 3.93 | 97.8 | 4.32 | 0.3 | 0.68 | 0.65 | 0.72 |
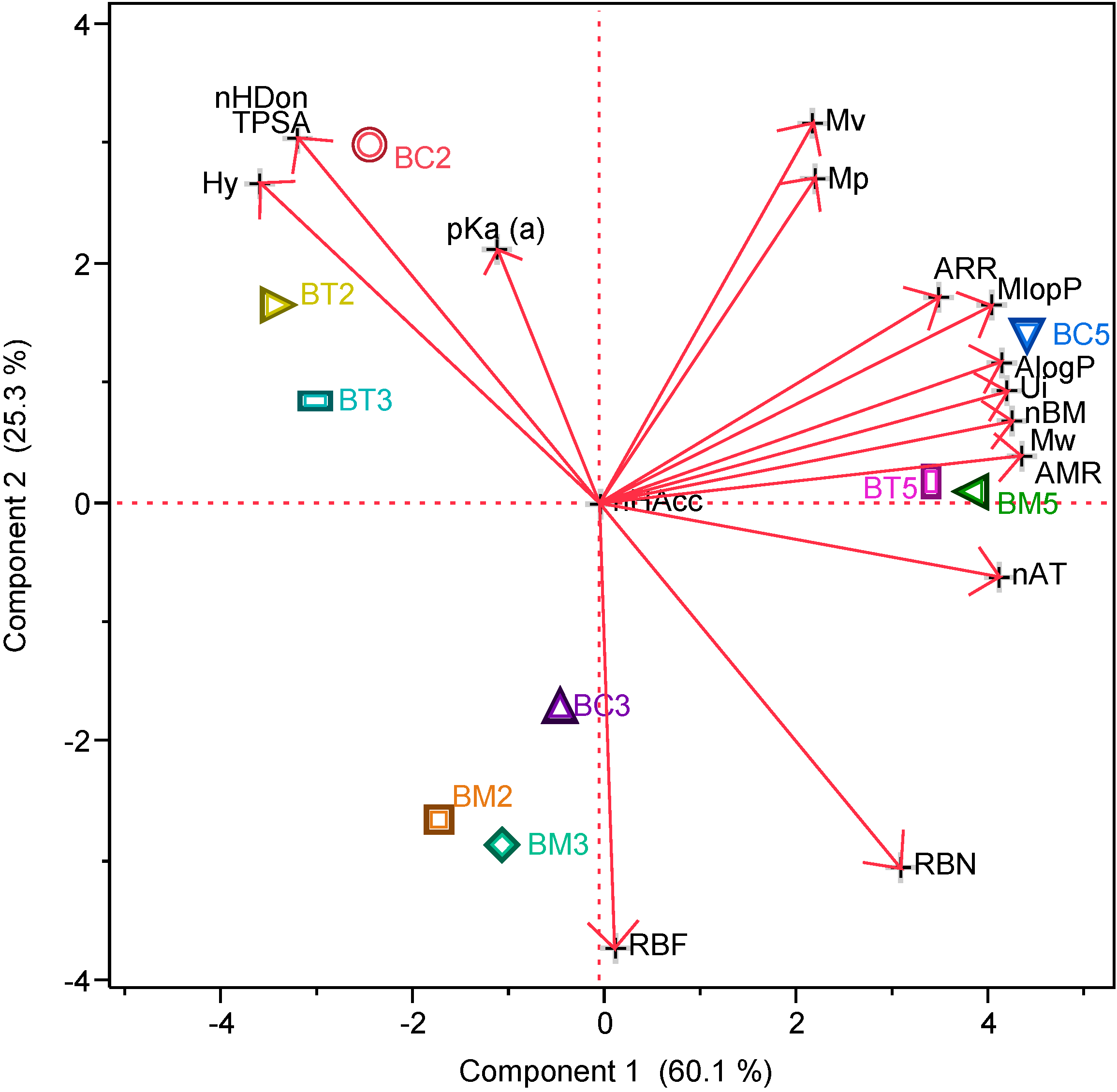
2.1.2. Drug-Like Indices
| Compound | LAI | GVWI * | Inflammat | Depressant | Psychotic | Hpertens | Hypnotic | Neoplasic | Infective |
|---|---|---|---|---|---|---|---|---|---|
| BC2 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 |
| BC3 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 |
| BC5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BM2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| BM3 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 1 |
| BM5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| BT2 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
| BT3 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 |
| BT5 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
2.2. Quantitative HPLC Analysis
| Compound | Slope ± SD | Intercept ± SD | R2 |
|---|---|---|---|
| BC2 | 45090 ± 602.1 | 7630 ± 2620 | 0.9991 |
| BC3 | 42700 ± 559.5 | −930.1 ± 2435 | 0.9991 |
| BC5 | 46360 ± 191.8 | 2016 ± 834.7 | 0.9999 |
| BM2 | 52610 ± 350.9 | 915.1 ± 1527 | 0.9998 |
| BM3 | 49990 ± 435.1 | 1287 ± 1894 | 0.9996 |
| BM5 | 48000 ± 676.5 | −2474 ± 2944 | 0.9990 |
| BT2 | 38650 ± 526.9 | 5702 ± 2293 | 0.9991 |
| BT3 | 36090 ± 243.6 | 5524 ± 1051 | 0.9998 |
| BT5 | 45620 ± 381.8 | 4301 ± 1647 | 0.9996 |

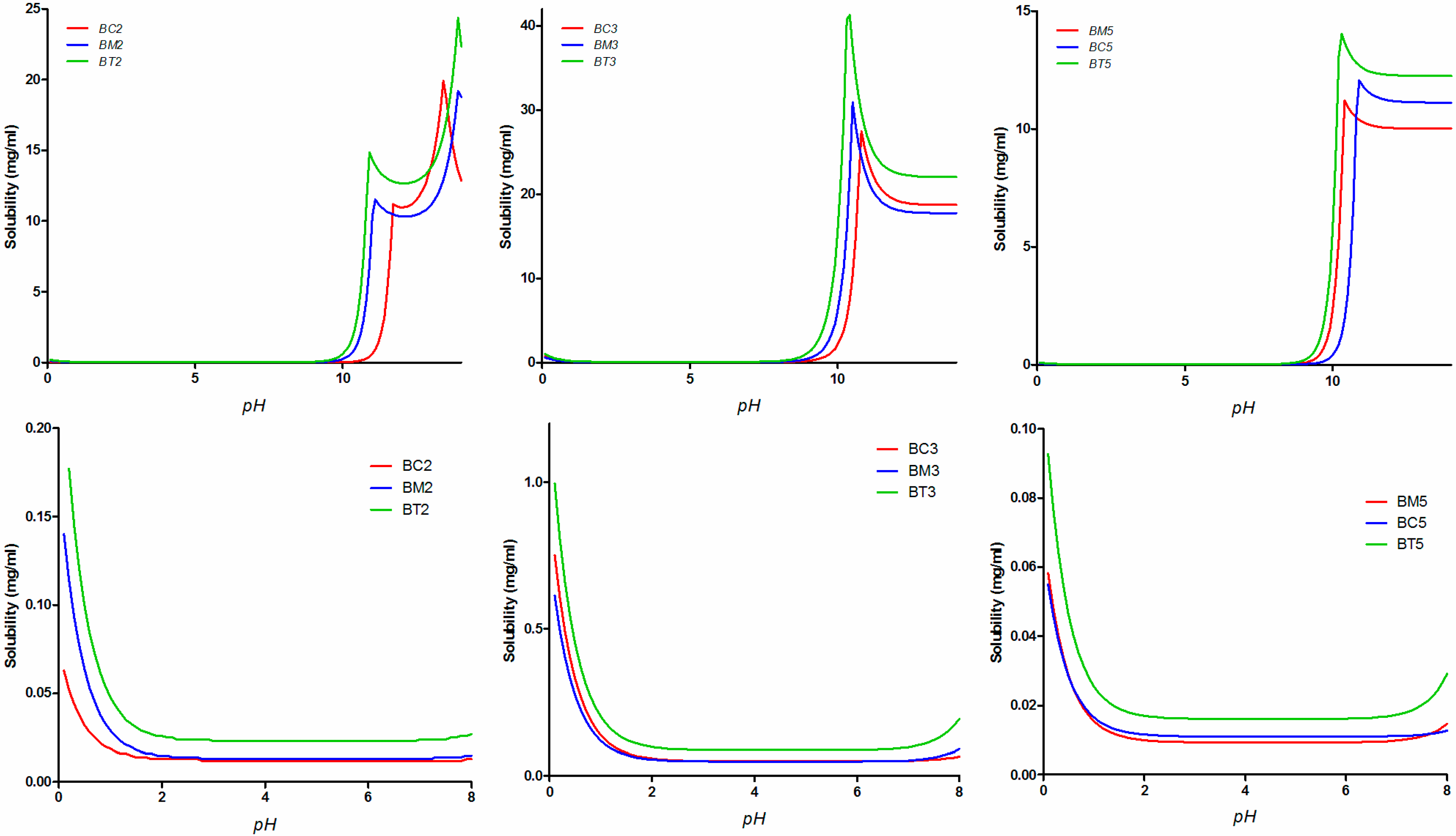
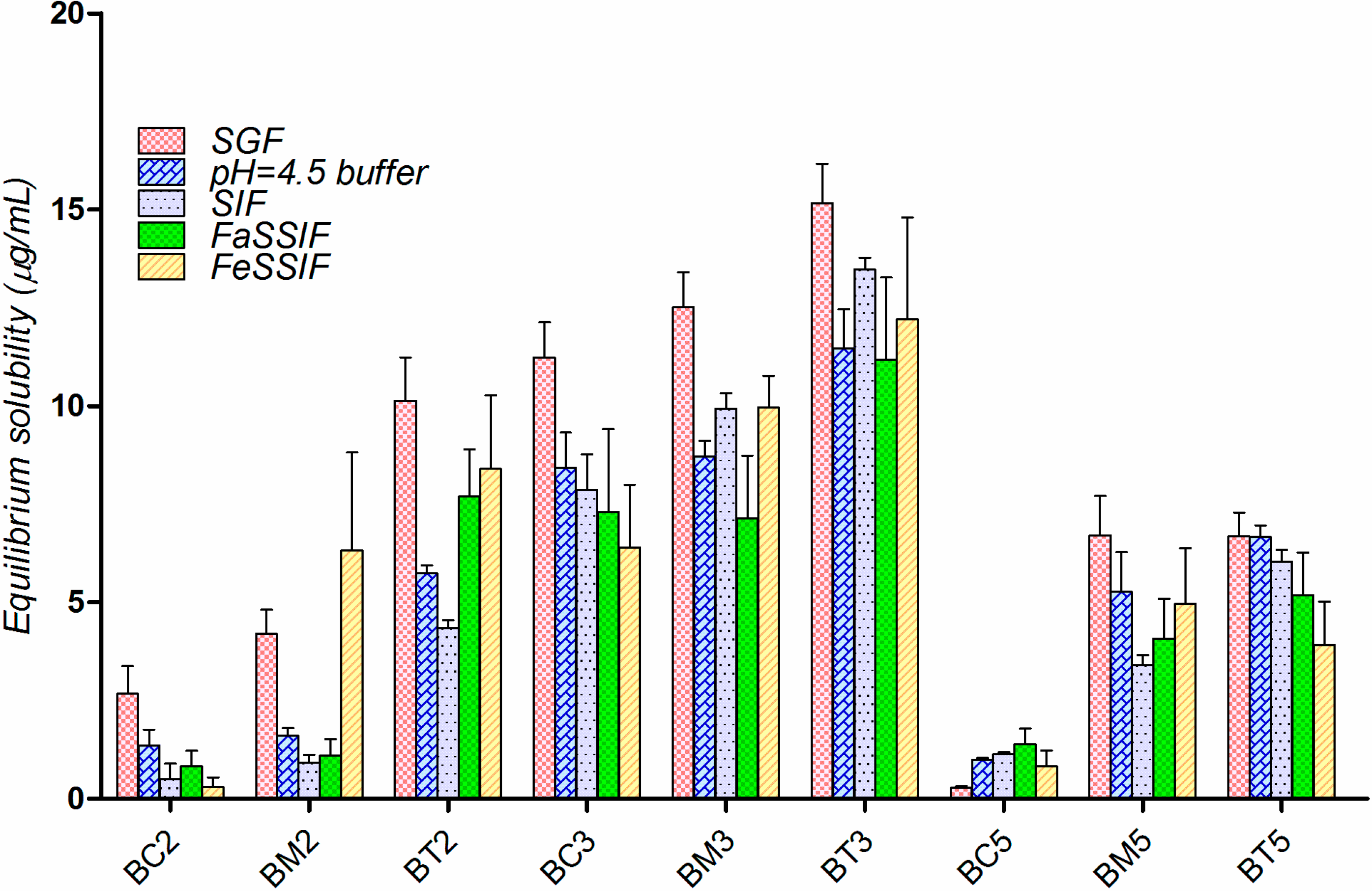
2.3. Equilibrium Solubility Study



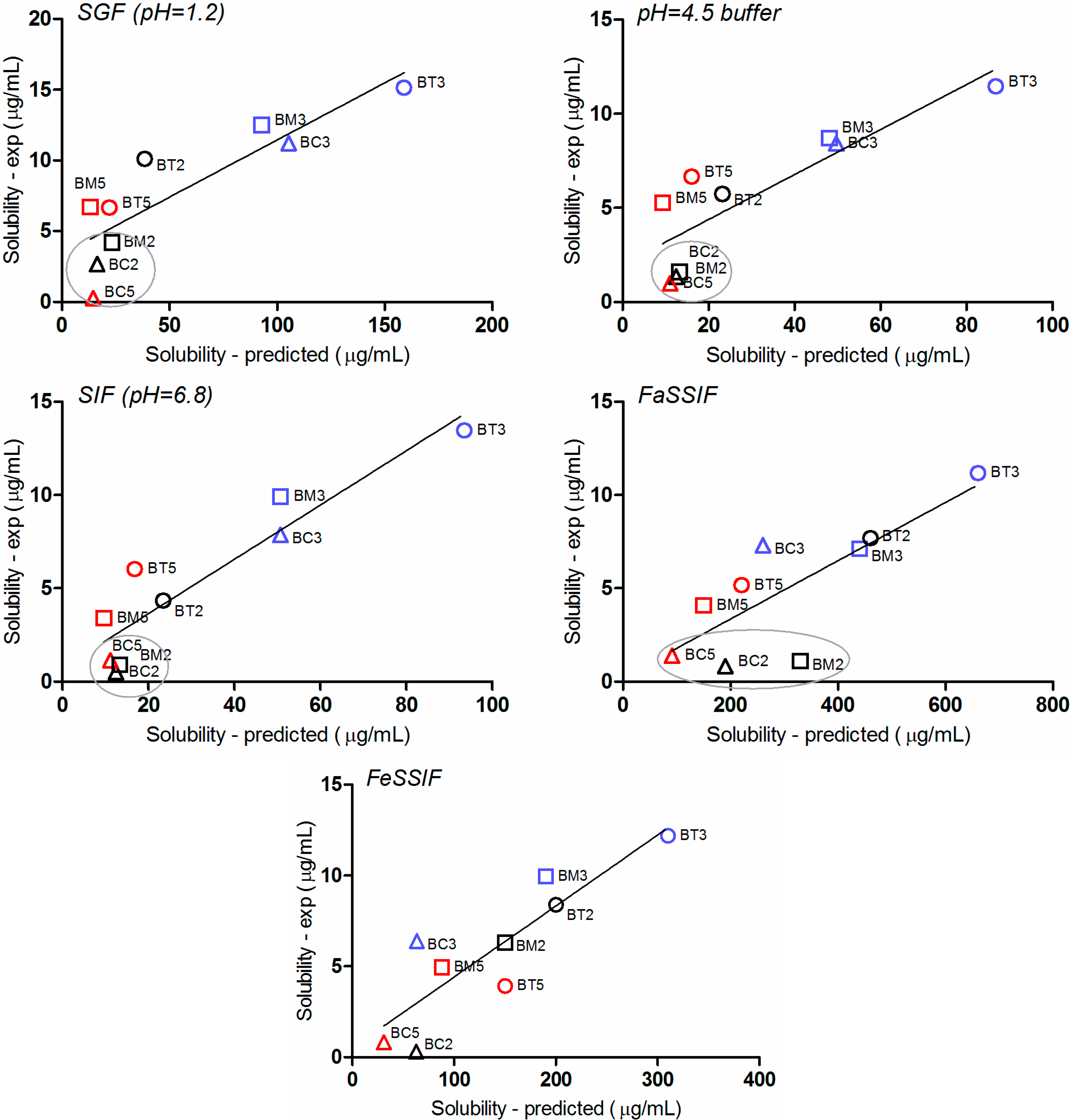
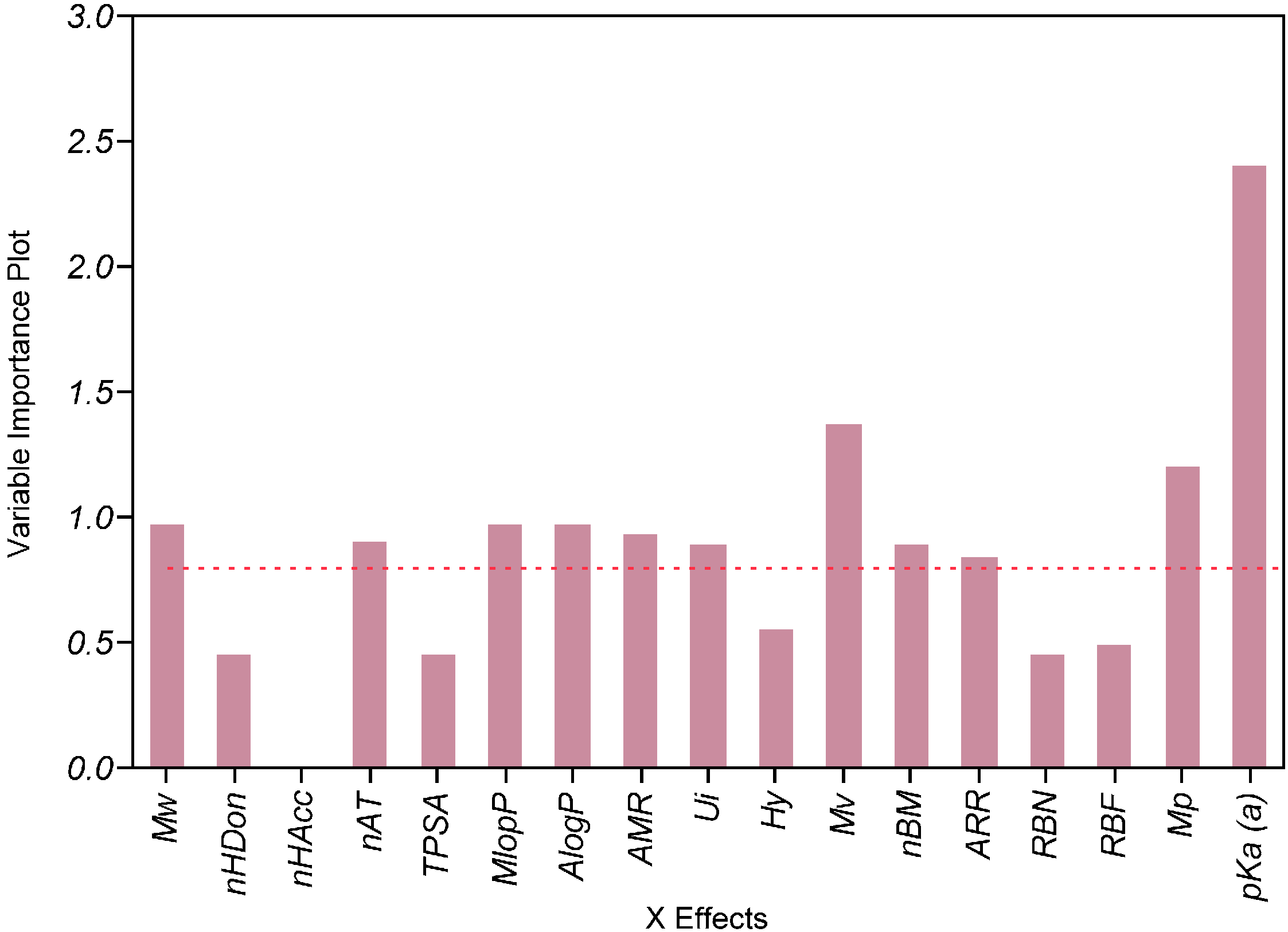
2.4. Impact of the Molecular Descriptors on Solubility in Different Media
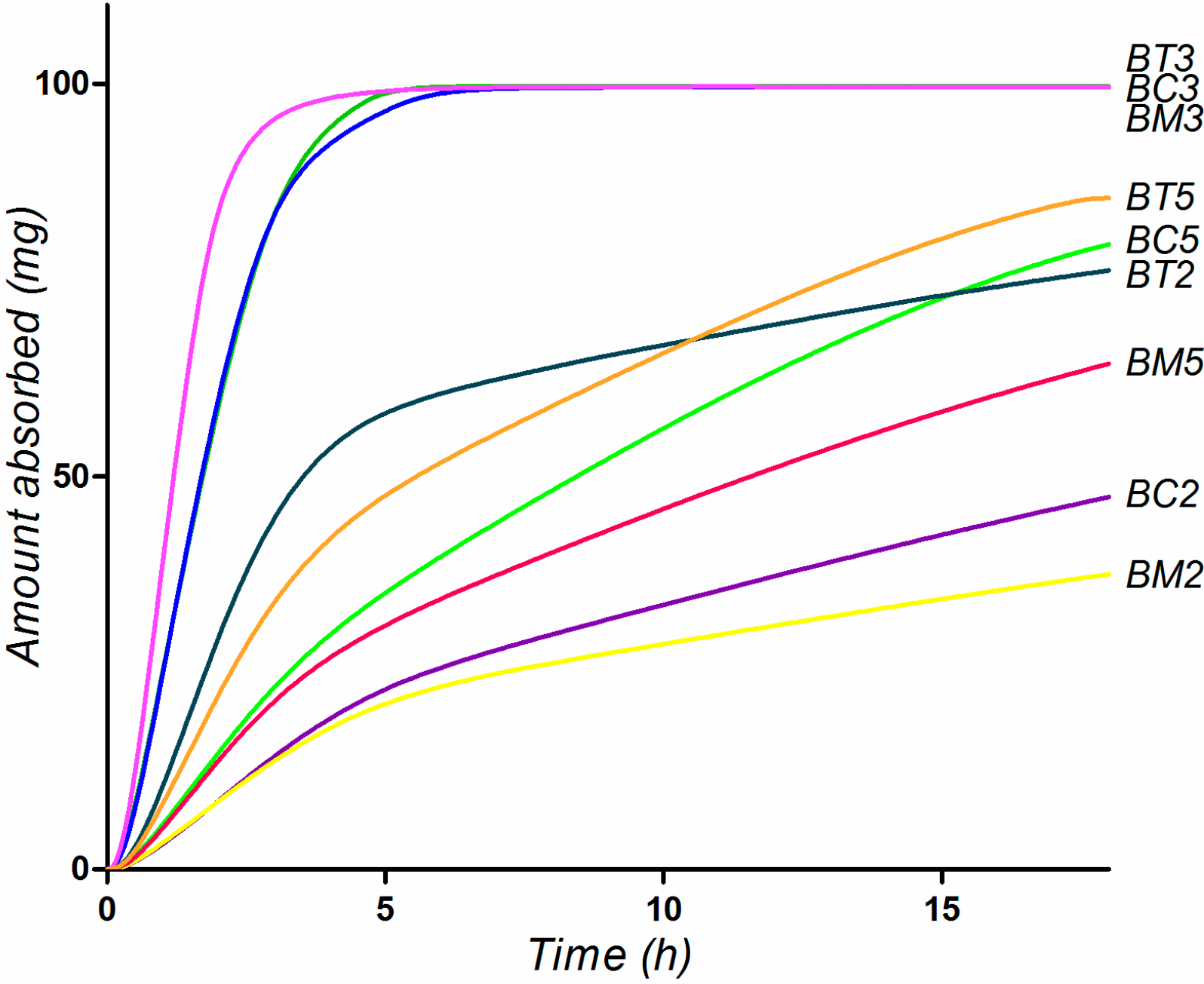
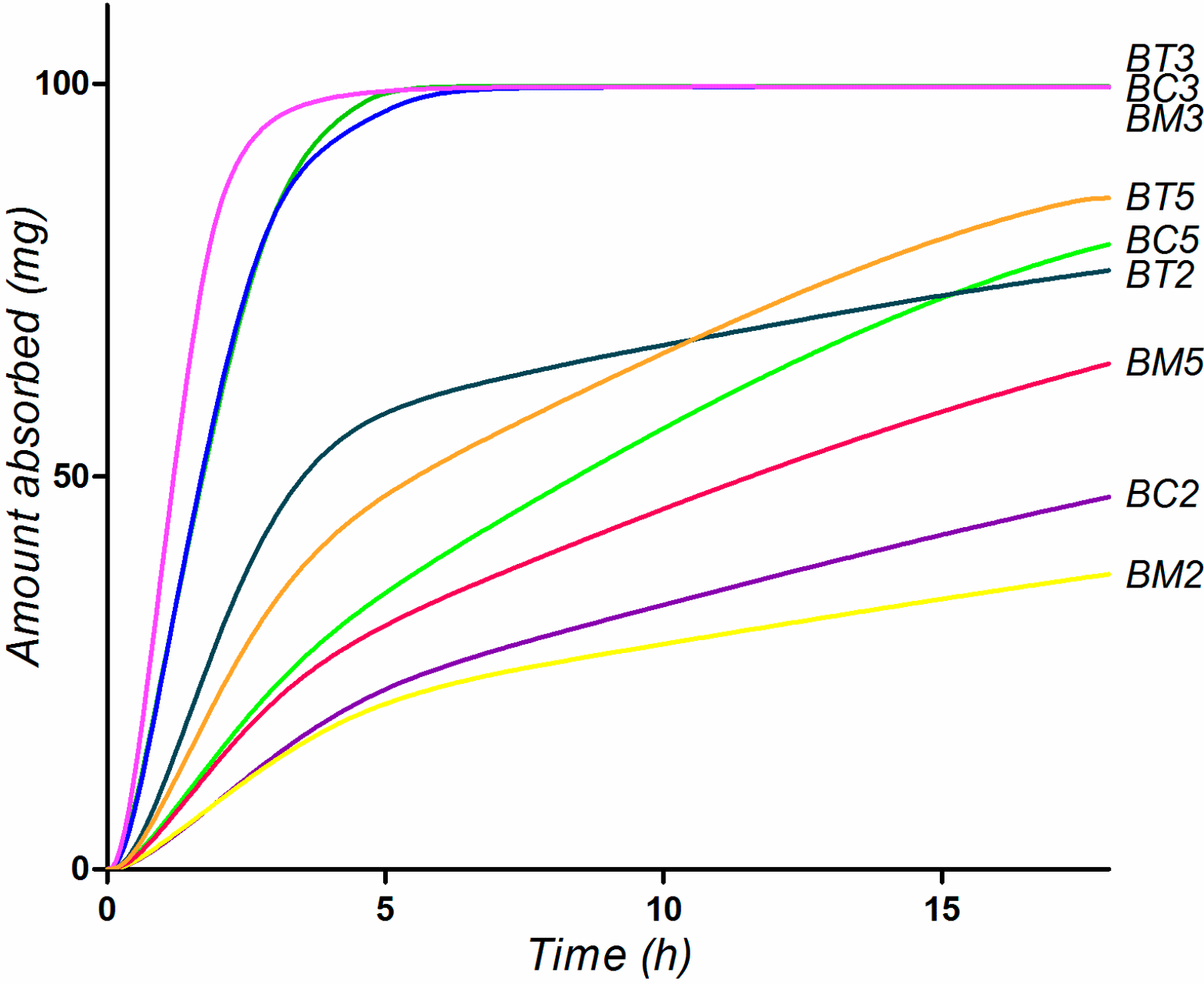
2.5. Biopharmaceutical in Silico Profiling
| Compound | logD | Ref pH a | Aq Sol b (µg/mL) | Diff Coef (cm2/s*105) | Peff (cm/s*104) | fp (%) | RBP | MAD (mg) | %Fa c |
|---|---|---|---|---|---|---|---|---|---|
| BC2 | 2.59 | 6.9 | 12.4 | 0.81 | 2.03 | 19.9 | 0.82 | 112.2 | 55.4 |
| BC3 | 2.64 | 6.14 | 49.9 | 0.78 | 3.29 | 16.03 | 0.78 | 744.9 | 99.6 |
| BC5 | 3.54 | 6.64 | 11.1 | 0.69 | 3.99 | 6.68 | 0.71 | 244.7 | 79.9 |
| BM2 | 2.22 | 6.58 | 13.3 | 0.8 | 1.79 | 24.68 | 0.88 | 101.6 | 42.9 |
| BM3 | 2.29 | 5.92 | 48.4 | 0.77 | 2.88 | 17.73 | 0.78 | 618.6 | 99.5 |
| BM5 | 3.19 | 6.4 | 9.4 | 0.69 | 3.47 | 7.3 | 0.75 | 165.6 | 72.4 |
| BT2 | 1.88 | 6.41 | 23.3 | 0.84 | 1.64 | 25.48 | 0.94 | 158.7 | 81.2 |
| BT3 | 1.89 | 5.71 | 87.3 | 0.8 | 2.56 | 17.05 | 0.82 | 972.8 | 99.6 |
| BT5 | 2.81 | 6.2 | 16.2 | 0.71 | 3.03 | 6.9 | 0.78 | 231.9 | 85.5 |

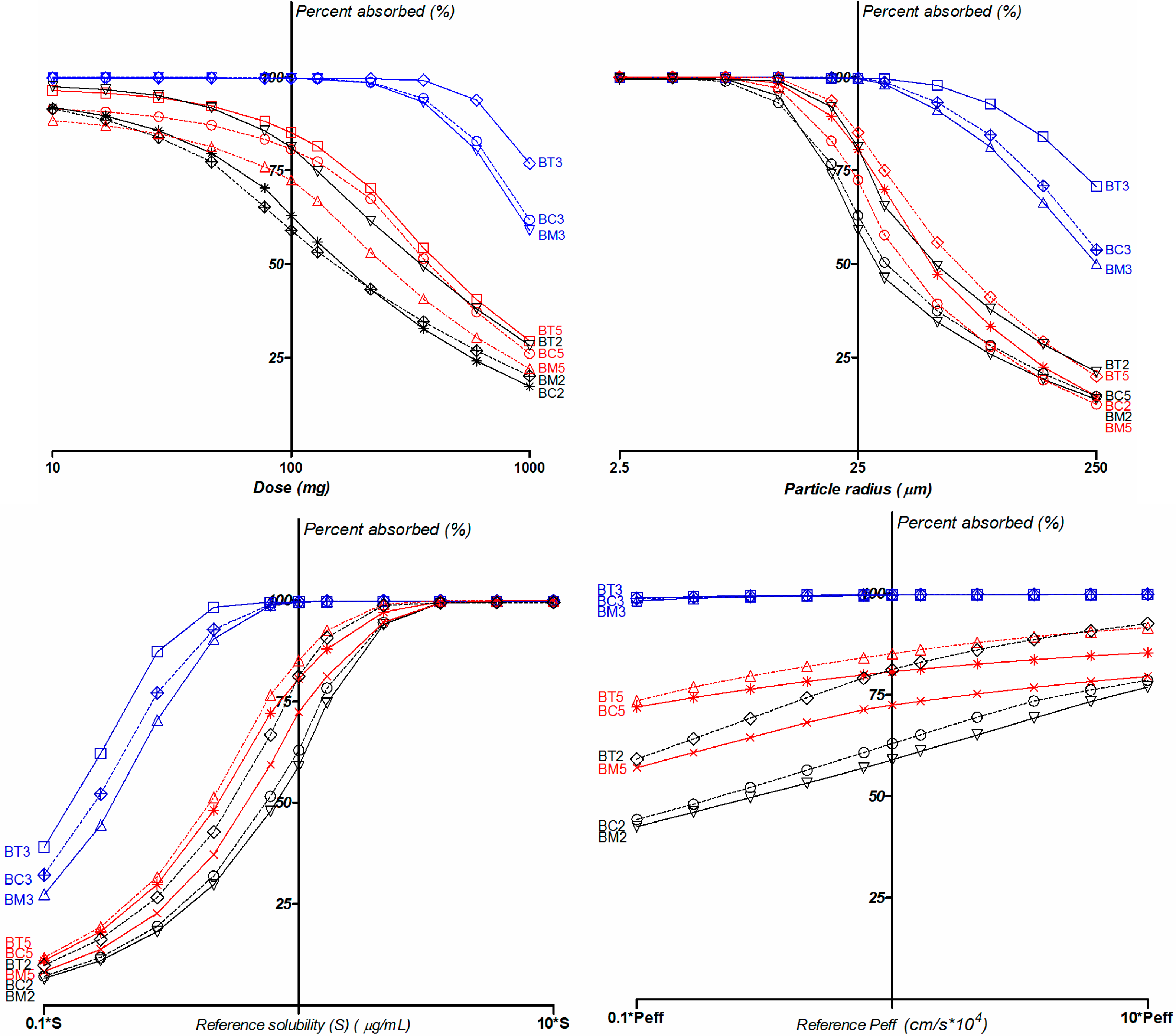
3. Experimental Section
3.1. Chemicals
3.2. Quantitative HPLC Analysis
3.3. Evaluation of Experimental Thermodynamic Solubility
3.4. In Silico Tools
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saeed, A.; Flörke, U.; Erben, M.F. A review on the chemistry, coordination, structure and biological properties of 1-(acyl/aroyl)-3-(substituted) thioureas. J. Sulfur Chem. 2014, 35, 318–355. [Google Scholar]
- Duan, L.P.; Xue, J.; Xu, L.L.; Zhang, H.B. Synthesis 1-acyl-3-(2'-aminophenyl) thioureas as anti-intestinal nematode prodrugs. Molecules 2010, 15, 6941–6947. [Google Scholar]
- Ibrahim, M.A.; Yusof, M.S.; Amin, N.M. Anti-amoebic properties of carbonyl thiourea derivatives. Molecules 2014, 19, 5191–5204. [Google Scholar]
- Saeed, A.; Shaheen, U.; Hameed, A.; Haider Naqui, S.Z. Synthesis, characterization and antimicrobial activity of some new 1-(fluorobenzoyl)-3-(fluorophenyl)thioureas. J. Fluor. Chem. 2009, 130, 1028–1034. [Google Scholar]
- Dobrikov, G.M.; Valcheva, V.; Nikolova, Y.; Ugrinova, I.; Pasheva, E.; Dimitrov, V. Efficient synthesis of new (R)-2-amino-1-butanol derived ureas, thioureas and acylthioureas and in vitro evaluation of their antimycobacterial activity. Eur. J. Med. Chem. 2013, 63, 468–473. [Google Scholar]
- Limban, C.; Missir, A.V.; Chirita, I.C.; Nitulescu, G.M.; Ilie, C.; Caproiu, M.T. The synthesis and characterization of some new thioureides of 2-(4-methyl-phenoxymethyl)benzoic acid with antimicrobial activity. Rev. Chim.-Buchar. 2008, 59, 1245–1248. [Google Scholar]
- Nitulescu, G.M.; Draghici, C.; Chifiriuc, M.C.; Marutescu, L.; Bleotu, C.; Missir, A.V. Synthesis and antimicrobial screening of N-(1-methyl-1H-pyrazole-4-carbonyl)-thiourea derivatives. Med. Chem. Res. 2012, 21, 308–314. [Google Scholar]
- Nitulescu, G.M.; Draghici, C.; Chifiriuc, M.C.; Missir, A.V. Synthesis of isomeric N-(1-methyl-1H-pyrazole-4-carbonyl)-N'-(xylyl)-thiourea and their antimicrobial evaluation. Farmacia 2014, 57, 527–533. [Google Scholar]
- Wu, J.; Shi, Q.; Chen, Z.; He, M.; Jin, L.; Hu, D. Synthesis and bioactivity of pyrazole acyl thiourea derivatives. Molecules 2012, 17, 5139–5150. [Google Scholar]
- Koca, I.; Ozgur, A.; Coskun, K.A.; Tutar, Y. Synthesis and anticancer activity of acyl thioureas bearing pyrazole moiety. Bioorg. Med. Chem. 2013, 21, 3859–3865. [Google Scholar]
- Sun, J.; Lv, X.H.; Qiu, H.Y.; Wang, Y.T.; Du, Q.R.; Li, D.D.; Yang, Y.H.; Zhu, H.L. Synthesis, biological evaluation and molecular docking studies of pyrazole derivatives coupling with a thiourea moiety as novel CDKs inhibitors. Eur. J. Med. Chem. 2013, 68, 1–9. [Google Scholar]
- Nitulescu, G.M.; Draghici, C.; Missir, A.V. Synthesis of new pyrazole derivatives and their anticancer evaluation. Eur. J. Med. Chem. 2010, 45, 4914–4919. [Google Scholar]
- Nitulescu, G.M.; Draghici, C.; Olaru, O.T. New potential antitumor pyrazole derivatives: Synthesis and cytotoxic evaluation. Int. J. Mol. Sci. 2013, 14, 21805–21818. [Google Scholar]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar]
- Hann, M.M.; Keseru, G.M. Finding the sweet spot: The role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discov. 2012, 11, 355–365. [Google Scholar]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar]
- Mircioiu, C.; Mircioiu, I.; Voicu, V.; Miron, D. Dissolution—Bioequivalence non-correlations. Basic Clin. Pharmacol. 2005, 96, 262–264. [Google Scholar]
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution testing as a prognostic tool for oral drug absorption: Immediate release dosage forms. Pharm. Res. 1998, 15, 11–22. [Google Scholar]
- Galia, E.; Nicolaides, E.; Horter, D.; Lobenberg, R.; Reppas, C.; Dressman, J.B. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998, 15, 698–705. [Google Scholar]
- Vertzoni, M.; Dressman, J.; Butler, J.; Hempenstall, J.; Reppas, C. Simulation of fasting gastric conditions and its importance for the in vivo dissolution of lipophilic compounds. Eur. J. Pharm. Biopharm. 2005, 60, 413–417. [Google Scholar]
- Vertzoni, M.; Diakidou, A.; Chatzilias, M.; Soderlind, E.; Abrahamsson, B.; Dressman, J.B.; Reppas, C. Biorelevant media to simulate fluids in the ascending colon of humans and their usefulness in predicting intracolonic drug solubility. Pharm. Res. 2010, 27, 2187–2196. [Google Scholar]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar]
- Keller, T.H.; Pichota, A.; Yin, Z. A practical view of ‘druggability’. Curr. Opin. Chem. Biol. 2006, 10, 357–361. [Google Scholar]
- Butler, J.M.; Dressman, J.B. The developability classification system: Application of biopharmaceutics concepts to formulation development. J. Pharm. Sci. 2010, 99, 4940–4954. [Google Scholar]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar]
- Ritchie, T.J.; Ertl, P.; Lewis, R. The graphical representation of ADME-related molecule properties for medicinal chemists. Drug Discov. Today 2011, 16, 65–72. [Google Scholar]
- Ritchie, T.J.; Macdonald, S.J.; Young, R.J.; Pickett, S.D. The impact of aromatic ring count on compound developability: Further insights by examining carbo- and hetero-aromatic and -aliphatic ring types. Drug Discov. Today 2011, 16, 164–171. [Google Scholar]
- Agoram, B.; Woltosz, W.S.; Bolger, M.B. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 2001, 50, S41–S67. [Google Scholar]
- Darwich, A.S.; Neuhoff, S.; Jamei, M.; Rostami-Hodjegan, A. Interplay of metabolism and transport in determining oral drug absorption and gut wall metabolism: A simulation assessment using the ‘advanced dissolution, absorption, metabolism (ADAM)’ model. Curr. Drug Metab. 2010, 11, 716–729. [Google Scholar]
- Sugano, K. Computational oral absorption simulation for low-solubility compounds. Chem. Biodivers. 2009, 6, 2014–2029. [Google Scholar]
- Thelen, K.; Coboeken, K.; Willmann, S.; Burghaus, R.; Dressman, J.B.; Lippert, J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part 1: Oral solutions. J. Pharm. Sci. 2011, 100, 5324–5345. [Google Scholar]
- Thelen, K.; Coboeken, K.; Willmann, S.; Dressman, J.B.; Lippert, J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part II: Extension to describe performance of solid dosage forms. J. Pharm. Sci. 2012, 101, 1267–1280. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar]
- Kerns, E.H.; Di, L.; Carter, G.T. In vitro solubility assays in drug discovery. Curr. Drug Metab. 2008, 9, 879–885. [Google Scholar]
- Preda, I.A.; Mircioiu, I.; Mircioiu, C.; Corlan, G.; Pahomi, G.; Prasacu, I.; Anuta, V. Research concerning the development of a biorelevant dissolution test for formulations containing norfloxacin. I. Modelling of in vitro release kinetics. Farmacia 2012, 60, 675–687. [Google Scholar]
- Mircioiu, I.; Anuta, V.; Purcaru, S.O.; Radulescu, F.S.; Miron, D.S.; Dumitrescu, I.B.; Ibrahim, N.; Mircioiu, C. In vitro dissolution of poorly soluble drugs in the presence of surface active agents—In vivo pharmacokinetics correlations. II. Nimesulide. Farmacia 2013, 61, 88–102. [Google Scholar]
- Mircioiu, I.; Anuta, V.; Ibrahim, N.; Mircioiu, C. Dissolution of tamoxifen in biorelevant media. A two phase release model. Farmacia 2012, 60, 315–324. [Google Scholar]
- Fagerberg, J.H.; Tsinman, O.; Sun, N.; Tsinman, K.; Avdeef, A.; Bergstrom, C.A. Dissolution rate and apparent solubility of poorly soluble drugs in biorelevant dissolution media. Mol. Pharm. 2010, 7, 1419–1430. [Google Scholar]
- Ottaviani, G.; Gosling, D.J.; Patissier, C.; Rodde, S.; Zhou, L.; Faller, B. What is modulating solubility in simulated intestinal fluids? Eur. J. Pharm. Sci. 2010, 41, 452–457. [Google Scholar]
- Jain, P.; Yalkowsky, S.H. Prediction of aqueous solubility from SCRATCH. Int. J. Pharm. 2010, 385, 1–5. [Google Scholar]
- Ran, Y.; Jain, N.; Yalkowsky, S.H. Prediction of aqueous solubility of organic compounds by the general solubility equation (GSE). J. Chem. Inf. Comput. Sci. 2001, 41, 1208–1217. [Google Scholar]
- Jain, N.; Yalkowsky, S.H. Estimation of the aqueous solubility I: Application to organic nonelectrolytes. J. Pharm. Sci. 2001, 90, 234–252. [Google Scholar]
- Bergstrom, C.A.; Wassvik, C.M.; Johansson, K.; Hubatsch, I. Poorly soluble marketed drugs display solvation limited solubility. J. Med. Chem. 2007, 50, 5858–5862. [Google Scholar]
- Wassvik, C.M.; Holmen, A.G.; Draheim, R.; Artursson, P.; Bergstrom, C.A. Molecular characteristics for solid-state limited solubility. J. Med. Chem. 2008, 51, 3035–3039. [Google Scholar]
- Uivarosi, V.; Dinu-Pirvu, C.E.; Ghica, M.V.; Anuta, V. Preformulation studies using cosolvent systems to increase the solubility of a new enrofloxacin ruthenium (III) complex with biological activity. Farmacia 2013, 61, 127–142. [Google Scholar]
- Carey, M.C. Measurement of the physical-chemical properties of bile salt solutions. In Bile Acids in Gastroenterology; Barbara, L., Dowling, R.H., Hofmann, A.F., Roda, E., Eds.; MTP Press: Lancaster, UK, 1983; pp. 19–56. [Google Scholar]
- Pahomi, G.; Corlan, G.; Anuta, V.; Sandulovici, R.C.; Mircioiu, I. Study of tile influence of bile salts and lecithin on distribution of ketoconazole between plasma and methylene chloride. Farmacia 2012, 60, 809–821. [Google Scholar]
- Mahlin, D.; Ponnambalam, S.; Hockerfelt, M.H.; Bergstrom, C.A. Toward in silico prediction of glass-forming ability from molecular structure alone: A screening tool in early drug development. Mol. Pharm. 2011, 8, 498–506. [Google Scholar]
- Bergstrom, C.A.; Holm, R.; Jorgensen, S.A.; Andersson, S.B.; Artursson, P.; Beato, S.; Borde, A.; Box, K.; Brewster, M.; Dressman, J.; et al. Early pharmaceutical profiling to predict oral drug absorption: Current status and unmet needs. Eur. J. Pharm. Sci. 2014, 57, 173–199. [Google Scholar]
- Zur, M.; Gasparini, M.; Wolk, O.; Amidon, G.L.; Dahan, A. The low/high BCS permeability class boundary: Physicochemical comparison of metoprolol and labetalol. Mol. Pharm. 2014, 11, 1707–1714. [Google Scholar]
- Amidon, K.S.; Langguth, P.; Lennernas, H.; Yu, L.; Amidon, G.L. Bioequivalence of oral products and the biopharmaceutics classification system: science, regulation, and public policy. Clin. Pharmacol. Ther. 2011, 90, 467–470. [Google Scholar]
- Dressman, J.B.; Vertzoni, M.; Goumas, K.; Reppas, C. Estimating drug solubility in the gastrointestinal tract. Adv. Drug Deliv. Rev. 2007, 59, 591–602. [Google Scholar]
- ICH Harmonised Tripartite Guideline: Validation of analytical procedures: Text and methodology Q2(R1). In Proceedings of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Chicago, IL, USA, 5–10 November 2005.
- The United States Pharmacopeial Convention I. The United States Pharmacopeia 32—The National Formulary 27; United States Pharmacopeial Convention, Inc.: Rockville, MD, USA, 2009. [Google Scholar]
- Jogia, H.; Sola, S.P.; Garg, L.K.; Arutla, S.; Reddy, A.M.; Venkateswarlu, V. A simple, safe, and environmentally friendly method of FaSSIF and FeSSIF preparation without Methylene Chloride. Dissolution Technol. 2014, 21, 45–48. [Google Scholar]
- Tetko, I.V. Computing chemistry on the web. Drug Discov. Today 2005, 10, 1497–1500. [Google Scholar]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Willey-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Viswanadhan, V.N.; Ghose, A.K.; Revankar, G.R.; Robins, R.K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Comput. Sci. 1989, 29, 163–172. [Google Scholar]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple method of calculating octanol water partition coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar]
- Sun, D.; Yu, L.X.; Hussain, M.A.; Wall, D.A.; Smith, R.L.; Amidon, G.L. In vitro testing of drug absorption for drug ‘developability’ assessment: forming an interface between in vitro preclinical data and clinical outcome. Curr. Opin. Drug Discov. Dev. 2004, 7, 75–85. [Google Scholar]
- Sample Availability: Samples of the compounds BC2, BC3, BC5, BM2, BM3, BM5, BT2, BT3 and BT5 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anuta, V.; Nitulescu, G.M.; Dinu-Pîrvu, C.E.; Olaru, O.T. Biopharmaceutical Profiling of New Antitumor Pyrazole Derivatives. Molecules 2014, 19, 16381-16401. https://doi.org/10.3390/molecules191016381
Anuta V, Nitulescu GM, Dinu-Pîrvu CE, Olaru OT. Biopharmaceutical Profiling of New Antitumor Pyrazole Derivatives. Molecules. 2014; 19(10):16381-16401. https://doi.org/10.3390/molecules191016381
Chicago/Turabian StyleAnuta, Valentina, George Mihai Nitulescu, Cristina Elena Dinu-Pîrvu, and Octavian Tudorel Olaru. 2014. "Biopharmaceutical Profiling of New Antitumor Pyrazole Derivatives" Molecules 19, no. 10: 16381-16401. https://doi.org/10.3390/molecules191016381