N-Substituted 5-Amino-6-methylpyrazine-2,3-dicarbonitriles: Microwave-Assisted Synthesis and Biological Properties

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
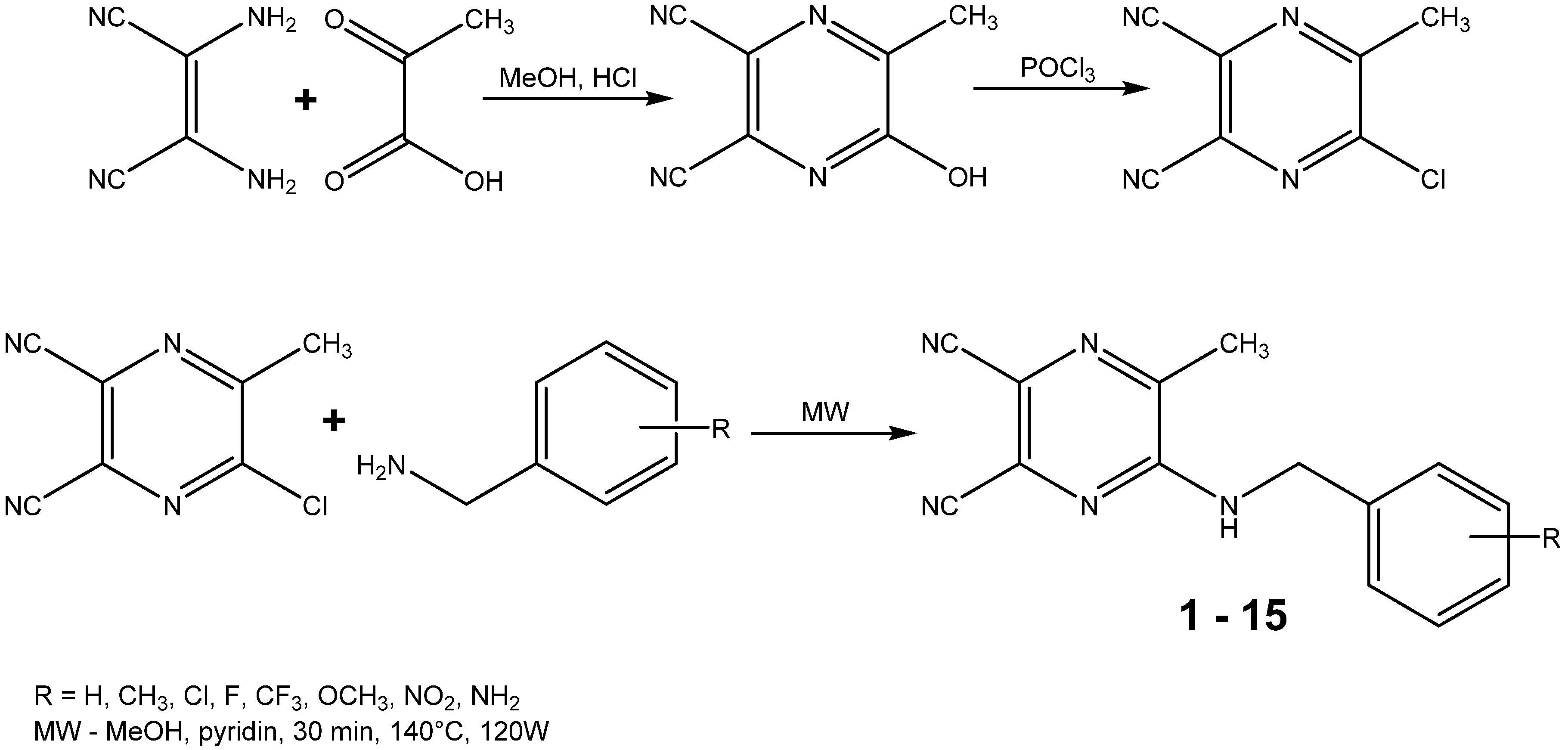
2.1. Chemistry
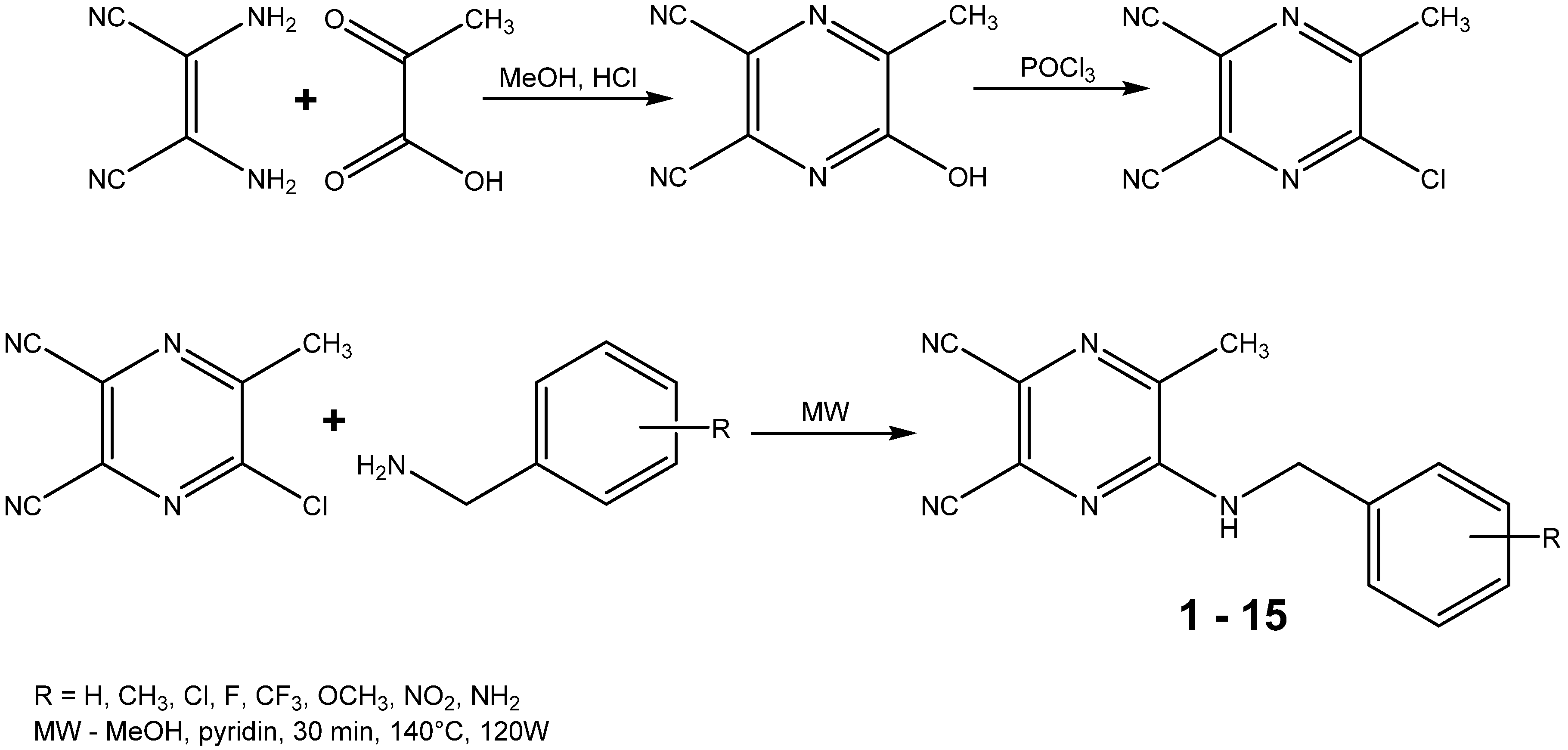

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | log P | log k | σ | π | IC50 [μmol/L] | MIC M.tuberculosis H37Rv [μg/mL] |
|---|---|---|---|---|---|---|---|
| 1 | 2-CH3 | 3.41 | 0.4668 | −0.17 | 0.1674 | 114.0 | >100 |
| 2 | 3-CF3 | 3.84 | 0.5369 | 0.43 | 0.2375 | 37.7 | 12.5 |
| 3 | 3,4-Cl | 4.04 | 0.7538 | 0.60 | 0.4544 | 16.4 | 6.25 |
| 4 | 4-CH3 | 3.41 | 0.5157 | −0.17 | 0.2163 | 104.7 | 25 |
| 5 | 4-OCH3 | 2.8 | 0.2820 | −0.27 | −0.0174 | 464.6 | >100 |
| 6 | H | 2.92 | 0.2994 | 0 | 0 | - | 25 |
| 7 | 4-NH2 | 2.12 | −0.2014 | −0.15 | −0.5008 | - | 25 |
| 8 | 3-Cl | 3.48 | 0.5172 | 0.37 | 0.2178 | 57.4 | 12.5 |
| 9 | 2-Cl | 3.48 | 0.4663 | 0.22 | 0.1669 | 79.0 | 6.25 |
| 10 | 2-F | 3.08 | 0.3042 | 0.06 | 0.0048 | 195.6 | 12.5 |
| 11 | 4-CF3 | 3.84 | 0.5626 | 0.51 | 0.2632 | 39.6 | 6.25 |
| 12 | 2-CF3 | 3.84 | 0.4865 | 0.51 | 0.1871 | 71.6 | 12.5 |
| 13 | 2,4-OCH3 | 2.67 | 0.3699 | −0.55 | 0.0705 | - | 25 |
| 14 | 3-NO2 | 2.71 | 0.1808 | 0.71 | −0.1186 | 487.4 | 12.5 |
| 15 | 4-Cl | 3.48 | 0.5384 | 0.23 | 0.2390 | 86.5 | 12.5 |
| PZA | - | 12.5 | |||||
| INH | - | 1.5625 | |||||
| DCMU | 1.9 | - |
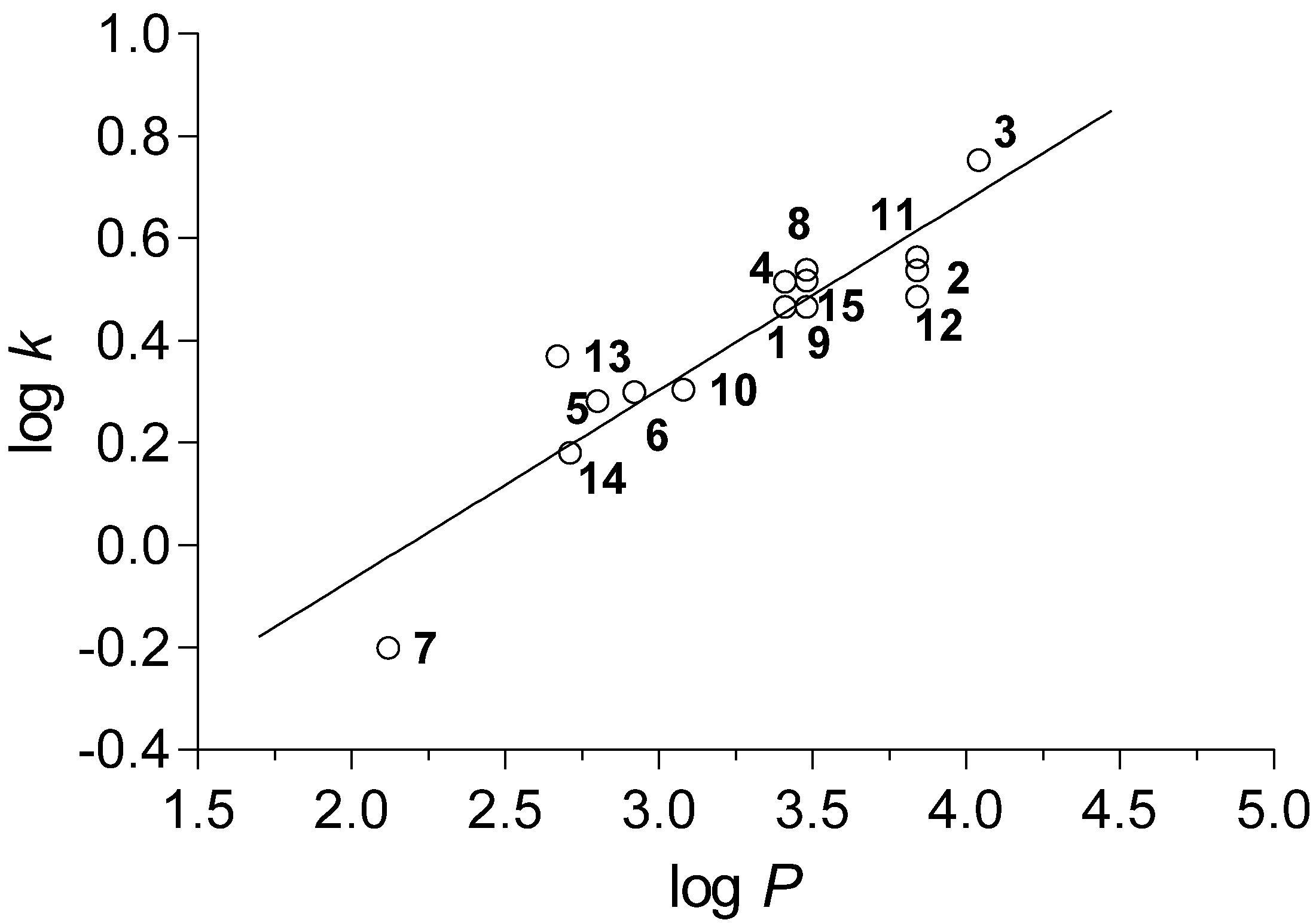
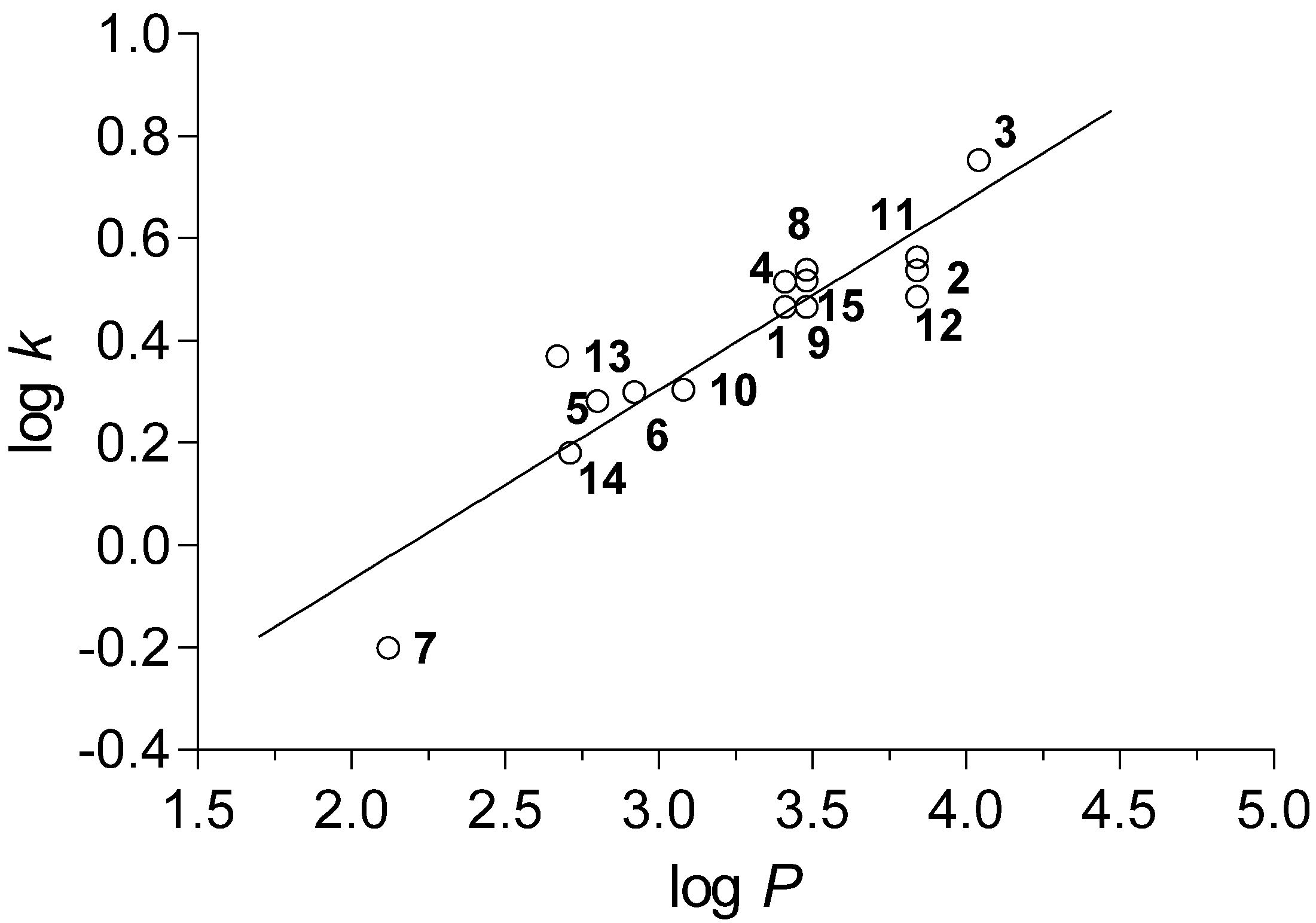
2.2. Calculated and Experimentally Set Lipophilicity
r = 0.915, s = 0.092, F = 66.9, n = 15
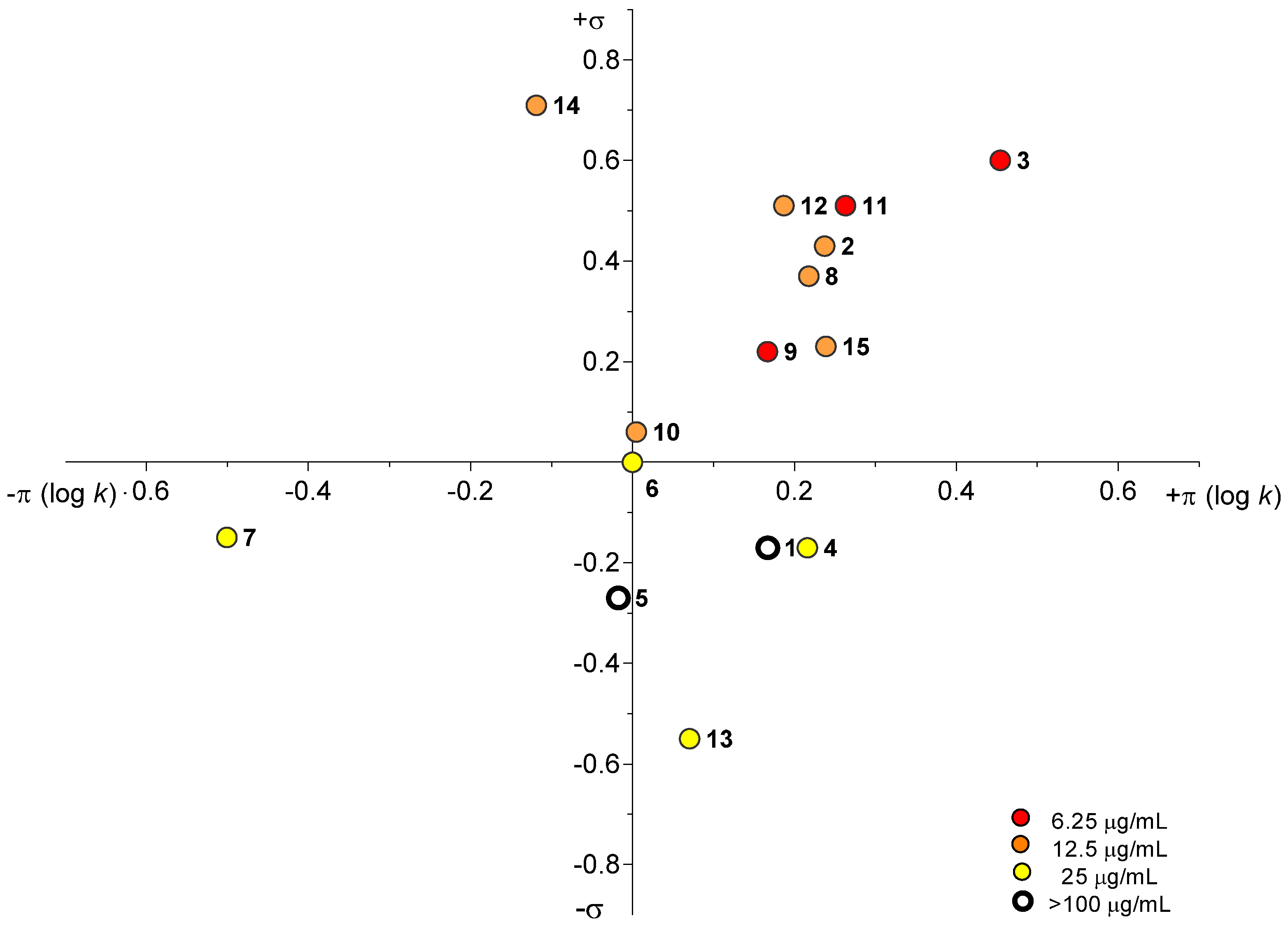
2.3. Biological Assays
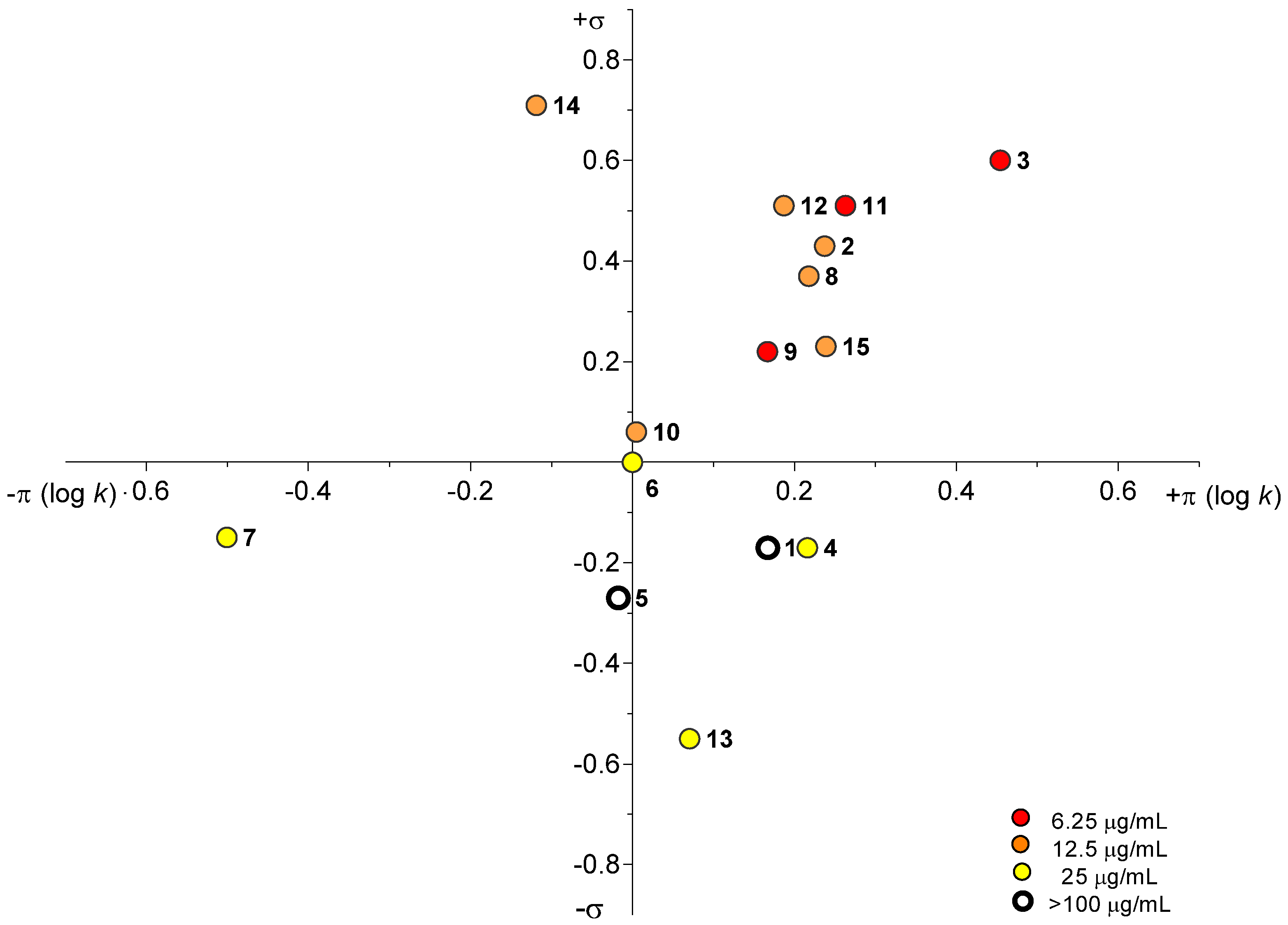
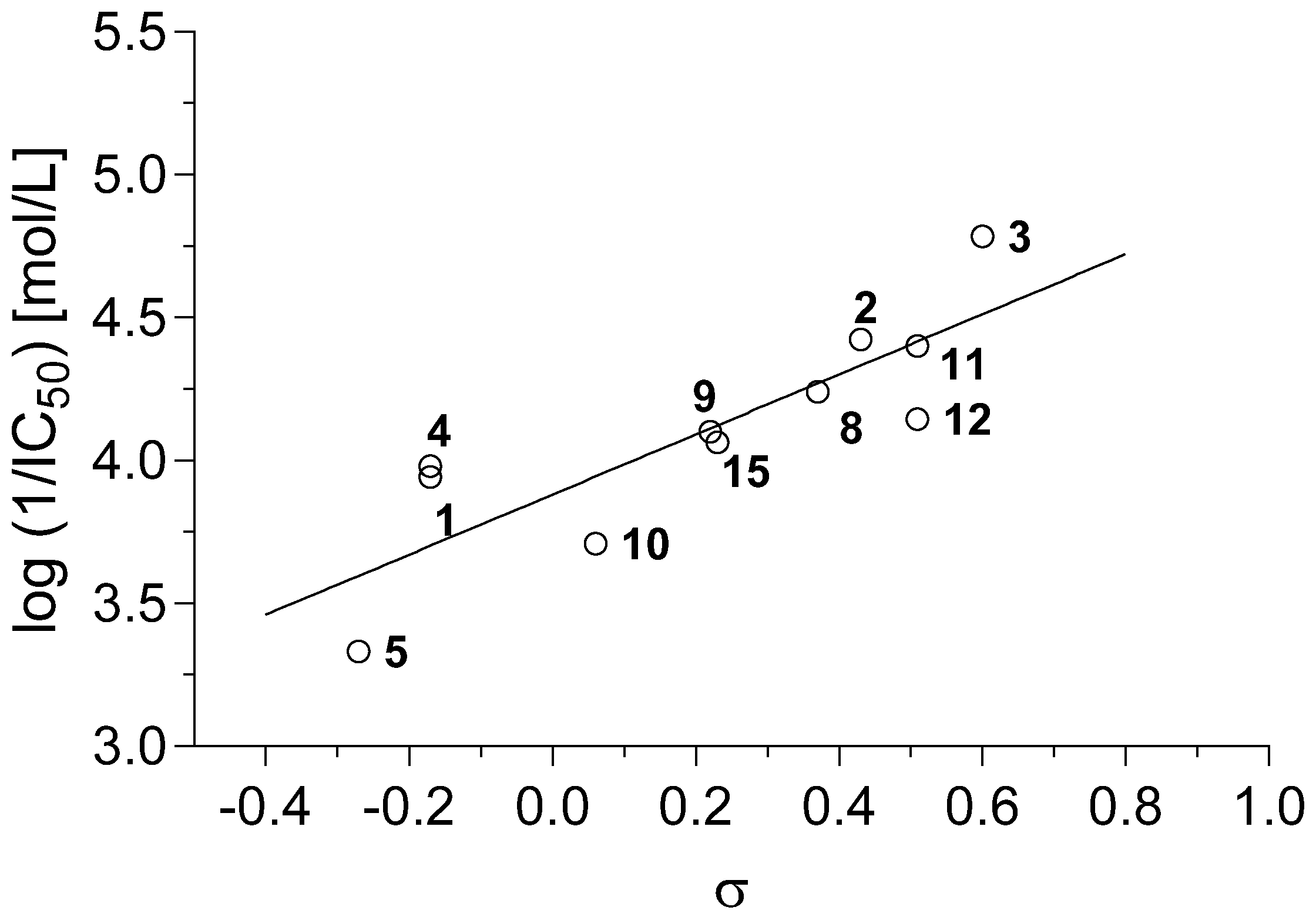
2.3.1. Antimycobacterial In Vitro Screening
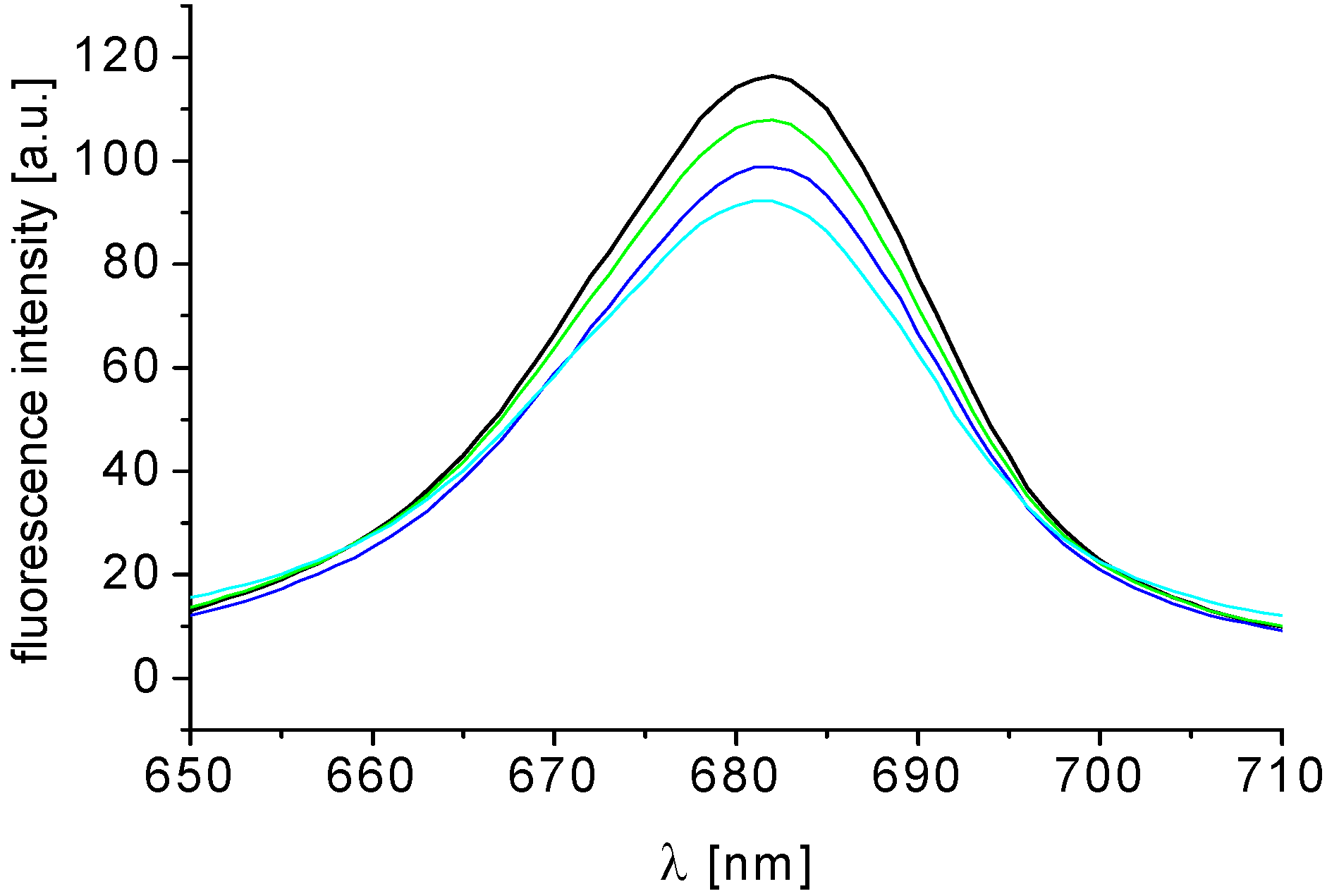
2.3.2. Antimycobacterial In Vitro Screening Against M. smegmatis
2.3.3. Antifungal and Antibacterial In Vitro Screening
2.3.4. Herbicidal Activity of Prepared Compounds
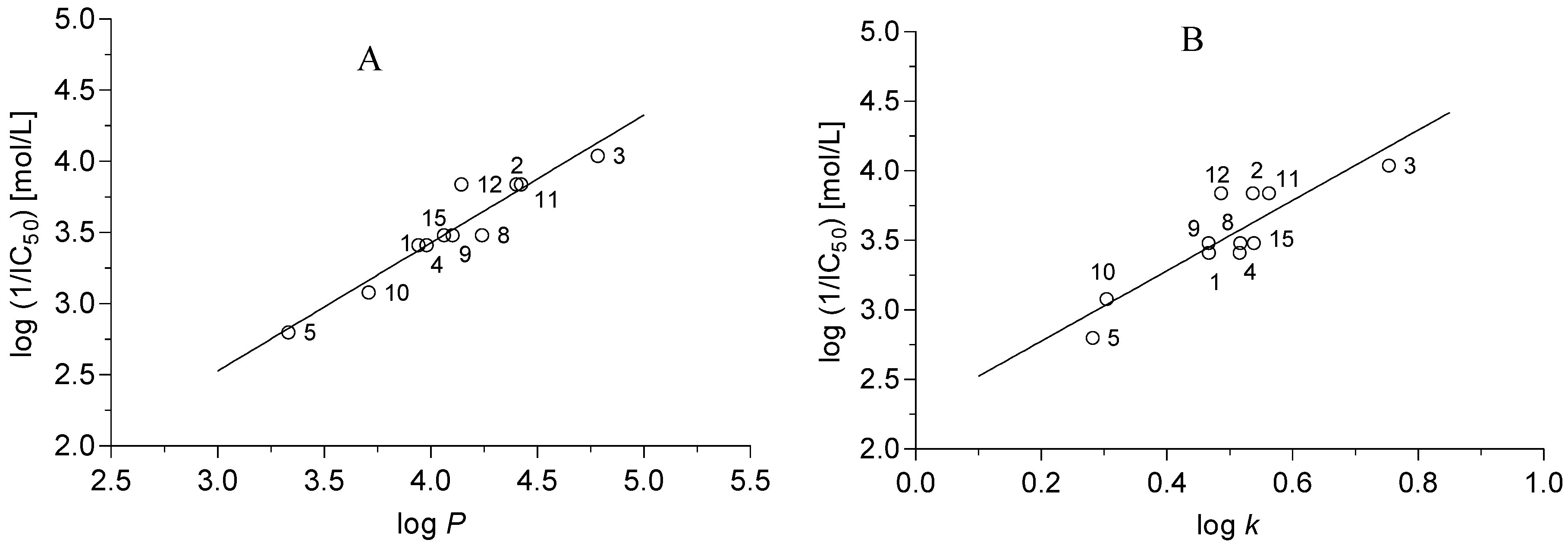
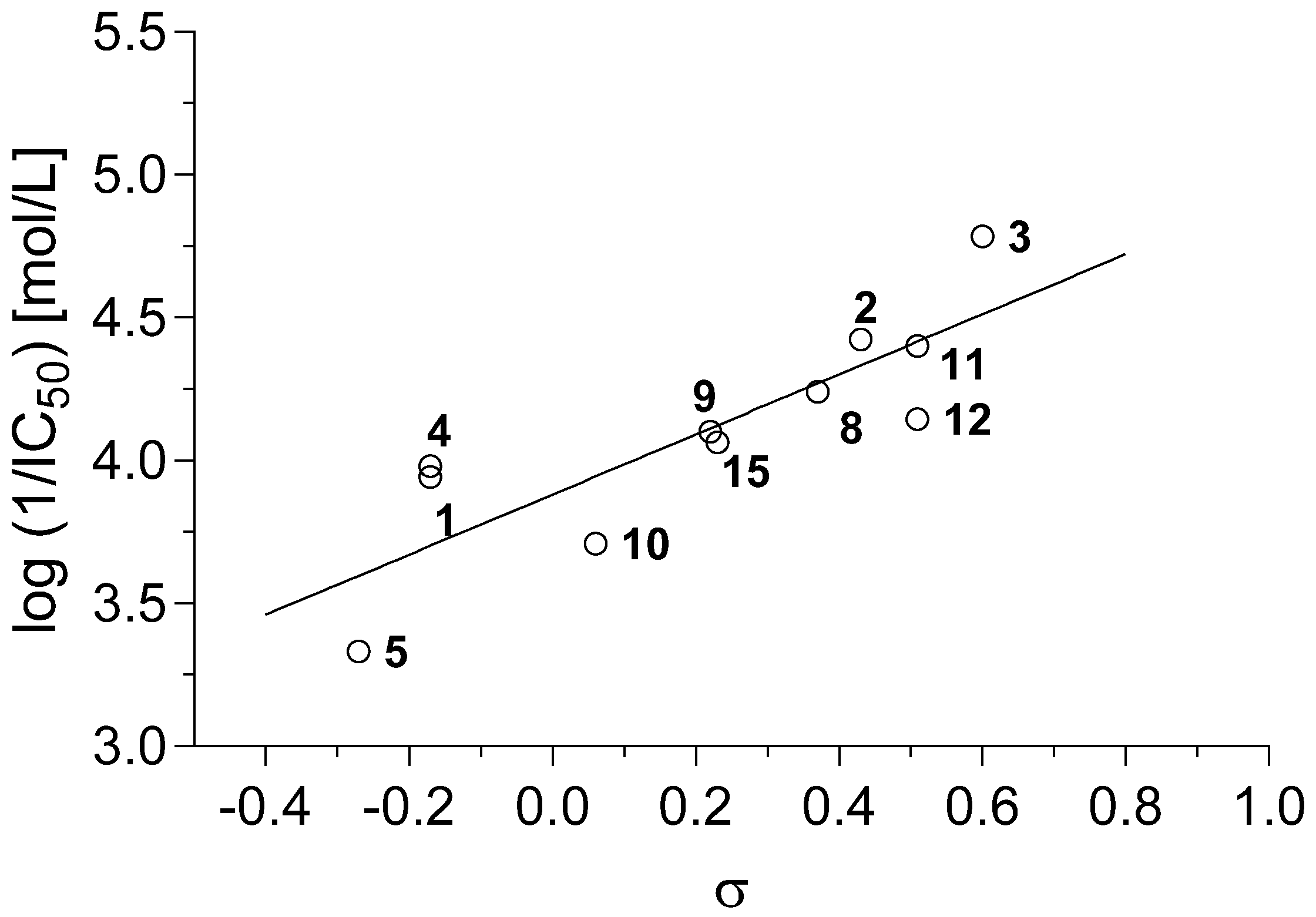
r = 0.950, s = 0.127, F = 82.5, n = 11
r = 0.844, s = 0.217, F = 22.34, n = 11
r = 0.952, s = 0.131, F = 38.6, n = 11
r = 0.934, s = 0.144, F = 61.8, n = 11
r = 0.976 s = 0.093 F = 80.2 n = 11

3. Experimental
3.1. General
3.2. Starting Compound and Final Products Synthesis
3.3. Analytical Data of the Prepared Compounds
3.4. Lipophilicity HPLC Determination and Calculations
3.5. Biological Assays
3.5.1. Antimycobacterial In Vitro Screening
3.5.2. Antimycobacterial In Vitro Screening Against M. smegmatis
3.5.3. Antifungal and Antibacterial In Vitro Screenings
3.5.4. Study of the Inhibition of Oxygen Evolution Rate in Spinach Chloroplasts
3.5.5. Study of Fluorescence of Chlorophyll a in Spinach Chloroplasts
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Conflicts of Interest
References and Notes
- Global Tuberculosis Report 2013 [online]. World Health Organization: France, 2013; pp. 1–27, ISBN 978 92 4 156465 6. Available online: http://apps.who.int/iris/bitstream/10665/91355/1/9789241564656_eng.pdf (accessed on 15 November 2013).
- Lima, C.H.S.; Bispo, M.L.F.; de Souza, M.V.N. Pirazinamida: Um farmaco essencial no tratamento da tuberculose. Rev. Virtual Quim. 2011, 3, 159–180. [Google Scholar]
- Zhang, Y.; Chiu Chang, K.; Leung, C.; Yew, W.W.; Gicquel, B.; Fallows, D.; Kaplan, G.; Chaisson, R.E.; Zhang, W. 'ZS-MDR-TB' versus 'ZR-MDR-TB': Improving treatment of MDR-TB by identifying pyrazinamide susceptibility. Emerg. Microbes Infect. 2012, 1, e5. [Google Scholar] [CrossRef]
- Velayati, A.A.; Masjedi, M.R.; Farnia, P.; Tabarsi, P.; Ghanavi, J.; Ziazarifi, A.H.; Hoffner, S.E. Emergence of new forms of totally drug-resistant tuberculosis bacilli: Super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest 2009, 136, 420–425. [Google Scholar] [CrossRef]
- Zhang, Y.; Mitchinson, D. The curious characteristics of pyrazinamide: A review. Int. J. Tubercul. Lung Dis. 2003, 7, 6–21. [Google Scholar]
- Zhang, Y.; Wade, M.M.; Scorpio, A. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef]
- Konno, K.; Feldmann, F.M.; McDermott, W. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am. Rev. Respir. Dis. 1967, 95, 461–469. [Google Scholar]
- Zhang, Y.; Scorpio, A.; Nikaido, H.; Sun, Z. Role of acid ph and deficient efflux of pyrazinoic acid in unique susceptibility of Mycobacterium tuberculosis to pyrazinamide. J. Bacteriol. 1999, 181, 2044–2049. [Google Scholar]
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef]
- Boshoff, H.I.; Mizrahi, V.; Barry, C.E., III. Effects of pyrazinamide on fatty acid synthesis by whole mycobacterial cells and purified fatty acid synthase I. J. Bacteriol. 2002, 184, 2167–2172. [Google Scholar] [CrossRef]
- Zimhony, O.; Cox, J.S.; Welch, J.T.; Vilcheze, C.; Jacobs, W.R. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FAS-I) of Mycobacterium tuberculosis. Nat. Med. 2000, 6, 1043–1047. [Google Scholar] [CrossRef]
- Zimhony, O.; Vilcheze, C.; Arai, M.; Welch, J.T.; Jacobs, W.R. Pyrazinoic acid and its n-propyl ester inhibit fatty acid synthase type I in replicating tubercle bacilli. Antimicrob. Agents Chemother. 2007, 51, 752–754. [Google Scholar] [CrossRef]
- Ngo, S.C.; Zimhony, O.; Chung, W.J.; Sayahi, H.; Jacobs, W.R., Jr.; Welch, J.T. Inhibition of isolated mycobacterium tuberculosis fatty acid synthase I by pyrazinamide analogs. Antimicrob. Agents Chemother. 2007, 51, 2430–2435. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, X.; Jiang, X.; Yuan, H.; Lee, J.S.; Barry, C.E.; Wang, H.; Zhang, W.; Zhang, Y. Pyrazinamide inhibits trans.-translation in Mycobacterium tuberculosis. Science 2011, 333, 1630–1632. [Google Scholar] [CrossRef]
- Dolezal, M.; Cmedlova, P.; Palek, L.; Vinsova, J.; Kunes, J.; Buchta, V.; Jampilek, J.; Kralova, K. Synthesis and antimycobacterial evaluation of substituted pyrazinecarboxamides. Eur. J. Med. Chem. 2008, 43, 1105–1113. [Google Scholar] [CrossRef]
- Dolezal, M. Biologically active pyrazines of natural and synthetic origin. Chem. Listy. 2006, 100, 959–966. [Google Scholar]
- Chaluvaraju, K.C.; Ishwar, B.K. Synthesis and antimicrobial activities of amino benzylated mannich bases of pyrazinamide. Int. J. Chem. Tech. Res. 2010, 2, 1368–1371. [Google Scholar]
- Dolezal, M.; Zitko, J.; Osicka, Z.; Kunes, J.; Buchta, V.; Vejsova, M.; Dohnal, J.; Jampilek, J.; Kralova, K. Synthesis, antimycobacterial, antifungal and photosynthesis-inhibiting activity of chlorinated N-phenylpyrazine-2-carboxamides. Molecules 2010, 15, 8567–8581. [Google Scholar] [CrossRef]
- Whitehead, R.P.; Unger, J.M.; Flaherty, L.E.; Kraut, E.H.; Mills, G.M.; Klein, C.E.; Chapman, R.A.; Doolittle, G.C.; Hammond, N.; Sondak, V.K. A phase II trial of pyrazine diazohydroxide in patients with disseminated malignant melanoma and no prior chemotherapy—southwest oncology group study. Invest. New Drug 2002, 20, 105–111. [Google Scholar] [CrossRef]
- Furuta, Y.; Takahashi, K.; Fukuda, Y.; Kuno, M.; Kamiyama, T.; Kozaki, K.; Nomura, N.; Egawa, H.; Minami, S.; Watanabe, Y.; et al. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. Agents Chemother. 2002, 46, 977–981. [Google Scholar] [CrossRef]
- Ritter, A.M.; Shaw, J.L.; Williams, W.M.; Travis, K.Z. Characterizing aquatic ecological risks from pesticides using a diquat bromide case study. I. Probabilistic exposure estimates. Environ. Toxicol. Chem. 2000, 19, 749–759. [Google Scholar]
- Dolezal, M.; Kralova, K. Synthesis and Evaluation of Pyrazine Derivatives with Herbicidal Activity. In Herbicides, Theory and Applications; Soloneski, S., Larramendy, M.L., Eds.; InTech: Vienna, Austria, 2011; pp. 581–610. [Google Scholar]
- Tamai, R.; Ito, M.; Kobayashi, M.; Mitsunari, T.; Nakano, Y. Oxopyrazine Derivative and Herbicide. US Patent Appl. US 2013/0137577, 30 May 2013. [Google Scholar]
- Reingruber, R.; Kraus, H.; Hutzler, J.; Newton, T.W.; Witschel, M.; Moberg, W.K.; Rapado, L.P.; Besong, G.; Rack, N.; van der Kloet, A.; et al. Substituted Pyrazines Having Herbicidal Activity. US Patent Appl. US 2013/0274109BA1, 17 October 2013. [Google Scholar]
- Nakamura, A.; Ataka, T.; Segawa, H.; Takeuchi, Y.; Takematsu, T. Studies on herbicidal 2,3-dicyanopyrazines. 2. Structure-activity relationships of herbicidal 5-ethylamino- and 5-propylamino-2,3-dicyanopyrazines. Agric. Biol. Chem. 1983, 47, 1561–1567. [Google Scholar] [CrossRef]
- Dolezal, M.; Tumova, L.; Kesetovicova, D.; Tuma, J.; Kralova, K. Substituted N-phenylpyrazine-2-carboxamides, their synthesis and evaluation as herbicides and abiotic elicitors. Molecules 2007, 12, 2589–2598. [Google Scholar] [CrossRef]
- Hosseini, S.; Monajjemi, M.; Rajaeian, E.; Haghgu, M.; Salari, A.; Gholami, M.R.A. Computational study of cytotoxicity of substituted amides of pyrazine-2-carboxylic acids using QSAR and DFT based molecular surface electrostatic potential. Iran. J. Pharm. Res. 2013, 12, 745–750. [Google Scholar]
- Hayes, B.L. Microwave Synthesis: Chemistry at the Speed of Light; CEM Publishing: Matthews, NC, USA, 2002. [Google Scholar]
- De la Hoz, A.; Diaz-Ortiz, A.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef]
- Takematsu, T.; Segawa, H.; Miura, T.; Ataka, T.; Chatani, M.; Nakamura, A. 2,3-Dicyanopyrazines. U.S. Patent 4.259.489, 1981. [Google Scholar]
- Lee, J.H.; Myung, P.K.; Kim, S.J.; Sun, N.D. Minimum structural requirements for herbicidal evaluation of 5-(R1)-6-(R2)-N-(R3-phenyl)-pyrazine-2-carboxamide analogues as new class potent herbicide. J. Korean Soc. Appl. Biol. Chem. 2010, 53, 440–445. [Google Scholar] [CrossRef]
- Prasad, R.K.; Sharma, R. 2D QSAR Analysis of pyrazinecarboxamides derivatives as an herbicidal agent. J. Comput. Method. Mol. Des. 2011, 1, 7–13. [Google Scholar]
- Dolezal, M.; Kralova, K.; Sersen, F.; Miletin, M. The site of action of pyrazine-2-carboxylic acids in the photosynthetic apparatus. Folia Pharm. Univ. Carol. 2001, 26, 13–20. [Google Scholar]
- Atal, N.; Saradhi, P.P.; Mohanty, P. Inhibition of the chloroplast photochemical reactions by treatment of wheat seedlings with low concentrations of cadmium: Analysis of electron transport activities and changes in fluorescence yields. Plant Cell Physiol. 1995, 32, 943–951. [Google Scholar]
- Servusova, B.; Eibinova, D.; Dolezal, M.; Kubicek, V.; Paterova, P.; Pesko, M.; Kralova, K. Substituted N-benzylpyrazine-2-carboxamides: Synthesis and biological evaluation. Molecules 2012, 17, 13183–13198. [Google Scholar] [CrossRef]
- Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Gonec, T.; Bobal, P.; Kauerova, T.; Oravec, M.; Kollar, P.; et al. Antibacterial and herbicidal activity of ring-substituted 3-hydroxynaphthalene-2-carboxanilides. Molecules 2013, 18, 7977–7997. [Google Scholar] [CrossRef]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Bobal, P.; Kollar, P.; Cizek, A.; Kralova, K.; et al. Antimycobacterial and herbicidal activity of ring-substituted 1-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2013, 21, 6531–6541. [Google Scholar] [CrossRef]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Govender, R.; Keltosova, S.; Chambel, B.; Pereira, D.; Kollar, P.; Imramovsky, A.; et al. Antibacterial and herbicidal activity of ring-substituted 2-hydroxynaphthalene-2-carboxanilides. Molecules 2013, 18, 9397–9419. [Google Scholar] [CrossRef]
- Otevrel, J.; Bobal, P.; Zadrazilova, I.; Govender, R.; Pesko, M.; Keltosova, S.; Koleckarova, P.; Marsalek, P.; Imramovsky, A.; Coffey, A.; et al. Antimycobacterial and photosynthetic electron transport inhibiting activity of ring-substituted 4-arylamino-7-chloroquinolinium chlorides. Molecules 2013, 18, 10648–10670. [Google Scholar] [CrossRef]
- Jones, R.N.; Barry, A.L. Optimal dilution susceptibility testing conditions, recommendations for MIC interpretation, and quality control guidelines for the ampicillin-sulbactam combination. J. Clin. Microbiol. 1987, 25, 1920–1925. [Google Scholar]
- National Committee for Clinical Laboratory Standards. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Proposed Standard M 27-P; National Committee for Clinical Laboratory Standards: Villanova, PA, USA, 1992. [Google Scholar]
- Masarovicova, E.; Kralova, K. Approaches to Measuring Plant Photosynthesis Activity. In Handbook of Photosynthesis, 2nd ed.; Pessarakli, M., Ed.; Taylor & Francis group: Boca Raton, FL, USA, 2005; pp. 617–656. [Google Scholar]
- Kralova, K.; Sersen, F.; Sidoova, E. Photosynthesis inhibition produced by 2-alkylthio-6-R-benzothiazoles. Chem. Pap. 1992, 46, 348–350. [Google Scholar]
- Fedke, C. Biochemistry and Physiology of Herbicide Action; Springer Verlag: Berlin, Germany, 1982. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jandourek, O.; Dolezal, M.; Paterova, P.; Kubicek, V.; Pesko, M.; Kunes, J.; Coffey, A.; Guo, J.; Kralova, K. N-Substituted 5-Amino-6-methylpyrazine-2,3-dicarbonitriles: Microwave-Assisted Synthesis and Biological Properties. Molecules 2014, 19, 651-671. https://doi.org/10.3390/molecules19010651
Jandourek O, Dolezal M, Paterova P, Kubicek V, Pesko M, Kunes J, Coffey A, Guo J, Kralova K. N-Substituted 5-Amino-6-methylpyrazine-2,3-dicarbonitriles: Microwave-Assisted Synthesis and Biological Properties. Molecules. 2014; 19(1):651-671. https://doi.org/10.3390/molecules19010651
Chicago/Turabian StyleJandourek, Ondrej, Martin Dolezal, Pavla Paterova, Vladimir Kubicek, Matus Pesko, Jiri Kunes, Aidan Coffey, Jiahui Guo, and Katarina Kralova. 2014. "N-Substituted 5-Amino-6-methylpyrazine-2,3-dicarbonitriles: Microwave-Assisted Synthesis and Biological Properties" Molecules 19, no. 1: 651-671. https://doi.org/10.3390/molecules19010651