Transition Metal Complexes and Radical Anion Salts of 1,10-Phenanthroline Derivatives Annulated with a 1,2,5-Tiadiazole and 1,2,5-Tiadiazole 1,1-Dioxide Moiety: Multidimensional Crystal Structures and Various Magnetic Properties

Abstract
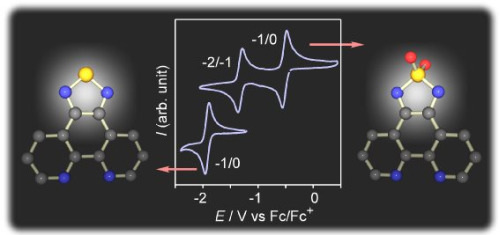
:
1. Introduction

2. Transition Metal Complexes of [1,2,5]thiadiazolo[3,4-f][1,10]phenanthroline
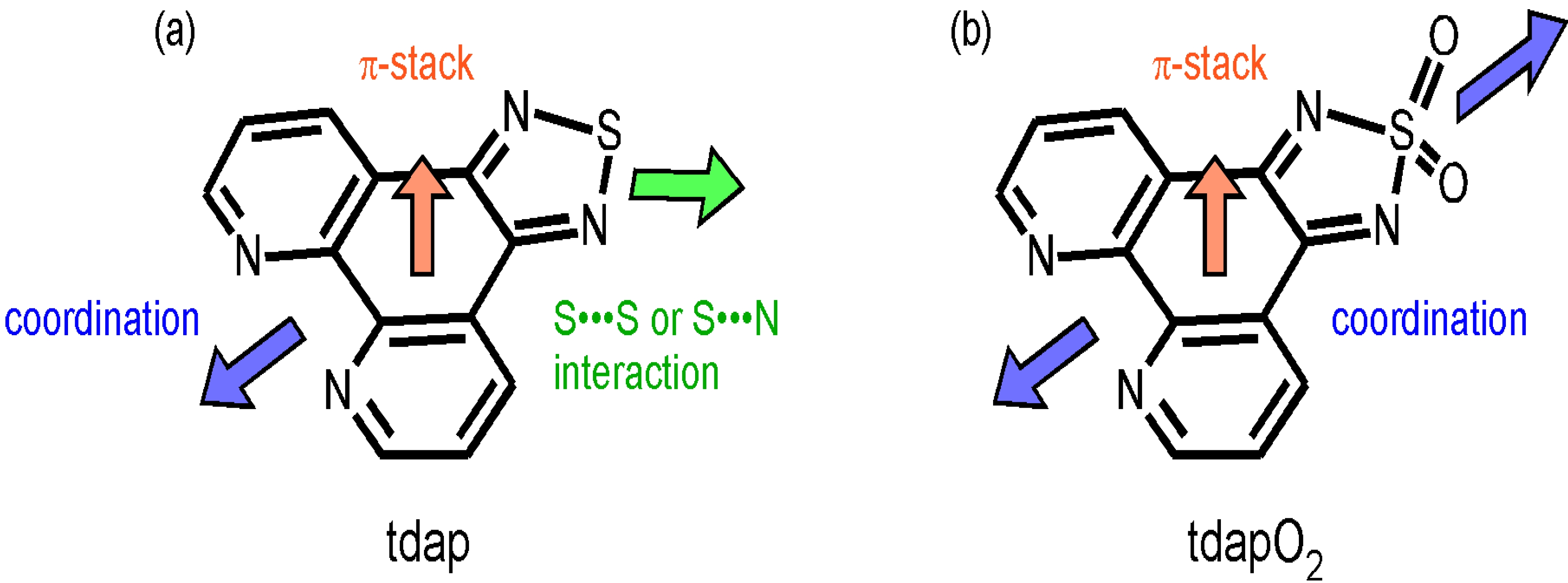
2.1. [1,2,5]thiadiazolo[3,4-f][1,10]phenanthroline (tdap)

2.2. Synthesis of Transition Metal Complexes of tdap
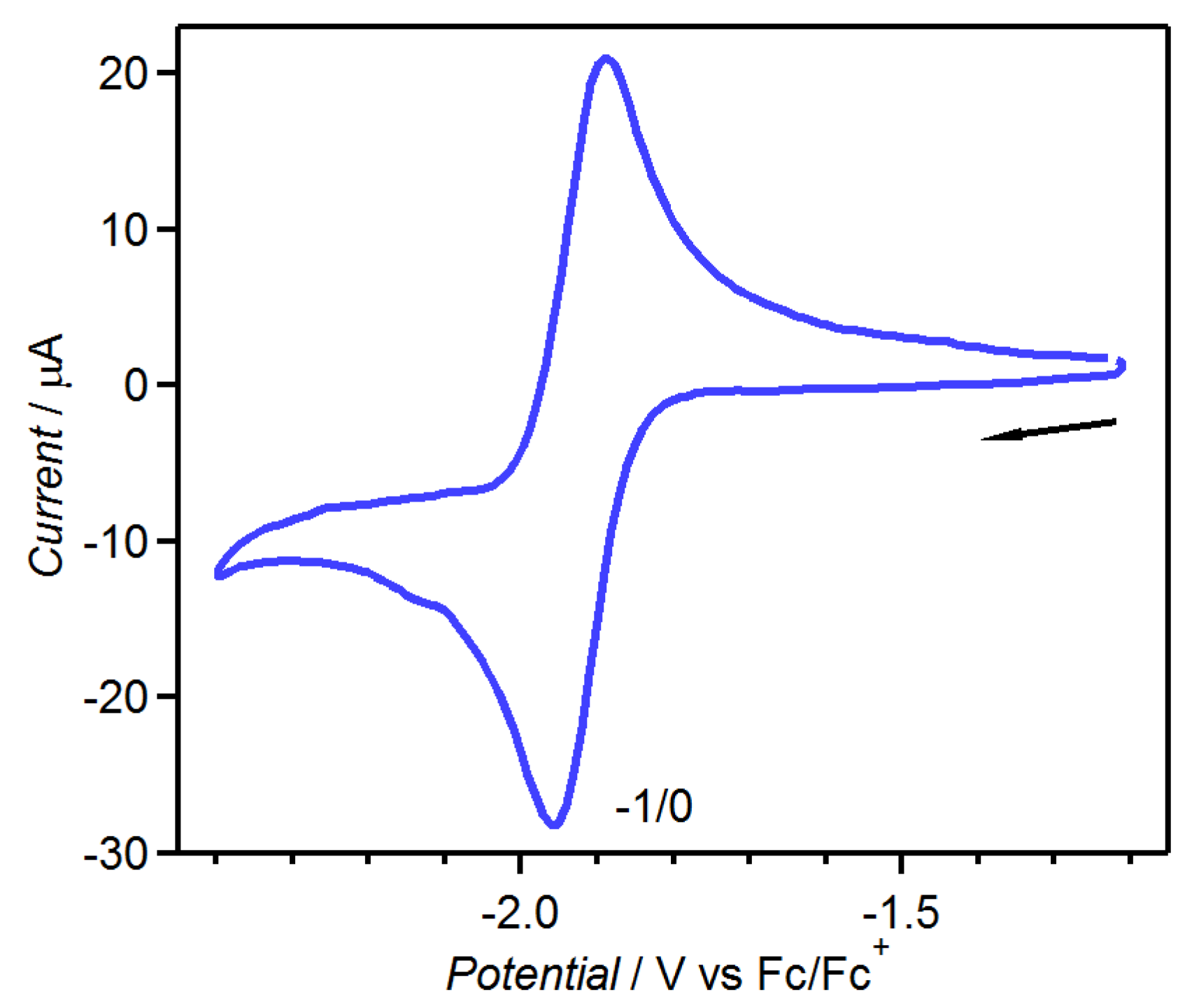
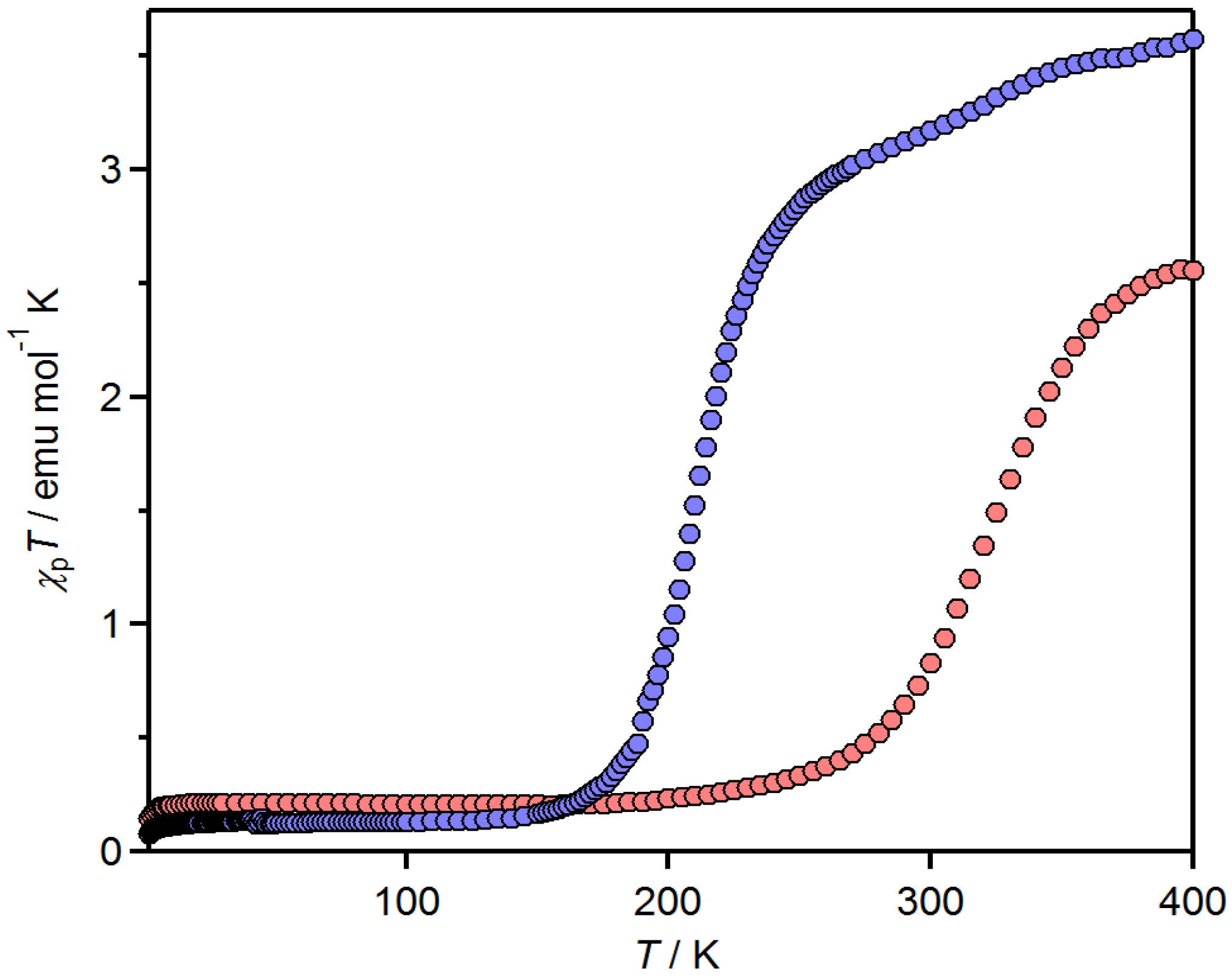
2.3. Spin-Crossover Complexes of tdap
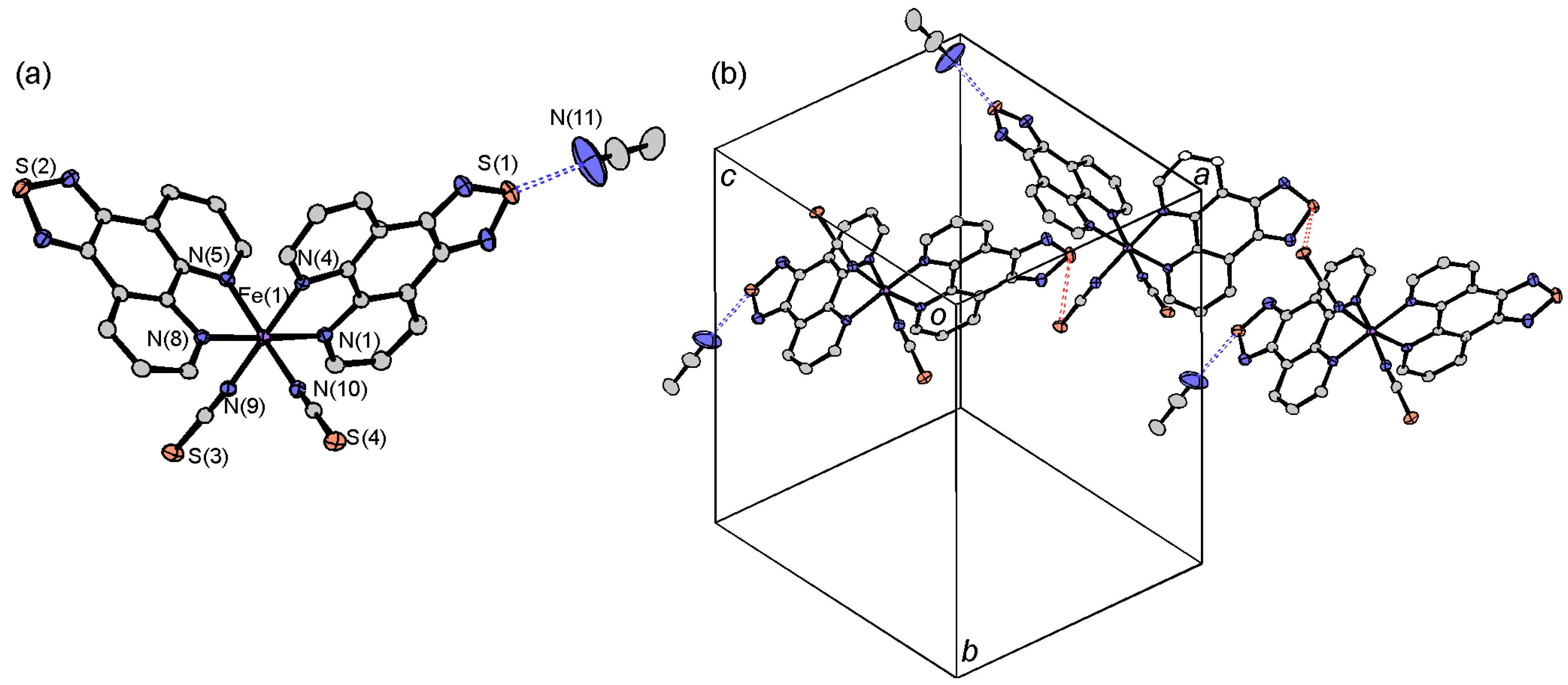
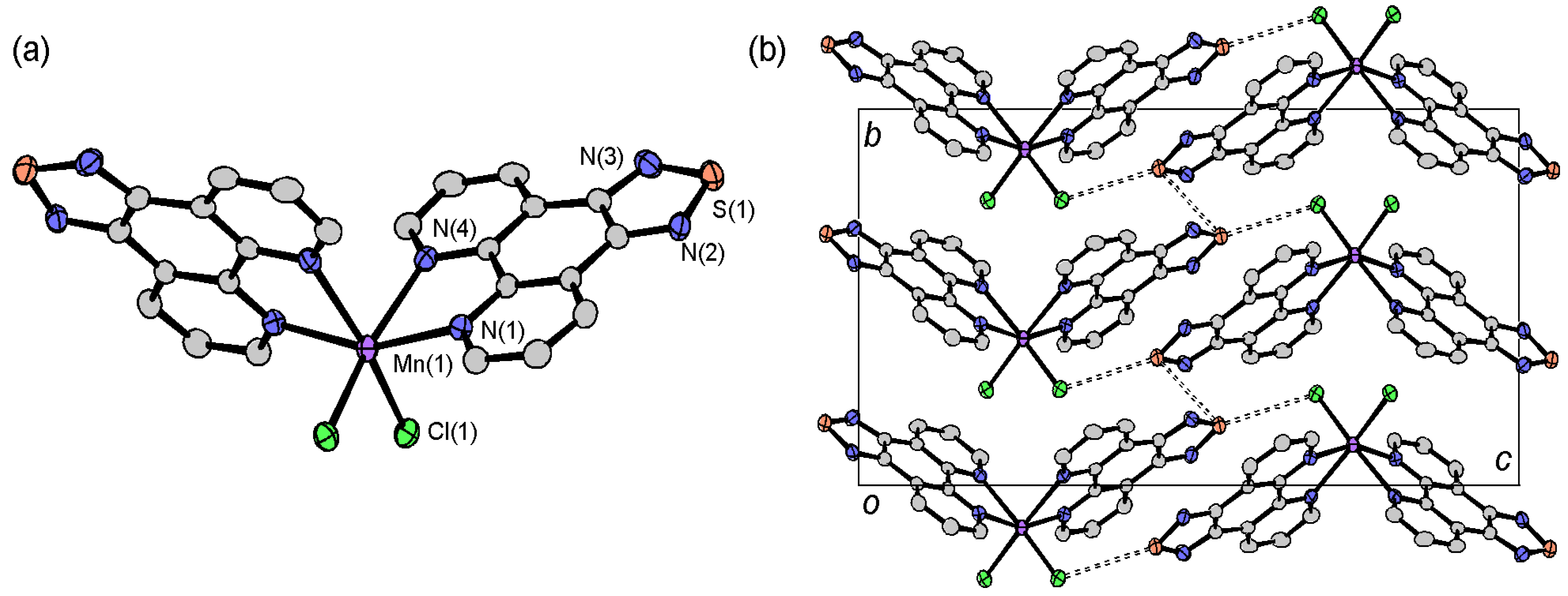
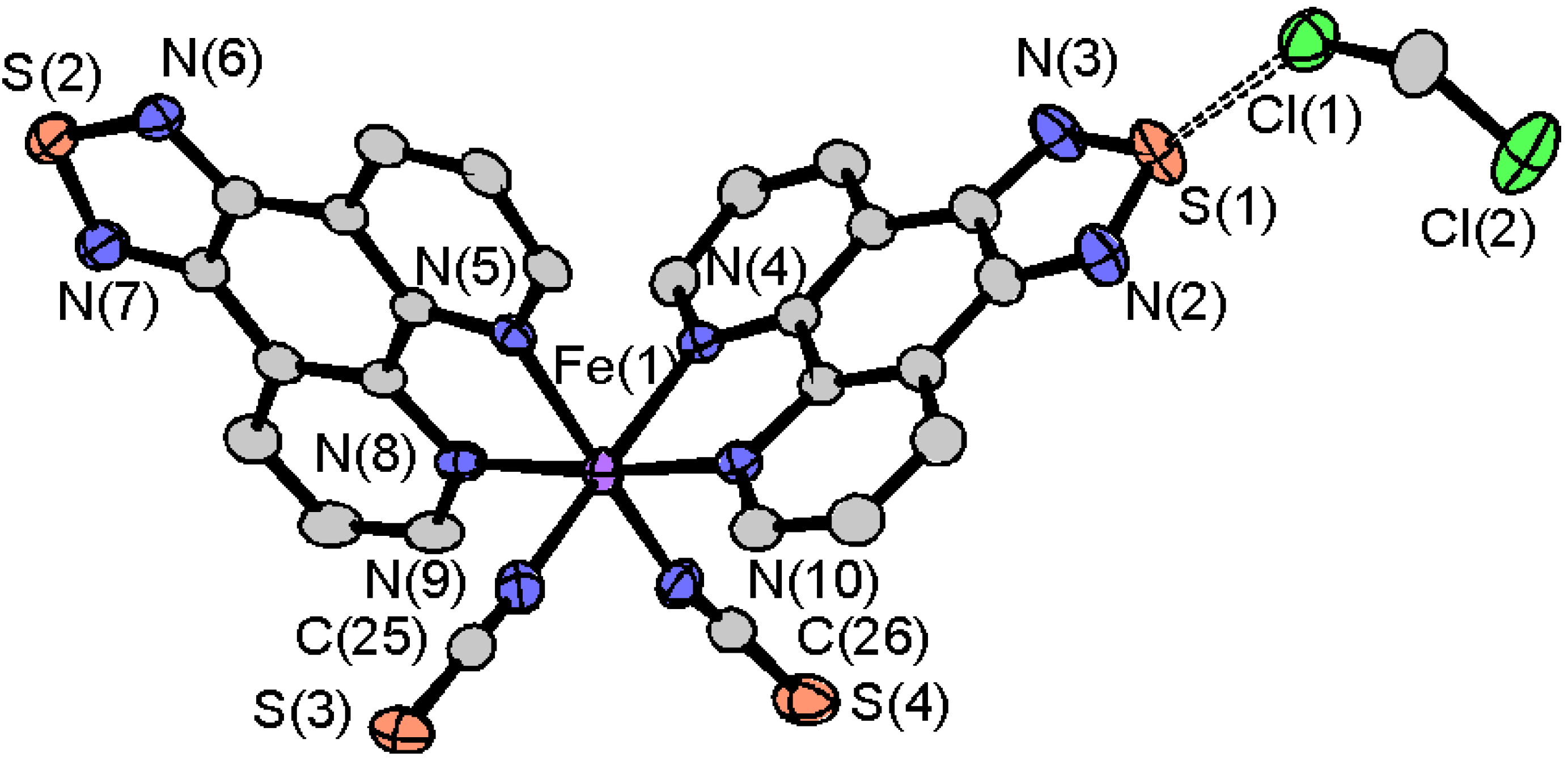
2.3.1. [Fe(tdap)2(NCS)2]•MeCN
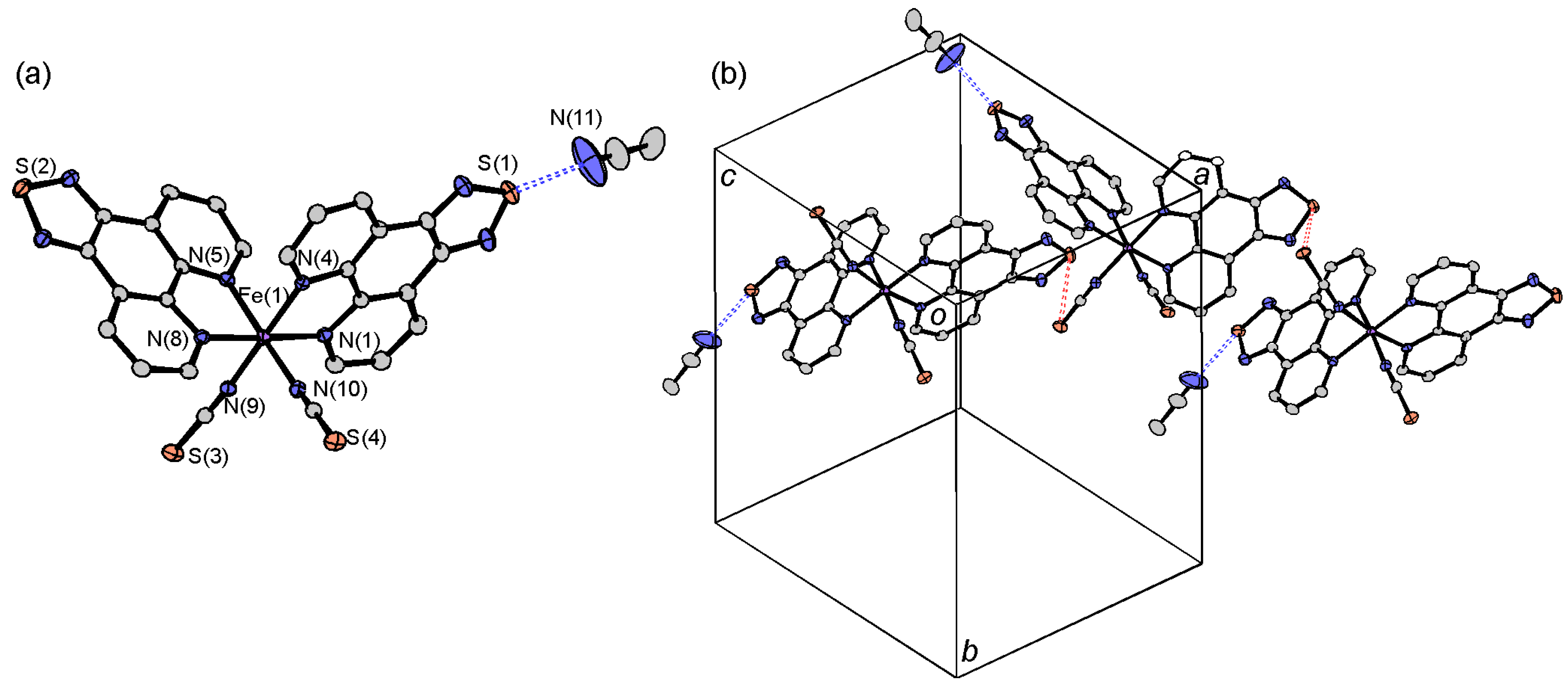

| [Fe(tdap)2(NCS)2]•MeCN | [Fe(tdap)2(NCS)2] | |||
|---|---|---|---|---|
| 320 K | 120 K | 400 K | 173 K | |
| Fe-Ntdap | 2.1838(15) | 1.9786(17) | 2.183(4) | 1.966(3) |
| 2.2212(14) | 1.9808(14) | 2.196(4) | 1.969(3) | |
| 2.1843(15) | 1.9815(16) | 2.197(4) | 1.978(2) | |
| 2.2244(15) | 1.9754(16) | 2.233(4) | 1.978(3) | |
| Fe-NCS | 2.0715(19) | 1.9538(17) | 2.092(6) | 1.940(3) |
| 2.0953(17) | 1.9608(16) | 2.112(7) | 1.948(3) | |
| N-CS | 1.157(2) | 1.164(2) | 1.117(9) | 1.159(5) |
| 1.153(2) | 1.161(2) | 1.055(11) | 1.159(5) | |

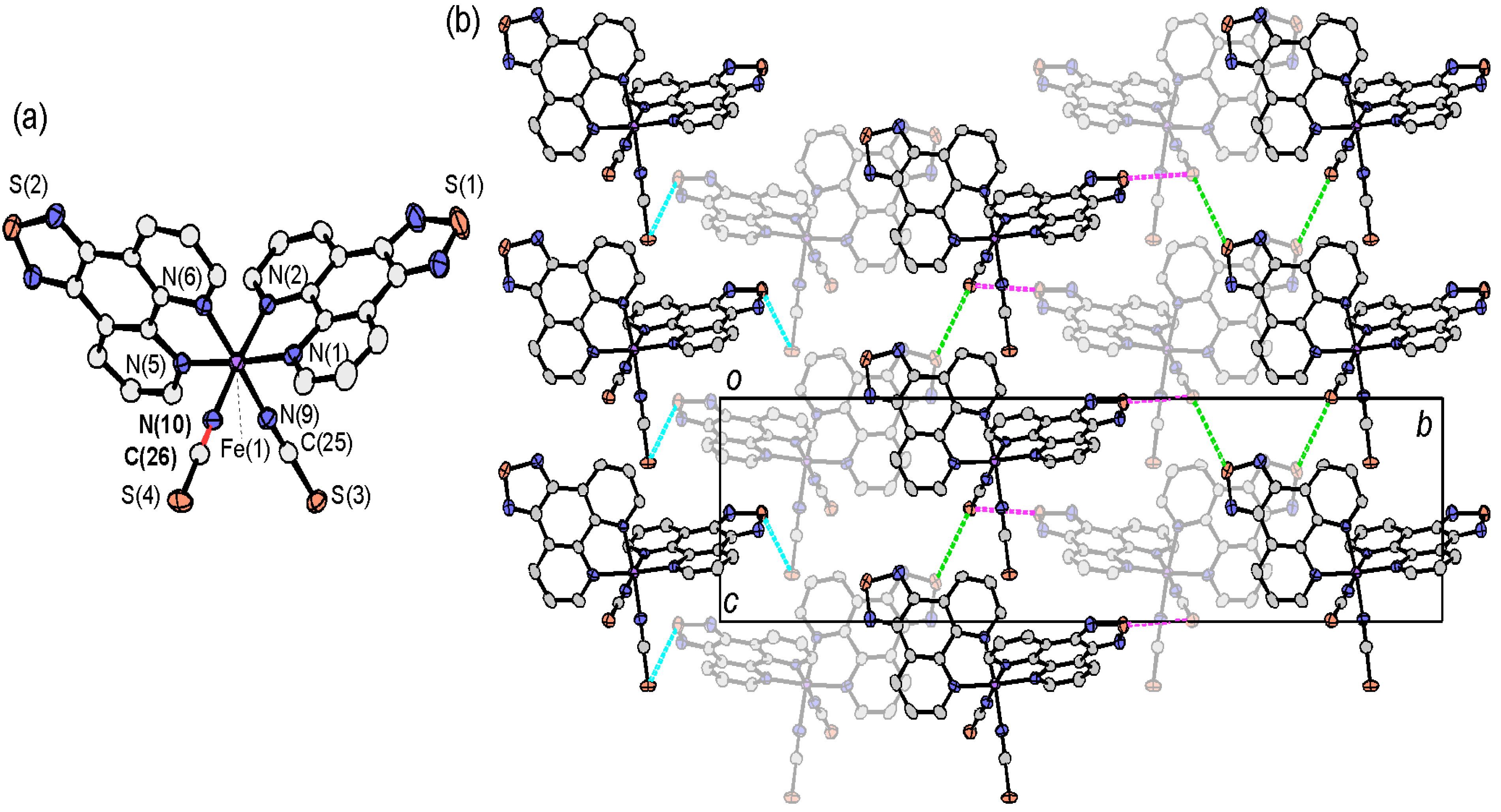
2.3.2. [Fe(tdap)2(NCS)2]
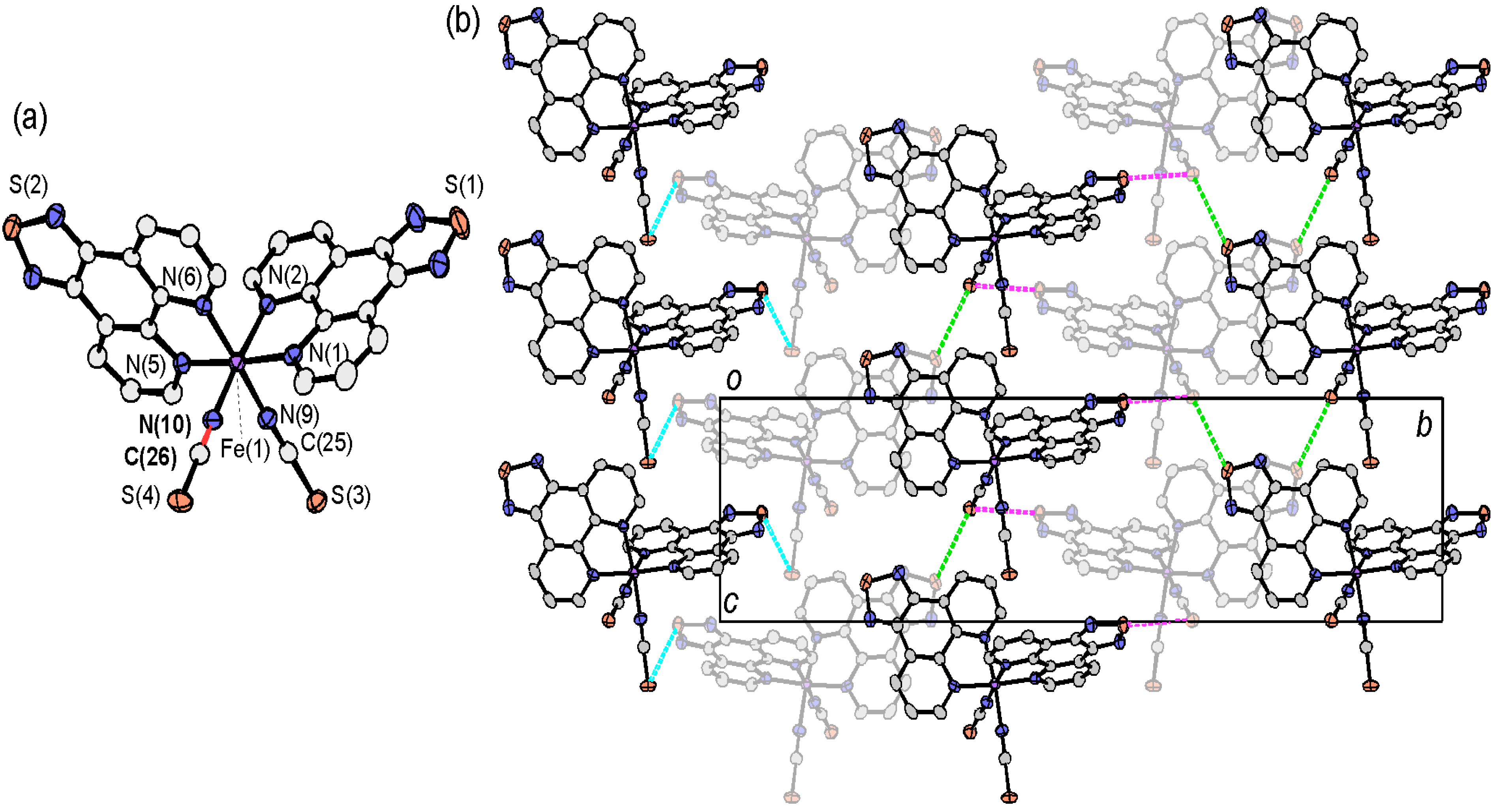

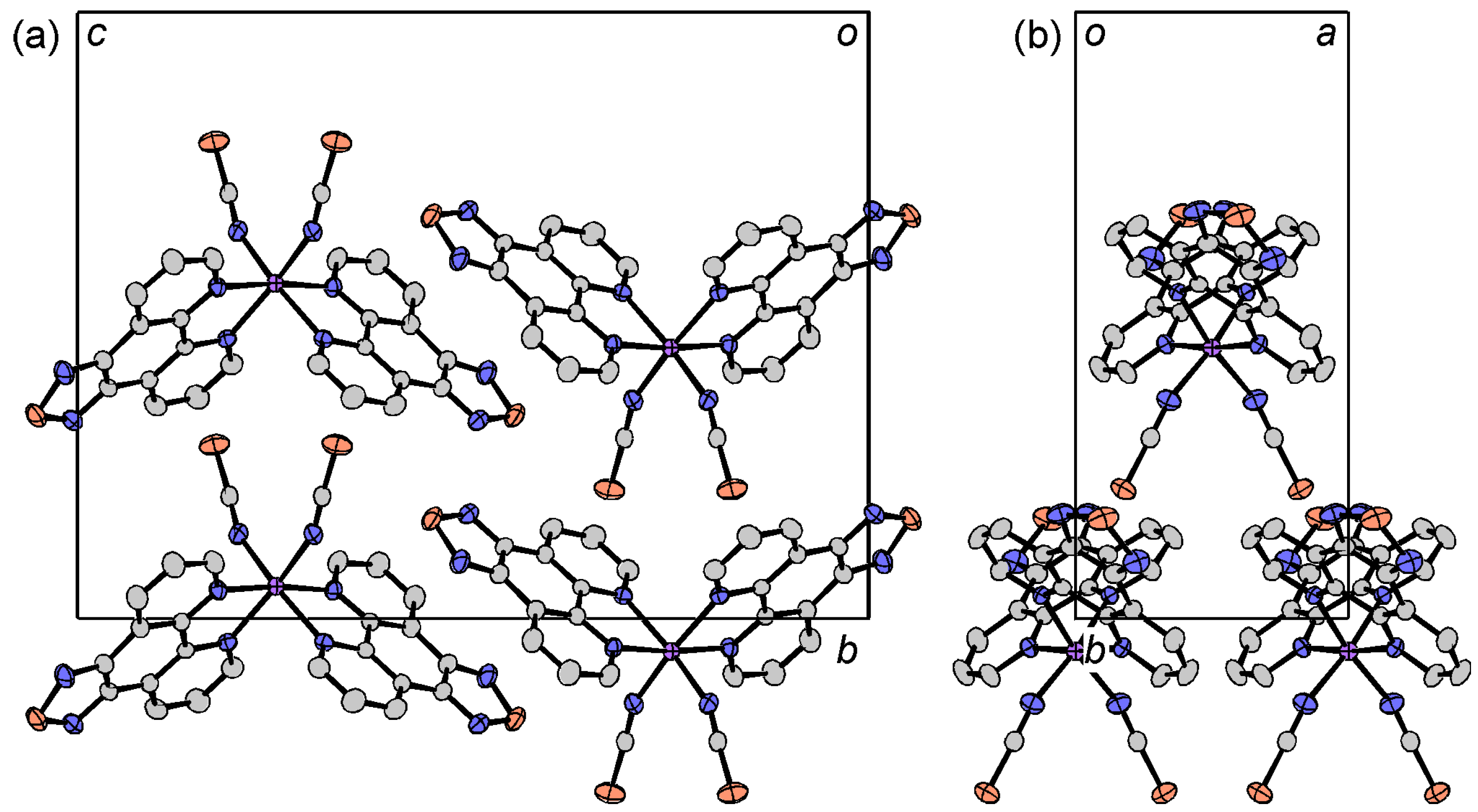
2.4. Crystal Structures of tdap Transition Metal Complexes
2.4.1. [M(tdap)2X2]


2.4.2. [M(tdap)2X2]•solv
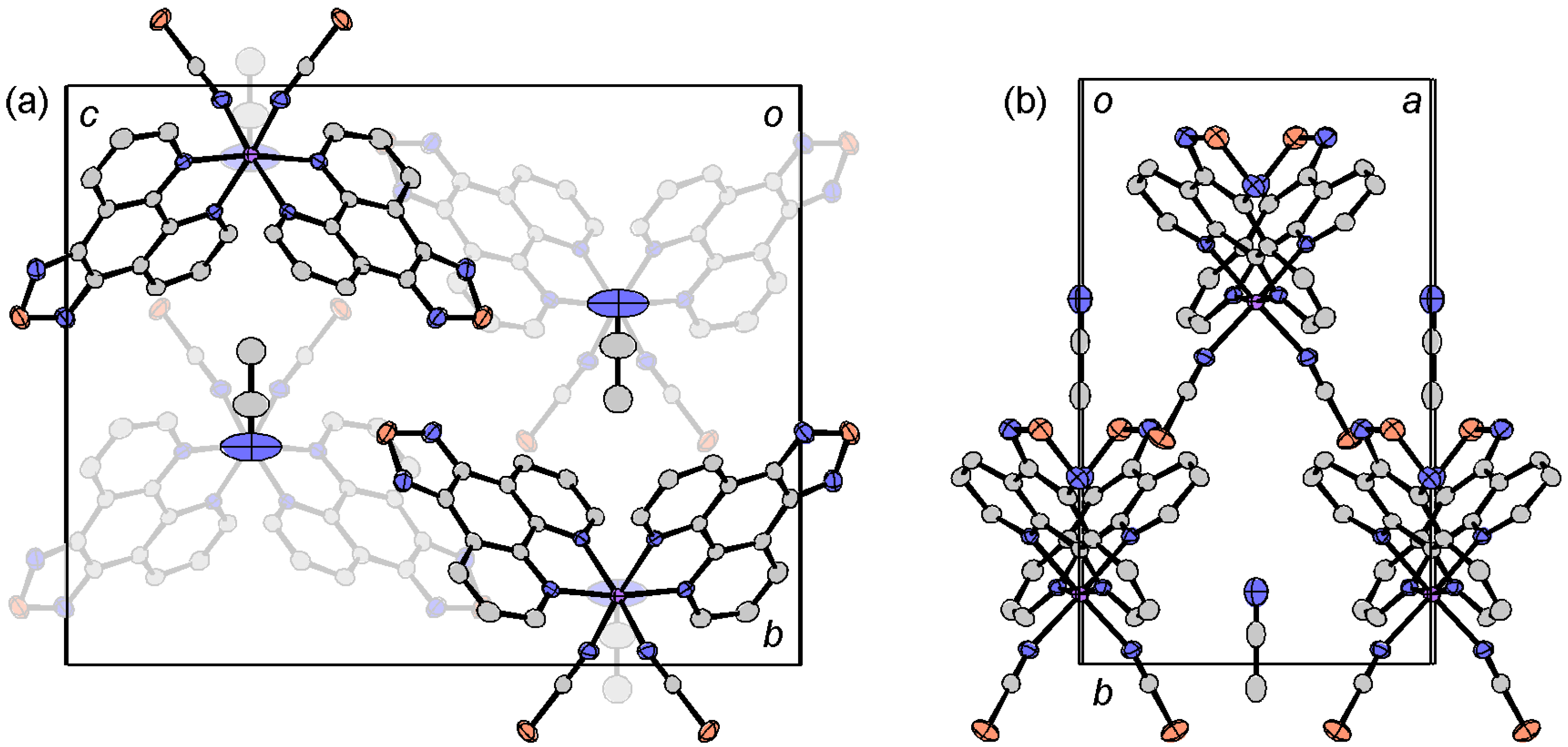

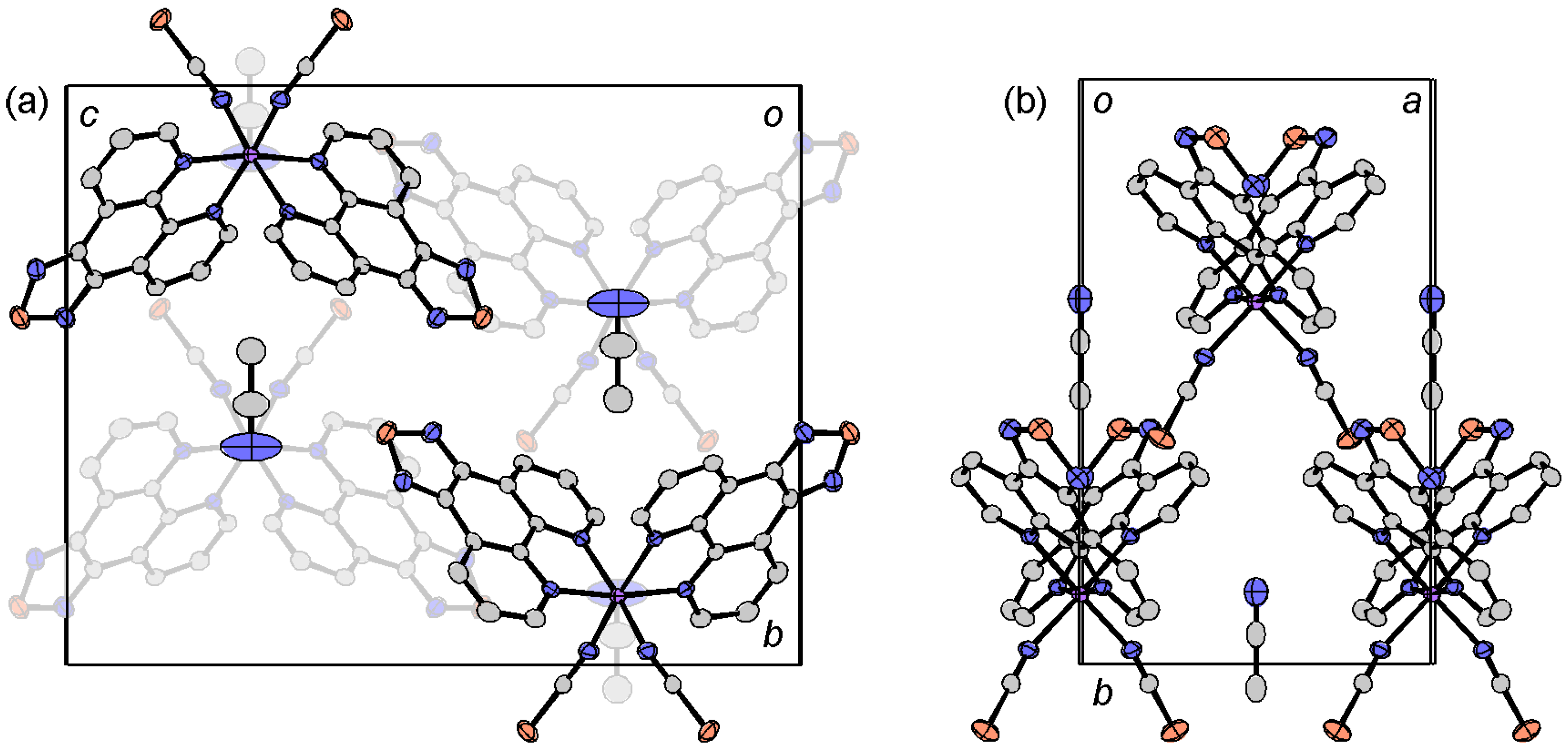
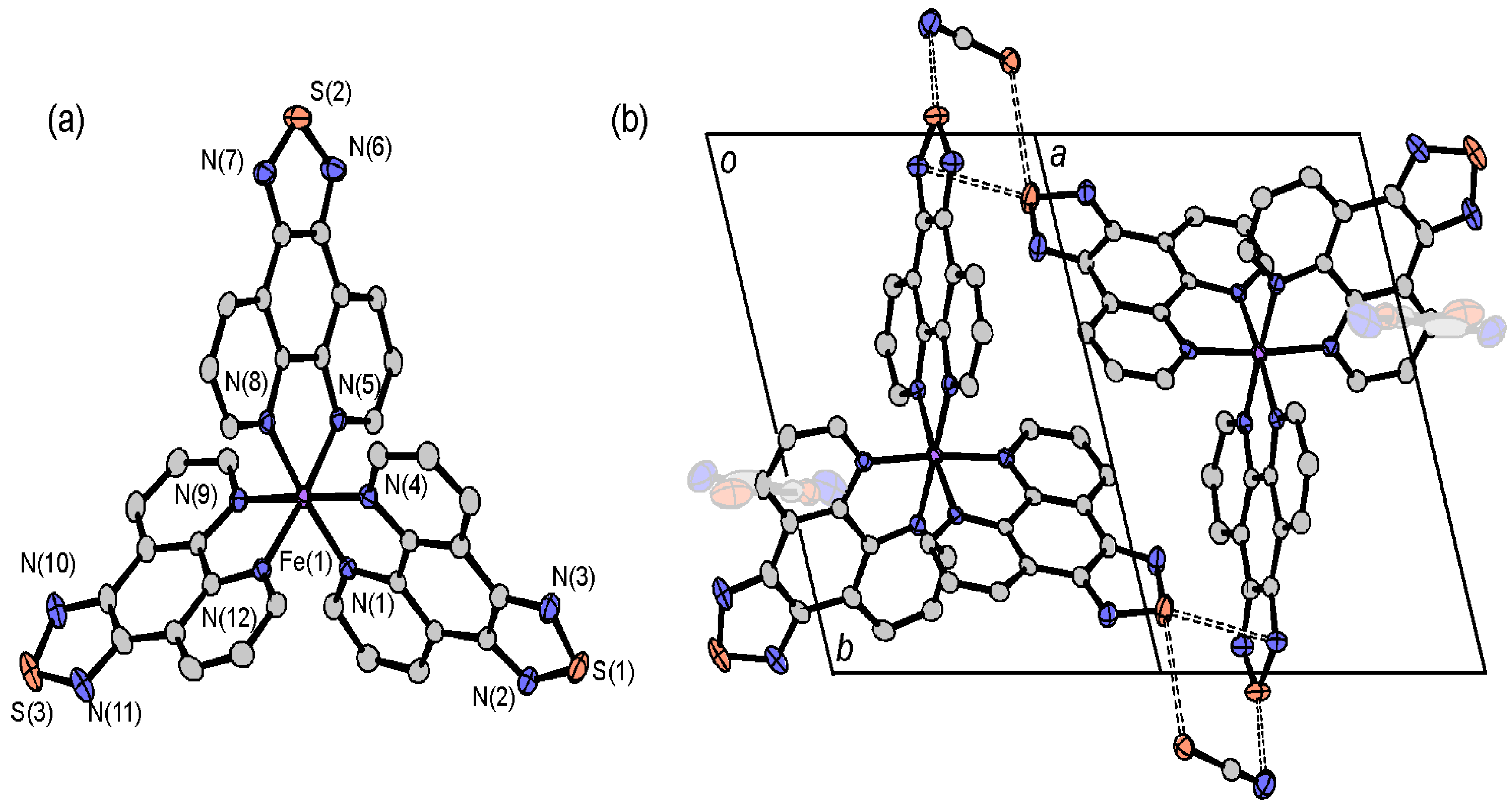
2.4.3. [Fe(tdap)3](NCS)2
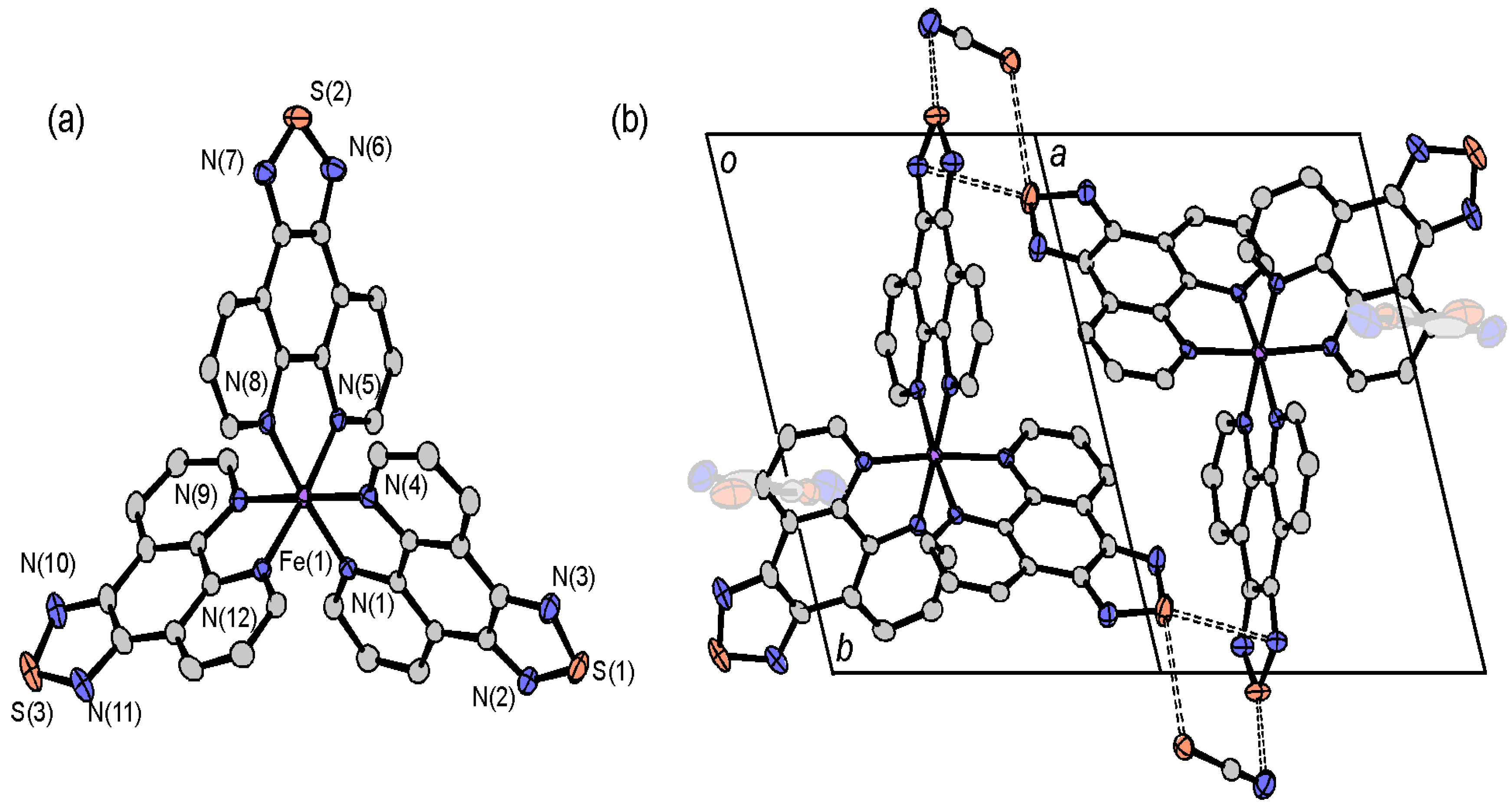
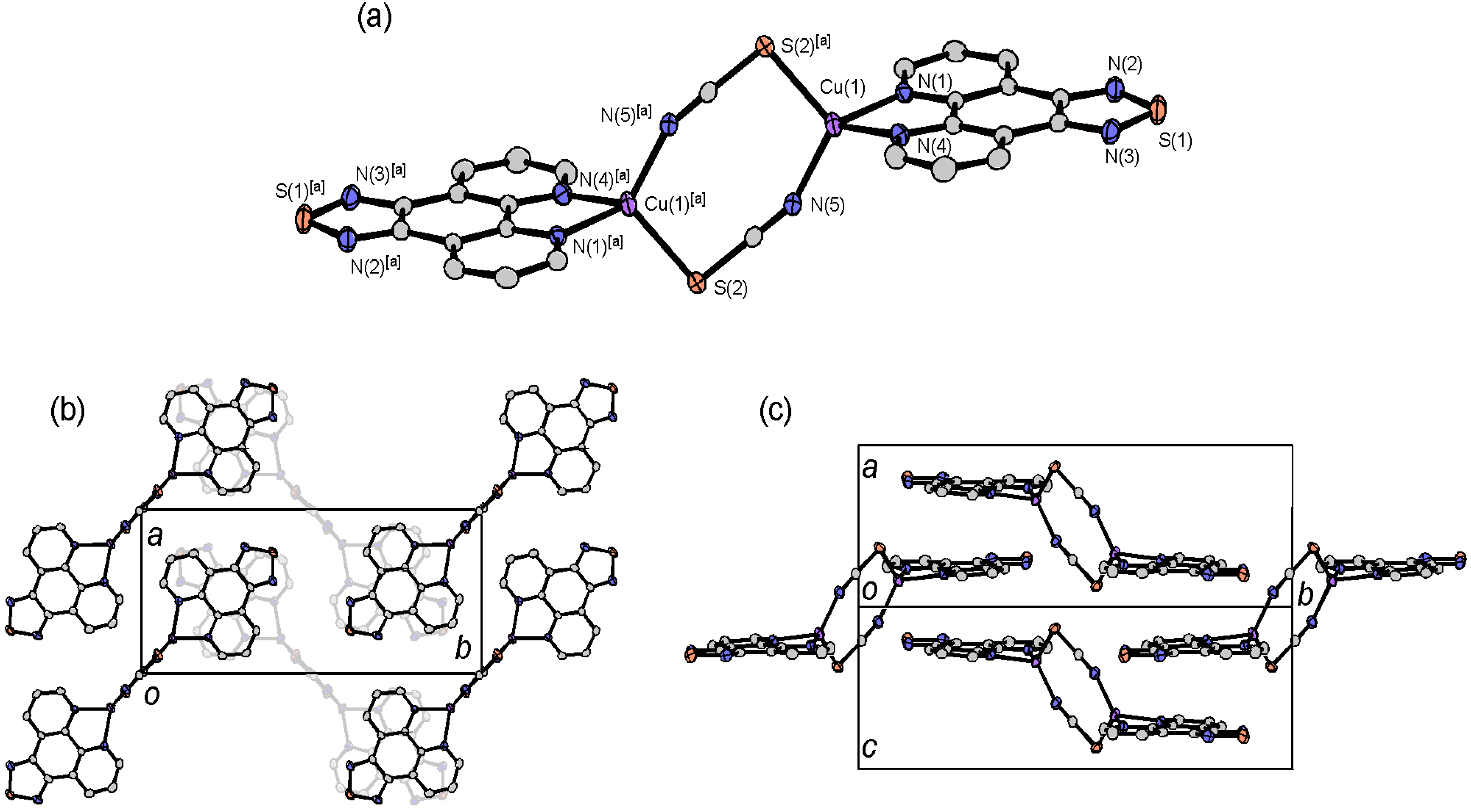
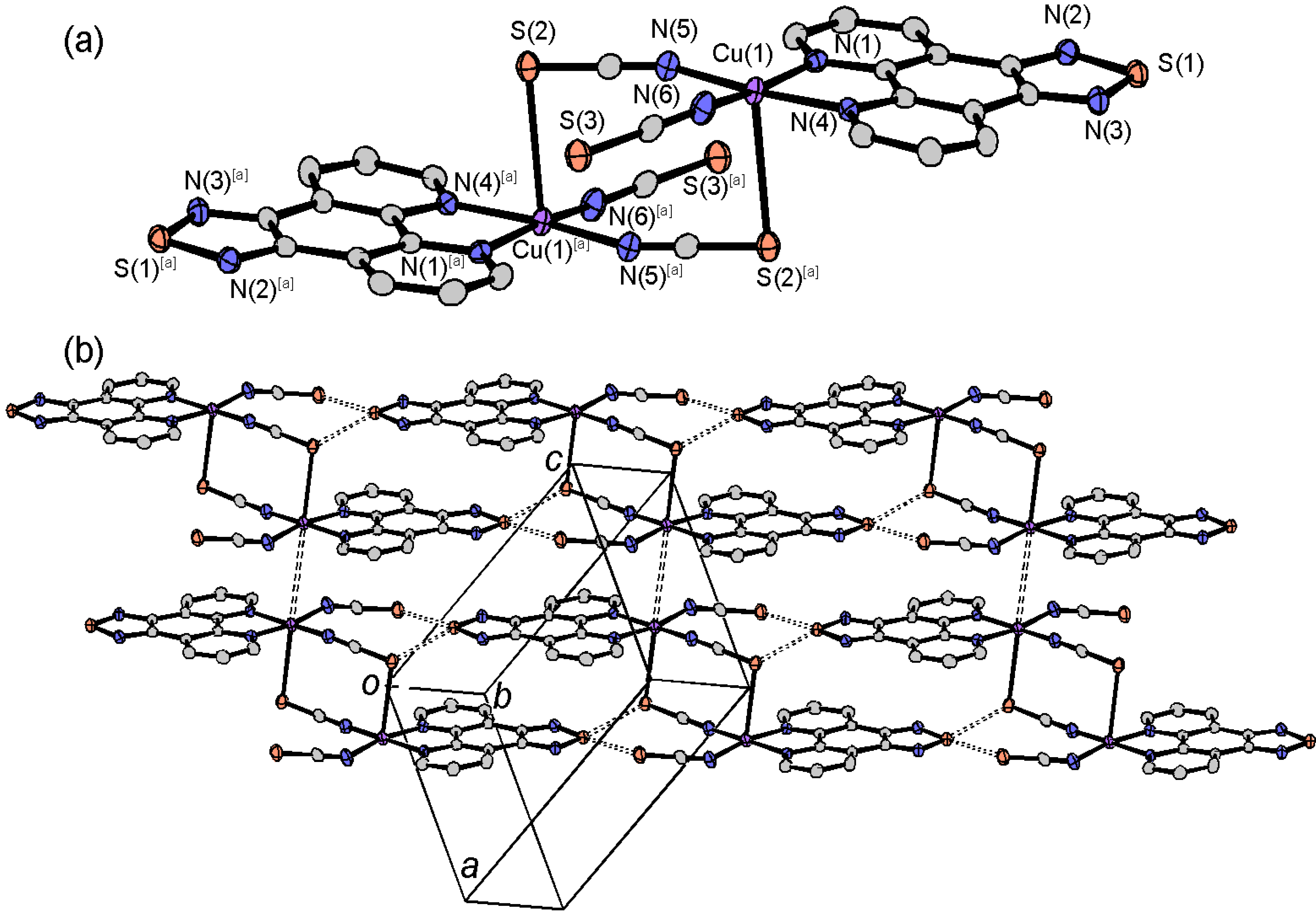
2.4.4. Dinuclear Copper Complexes


3. Radical Anion Salts of [1,2,5]thiadiazolo[3,4-f][1,10]phenanthroline 1,1-dioxide
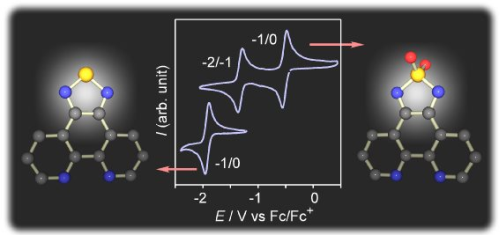
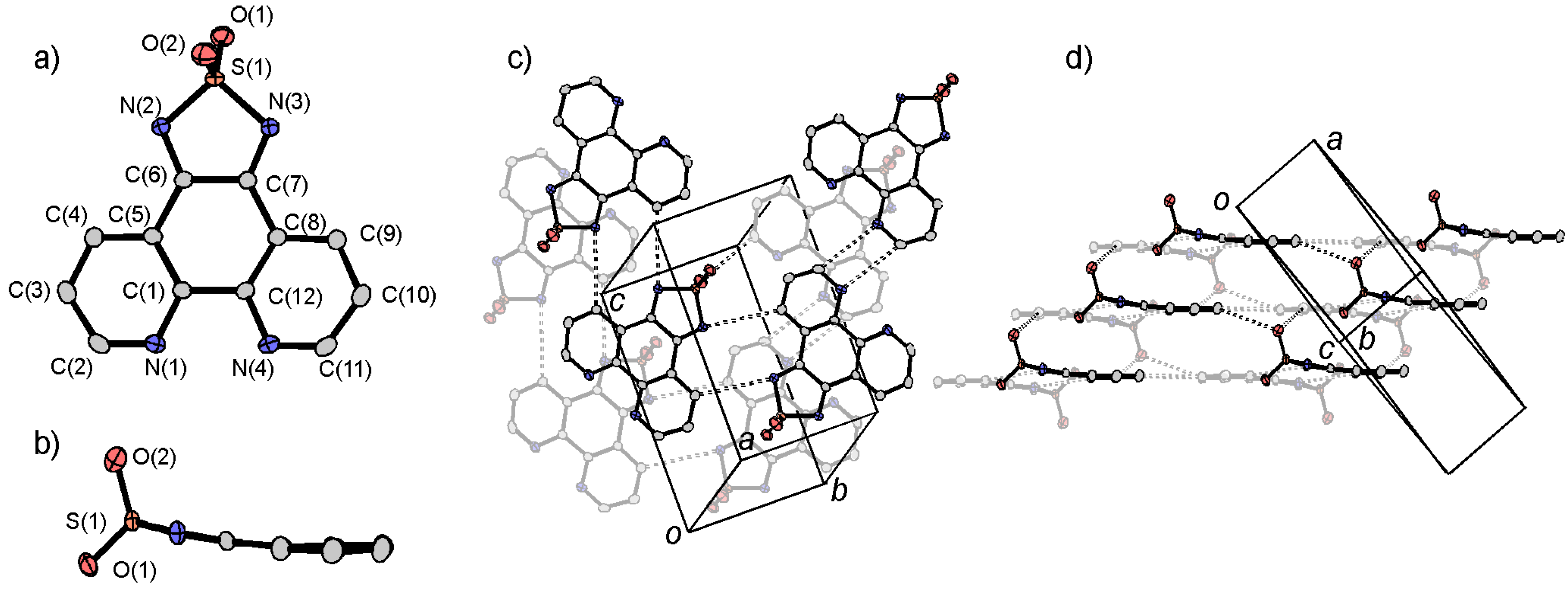
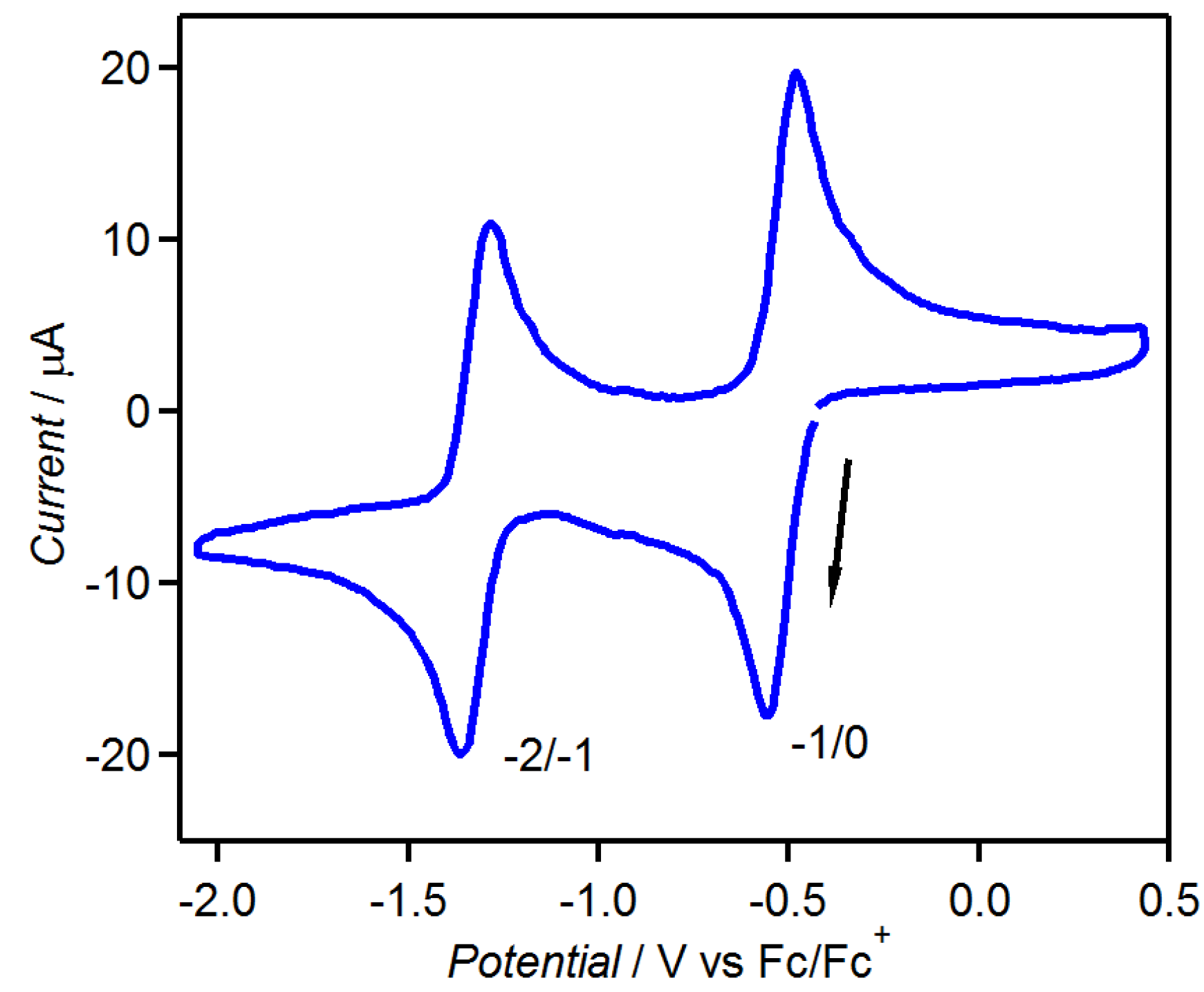
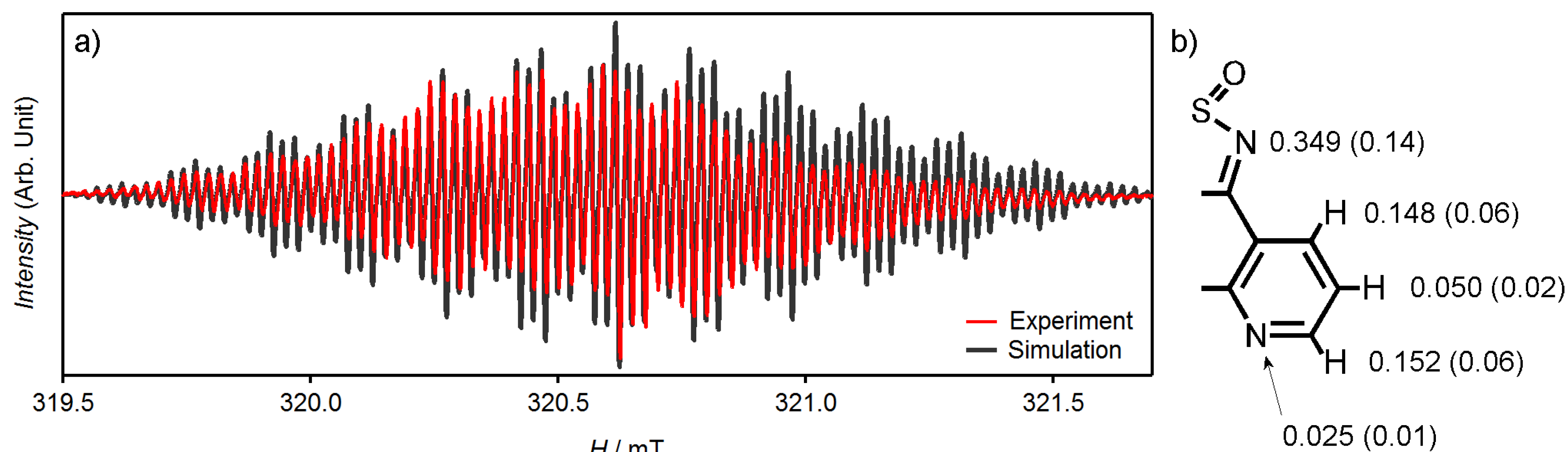
3.1. [1,2,5]thiadiazolo[3,4-f][1,10]phenanthlorine 1,1-dioxide (tdapO2)
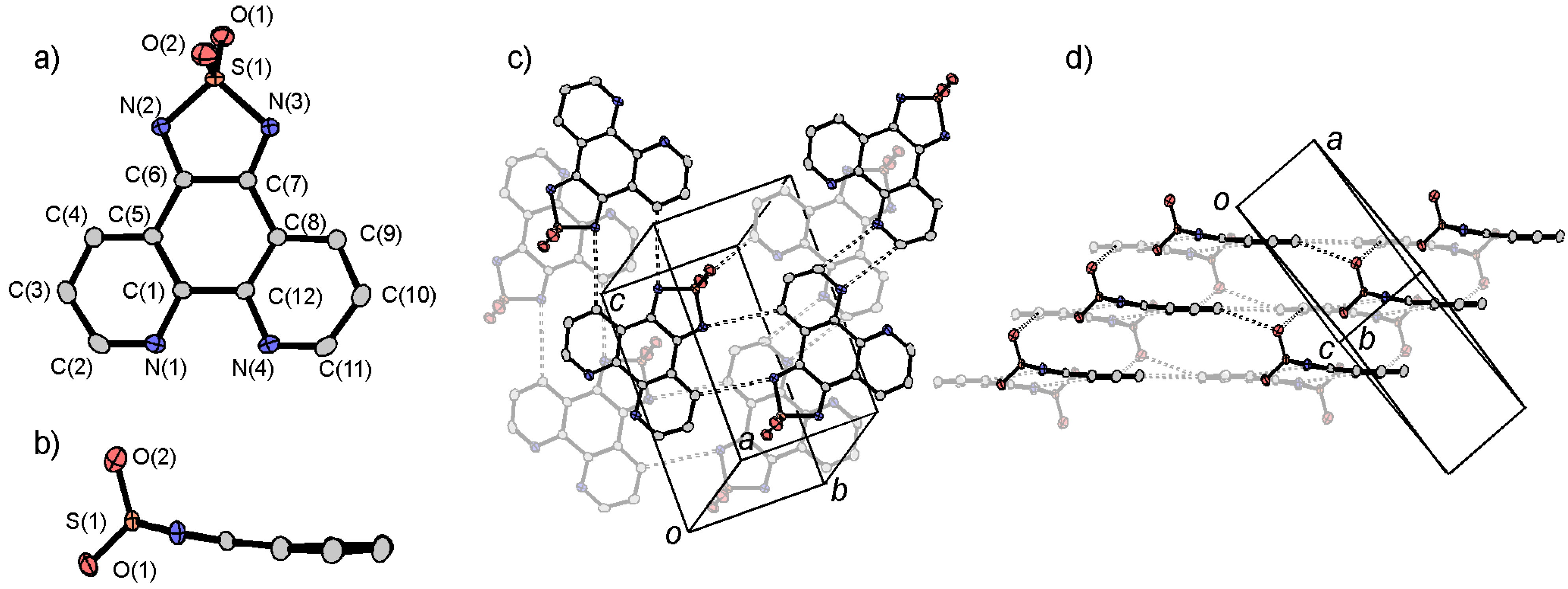
3.2. Synthesis of Intermolecular Compounds and Anion Salts of tdapO2
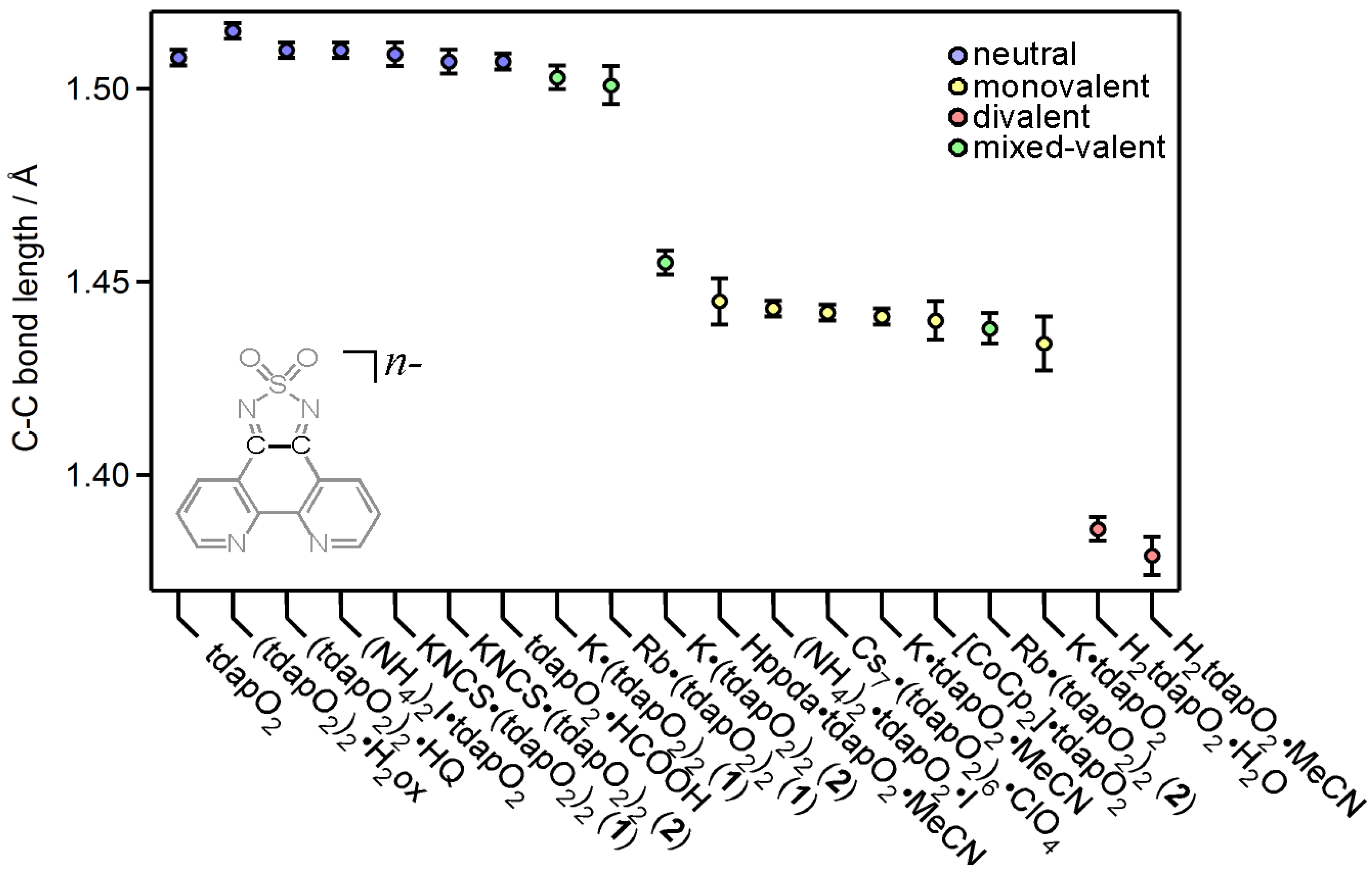
3.3. Crystal Structures and Magnetic Properties of Radical Anion Salts
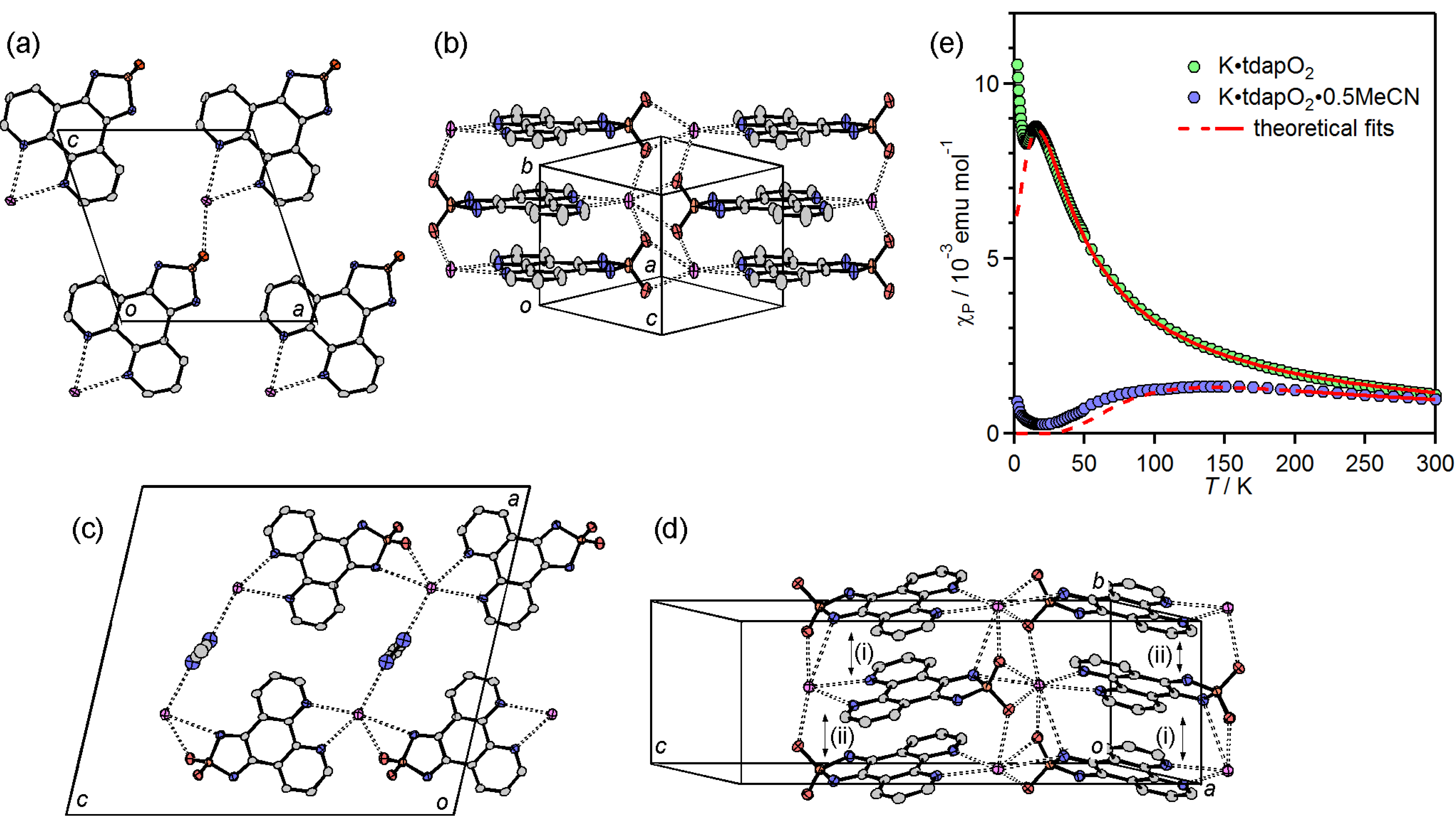
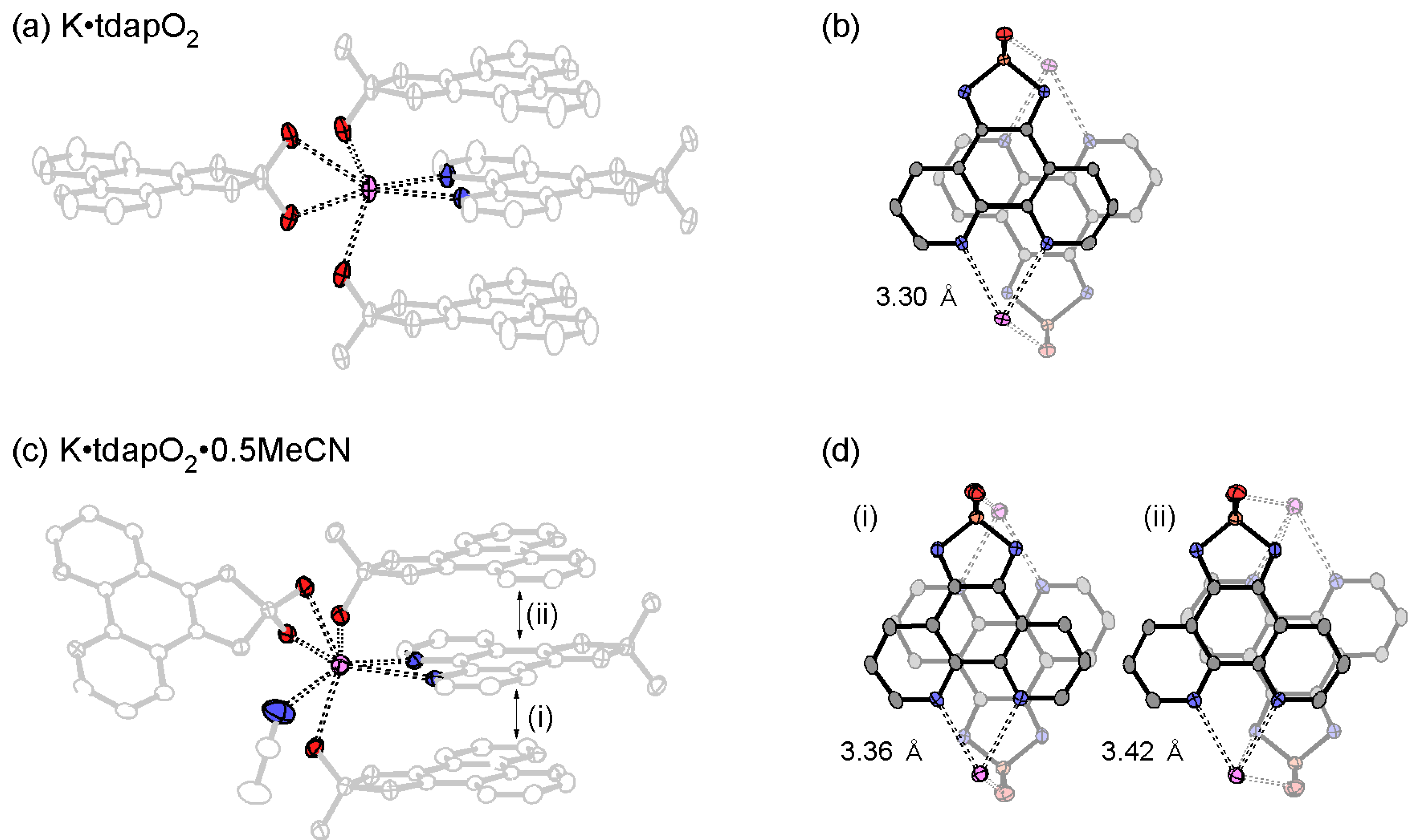
3.3.1. Nonsolvated and Solvated Potassium Salts: K•tdapO2 and KtdapO2•0.5MeCN
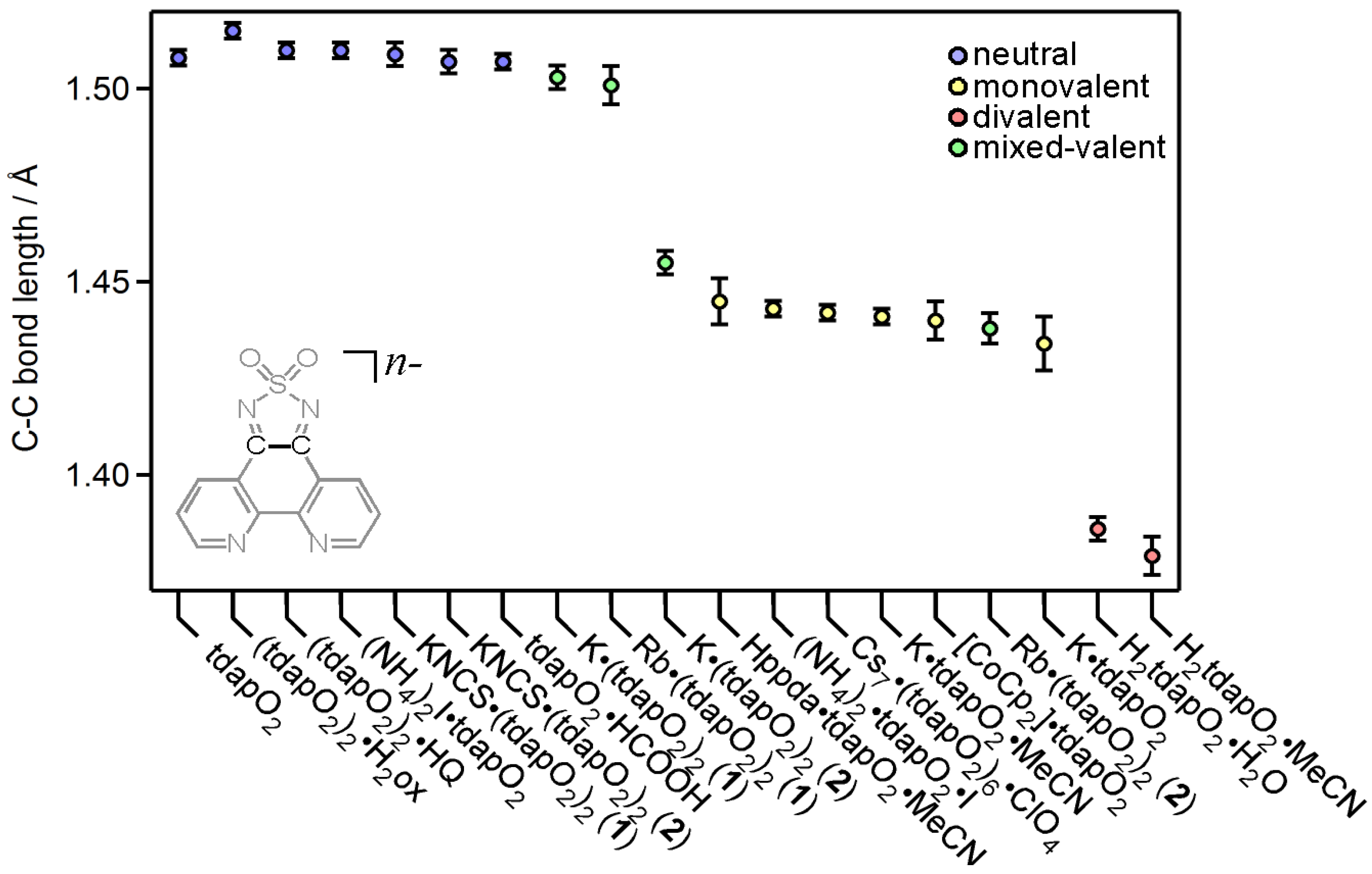
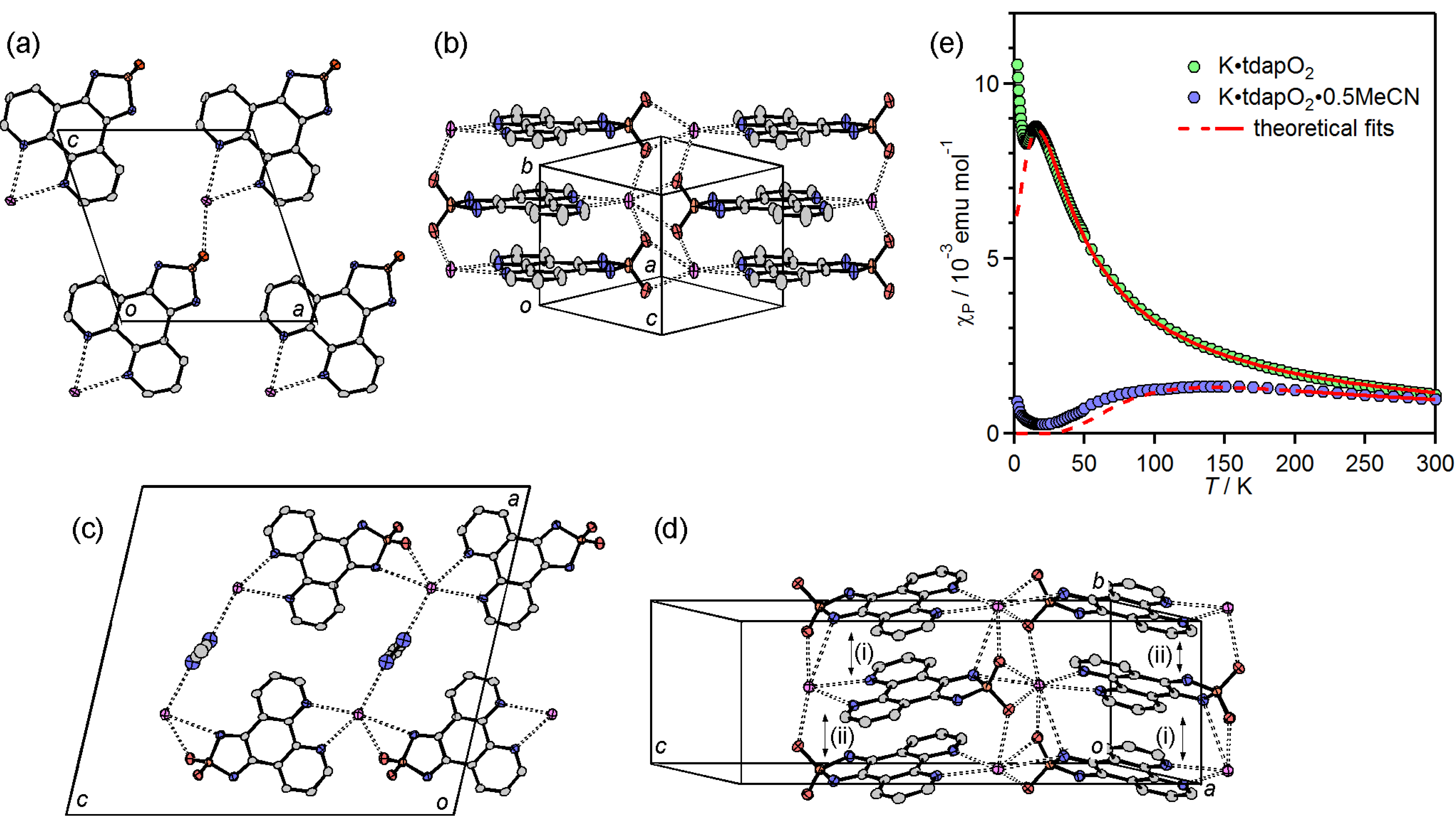
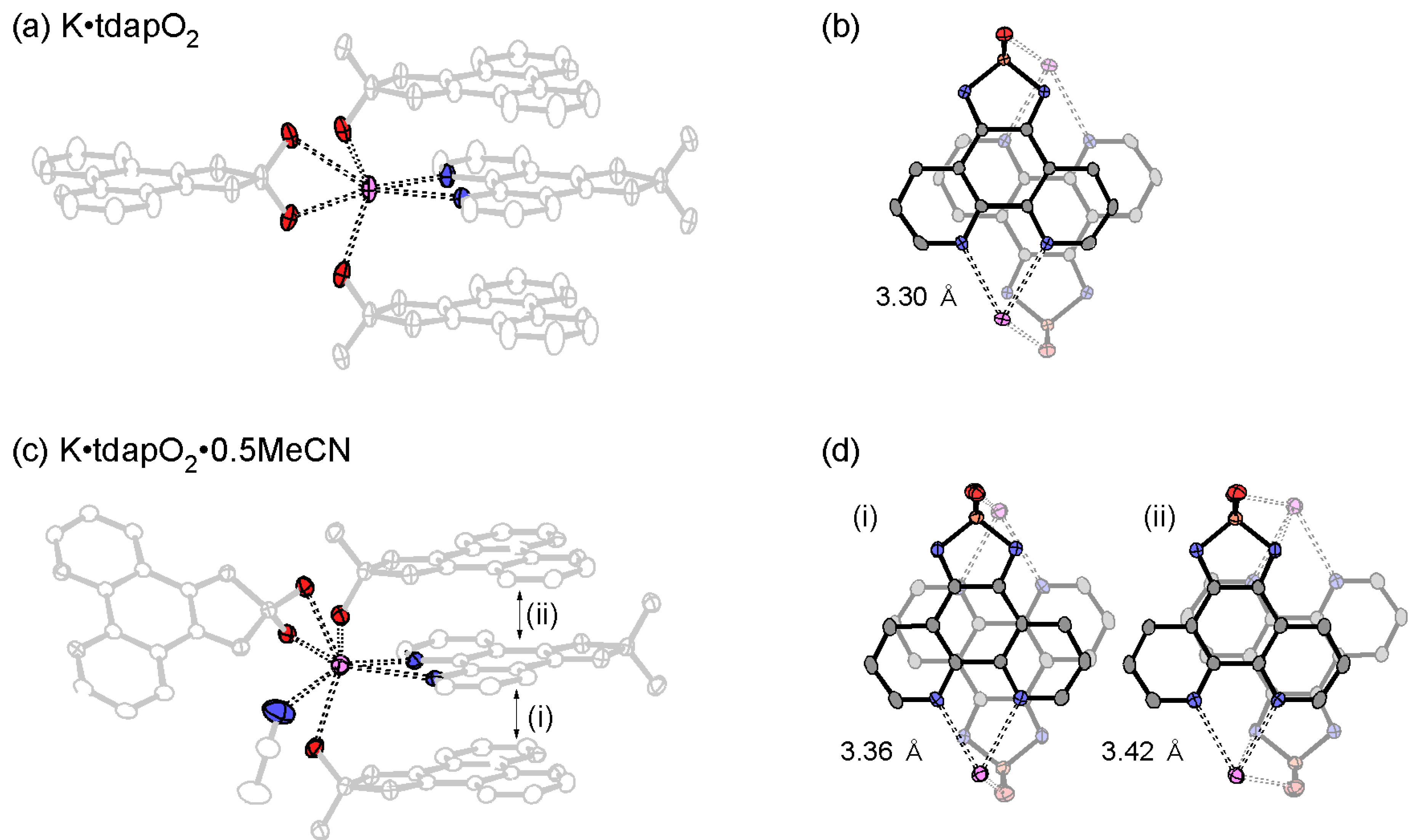
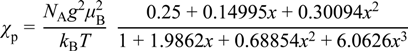
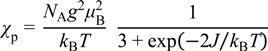
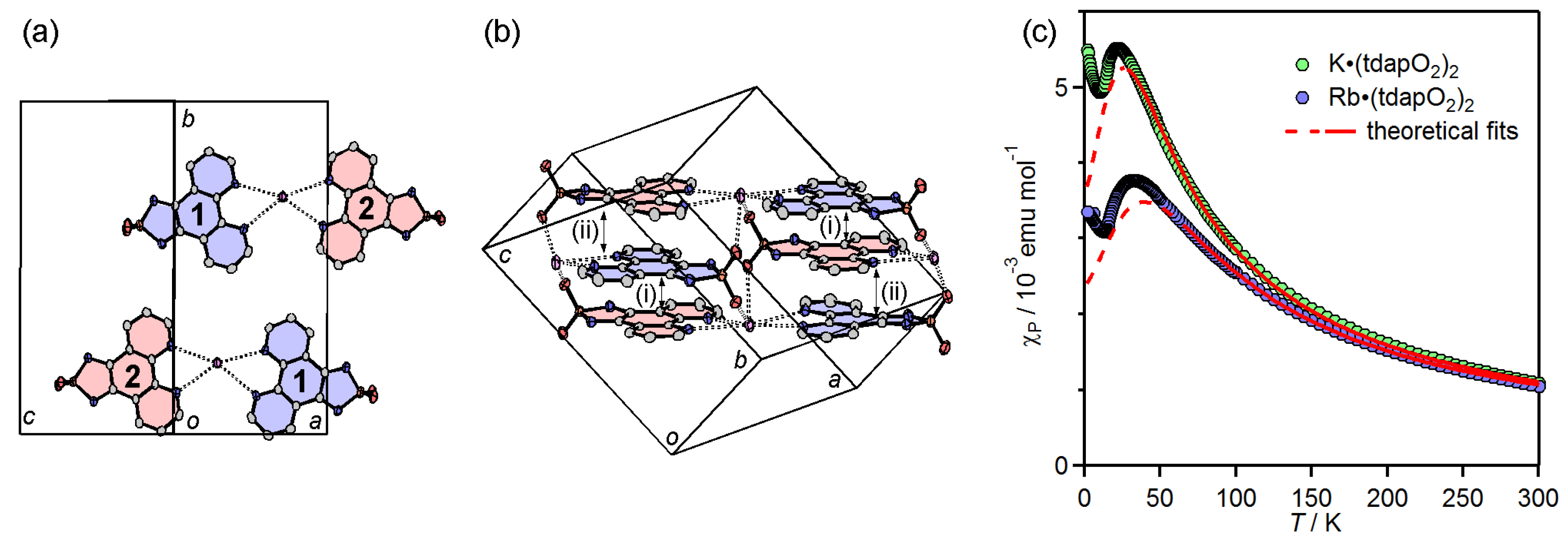
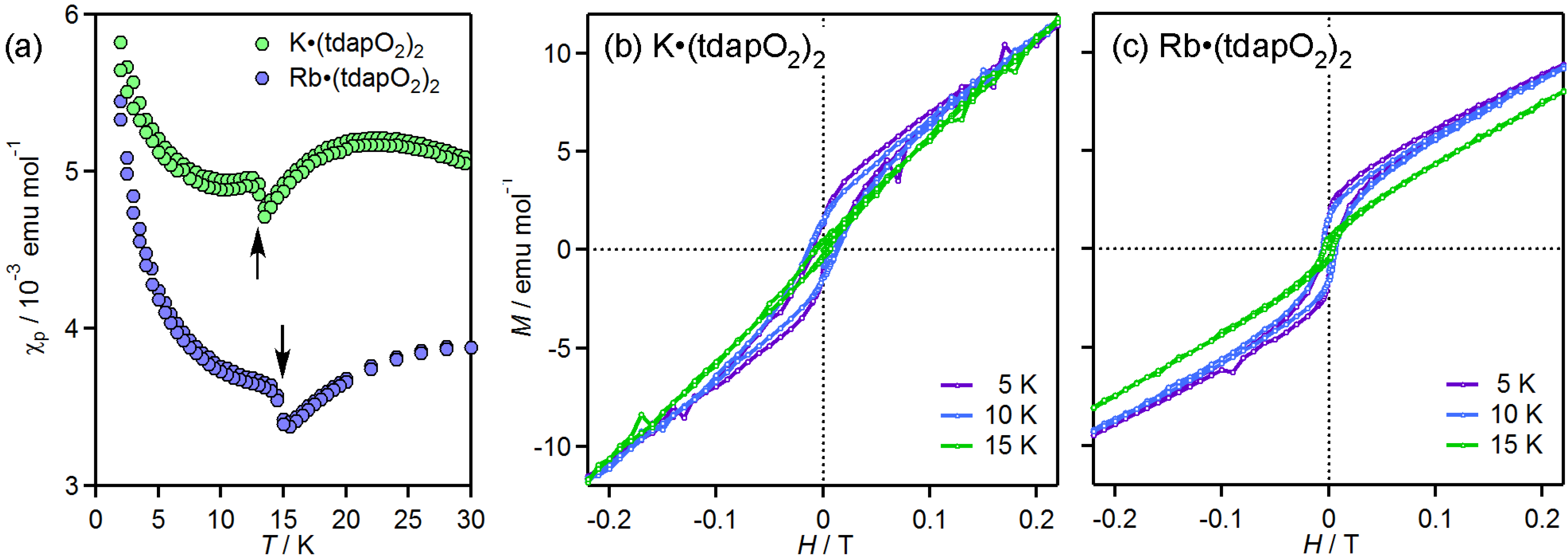
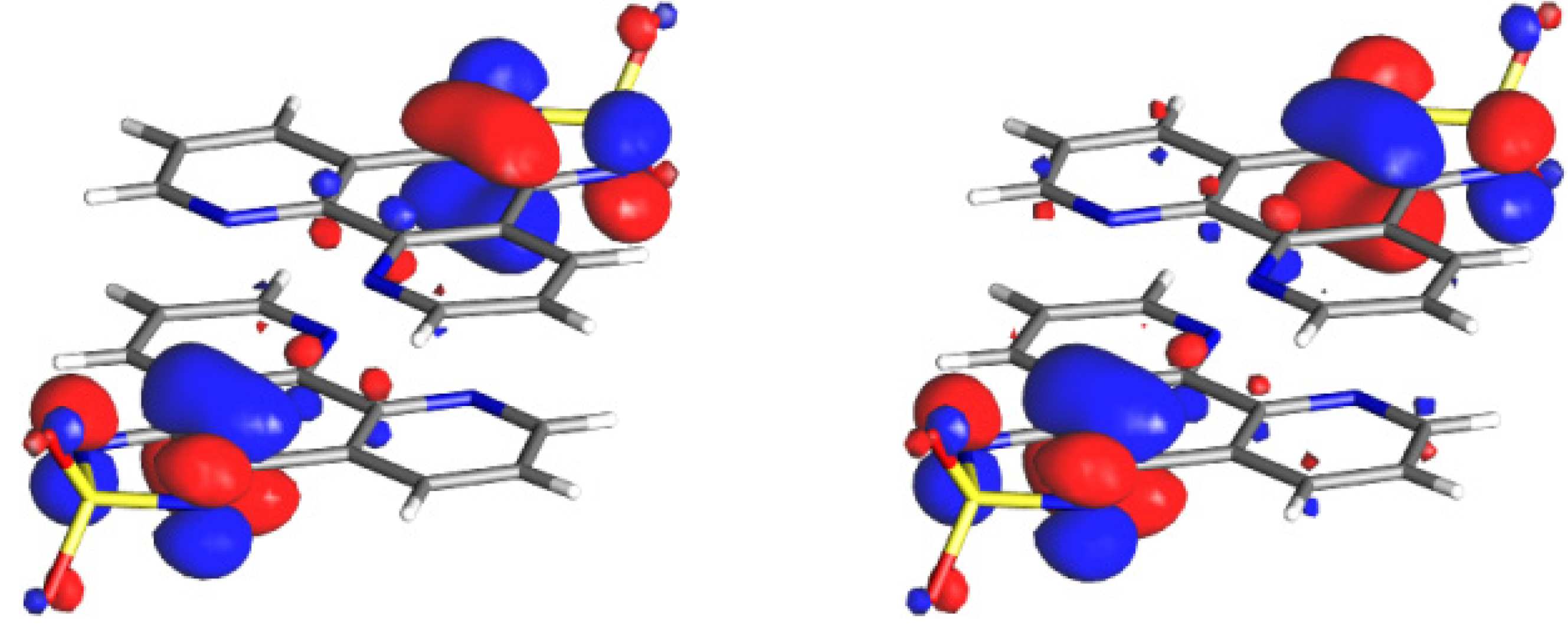
3.3.2. Mixed Valent Salts: K•(tdapO2)2 and Rb•(tdapO2)2
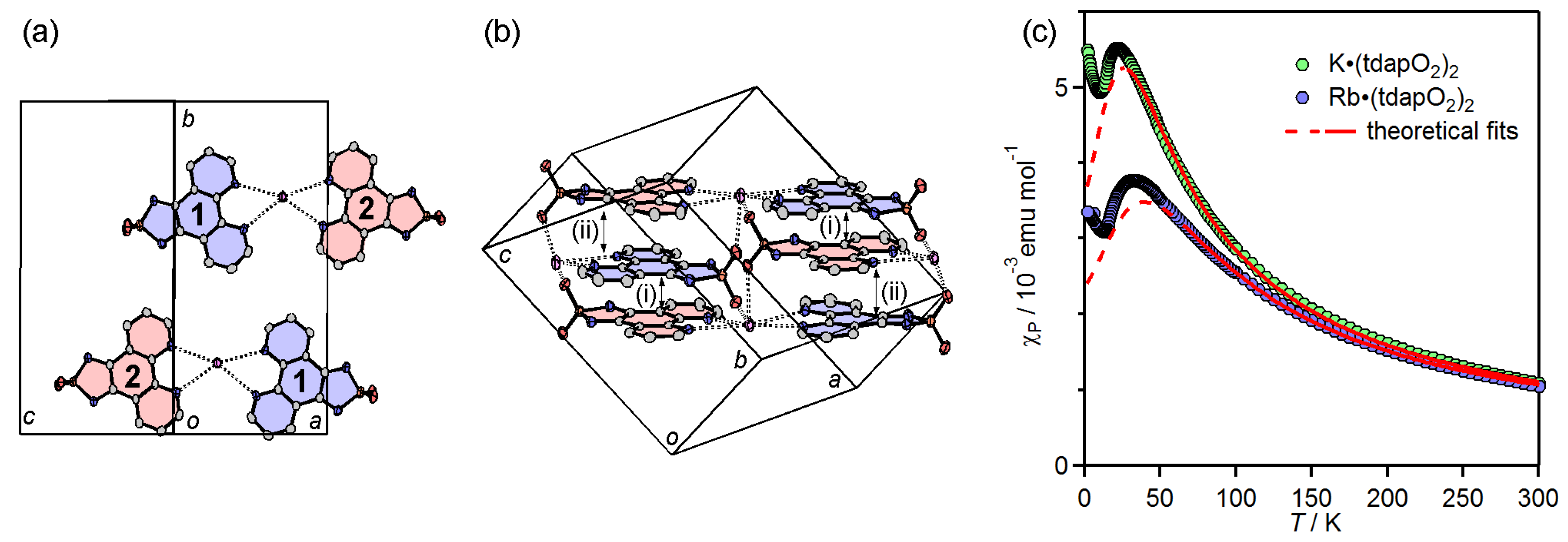
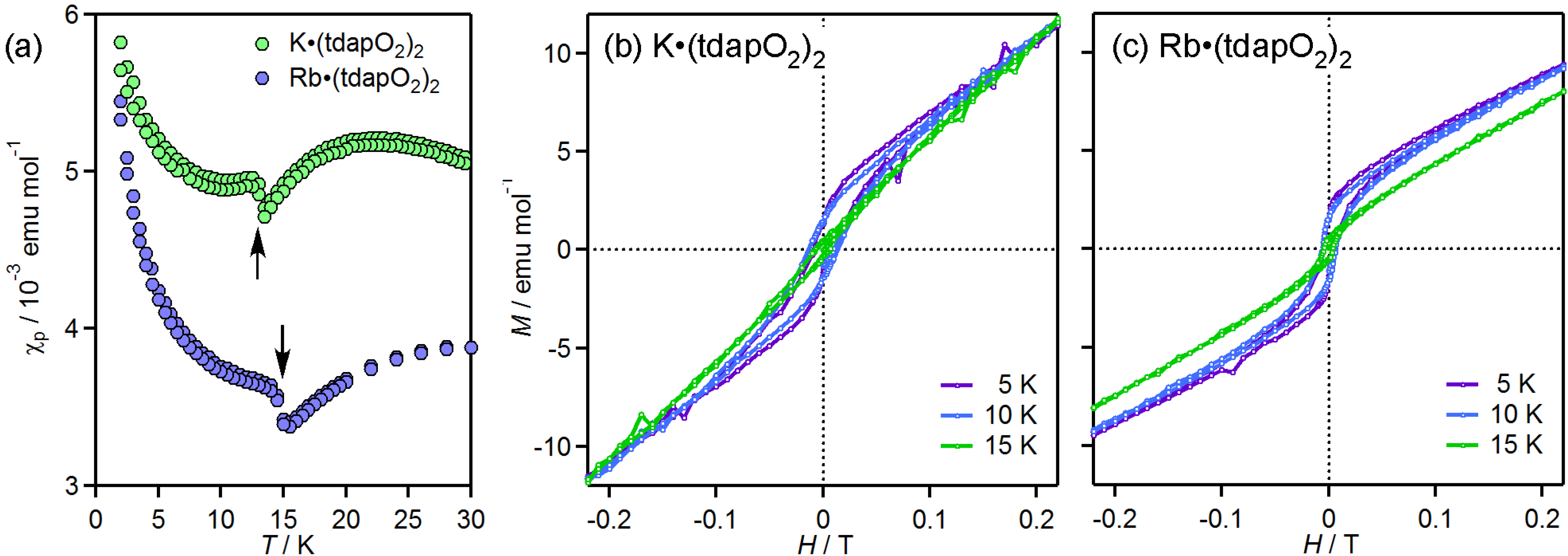
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
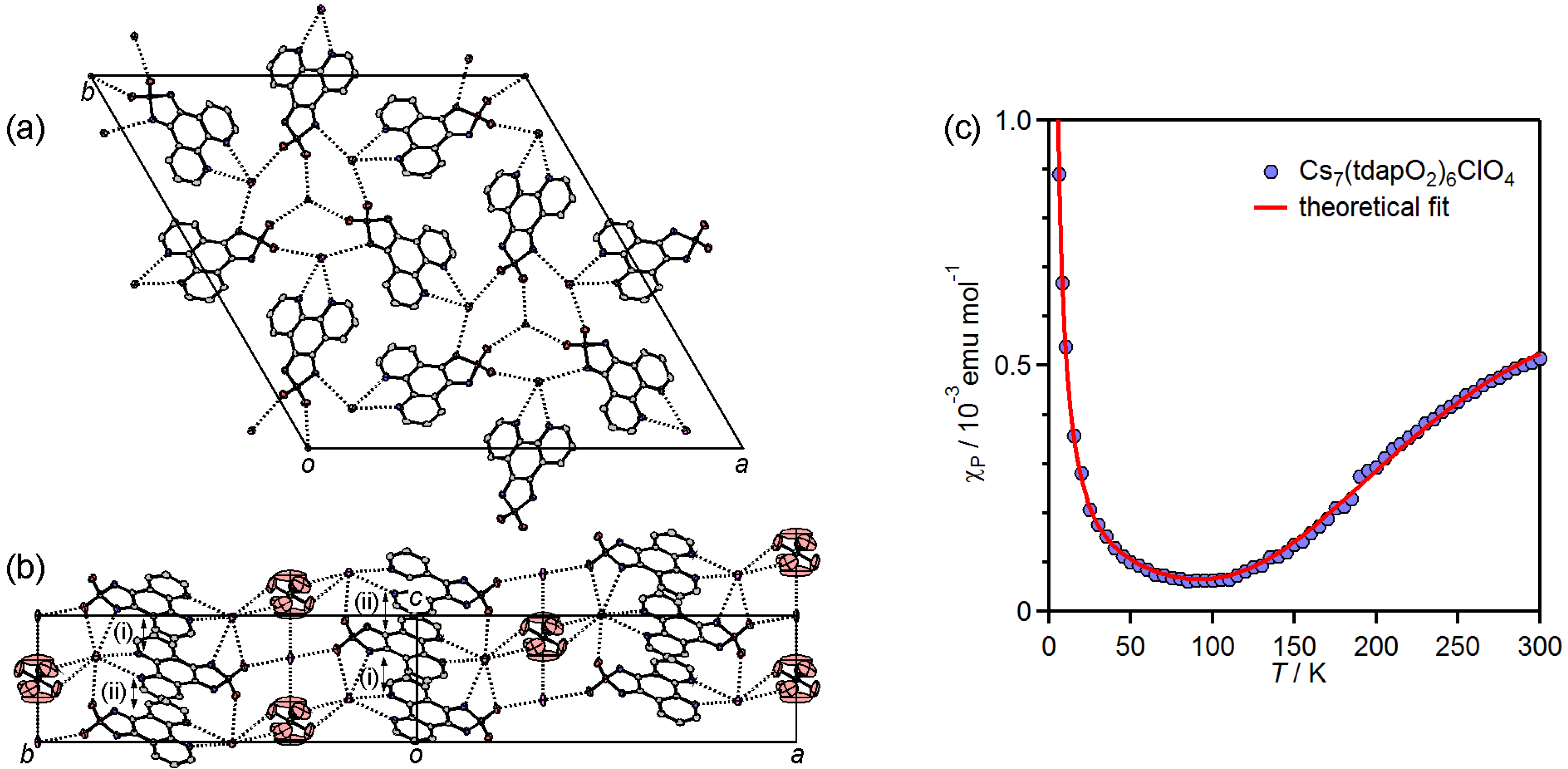
3.3.3. Kagome Lattice Formed by Coordination Bonding: Cs7•(tdapO2)6•ClO4

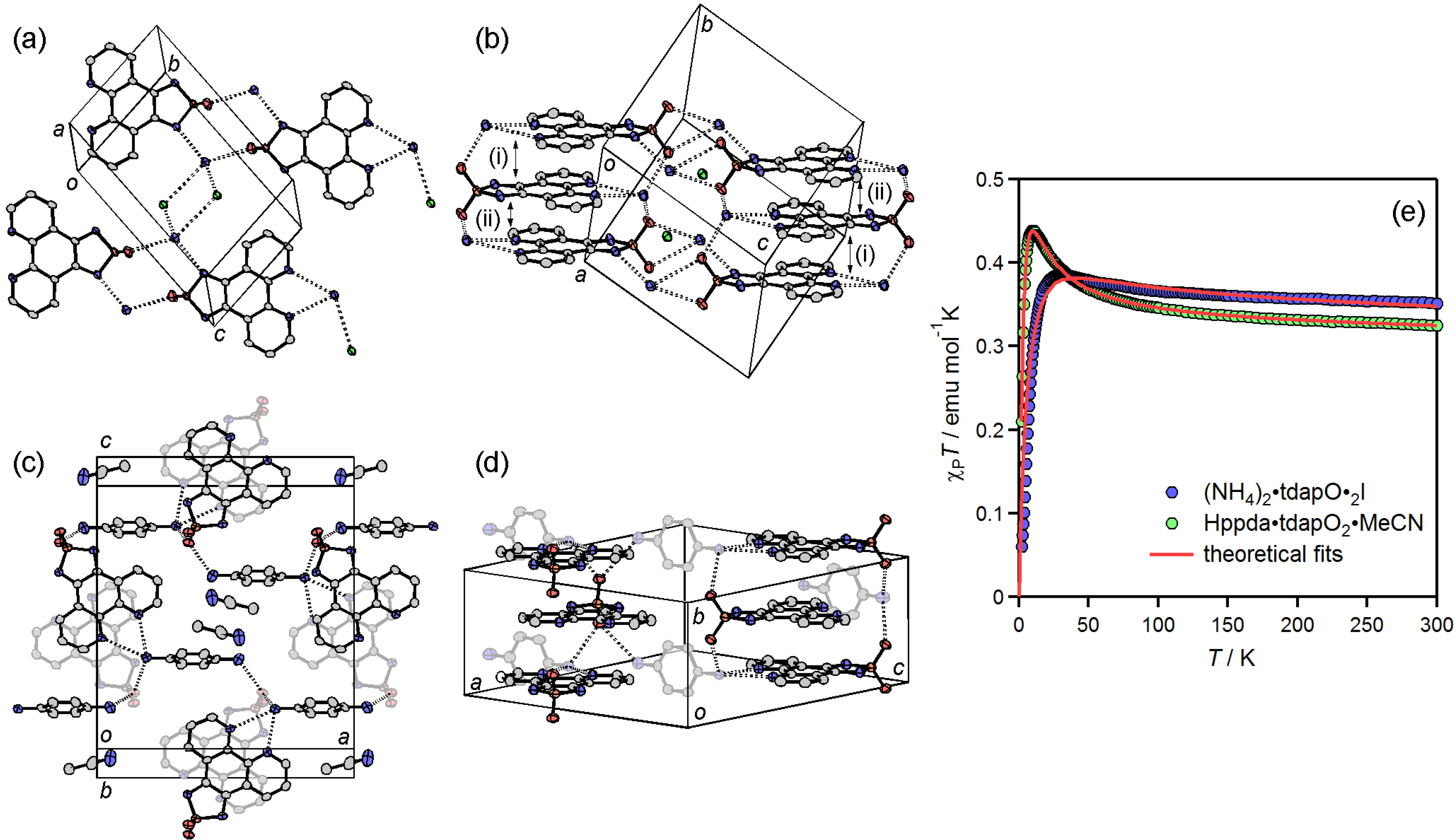
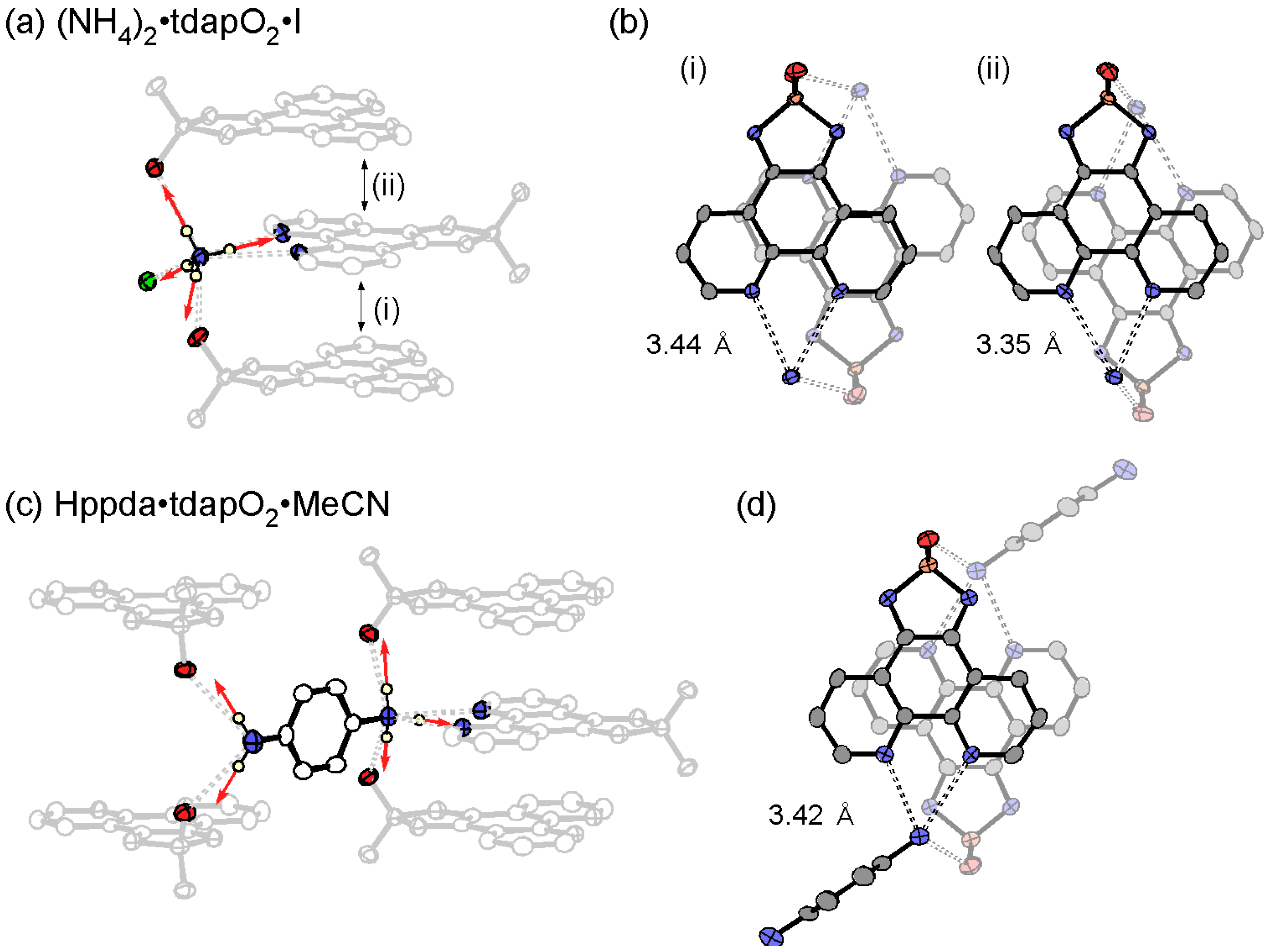
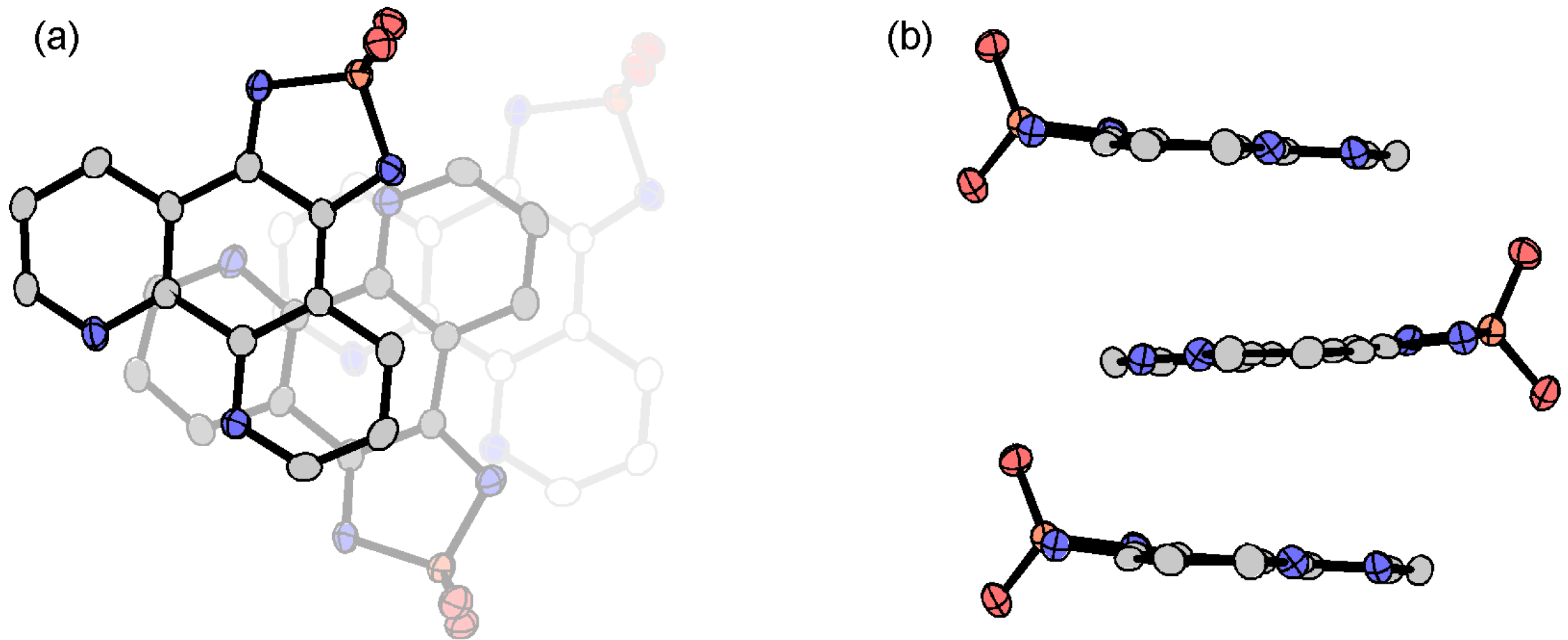
3.3.4. Ammonium Salts: (NH4)2•tdapO2•I and Hppda•tdapO2•MeCN
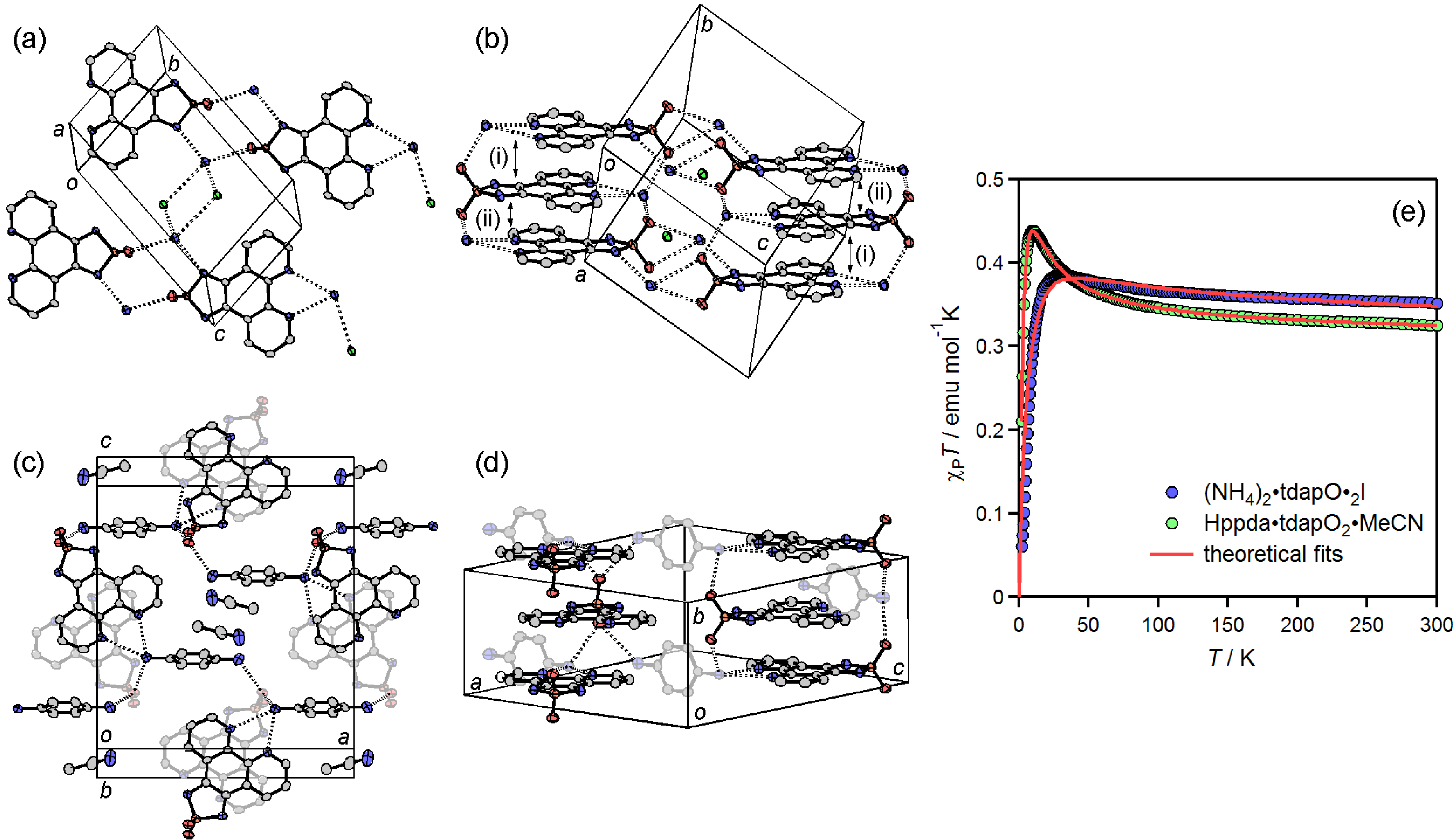
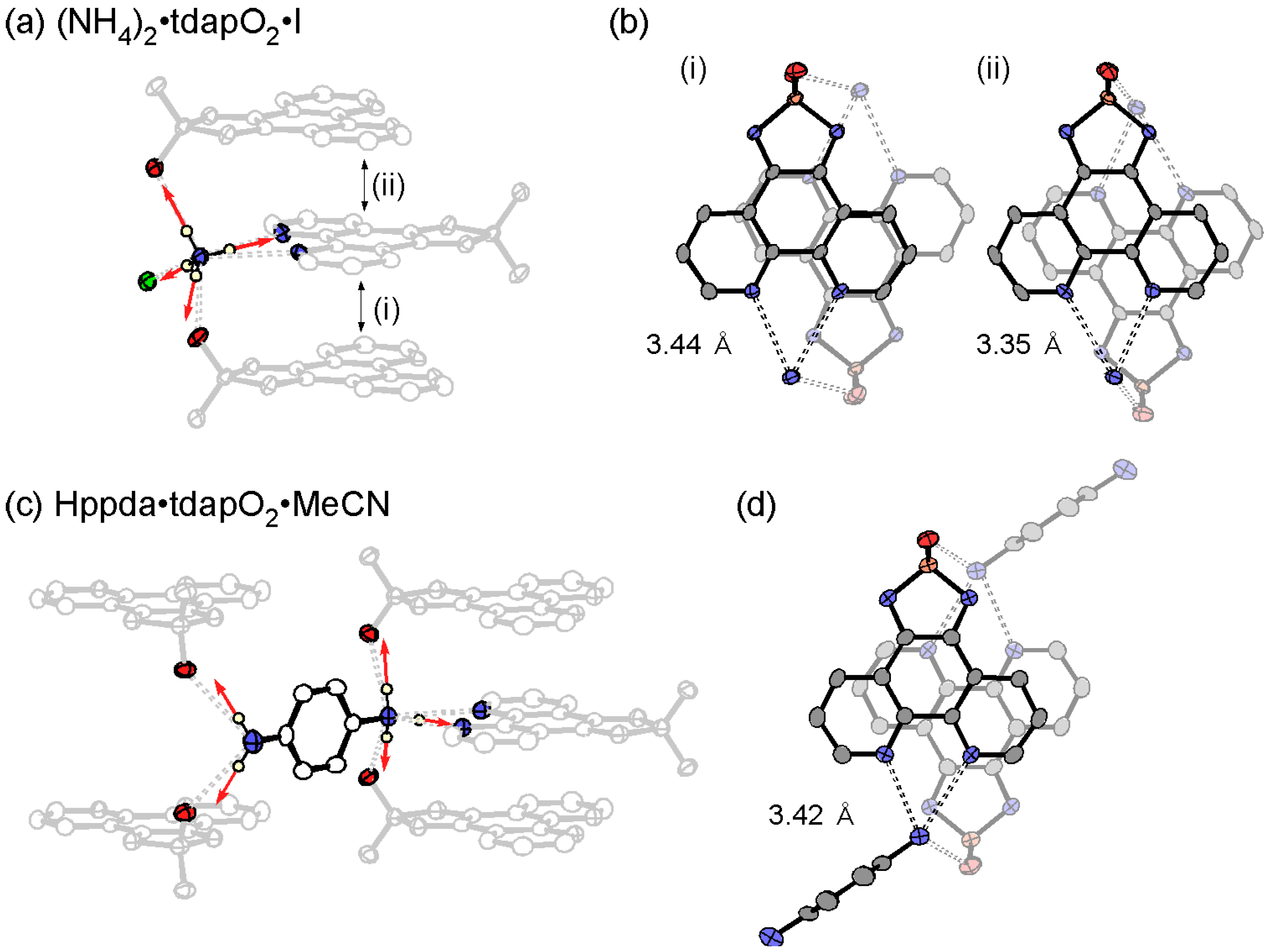
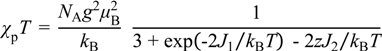
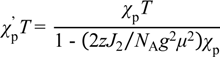
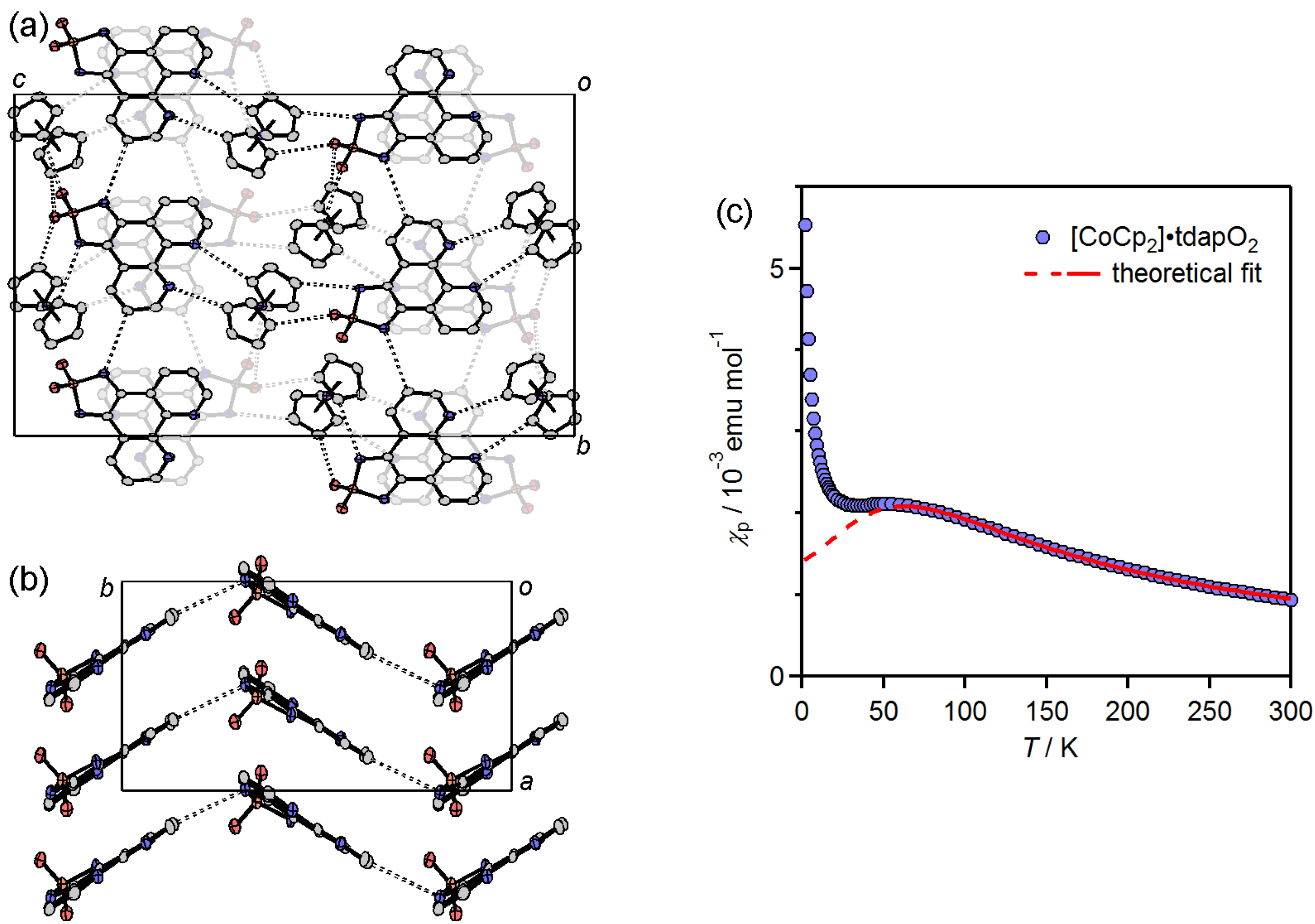
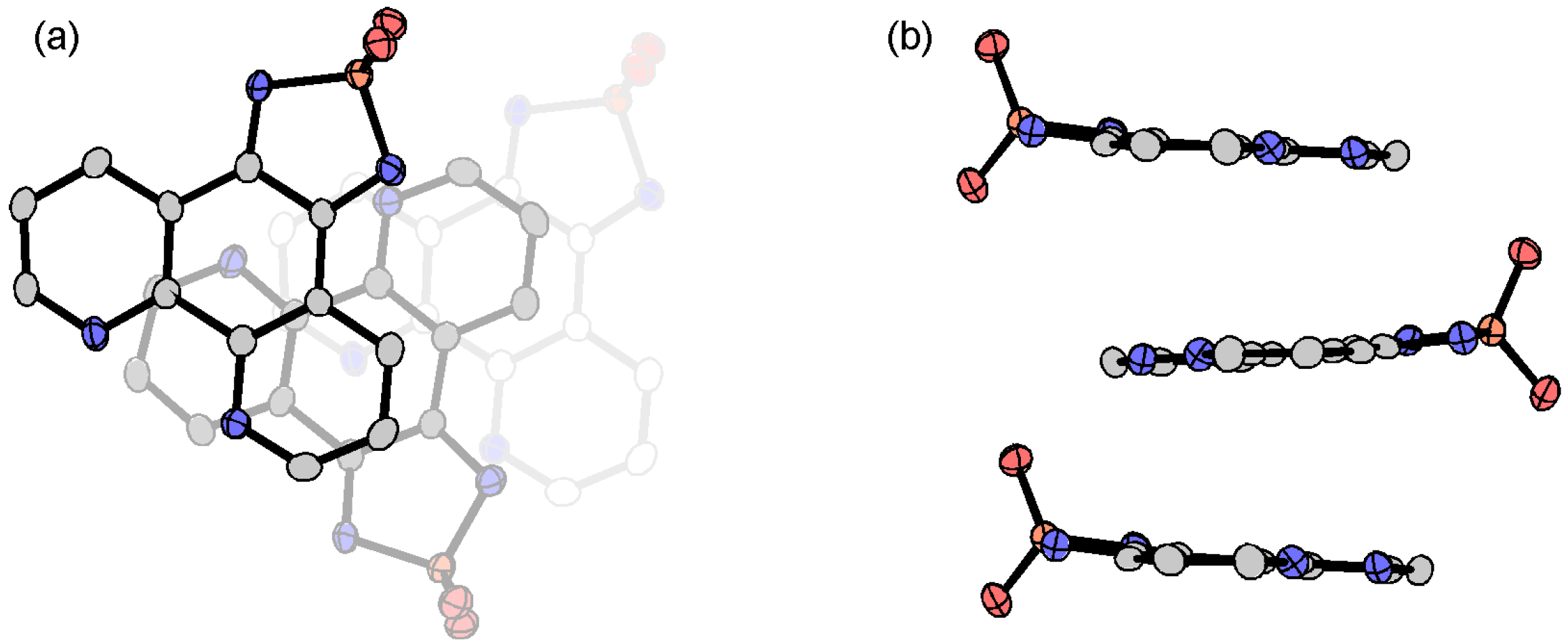
3.3.5. Cobaltocenium Salt: [CoCp2]•tdapO2
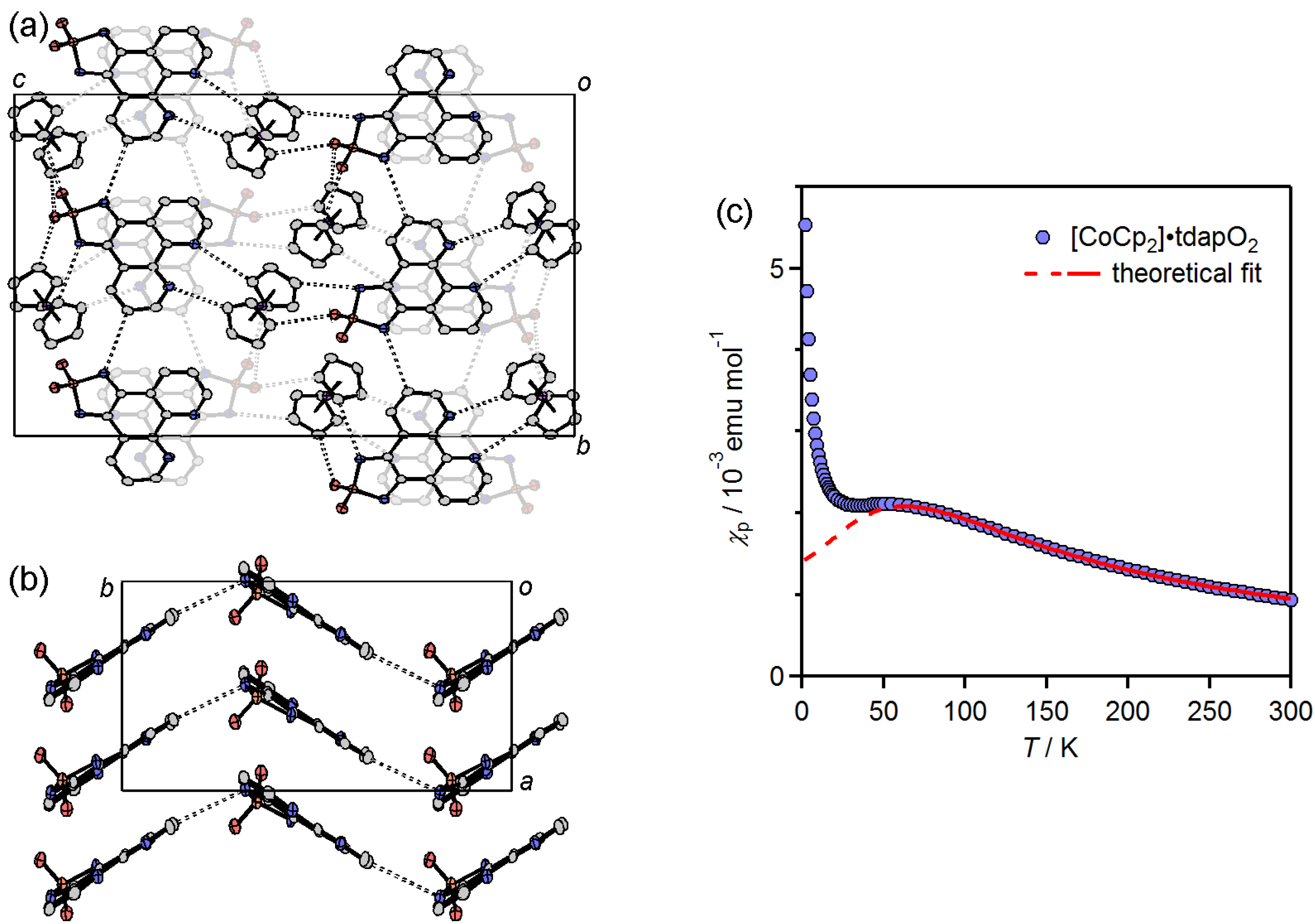
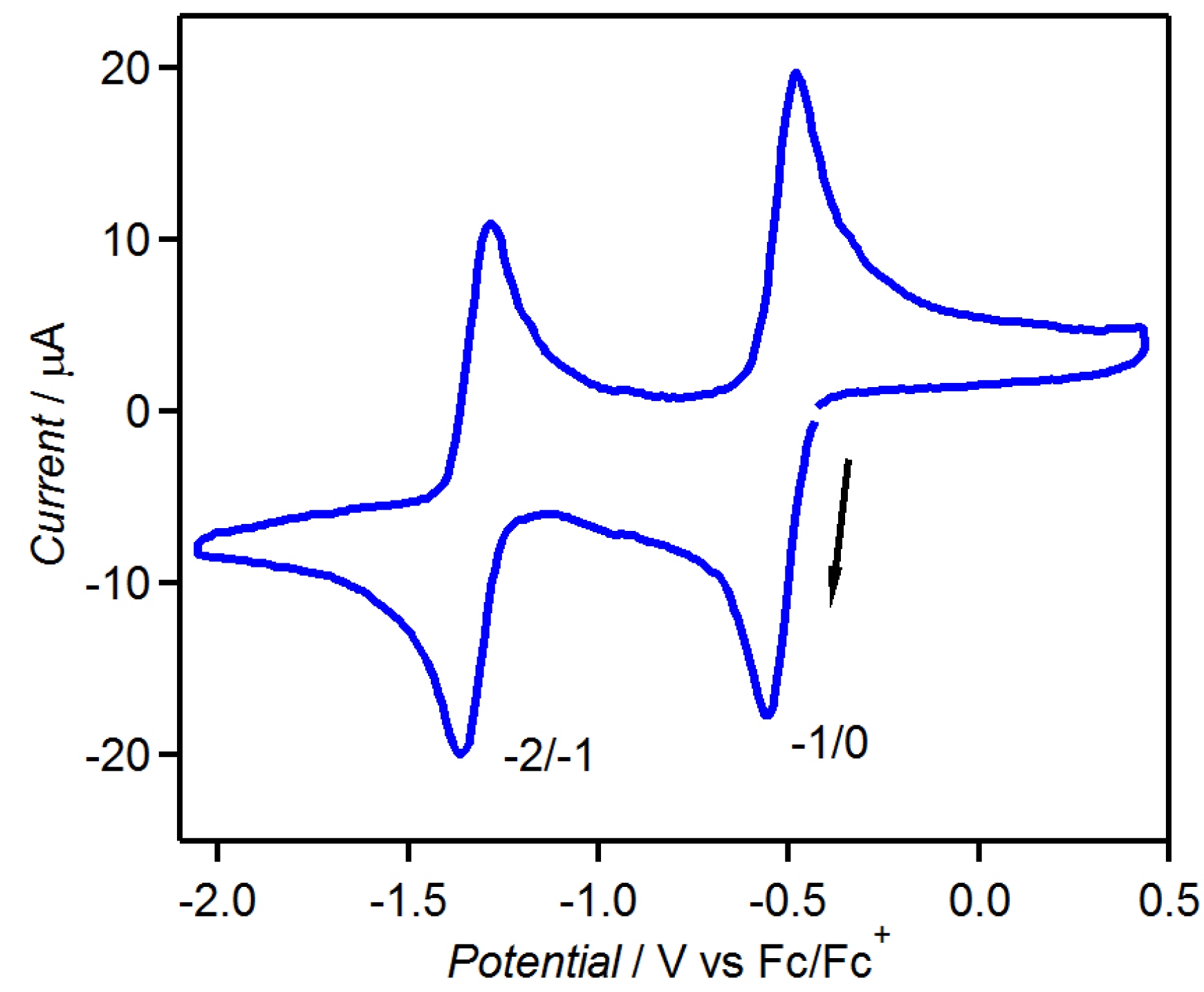
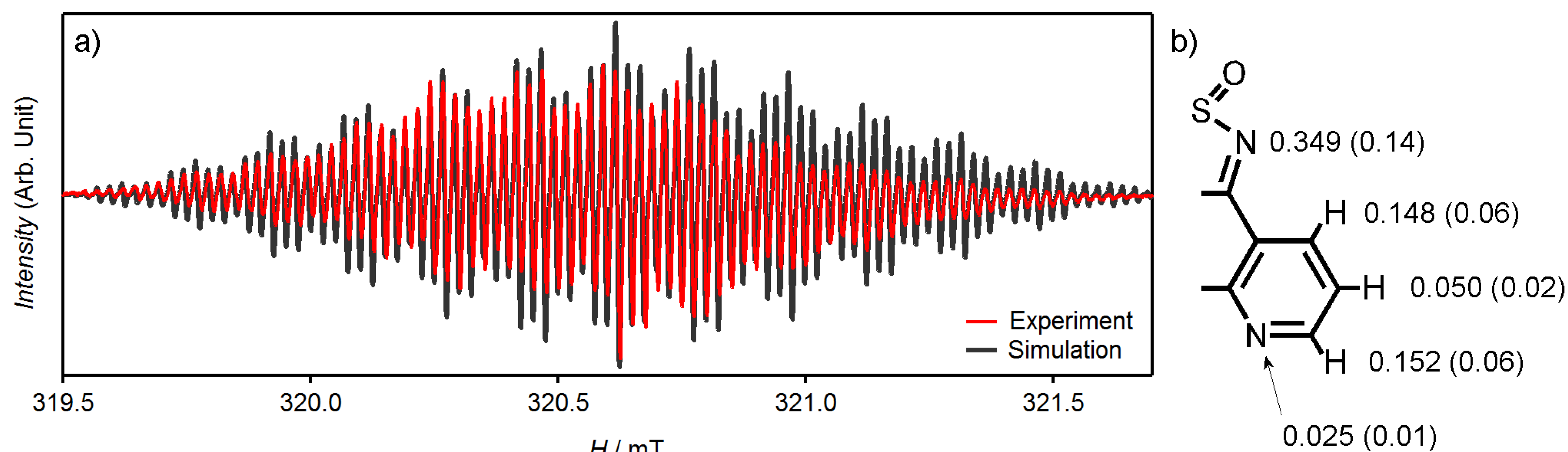
3.4. Theoretical Calculations
| Experimental data | Computational calculation | ||||||
|---|---|---|---|---|---|---|---|
| dint/Å | J1kB−1/K (zJ2kB−1/K) | JcalckB−1/K | tkB−1/K | KkB−1/K | UkB−1/K | ||
| K•tdapO2 | 3.30 | −13 | 17 | 72 | 23 | 30522 | |
| K•tdapO2•0.5MeCN | (i) | 3.36 | −110 | −100 | 1352 | 26 | 29355 |
| (ii) | 3.42 | 8 | 237 | 13 | 30118 | ||
| K•(tdapO2)2 | (i) | 3.29 | −20 | ||||
| (ii) | 3.29 | ||||||
| Rb•(tdapO2)2 | (i) | 3.29 | −29 | ||||
| (ii) | 3.31 | ||||||
| Cs7•(tdapO2)6•ClO4 | (i) | 3.13 | −310 | ||||
| (ii) | 3.36 | ||||||
| (NH4)2•tdapO2•I | (i) | 3.44 | 24 | 13 | 142 | 17 | 30709 |
| (ii) | 3.35 | (−7.9) | 6 | 208 | 13 | 30876 | |
| Hppda•tdapO2•MeCN | 3.42 | 5.6 | 13 | 85 | 17 | 30745 | |
| (−2.2) | |||||||
| Hppda•tdap•MeCN | Hypothetical system | −4 | 791 | 32 | 34533 | ||
| [CoCp2]•tdapO2 | −49 | ||||||

4. Summary
Supplementary Materials
Acknowledgments
Conflicts of Interest
References and Notes
- Akamatu, H.; Inokuchi, H.; Matsunaga, Y. Electrical conductivity of the perylene-brimine complex. Nature 1954, 173, 168–169. [Google Scholar] [CrossRef]
- Ferraris, J.; Cowan, D.O.; Walatka, V., Jr.; Perlsteri, J.H. Electron transfer in a new highly conducting donor-acceptor complex. J. Am. Chem. Soc. 1973, 95, 948–949. [Google Scholar] [CrossRef]
- Cohen, M.J.; Coleman, L.B.; Garito, A.F.; Heeger, A. Electrical conductivity of tetrathiofulvalinium tetracyanoquinodimethan (TTF) (TCNQ). J. Phys. Rev. B 1974, 10, 1298–1307. [Google Scholar] [CrossRef]
- Saito, G.; Enoki, T.; Toriumi, K.; Inokuchi, H. Two-dimensionality and suppression of metal-semiconductor transition in a new organic metal with Alkylthio substituted TTF and Perchlorate. Solid State Commun. 1982, 42, 557–560. [Google Scholar] [CrossRef]
- Tanaka, H.; Okano, Y.; Kobayashi, H.; Suzuki, W.; Kobayashi, A. A Three-dimensional synthetic metallic crystal composed of single-component molecules. Science 2001, 291, 285–287. [Google Scholar] [CrossRef]
- Kobayashi, H.; Kobayashi, A.; Tajima, H. Studies on molecular conductors: From organic semiconductors to molecular metals and superconductors. Chem. Asian J. 2011, 6, 1688–1704. [Google Scholar] [CrossRef]
- Isono, T.; Kamo, H.; Ueda, A.; Takahashi, K.; Nakao, A.; Kumai, R.; Nakao, H.; Kobayashi, K.; Murakami, Y.; Mori, H. Hydrogen bond-promoted metallic state in a purely organic single-component conductor under pressure. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Saito, G.; Yoshida, Y. Organic superconductors. Chem. Rec. 2011, 11, 124–145. [Google Scholar] [CrossRef]
- Jerome, D.; Mazaud, A.; Ribault, M.; Bechgaard, K. Superconductivity in a synthetic organic conductor (TMSF)2PF6. J. Phys. Lett. 1980, 41, 95–98. [Google Scholar] [CrossRef]
- Bechgaard, K.; Carneiro, K.; Rasmussen, F.B.; Olsen, M.; Rindorf, G.; Jacobsen, C.S.; Pedersen, H.J.; Scott, J.C. Superconductivity in an organic solid. Synthesis, Structure, and conductivity of Bis(tetramethyltetraselenafulvalenium) Perchlorate (TMTSF)2ClO4. J. Am. Chem. Soc. 1981, 103, 2440–2442. [Google Scholar] [CrossRef]
- Parkin, S.S.P.; Ribault, M.; Jerome, D.; Bechgaard, K. Superconductivity in the family of organic salts based on the Tetramethyltetraselenafulvalene (TMTSF) molecule: (TMTSF)2X (X = ClO4, PF6, AsF6, SbF6, TaF6). J. Phys. C 1981, 14, 5305–5326. [Google Scholar] [CrossRef]
- Tanigaki, K.; Ebbesen, T.W.; Saito, S.; Mizuki, J.; Tsai, J.S.; Kubo, Y.; Kuroshima, S. Superconductivity at 33 K in CsxRbyC60. Nature 1991, 352, 222–223. [Google Scholar] [CrossRef]
- Stephens, P.W.; Mihaly, L.; Lee, P.L.; Whetten, R.L.; Huang, S.M.; Kaner, R.; Deiderich, F.; Holczer, K. Structure of single-phase superconductiong K3C60. Nature 1991, 351, 632–634. [Google Scholar] [CrossRef]
- Hebard, A.F.; Rosseinsky, M.J.; Haddon, R.C.; Murphy, D.W.; Glarum, S.H.; Palstra, T.T.M.; Ramirez, A.P.; Kortan, A.R. Superconductivity at 18 K in Potassium-Doped C60. Nature 1991, 350, 600–601. [Google Scholar] [CrossRef]
- Kinoshita, M.; Turek, P.; Tamura, M.; Nozawa, K.; Shiomi, D.; Nakazawa, Y.; Ishikawa, M.; Takahashi, M.; Awaga, K.; Inabe, T.; et al. An organic radical ferromagnet. Chem. Lett. 1991, 20, 1225–1228. [Google Scholar]
- Takahashi, M.; Turek, P.; Nakazawa, Y.; Tamura, M.; Nozawa, K.; Shiomi, D.; Ishikawa, M.; Kinoshita, M. Discovery of a quasi-1D organic ferromagnet, p-NPNN. Phys. Rev. Lett. 1991, 67, 746–748. [Google Scholar] [CrossRef]
- Turek, P.; Nozawa, K.; Shiomi, D.; Awaga, K.; Inabe, T.; Maruyama, Y.; Kinoshita, M. Ferromagnetic coupling in a new phase of the p-Nitrophenyl Nitronyl Nitroxide radical. Chem. Phys. Lett. 1991, 180, 327–331. [Google Scholar] [CrossRef]
- Deumal, M.; Rawson, J.M.; Goeta, A.E.; Howard, J.A.K.; Copley, R.C.B.; Robb, M.A.; Novoa, J.J. Studying the origin of the antiferromagnetic to spin-canting transition in the β-p-NCC6F4CNSSN• molecular magnet. Chem. Eur. J. 2010, 16, 2741–2750. [Google Scholar]
- Kohler, C.P.; Jakobi, R.; Meissner, E.; Wiehl, L.; Spiering, H.; Gutlich, P. Nature of the phase transition in spin crossover compounds. J. Phys. Chem. Solids 1990, 51, 239–247. [Google Scholar] [CrossRef]
- Adams, D.M.; Dei, A.; Rheingold, A.L.; Hendrickson, D.N. Bistability in the [CoII(semiquinonate)2] to [CoIII(catecholate)(semiquinonate)] Valence-Tautomeric conversion. J. Am. Chem. Soc. 1993, 115, 8221–8229. [Google Scholar] [CrossRef]
- Sato, O.; Tao, J.; Zhang, Y. Control of magnetic properties through external stimuli. Angew. Chem. Int. 2007, 46, 2152–2187. [Google Scholar]
- Dunbar, K.R.; Heintz, R.A. Chemistry of transition metal cyanide compounds: Modern perspectives. Prog. Inorg. Chem. 1997, 45, 283–391. [Google Scholar]
- Ohba, M.; Okawa, H. Synthesis and magnetism of multi-dimensional cyanide-bridged bimetallic assemblies. Coord. Chem. Rev. 2000, 198, 313–328. [Google Scholar] [CrossRef]
- Clement, R.; Decurtins, S.; Gruselle, M.; Train, C. Polyfunctional two- (2D) and three- (3D) dimensional oxalate bridged bimetallic magnets. Monatsh. Chem. 2003, 134, 117–135. [Google Scholar] [CrossRef]
- Miller, J.S. Magnetically ordered molecule-based materials. Chem. Soc. Rev. 2011, 40, 3266–3296. [Google Scholar] [CrossRef]
- Clemente-Leon, M.; Coronado, E.; Marti-Gastaldo, C.; Romero, F.M. Multifunctionality in hybrid magnetic materials based on bimetallic oxalate complexes. Chem. Soc. Rev. 2011, 40, 473–497. [Google Scholar] [CrossRef]
- Gatteschi, D.; Sessoli, R. Quantum tunneling of magnetization and related phenomena in molecular materials. Angew. Chem. Int. 2003, 42, 268–297. [Google Scholar]
- Awaga, K.; Nomura, K.; Kishida, H.; Fujita, W.; Yoshikawa, H.; Matsushita, M.M.; Hu, L.; Shuku, Y.; Suizu, R. Electron transfer processes in highly-correlated electron systems of Thiazyl radicals. Bull. Chem. Soc. Jpn. 2014, in press. [Google Scholar]
- Oakley, R.T. Cyclic and heterocyclic Thiazenes. Prog. Inorg. Chem. 1988, 36, 299–391. [Google Scholar] [CrossRef]
- Rawson, J.M.; Banister, A.J.; Lavender, I. The chemistry of dithiadiazolylium and dithiadiazolyl rings. Adv. Heterocycl. Chem. 1995, 62, 137–247. [Google Scholar]
- Rawson, J.M.; McManus, G.D. Benzo-fused Dithiazolyl radicals: From chemical curiosities to materials chemistry. Coord. Chem. Rev. 1999, 189, 135–168. [Google Scholar] [CrossRef]
- Rawson, J.M.; Palacio, F. Magnetic properties of Thiazyl radicals. Struct. Bond. 2001, 100, 93–128. [Google Scholar] [CrossRef]
- Awaga, K.; Tanaka, T.; Shirai, T.; Fujimori, M.; Suzuki, Y.; Yoshikawa, H.; Fujita, W. Multi-dimensional crystal structures and unique solid-state properties of Heterocyclic Thiazyl radicals and related materials. Bull. Chem. Soc. Jpn. 2006, 79, 25–34. [Google Scholar] [CrossRef]
- Conte, G.; Bortoluzzi, A.J.; Gallardo, H. [1,2,5]Thiadiazolo[3,4-f][1,10]phenanthroline as a building block for organic materials. Synthesis 2006, 2006, 3945–3947. [Google Scholar] [CrossRef]
- Castellano, E.E.; Piro, O.E.; Caram, J.A.; Mirifico, M.V.; Aimone, S.L.; Vasini, E.J.; Glossman, M.D. Crystallographic study and molecular orbital calculations of 1,2,5-Thiadiazole 1,1-dioxide derivatives. J. Phys. Org. Chem. 1998, 11, 91–100. [Google Scholar] [CrossRef]
- Mirifico, M.V.; Caram, J.A.; Gennaro, A.M.; Cobos, C.J.; Vasini, E.J. Radical anions containing the Dioxidated 1,2,5-Thiadiazole heterocycle. J. Phys. Org. Chem. 2009, 22, 964–970. [Google Scholar] [CrossRef]
- Miller, J.S.; Epstein, A.J. Organometallic magnets. Coord. Chem. Rev. 2000, 206–207, 651–660. [Google Scholar] [CrossRef]
- Miller, J.S. Tetracyanoethylene (TCNE). The characteristic geometries and vibrational absorptions of its numerous structures. Angew.Chem. Int. 2006, 45, 2508–2525. [Google Scholar]
- Heintz, R.A.; Zhao, H.; Ouyang, X.; Grandinetti, G.; Cowen, J.; Dunbar, K.R. New insight into the nature of Cu(TCNQ): Solution routes to two distinct polymorphs and their relationship to Crystalline films that display bistable switching behavior. Inorg. Chem. 1999, 38, 144–156. [Google Scholar] [CrossRef]
- O’Kane, S.A.; Clerac, R.; Zhao, H.; Ouyang, X.; Galan-Mascaros, J.R.; Heintz, R.; Dunbar, K.R. New Crystalline Polymers of Ag(TCNQ) and Ag(TCNQF4): Structures and magnetic properties. J. Solid State Chem. 2000, 152, 159–173. [Google Scholar] [CrossRef]
- Clerac, R.; O’Kane, S.; Cowen, J.; Ouyang, X.; Heintz, R.; Zhao, H.; Bazile, M.J., Jr.; Dunbar, K.R. Glassy magnets composed of metals coordinated to 7,78,8-Tetracyanoquinodimethane: M(TCNQ)2 (M = Mn, Fe, Co, Ni). Chem. Mater. 2003, 15, 1840–1850. [Google Scholar] [CrossRef]
- Kato, R. Conductive copper salts of 2,5-disubstituted n,n’-dicyanobenzoquinonediimines (dcnqis): Structural and physical properties. Bull. Chem. Soc. Jpn. 2000, 73, 515–534. [Google Scholar] [CrossRef]
- Pierpont, C.G.; Buchanan, R.M. Transition metal complexes of O-benzoquinone, O-semiquinone, and catecholate ligands. Coord. Chem. Rev. 1981, 38, 45–87. [Google Scholar] [CrossRef]
- Pierpont, C.G. Unique properties of transition metal Quinone complexes of the MQ3 series. Coord. Chem. Rev. 2001, 219–221, 415–433. [Google Scholar] [CrossRef]
- Evangelio, E.; Ruiz-Molina, D. Valence tautomerism: New challenges for electroactive ligands. Eur. J. Inorg. Chem. 2005, 2005, 2957–2971. [Google Scholar] [CrossRef]
- Manriquez, J.M.; Yee, G.T.; McLean, R.S.; Epstein, A.J.; Miller, J.S. A room-temperature molecular/organic-based magnet. Science 1991, 252, 1415–1417. [Google Scholar]
- Shuku, Y.; Suizu, R.; Awaga, K. Monovalent and mixed-valent potassium salts of [1,2,5]Thiadiazolo[3,4-f][1,10]phenanthroline 1,1-Dioxide: A radical anion for multidimensional network structures. Inorg. Chem. 2011, 50, 11859–11861. [Google Scholar] [CrossRef]
- Shuku, Y.; Suizu, R.; Domingo, A.; Calzado, C.J.; Robert, V.; Awaga, K. Multidimensional network structures and versatile magnetic properties of intermolecular compounds of a radical–anion ligand, [1,2,5]Thiadiazolo[3,4-f][1,10]phenanthroline 1,1-Dioxide. Inorg. Chem. 2013, 52, 9921–9930. [Google Scholar] [CrossRef]
- Gallardo, H.; Conte, G.; Tuzimoto, P.; Bortoluzzi, A.; Peralta, R.A.; Neves, A. Synthesis, crystal structure and luminescent properties of new Tris-β-diketonate Eu(III) complexes with Thiadiazolophenanthroline derivative ligand. Inorg. Chem. Commun. 2008, 11, 1292–1296. [Google Scholar] [CrossRef]
- Gallardo, H.; Conte, G.; Bortoluzzi, A.J.; Bechtold, I.H.; Pereira, A.; Quirino, W.G.; Legnani, C.; Cremona, M. Synthesis, structural characterization, and photo and electroluminescence of a novel Terbium (III) complex: {Tris (acetylacetonate) [1,2,5]Thiadiazolo[3,4-f][1,10] phaenanthroline} terbium(III). Inorg. Chim. Acta 2011, 365, 152–158. [Google Scholar] [CrossRef]
- De Souza, B.; Xavier, F.R.; Peralta, R.A.; Bortoluzzi, A.J.; Conte, G.; Gallardo, H.; Fischer, F.L.; Bussi, G.; Terenzi, H.; Neves, A. Oxygen-independent photonuclease activity of a new Iron(II) COMPLEX. Chem. Commun. 2010, 46, 3375–3377. [Google Scholar] [CrossRef]
- Crystallographic data for the compounds discussed in this review that have not previously been published are available through Cambridge Crystallographic Data Centre (CCDC): CCDC 969113 ([Co(tdap)2(NCS)2]), CCDC 969114 ([Mn(tdap)2Cl2]), CCDC 969115 ([Mn(tdap)2(NCS)2]•MeCN), CCDC 969116 ([Fe(tdap)2(NCS)2]•MeCN at 120 K), CCDC 969117 ([Fe(tdap)2(NCS)2]•MeCN at 320 K), CCDC 969118 ([Co(tdap)2(NCS)2]•MeCN), CCDC 969119 ([Ni(tdap)2(NCS)2]•MeCN), CCDC 969120 ([Cu(tdap)2(NCS)2]•MeCN), CCDC 969121 ([Zn(tdap)2(NCS)2]•MeCN), CCDC 969122 ([Cu2(tdap)2(NCS)2]), CCDC 969123 ([Cu2(tdap)2(NCS)4]), and CCDC 9691224 ([CoCp]2•tdapO2). These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]).
- Shuku, Y.; Suizu, R.; Awaga, K.; Sato, O. Fe(II) spincrossover complex of [1,2,5]Thiadiazolo[3,4-f][1,10]phenanthroline. Cryst. Eng. Comm. 2009, 11, 2065–2068. [Google Scholar] [CrossRef]
- Gallois, B.; Real, J.A.; Hauw, C.; Zarembowich, J. Structural changes associated with the spin transition in Fe(phen)2(NCS)2: A single-crystal X-ray investigation. Inorg. Chem. 1990, 29, 1152–1158. [Google Scholar] [CrossRef]
- Guionneau, P.; Marchivie, M.; Bravic, G.; Letard, J.F.; Chasseau, D. Structural aspects of spin crossover example of the [Fe(II)Ln(NCS)2] complexes. Top. Curr. Chem. 2004, 234, 97–128. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Caram, J.A.; Mirifico, M.V.; Vasini, E.J. Electrochemistry of 3,4-Diphenyl-1,2,5-thiadiazole 1, 1-dioxide (1) and its derivatives in ethanol-acetonitrile solutions and interactions of the dianion of 1 with metal cations. Electrochim. Acta 1994, 39, 939–945. [Google Scholar] [CrossRef]
- Caram, J.A.; Mirifico, M.V.; Aimone, S.L.; Vasini, E.J. 3,4-Disubstituted derivatives of 1,2,5-thiadiazole 1,1-Dioxide. Ethanol addition reactions and electroreduction of 3-Methyl-4-phenyl and 3,4-Dimethyl derivatives in acetonitrile and ethanol solvents. Can. J. Chem. 1996, 74, 1564–1571. [Google Scholar] [CrossRef]
- Caram, J.A.; Mirifico, M.V.; Aimone, S.L.; Piro, O.E.; Castellano, E.E.; Vasini, E.J. The addition reaction of diamides to 1,2,5-Thiadiazole 1,1-Dioxide Derivatives. J. Phys. Org. Chem. 2004, 17, 1091–1098. [Google Scholar] [CrossRef]
- Yamada, M.; Tanaka, Y.; Yoshimoto, Y.; Kuroda, S.; Shimao, I. Synthesis and properties of diamino-substituted Dipyrido [3,2-a: 2’,3’-c]Phenazine. Bull. Chem. Soc. Jpn. 1992, 65, 1006–1011. [Google Scholar]
- Chambers, J.Q. Electrochemistry of Quinones. In The Chemistry of the Quinonoid Compounds; Patai, S., Ed.; John Wiley & Sons: New York, NY, USA, 1974; pp. 737–791. [Google Scholar]
- Peover, M.E. A polarographic investigation into the redox behavior of quinones: The roles of electron affinity and solvent. J. Chem. Soc. 1962, 4540–4549. [Google Scholar] [CrossRef]
- Davis, K.M.; Hammond, P.R.; Peover, M.E. Electron affinities of monosubstituted benzoquinones. Trans. Faraday Soc. 1965, 61, 1516–1522. [Google Scholar] [CrossRef]
- Ryba, O.; Pilar, J.; Petranek, J. Polarographic and electron paramagnetic resonance study of one electron reduction of Alkylated Benzoquinones. Collect. Czech. Chem. Commun. 1968, 33, 26–34. [Google Scholar] [CrossRef]
- McConnell, H.M. Spin density matrices for paramagnetic molecules. J. Chem. Phys. 1958, 28, 1188–1192. [Google Scholar] [CrossRef]
- McConnell, H.M.; Stathdee, J. Theory of anisotropic hyperfine interactions in π-electron radicals. Mol. Phys. 1959, 2, 129–138. [Google Scholar] [CrossRef]
- Talcott, C.L.; Myers, R.J. Electron spin resonance spectra of the radical anions of pyridine and related nitrogen heterocyclics. Mol. Phys. 1967, 12, 549–567. [Google Scholar] [CrossRef]
- Carrington, A.; dos Santos-Veiga, J. Electron spin resonance spectra of Nitrogen Heterocyclic radical Ions. Mol. Phys. 1962, 5, 21–29. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism; Wiley-VHC: Weinheim, Germany, 1993. [Google Scholar]
- Bonner, J.C.; Fisher, M.E. Linear magnetic chains with anisotropic coupling. Phys. Rev. A 1964, 135, A640–A658. [Google Scholar] [CrossRef]
- Estes, W.E.; Gavel, D.P.; Hatfield, W.E.; Hodgson, D.J. Magnetic and structural characterization of Dibromo- and Dichlorobis(thiazole)copper(II). Inorg. Chem. 1978, 17, 1415–1421. [Google Scholar] [CrossRef]
- Bleaney, B.; Bowers, K.D. Anomalous paramagnetism of copper acetate. Proc. R. Soc. 1952, 214, 451–465. [Google Scholar] [CrossRef]
- Robin, M.B.; Day, P. Mixed valence chemistry—a survey and classification. Adv. Inorg. Chem. Radiochem. 1967, 10, 247–422. [Google Scholar]
- Kimura, S.; Suzuki, H.; Maejima, T.; Mori, H.; Yamaura, J.; Kakiuchi, T.; Sawa, H.; Moriyama, H. Checkerboard-type charge-ordered state of a pressure-induced superconductor, β-(meso-DMBEDT-TTF)2PF6. J. Am. Chem. Soc. 2006, 128, 1456–1457. [Google Scholar] [CrossRef]
- Ramirez, A.P. Strongly geometrically frustrated magnets. Annu. Rev. Mater. Sci. 1994, 24, 453–480. [Google Scholar] [CrossRef]
- Awaga, K.; Okuno, T.; Yamaguchi, A.; Hasegawa, M.; Inabe, T.; Maruyama, Y.; Wada, N. Variable magnetic interactions in an organic radical system of (m-N-Methylpyridinium α-Nitronyl Nitroxide)•X−: A possible Kagomé antiferromagnet. Phys. Rev. B 1994, 49, 3975–3981. [Google Scholar] [CrossRef]
- Baker, G.A.; Rushbrooke, G.S.; Gilbert, H.E. High-temperature series expansions for the spin−1/2 heisenberg model by the method of irreducible representations of the symmetric group. Phys. Rev. 1964, 135, A1272–A1277. [Google Scholar] [CrossRef]
- Rota, J.B.; le Guennic, B.; Robert, V. Toward Verdazyl radical-based materials: Ab Initio inspection of potential organic candidates for spin-crossover phenomenon. Inorg. Chem. 2010, 49, 1230–1237. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shuku, Y.; Awaga, K. Transition Metal Complexes and Radical Anion Salts of 1,10-Phenanthroline Derivatives Annulated with a 1,2,5-Tiadiazole and 1,2,5-Tiadiazole 1,1-Dioxide Moiety: Multidimensional Crystal Structures and Various Magnetic Properties. Molecules 2014, 19, 609-640. https://doi.org/10.3390/molecules19010609
Shuku Y, Awaga K. Transition Metal Complexes and Radical Anion Salts of 1,10-Phenanthroline Derivatives Annulated with a 1,2,5-Tiadiazole and 1,2,5-Tiadiazole 1,1-Dioxide Moiety: Multidimensional Crystal Structures and Various Magnetic Properties. Molecules. 2014; 19(1):609-640. https://doi.org/10.3390/molecules19010609
Chicago/Turabian StyleShuku, Yoshiaki, and Kunio Awaga. 2014. "Transition Metal Complexes and Radical Anion Salts of 1,10-Phenanthroline Derivatives Annulated with a 1,2,5-Tiadiazole and 1,2,5-Tiadiazole 1,1-Dioxide Moiety: Multidimensional Crystal Structures and Various Magnetic Properties" Molecules 19, no. 1: 609-640. https://doi.org/10.3390/molecules19010609