EGF Receptor-Dependent Mechanism May be Involved in the Tamm–Horsfall Glycoprotein-Enhanced PMN Phagocytosis via Activating Rho Family and MAPK Signaling Pathway

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
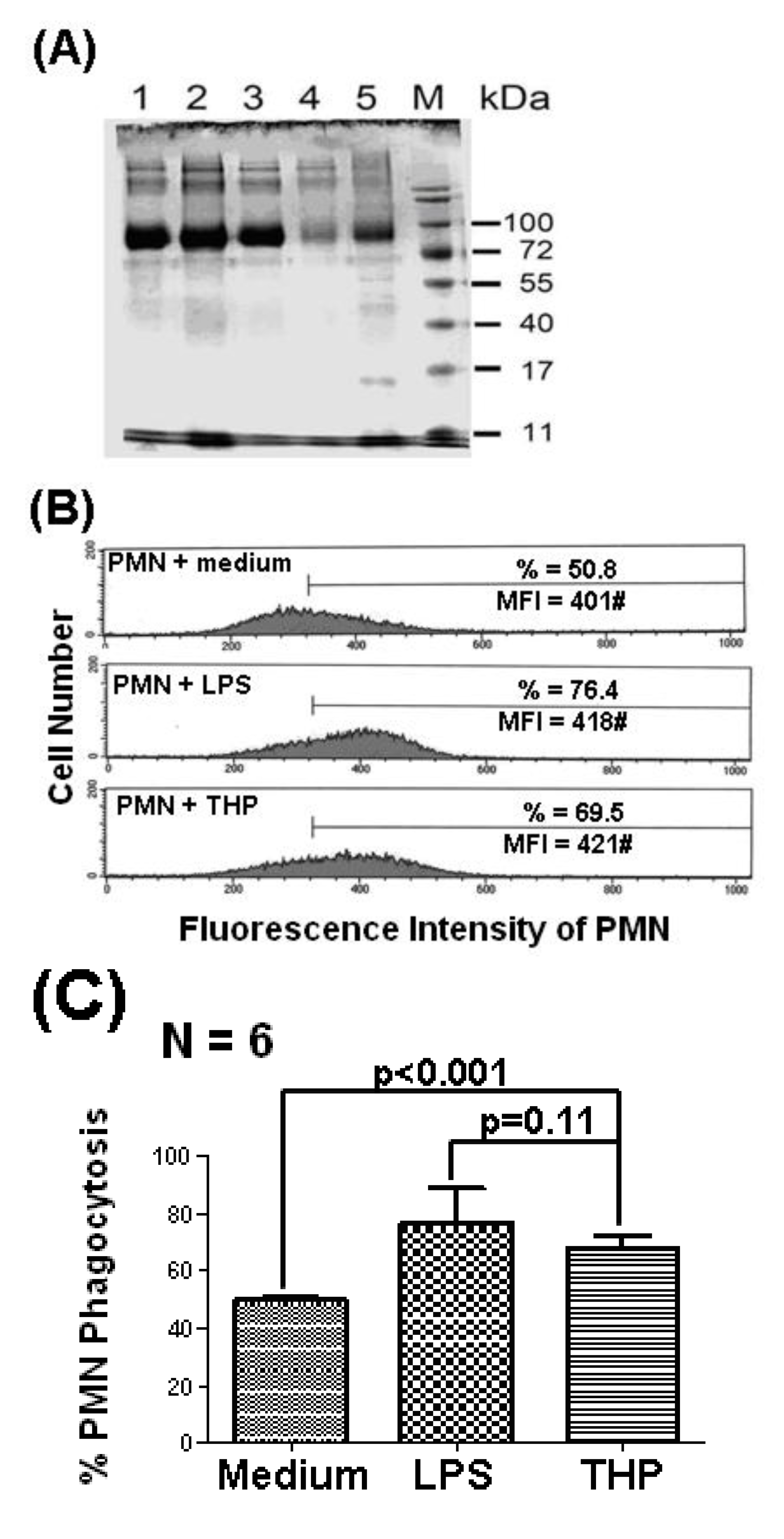
2.1. THP Enhances PMN Phagocytosis
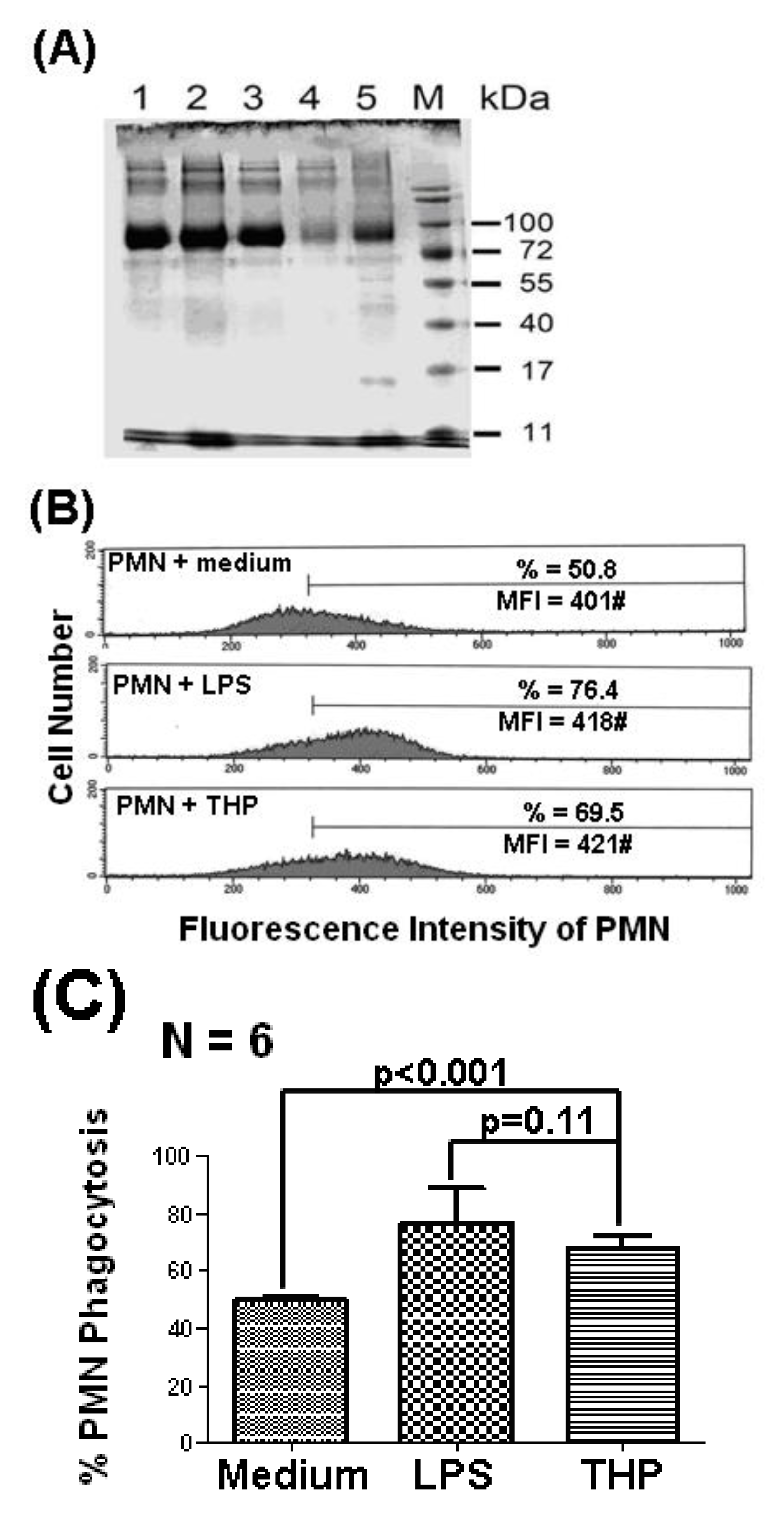
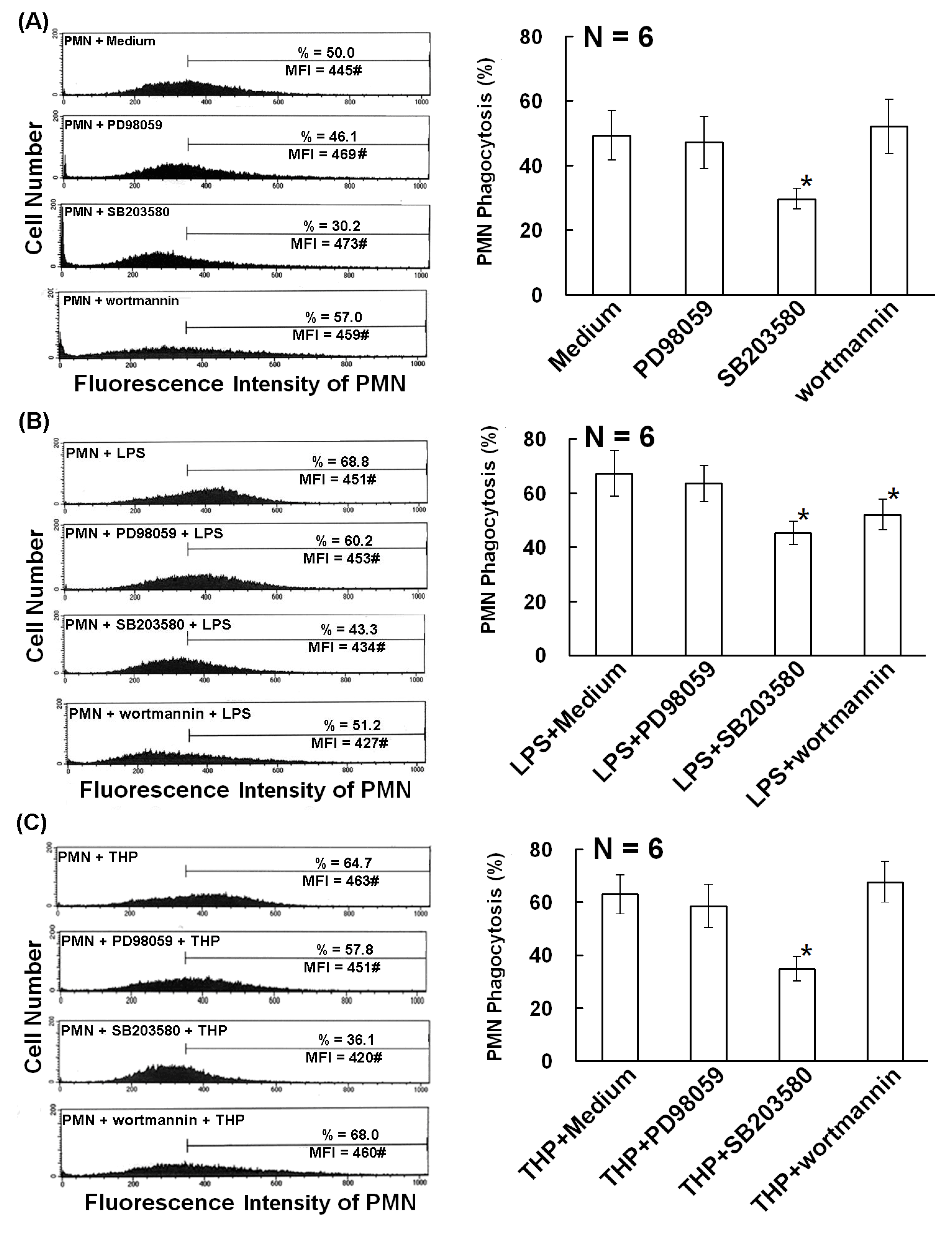
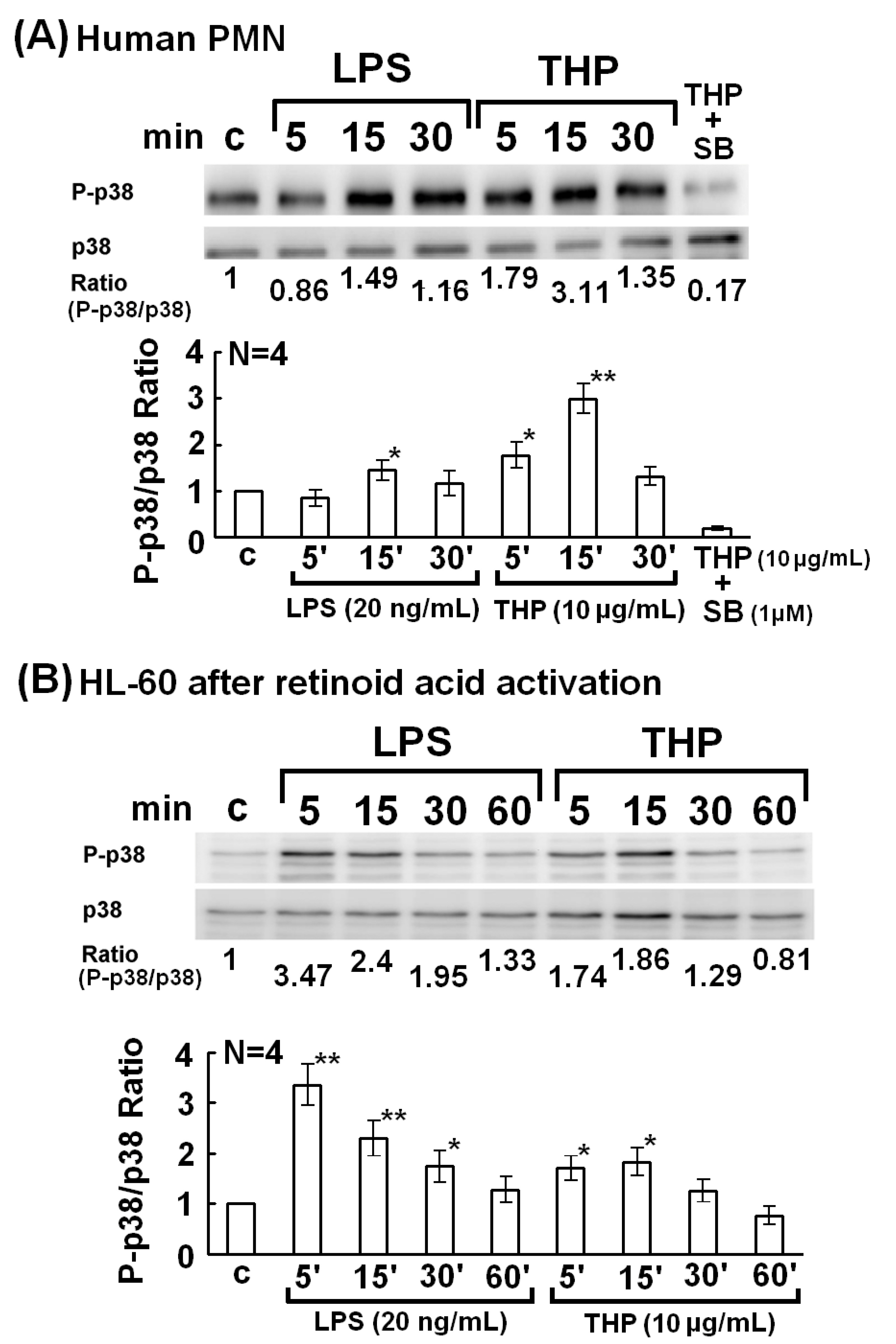
2.2. The p38 MAP Kinase Signal Pathway is Involved in Spontaneous, Bacterial Lipopolysaccharide- Enhanced, and THP-Enhanced PMN Phagocytosis

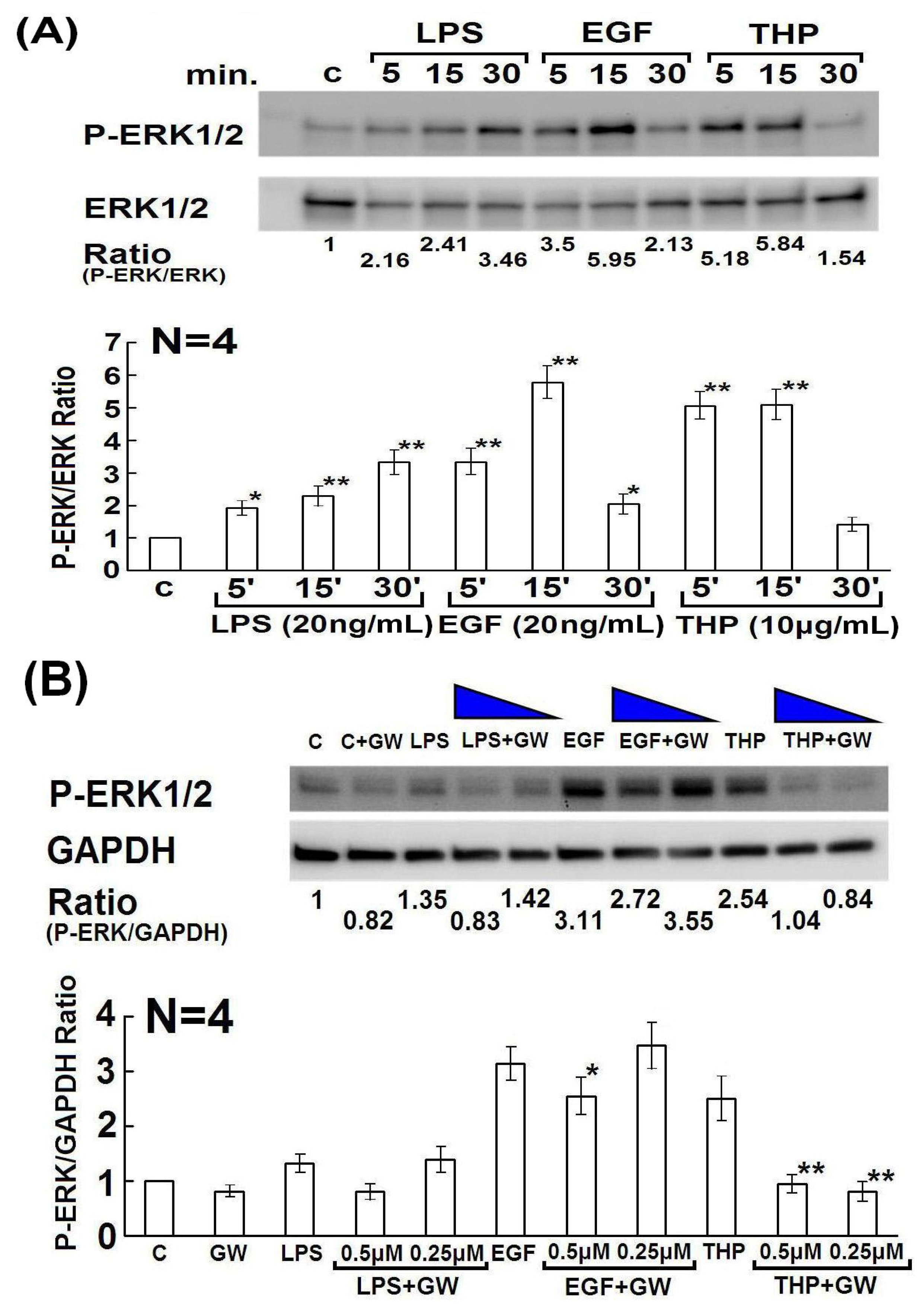
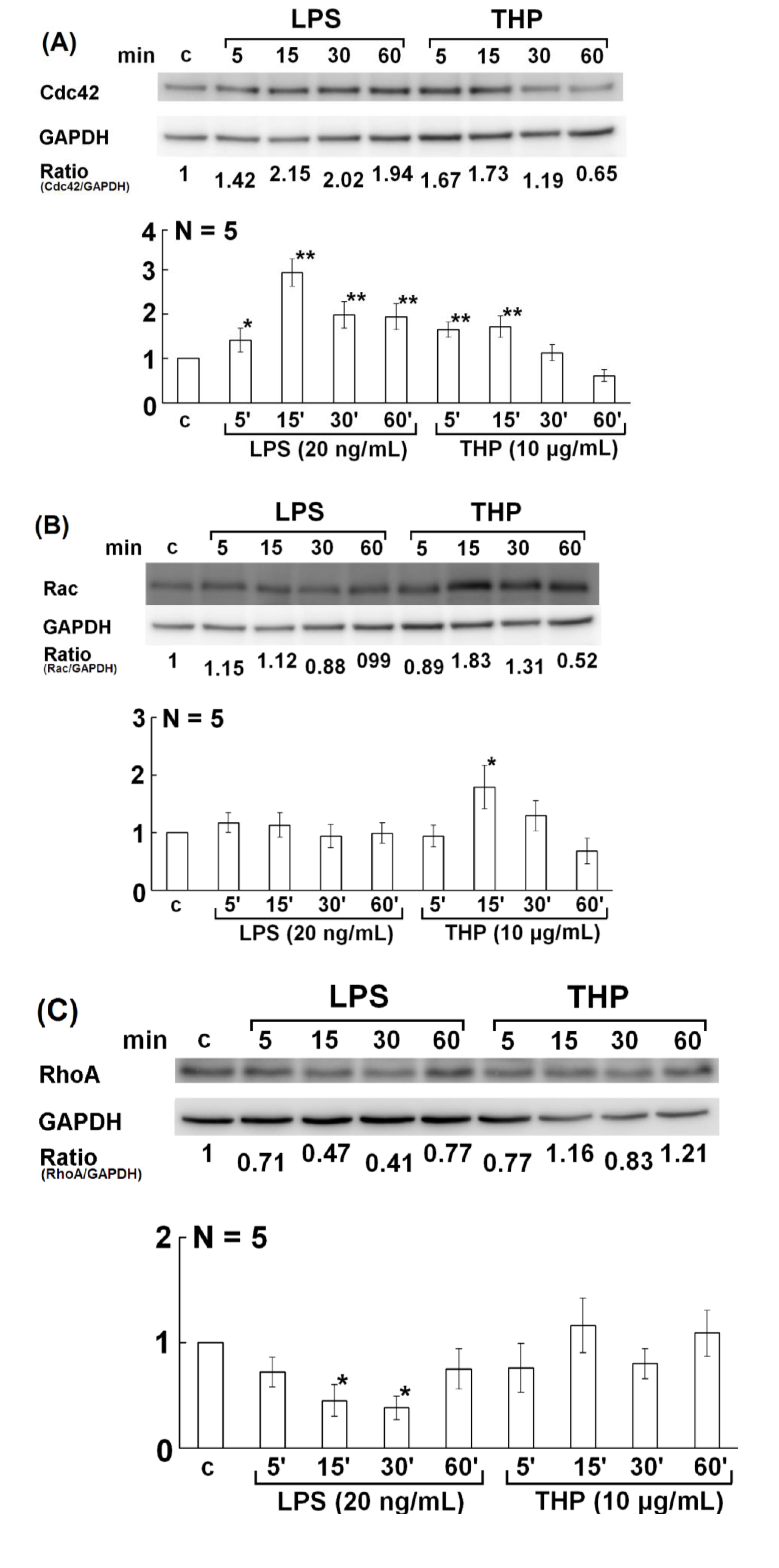
2.3. Involvement of Rho Family GTPase Molecules in THP-Enhanced PMN Phagocytosis

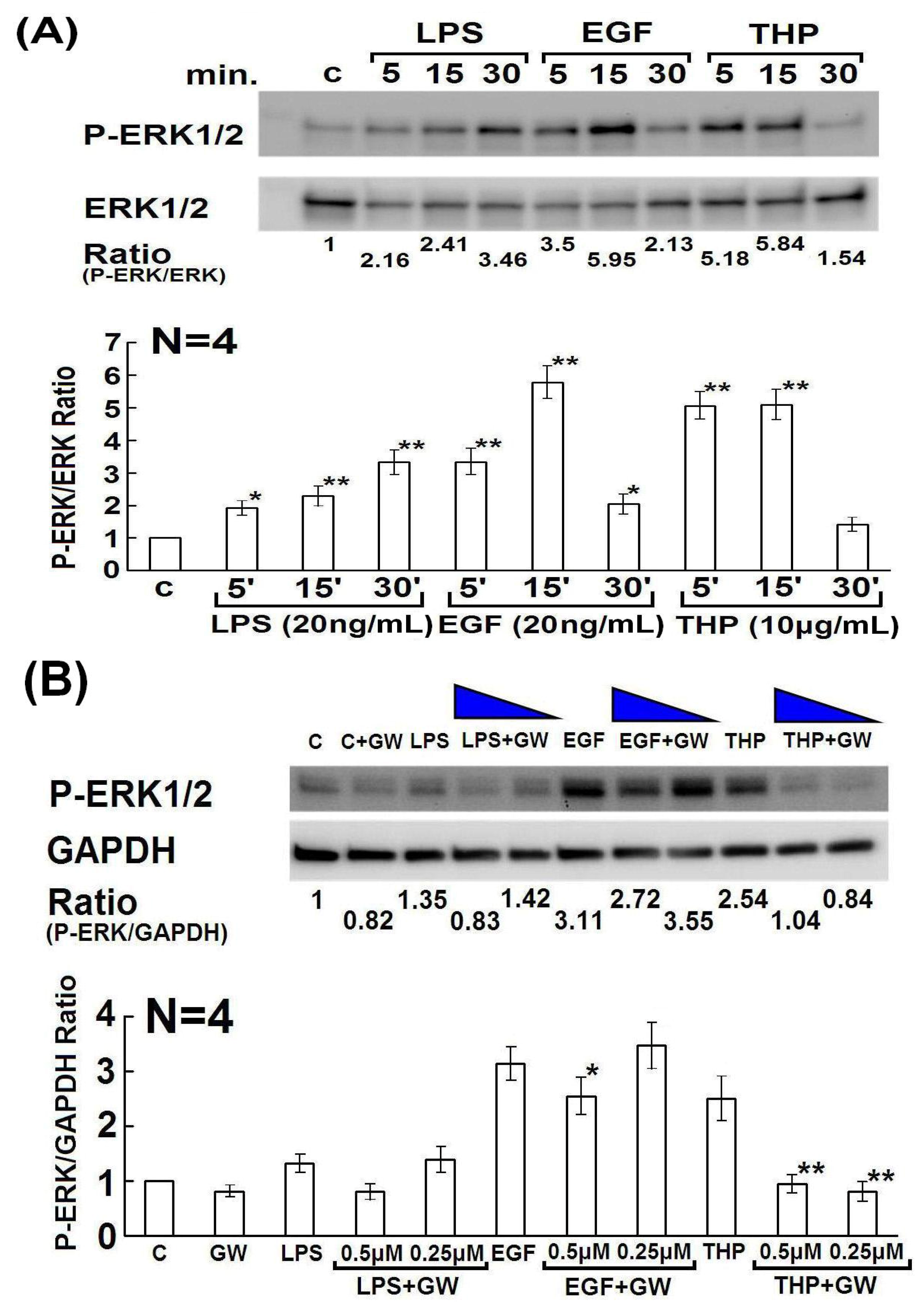
2.4. Determination of Domain Structure(s) in the THP Protein Core Responsible for Enhancing PMN Phagocytosis and Their Signaling Pathways
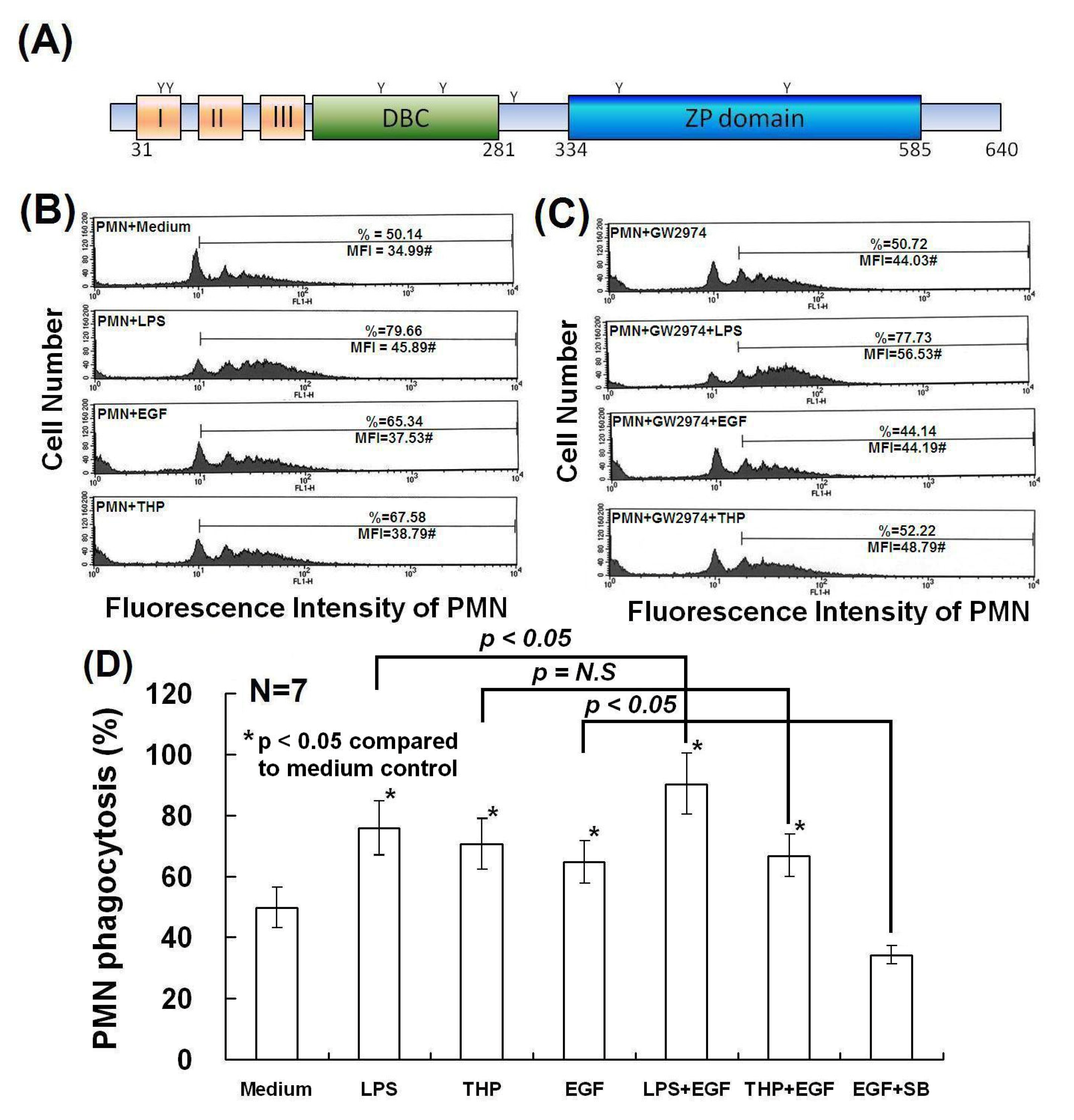
3. Experimental
3.1. Reagents and Antibodies
3.2. Purification of THP from Pooled Normal Human Urine
3.3. Isolation of PMNs from Normal Human Peripheral Blood
3.4. Detection of PMN Phagocytosis by Flow Cytometry
3.5. Preparation of Whole Cell Lysates
3.6. Western Immunoblotting
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bachmann, S.; Metzger, R.; Bunnemann, B. Tamm–Horsfall protein mRNA synthesis is localized to the thick ascending limb of Henle’s loop in rat kidney. Histochemistry 1990, 94, 517–523. [Google Scholar] [CrossRef]
- Serafini-Cessi, F.; Malagolini, N.; Cavallone, D. Tamm–Horsfall glycoprotein: Biology and clinical relevance. Am. J. Kidney Dis. 2003, 42, 658–676. [Google Scholar] [CrossRef]
- Pak, J.; Pu, Y.; Zhang, Z.T.; Hasty, D.L.; Wu, X.R. Tamm–Horsfall protein binds to type 1 fimbriated Escherichia coli and prevents E. coli from binding to uroplakin Ia and Ib receptors. J. Biol. Chem. 2001, 276, 9924–9930. [Google Scholar]
- Raffi, H.S.; Bates, J.M., Jr.; Laszik, Z.; Kumar, S. Tamm–Horsfall protein protects against urinary tract infection by Proteus mirabilis. J. Urol. 2009, 181, 2332–2338. [Google Scholar]
- Kokot, F.; Dulawa, J. Tamm–Horsfall protein updated. Nephron 2000, 85, 97–102. [Google Scholar] [CrossRef]
- Berke, E.S.; Mayrer, A.R.; Miniter, P.; Andriole, V.T. Tubulointerstitial nephritis in rabbits challenged with homologous Tamm–Horsfall protein: The role of endotoxin. Clin. Exp. Immunol. 1983, 53, 562–572. [Google Scholar]
- Scolari, F.; Caridi, G.; Rampoldi, L.; Tandanico, R.; Izzi, C.; Oirulli, D.; Amoroso, A.; Casari, G.; Ghiggeri, G.M. Uromodulin storage diseases: Clinical aspects and mechanisms. Am. J. Kidney Dis. 2004, 44, 987–999. [Google Scholar] [CrossRef]
- Tinschert, S.; Ruf, N.; Bernascone, I.; Sacherer, K.; Lamorte, H.-H.; Nurnberg, P.; Luft, F.C.; Rampoldi, L. Functional consequences of a novel uromodulin mutation in a family with familial juvenile hyperuricemic nephropathy. Nephrol. Dial. Transplant. 2004, 19, 3150–3154. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Hart, T.C. Familial juvenile hyperuricaemic nephropathy. QJM 2003, 96, 867–868. [Google Scholar] [CrossRef]
- Fletcher, A.P.; Neuberger, A.; Ratcliffe, W.A. Tamm–Horsfall urinary glycoprotein. The subunit structure. Biochem. J. 1970, 120, 425–432. [Google Scholar]
- Santambrogio, S.; Cattaneo, A.; Bernascone, I.; Schwend, T.; Jovine, L.; Bachi, A.; Rampoldi, L. Urinary uromodulin carries an intact ZP domain generated by a conserved C-terminal proteolytic cleavage. Biochem. Biophys. Res. Commun. 2008, 370, 410–413. [Google Scholar] [CrossRef]
- Afonso, A.M.M.; Marshall, R.D. Observations on the structure of the carbohydrate moieties of the Tamm–Horsfall glycoprotein. Biochem. Soc. Trans. 1979, 7, 170–173. [Google Scholar]
- Van Rooijen, J.J.; Voskamp, A.F.; Kamerling, J.P.; Vliegenthart, J.F. Glycosylation sites and site-specific glycosylation in human Tamm–Horsfall glycoprotein. Glycobiology 1999, 9, 21–30. [Google Scholar] [CrossRef]
- Firon, N.; Ofek, I.; Sharon, N. Carbohydrate-binding sites of the mannose-specific fimbrial lectins of enterobacteria. Infect. Immun. 1984, 43, 1088–1090. [Google Scholar]
- Easton, R.L.; Patankar, M.S.; Clark, G.F.; Morris, H.R.; Dell, A. Pregnancy-associated changes in the glycosylation of Tamm–Horsfall glycoprotein. Expression of sialyl Lewisx sequences on core 2 type O-glycans derived from uromodulin. J. Biol. Chem. 2000, 275, 21928–21938. [Google Scholar]
- Muchmore, A.V.; Decker, J.M. Evidence that recombinant IL-1 alpha exhibits lectin-like specificity and binds to homogenous uromodulin via N-linked oligosaccharides. J. Immunol. 1987, 138, 2541–2546. [Google Scholar]
- Sherblom, A.P.; Decker, J.M.; Muchmore, A.V. The lectin-like interaction between recombinant tumor necrosis factor and uromodulin. J. Biol. Chem. 1988, 263, 5418–5424. [Google Scholar]
- Rhodes, D.C.; Hinsman, E.J.; Rhodes, J.A. Tamm–Horsfall glycoprotein binds IgG with high affinity. Kidney Int. 1993, 44, 1014–1021. [Google Scholar] [CrossRef]
- Rhodes, D.C. Binding of Tamm–Horsfall protein to complement 1q and complement 1, including influence of hydrogen-ion concentration. Immunol. Cell. Biol. 2002, 80, 558–566. [Google Scholar] [CrossRef]
- Thomas, D.B.; Davies, M.; Peters, J.R.; Williams, J.D. Tamm–Horsfall protein binds to a single class of carbohydrate specific receptors on human neutrophils. Kidney Int. 1993, 44, 423–429. [Google Scholar] [CrossRef]
- Wimmer, T.; Cohen, G.; Saemann, M.D.; Horl, W.H. Effects of Tamm–Horsfall protein on polymorphonuclear leukocyte function. Nephrol. Dial. Transplant. 2004, 19, 2192–2197. [Google Scholar] [CrossRef]
- Yu, C.L.; Lin, W.M.; Liao, T.S.; Tsai, C.Y.; Sun, K.H.; Chen, K.H. Tamm–Horsfall glycoprotein (THG) purified from normal human pregnancy urine increases phagocytosis, complement receptor expressions and arachidonic acid metabolism of polymorphonuclear neutrophils. Immunopharmacology 1992, 24, 181–190. [Google Scholar] [CrossRef]
- Saemann, M.D.; Weichhart, T.; Zeyda, M.; Staffler, G.; Schunn, M.; Stuhlmeier, K.N.; Sobanov, Y.; Stulnig, T.M.; Akira, S.; von Gabain, A.; et al. Tamm–Horsfall glycoprotein links innate immune cell activation with adaptive immunity via a Toll-like receptor-4-dependent mechanism. J. Clin. Invest. 2005, 115, 468–475. [Google Scholar]
- Siao, S.C.; Li, K.J.; Hsieh, S.C.; Wu, C.H.; Lu, M.C.; Tsai, C.Y.; Yu, C.L. Tamm–Horsfall glycoprotein enhances PMN phagocytosis by binding to cell surface-expressed lactoferrin and cathepsin G that activates MAP kinase pathway. Molecules 2011, 16, 2119–2134. [Google Scholar] [CrossRef]
- Wu, T.-H.; Hsieh, S.-C.; Tsai, C.-Y.; Lee, Y.-F.; Yu, C.-L. Intact protein core structure is essential for protein-binding, mononuclear cell proliferation, and neutrophil phagocytosis-enhancing activities of normal human Tamm–Horsfall glycoprotein. Int. Immunopharmacol. 2008, 8, 90–99. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, C.; Li, Z.; Guo, W.; Gegner, J.A.; Lin, S.; Han, J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J. Biol. Chem. 1996, 271, 17920–17926. [Google Scholar]
- Hoppe, A.D.; Swanson, J.A. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol. Biol. Cell. 2004, 15, 3509–3519. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, J.; Bu, X.; Cushion, M.; Kinanem, B.; Avraham, H.; Koziel, H. Cdc42 and RhoB activation are required for mannose receptor-mediated phagocytosis by human alveolar macrophages. Mol. Biol. Cell. 2005, 16, 824–834. [Google Scholar]
- Lerm, M.; Brodin, V.P.; Ruishalme, I.; Stendahl, O.; Saerndahl, E. Inactivation of Cdc42 is necessary for depolymerization of phagosomal F-actin and subsequent phagosomal maturation. J. Immunol. 2007, 178, 7357–7365. [Google Scholar]
- Beemiller, P.; Zhang, Y.; Mohan, S.; Levinsohn, E.; Gaeto, I.; Hoppe, A.D.; Swanson, J.A. A Cdc42 activation cycle coordinated by PI3-kinase during Fc receptor-mediated phagocytosis. Mol. Biol. Cell 2010, 21, 470–480. [Google Scholar] [CrossRef]
- Schmidt, M.H.; Bicker, F.; Nikolic, I.; Meister, J.; Babuke, T.; Picuric, S.; Mueller-Esterl, W.; Plate, K.H.; Dikic, I. Epidermal growth factor-like domain 7 (EGFL7) modulates Notch signalling and affects neural stem cell renewal. Nat. Cell. Biol. 2009, 11, 873–880. [Google Scholar] [CrossRef]
- Kansas, G.S.; Saunders, K.; Ley, K.; Zakrzewicz, A.; Gibson, M.; Furie, B.C.; Furie, B.; Tedder, T.F. A role for the epidermal growth factor-like domain of P-selectin in ligand recognition and cell adhesion. J. Cell. Biol. 1994, 124, 609–618. [Google Scholar] [CrossRef]
- Lewkowicz, P.; Tchorzewski, H.; Dytnerska, K.; Banasik, M.; Lewkowicz, N. Epidermal growth factor enhances TNF-alpha-induced priming of human neutrophils. Immunol. Lett. 2005, 96, 203–210. [Google Scholar] [CrossRef]
- Li, S.; Wang, Q.; Wang, Y.; Chen, X.; Wang, Z. PLC-gamma1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol. Endocrinol. 2009, 23, 901–913. [Google Scholar] [CrossRef]
- Park, S.; Jung, H.H.; Park, Y.H.; Ahn, J.S.; Im, Y.H. ERK/MAPK pathways play critical roles in EGFR ligands-induced MMP1 expression. Biochem. Biophys. Res. Commun. 2011, 407, 680–686. [Google Scholar] [CrossRef]
- Petecchia, L.; Sabatini, F.; Usai, C.; Caci, F.; Varesio, L.; Rossi, G.A. Cytokines induce tight junction dissembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab. Invest. 2012, 92, 1140–1148. [Google Scholar] [CrossRef]
- Hunt, J.S.; McGiven, A.R. Stimulation of human peripheral blood lymphocytes by Tamm–Horsfall glycoprotein. Immunology 1978, 35, 391–395. [Google Scholar]
- Sample Availability: Normal human urinary THP is available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, K.-J.; Siao, S.-C.; Wu, C.-H.; Shen, C.-Y.; Wu, T.-H.; Tsai, C.-Y.; Hsieh, S.-C.; Yu, C.-L. EGF Receptor-Dependent Mechanism May be Involved in the Tamm–Horsfall Glycoprotein-Enhanced PMN Phagocytosis via Activating Rho Family and MAPK Signaling Pathway. Molecules 2014, 19, 1328-1343. https://doi.org/10.3390/molecules19011328
Li K-J, Siao S-C, Wu C-H, Shen C-Y, Wu T-H, Tsai C-Y, Hsieh S-C, Yu C-L. EGF Receptor-Dependent Mechanism May be Involved in the Tamm–Horsfall Glycoprotein-Enhanced PMN Phagocytosis via Activating Rho Family and MAPK Signaling Pathway. Molecules. 2014; 19(1):1328-1343. https://doi.org/10.3390/molecules19011328
Chicago/Turabian StyleLi, Ko-Jen, Sue-Cien Siao, Cheng-Han Wu, Chieh-Yu Shen, Tsai-Hung Wu, Chang-Youh Tsai, Song-Chou Hsieh, and Chia-Li Yu. 2014. "EGF Receptor-Dependent Mechanism May be Involved in the Tamm–Horsfall Glycoprotein-Enhanced PMN Phagocytosis via Activating Rho Family and MAPK Signaling Pathway" Molecules 19, no. 1: 1328-1343. https://doi.org/10.3390/molecules19011328