Construction of the 1,2-Dialkenylcyclohexane Framework via Ireland-Claisen Rearrangement and Intramolecular Barbier Reaction: Application to the Synthesis of (±)-Geijerone and a Diastereoisomeric Mixture with Its 5-Epimer

Abstract
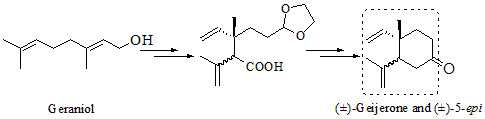
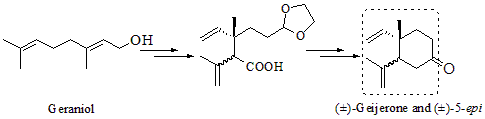
:
1. Introduction
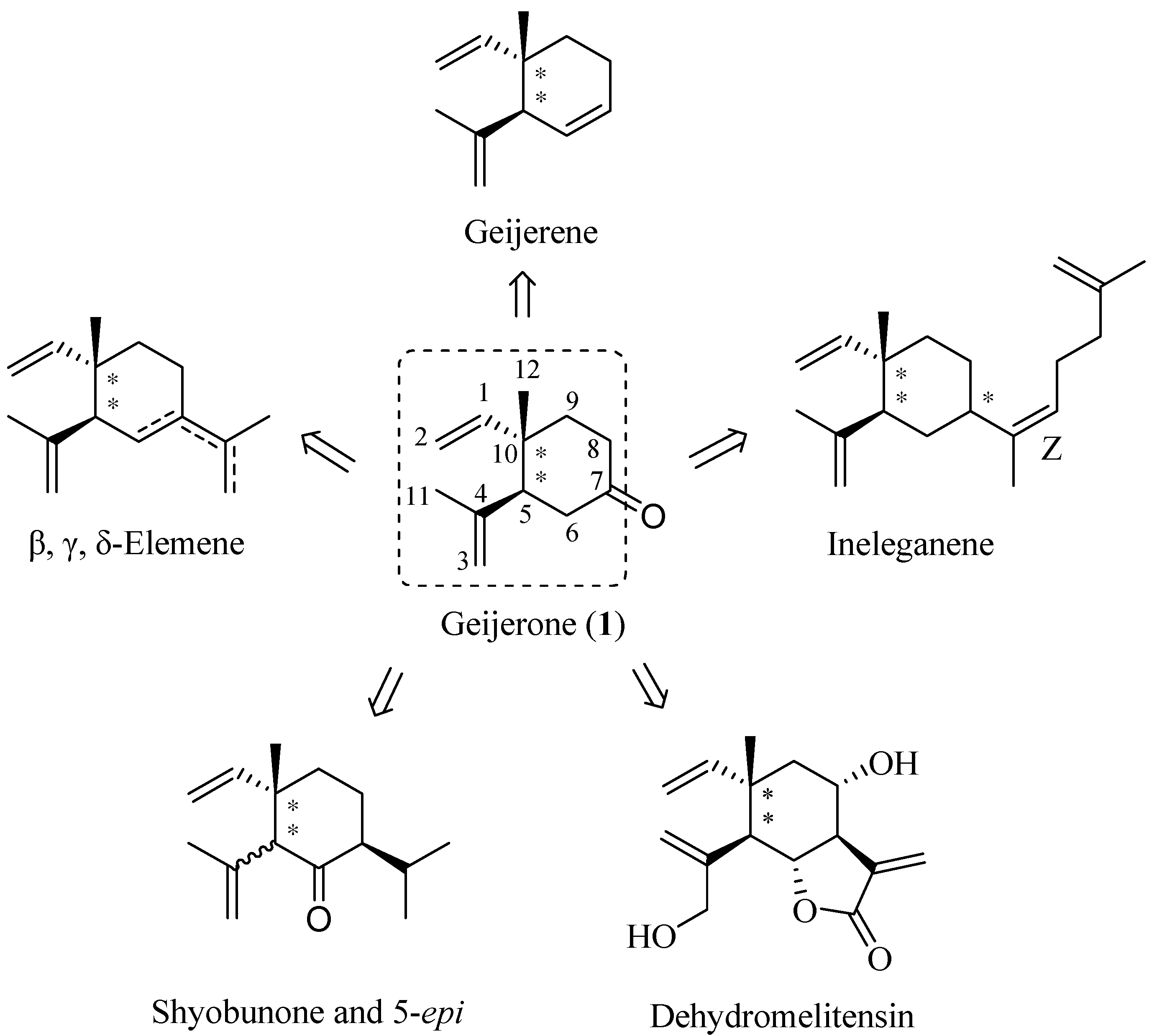
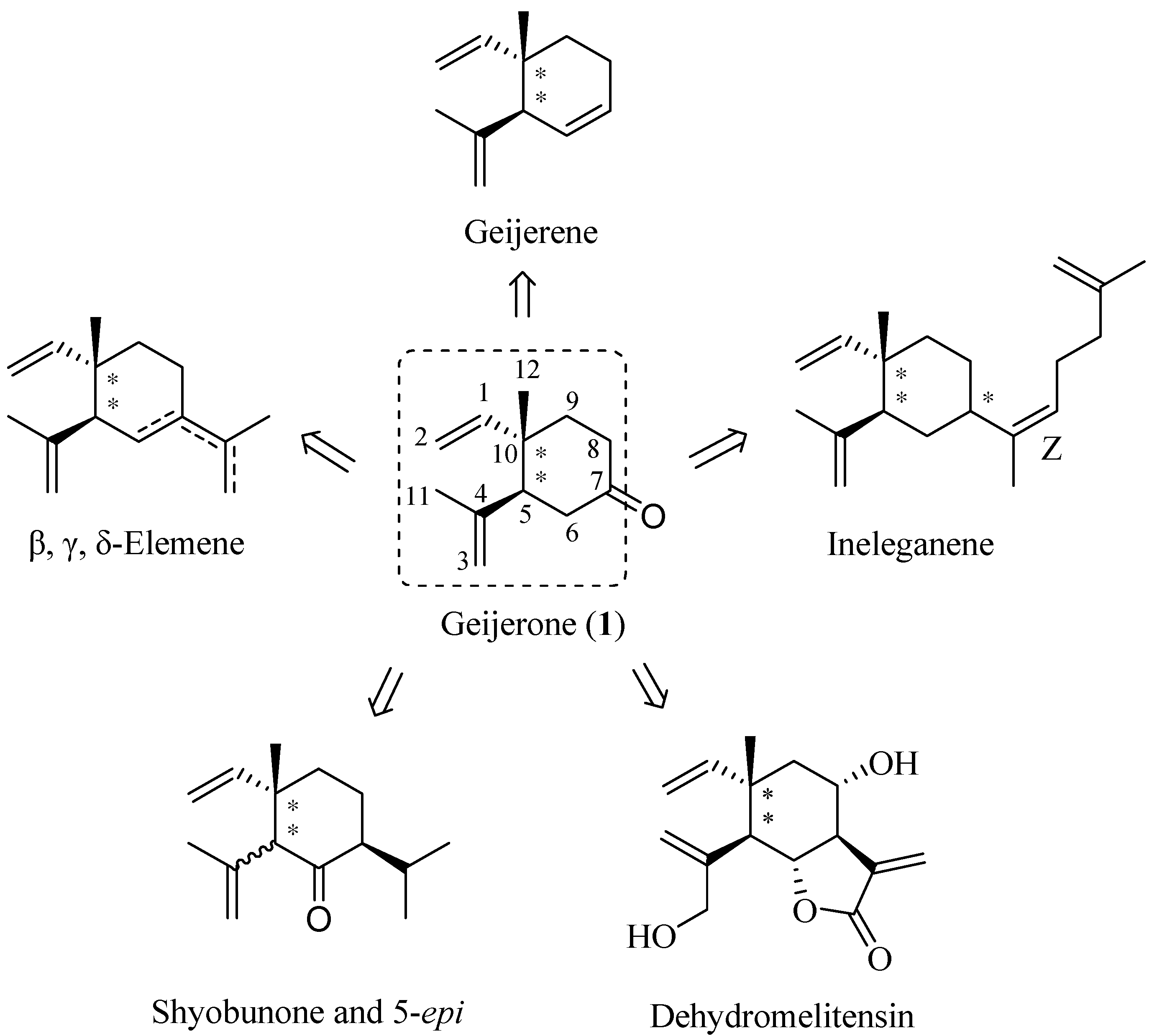
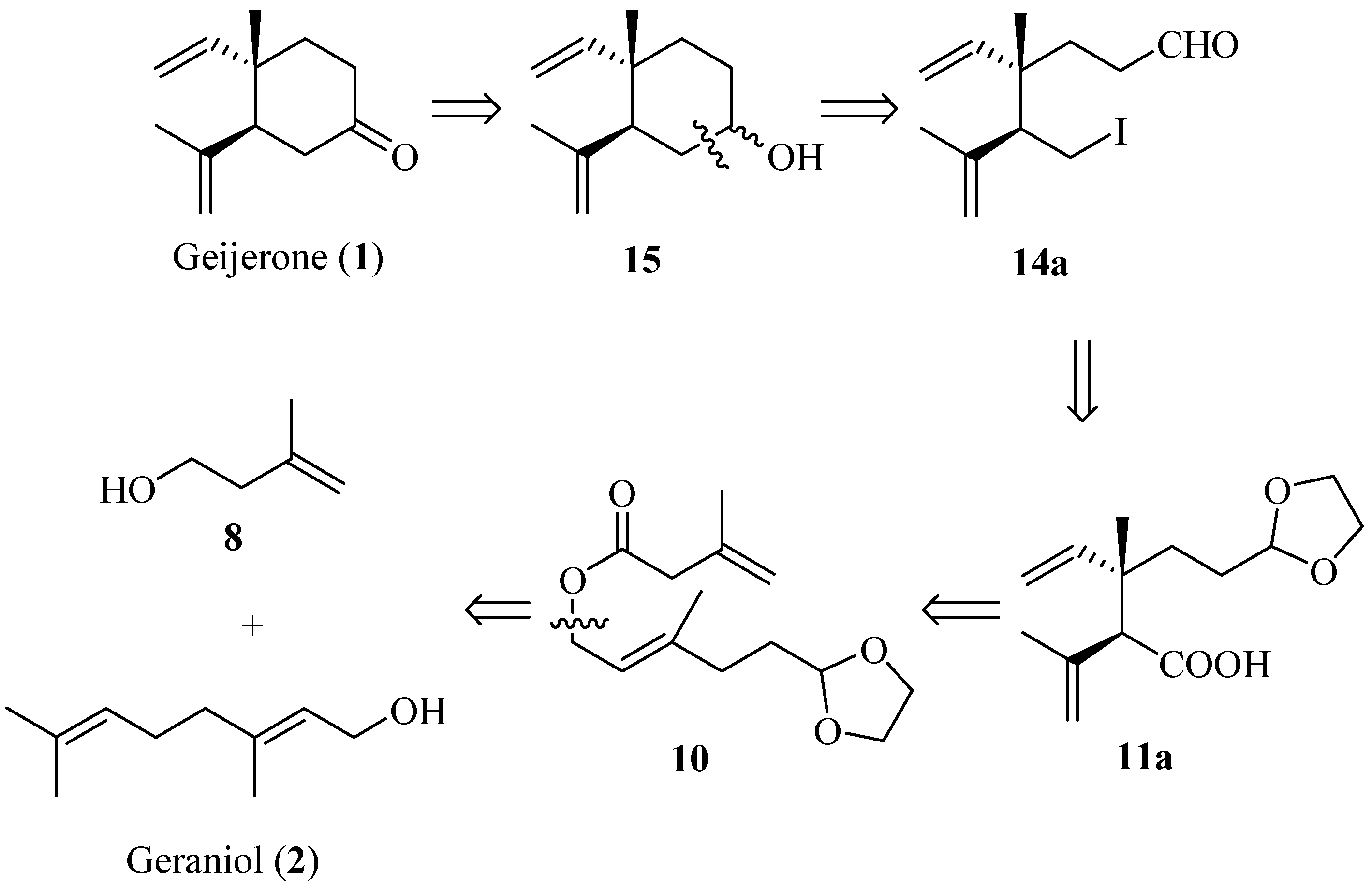
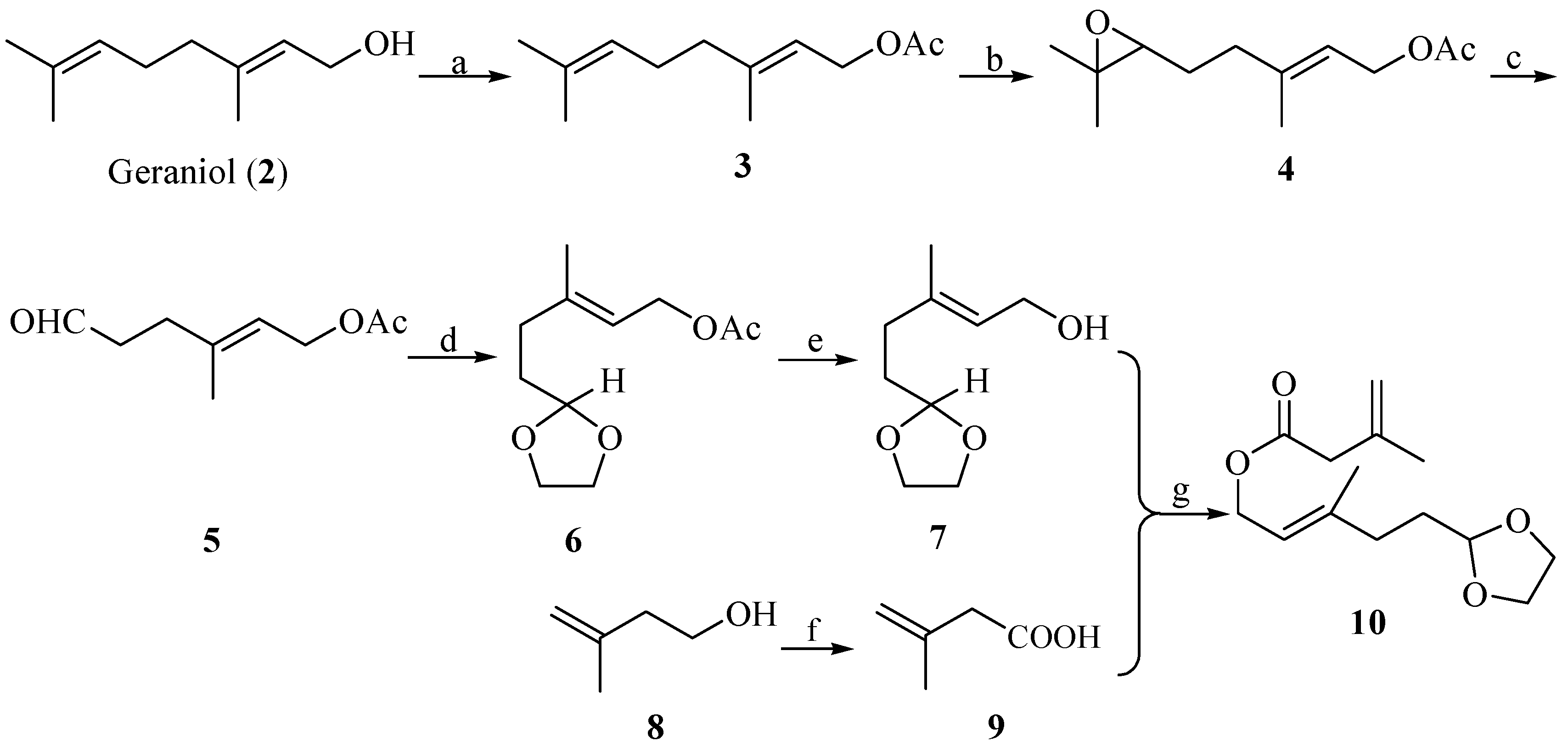
2. Results and Discussion
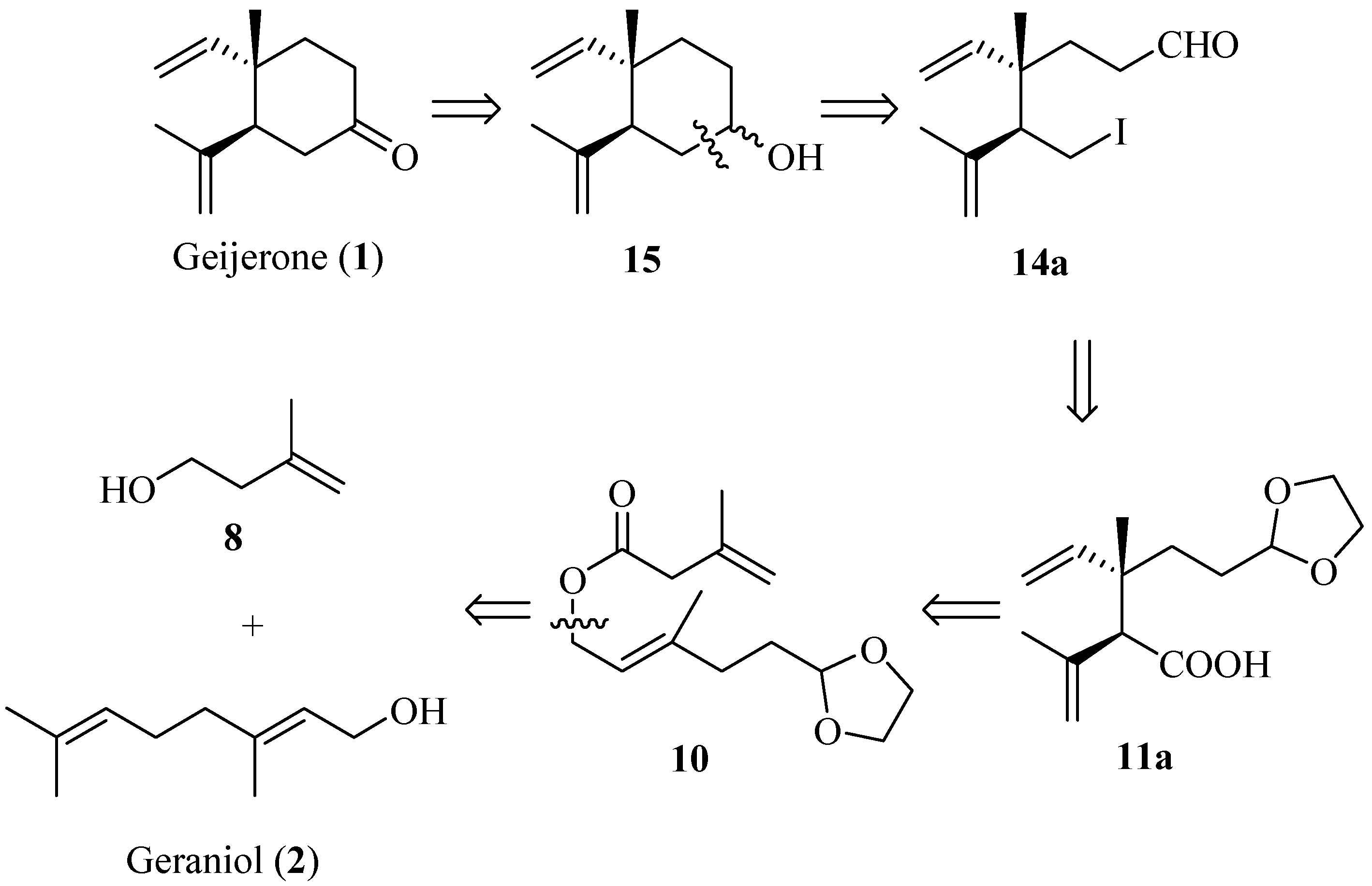

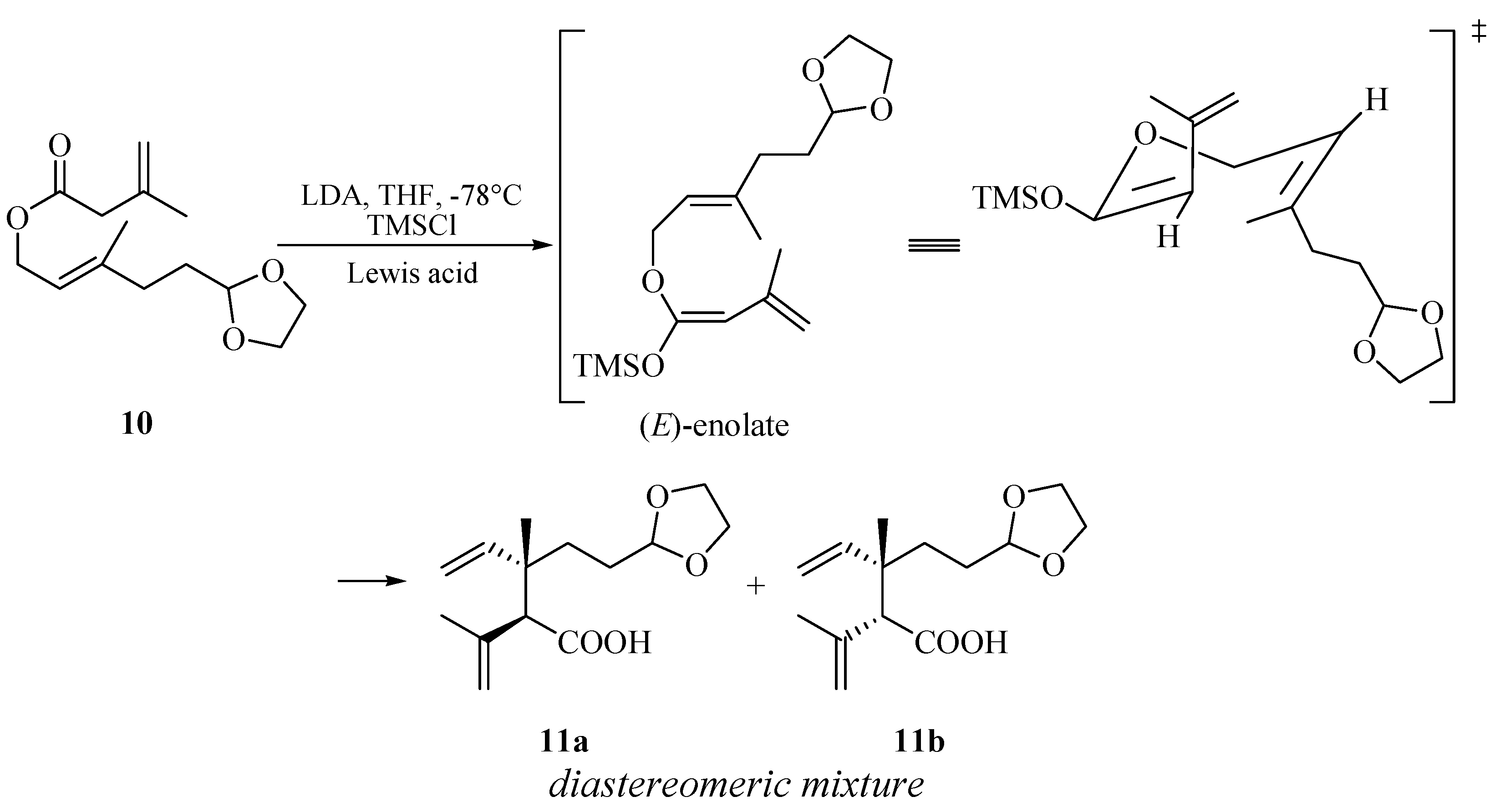

{kind=link}
{kind=link}
{kind=link}
{kind=link}
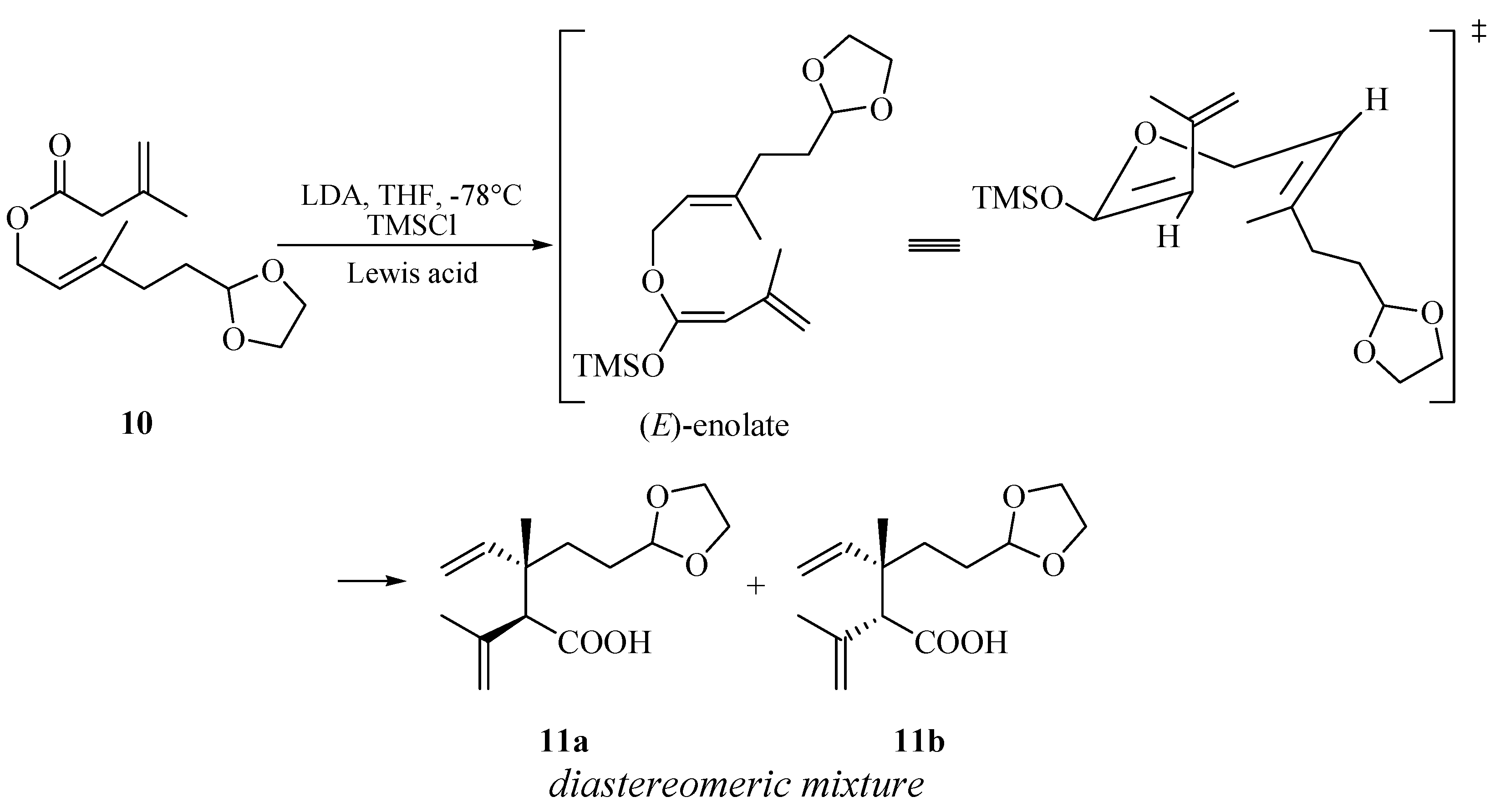{kind=link}
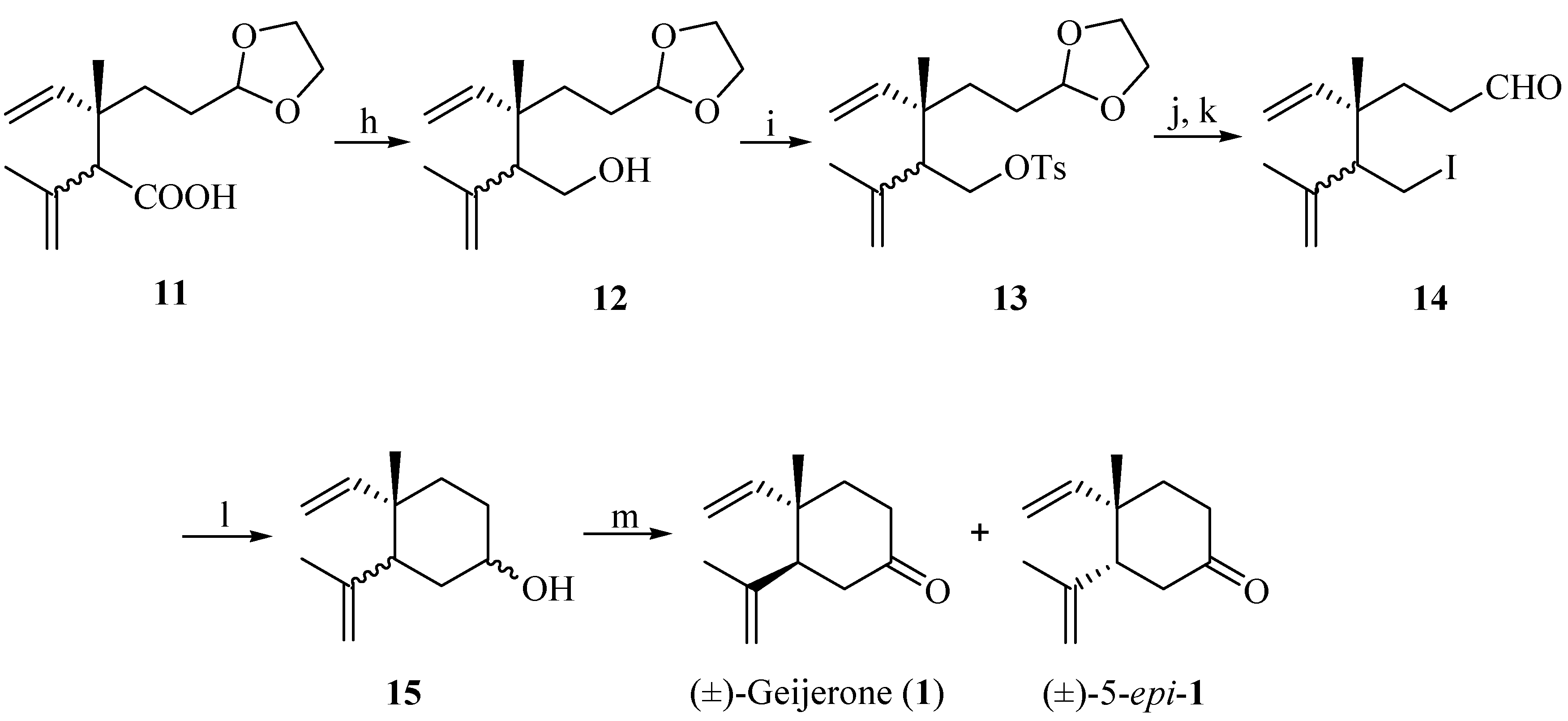{kind=link}
| Entry a | Lewis acid | Yield(%) b | dr c |
|---|---|---|---|
| 1 | None | 72% | 2:1 |
| 2 | TMSOTf | 94% | 1:1 |
| 3 | BF3-Et2O | 24% | 1:1 |
| 4 | ZnCl2 | 75% | 1:1 |
| 5 | SnCl4 | 91% | 1:1 |

3. Experimental
General Information
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Newman, D.J. Natural products as leads to potential drugs: An old process or the new hope for drug discovery? J. Med. Chem. 2008, 51, 2589–2599. [Google Scholar] [CrossRef]
- Choi, H.S.; Sawamura, M. Effects of storage conditions on the composition of Citrus tamurana Hort. ex Tanaka (hyuganatsu) essential oil. Biosci. Biotechnol. Biochem. 2002, 66, 439–443. [Google Scholar] [CrossRef]
- Kweka, E.J.; Nyindo, M.; Mosha, F.; Silva, A.G. Insecticidal activity of the essential oil from fruits and seeds of Schinus terebinthifolia Raddi against African malaria vectors. Parasite. Vector. 2011, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Djabou, N.; Allali, H.; Battesti, M.J.; Tabti, B.; Costa, J.; Muselli, A.; Varesi, L. Chemical and genetic differentiation of two Mediterranean subspecies of Teucrium scorodonia L. Phytochemistry 2011, 74, 123–132. [Google Scholar]
- Birch, A.J.; Grimshaw, J.; Penfold, A.R.; Sheppard, N.; Speake, R.N. An independent confirmation of the structure of geijerene by physical methods. J. Chem. Soc. 1961, 2286–2291. [Google Scholar] [CrossRef]
- Chai, M.C.; Wang, S.K.; Dai, C.F.; Duh, C.Y. A cytotoxic lobane diterpene from the formosan soft coral Sinularia inelegans. J. Nat. Prod. 2000, 63, 843–844. [Google Scholar] [CrossRef]
- Terada, Y.; Yamamura, S. Stereochemical studies on germacrenes: An application of molecular mechanics calculations. Tetrahedron Lett. 1979, 35, 3303–3306. [Google Scholar] [CrossRef]
- Terada, Y.; Yamamura, S. An Application of molecular mechanics calculations on thermal reactions of ten-membered ring Sesquit. Bull. Chem. Soc. Jpn. 1982, 55, 2495–2499. [Google Scholar] [CrossRef]
- Machado, F.B.; Yamamoto, R.E.; Zanoli, K.; Nocchi, S.R.; Novello, C.R.; Schuquel, I.T.A.; Sakuragui, C.M.; Luftmann, H.; Ueda-Nakamura, T.; Nakamura, C.V.; et al. Evaluation of the antiproliferative activity of the leaves from Arctium lappa by a bioassay-guided fractionation. Molecules 2012, 17, 1852–1859. [Google Scholar] [CrossRef]
- Yang, H.; Wang, X.S.; Yu, L.L.; Zheng, S. The antitumor activity of elemene is associated with apoptosis. Chin. J. Oncol. 1996, 18, 169–172. [Google Scholar]
- Srivastava, S.K.; Abraham, A.; Bhat, B.; Jaggi, M.; Singh, A.T.; Sanna, V.K.; Singh, G.; Agarwal, S.K.; Mukherjee, R.; Burman, A.C. Synthesis of 13-amino costunolide derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2006, 16, 4195–4199. [Google Scholar] [CrossRef]
- Choi, B.G.; Kwak, E.Y.; Chung, B.H.; Cho, W.J.; Cheon, S.H. Synthesis of sesquiterpene derivatives as potential antitumor agents; elemane derivatives. Arch. Pharm. Res. 1999, 22, 575–578. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Qu, J.L.; Xu, L.; Hou, K.Z.; Zhang, J.D.; Qu, X.J.; Liu, Y.P. â-Elemene-induced autophagy protects human gastric cancer cells from undergoing apoptosis. BMC Cancer 2011, 11, 183. [Google Scholar] [CrossRef]
- Chen, W.; Lu, Y.; Wu, J.; Gao, M.; Wang, A.; Xu, B. Beta-elemene inhibits melanoma growth and metastasis via suppressing vascular endothelial growth factor-mediated angiogenesis. Cancer Chemoth. Pharmacol. 2011, 67, 799–808. [Google Scholar] [CrossRef]
- Xie, C.Y.; Yang, W.; Ying, J.; Ni, Q.C.; Pan, X.D.; Dong, J.H.; Li, K.; Wang, X.S. B-Cell Lymphoma-2 over-expression protects ä-elemene-induced apoptosis in human lung carcinoma mucoepidermoid cells via a nuclear factor Kappa B-related pathway. Biol. Pharm. Bull. 2011, 34, 1279–1286. [Google Scholar] [CrossRef]
- Wang, X.W. Elemene: Antineoplastic. Drugs Future 1998, 23, 266–270. [Google Scholar] [CrossRef]
- Li, D.J.; Shao, J.L.; Zhang, Z.L.; Ao, J.H.; Zhang, Y.; Gu, M.; Chen, T. Pharmacological studies on elemene and the clinical application. Lishizhen Med. Mater. Med. Res. 2001, 12, 1123–1124. [Google Scholar]
- Zhao, R.B.; Zhao, Y.F.; Song, G.Q.; Wu, Y.L. Double michael reaction of carvone and its derivatives. Tetrahedron Lett. 1990, 31, 3559–3562. [Google Scholar]
- Zhao, Y.F.; Zhao, R.B.; Wu, Y.L. Double michael reaction of carvone and its utilization in chiral synthesis of natural products. Huaxue Xuebao 1994, 52, 823–830. [Google Scholar]
- Patil, L.J.; Rao, A.S. Synthesis of â-elemene and elemol. Tetrahedron Lett. 1967, 8, 2273–2275. [Google Scholar] [CrossRef]
- McMurry, J.E.; Kocovsky, P. Synthesis of helminthogermacrene and â-elemene. Tetrahedron Lett. 1985, 26, 2171–2172. [Google Scholar] [CrossRef]
- Corey, E.J.; Roberts, B.E.; Dixon, B.R. Enantioselective total synthesis of â-elemene and fuscol based on enantiocontrolled Ireland-Claisen rearrangement. J. Am. Chem. Soc. 1995, 117, 193–196. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.; Chang, J.; Kim, S. Stereoselective synthesis of (±)-â-elemene by a doubly diastereodifferentiating internal alkylation: A remarkable difference in the rate of enolization between syn and anti esters. Tetrahedron 2001, 57, 1247–1252. [Google Scholar] [CrossRef]
- Barrero, A.F.; Herrador, M.M.; Quílez del Moral, J.F.; Arteaga, P.; Meine, N.; Pérez-Morales, M.C.; Catalán, J.V. Efficient synthesis of the anticancer â-elemene and other bioactive elemanes from sustainable germacrone. Org. Biomol. Chem. 2011, 9, 1118–1125. [Google Scholar]
- Kim, D.; Kim, H.S. Stereoselective construction of functionalized cis-1,2-dialkyl-cyclo-hexanecarboxylates: A novel synthesis of (±)-geijerone and ã-elemene. J. Org. Chem. 1987, 52, 4633–4634. [Google Scholar] [CrossRef]
- Kato, M.; Kurihara, H.; Yoshikoshi, A. Total synthesis of racemic geijerone and ã-elemene. J. Chem. Soc. Perkin Trans. 1 1979, 2740–2743. [Google Scholar]
- Kodd, D.S.; Oehlschlager, A.C.; Georgopapadakou, N.H.; Polak, A.M.; Hartman, P.G. Synthesis of inhibitors of 2,3-oxidosqualene-lanosterol cyclase. 2. Cyclocondensation of ã-, ä-unsaturated â-keto esters with imines. J. Org. Chem. 1992, 57, 7226–7234. [Google Scholar] [CrossRef]
- Davis, C.E.; Bailey, J.L.; Lockner, J.W.; Coates, R.M. Regio- and stereoselectivity of diethylaluminum azide opening of trisubstituted epoxides and conversion of the 3° azidohydrin adducts to isoprenoid aziridines. J. Org. Chem. 2003, 68, 75–82. [Google Scholar] [CrossRef]
- Ireland, R.E.; Mueller, R.H.; Willard, A.K. The ester enolate Claisen rearrangement. Stereochemical control through stereoselective enolate formation. J. Am. Chem. Soc. 1976, 98, 2868–2877. [Google Scholar] [CrossRef]
- Koch, G.; Janser, P.; Kottirsch, G.; Romero-Giron, E. Highly diastereoselective Lewis acid promoted Claisen-Ireland rearrangement. Tetrahedron Lett. 2002, 43, 4837–4840. [Google Scholar] [CrossRef]
- Cooke, M.P., Jr.; Houpis, I.N. Metal-halogen exchange-initiated cyclization of iodo carbonyl compounds. Tetrahedron Lett. 1985, 26, 4987–4990. [Google Scholar] [CrossRef]
- Kihara, M.; Kashimoto, M.; Kobayashi, Y.; Kobayashi, S. A new intramolecular Barbier reaction of N-(2-iodobenzyl)phenacylamines: A convenient synthesis of 1,2,3,4-tetrahydroisoquinomn-4-ols. Tetrahedron Lett. 1990, 31, 5347–5348. [Google Scholar]
- Zhang, W.; Dowd, P. Intramolecular Barbier reaction of a 3-(3'-bromopropyl) bicyclo[2.2.2]oct-5-en-2-one. Tetrahedron Lett. 1993, 34, 2095–2098. [Google Scholar] [CrossRef]
- Ramón, D.J.; Yus, M. Carbamoyl and thiocarbamoyl lithium: A new route by naphthalene-catalysed chlorine-lithium exchange. Tetrahedron Lett. 1993, 34, 7115–7118. [Google Scholar] [CrossRef]
- Ennis, D.S.; Lathbury, D.C.; Wanders, A.; Watts, D. Scale-up of an intermolecular Barbier Reaction. Org. Process. Res. Dev. 1998, 2, 287–289. [Google Scholar] [CrossRef]
- Saito, T.; Takeuchi, T.; Matsuhashi, M.; Nakata, T. Chromone derivatives from the leaves of Nicotiana tabacum and their anti-Tobacco Mosaic Virus Activities. Heterocycles 2007, 72, 151–156. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1, 3–7, 9–12 and 14 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liang, D.; Gao, N.; Liu, W.; Dong, J. Construction of the 1,2-Dialkenylcyclohexane Framework via Ireland-Claisen Rearrangement and Intramolecular Barbier Reaction: Application to the Synthesis of (±)-Geijerone and a Diastereoisomeric Mixture with Its 5-Epimer. Molecules 2014, 19, 1238-1249. https://doi.org/10.3390/molecules19011238
Liang D, Gao N, Liu W, Dong J. Construction of the 1,2-Dialkenylcyclohexane Framework via Ireland-Claisen Rearrangement and Intramolecular Barbier Reaction: Application to the Synthesis of (±)-Geijerone and a Diastereoisomeric Mixture with Its 5-Epimer. Molecules. 2014; 19(1):1238-1249. https://doi.org/10.3390/molecules19011238
Chicago/Turabian StyleLiang, Dawei, Nana Gao, Wei Liu, and Jinhua Dong. 2014. "Construction of the 1,2-Dialkenylcyclohexane Framework via Ireland-Claisen Rearrangement and Intramolecular Barbier Reaction: Application to the Synthesis of (±)-Geijerone and a Diastereoisomeric Mixture with Its 5-Epimer" Molecules 19, no. 1: 1238-1249. https://doi.org/10.3390/molecules19011238