Advances in Kinetic Isotope Effect Measurement Techniques for Enzyme Mechanism Study

Abstract
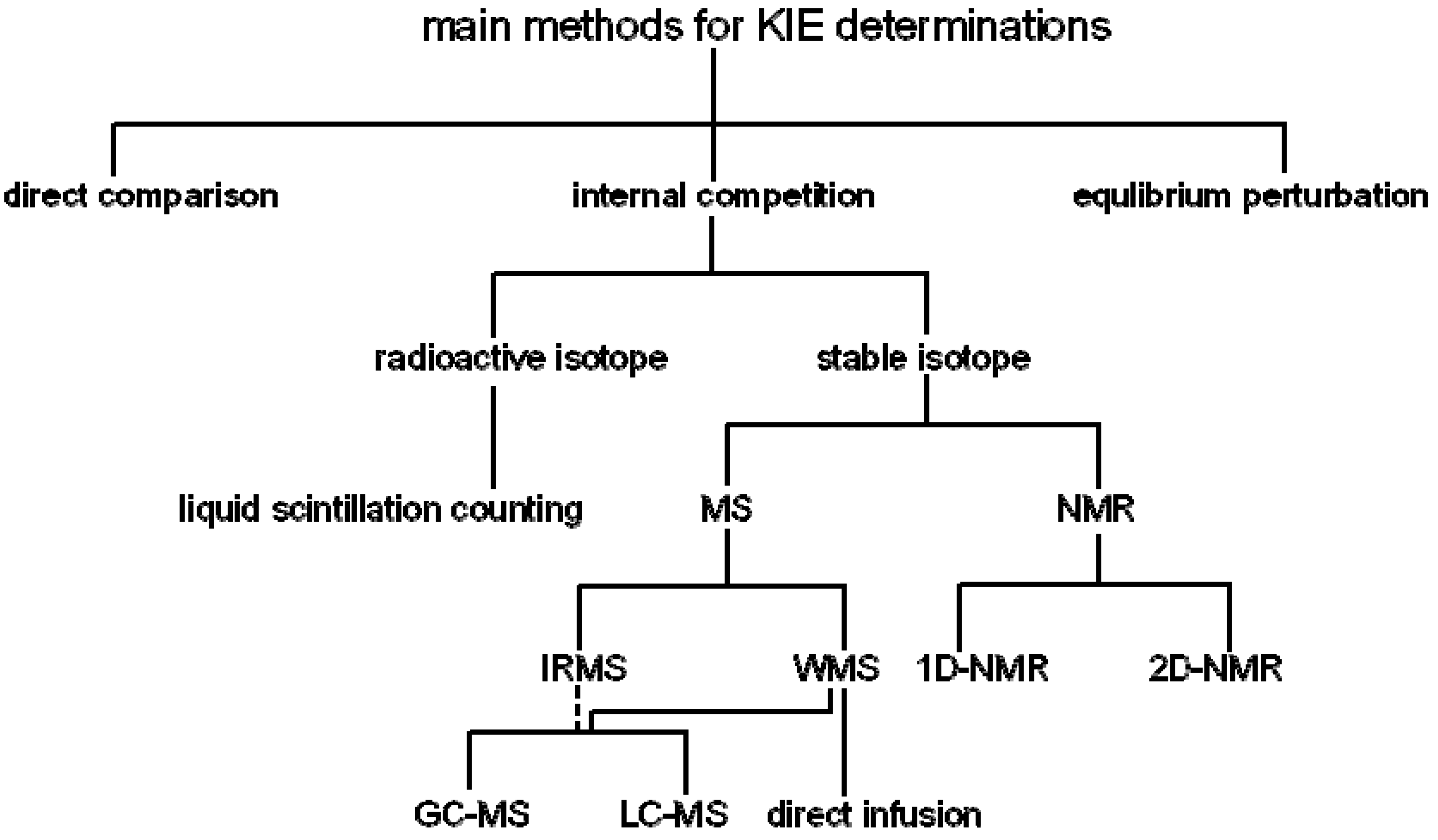
:1. Introduction
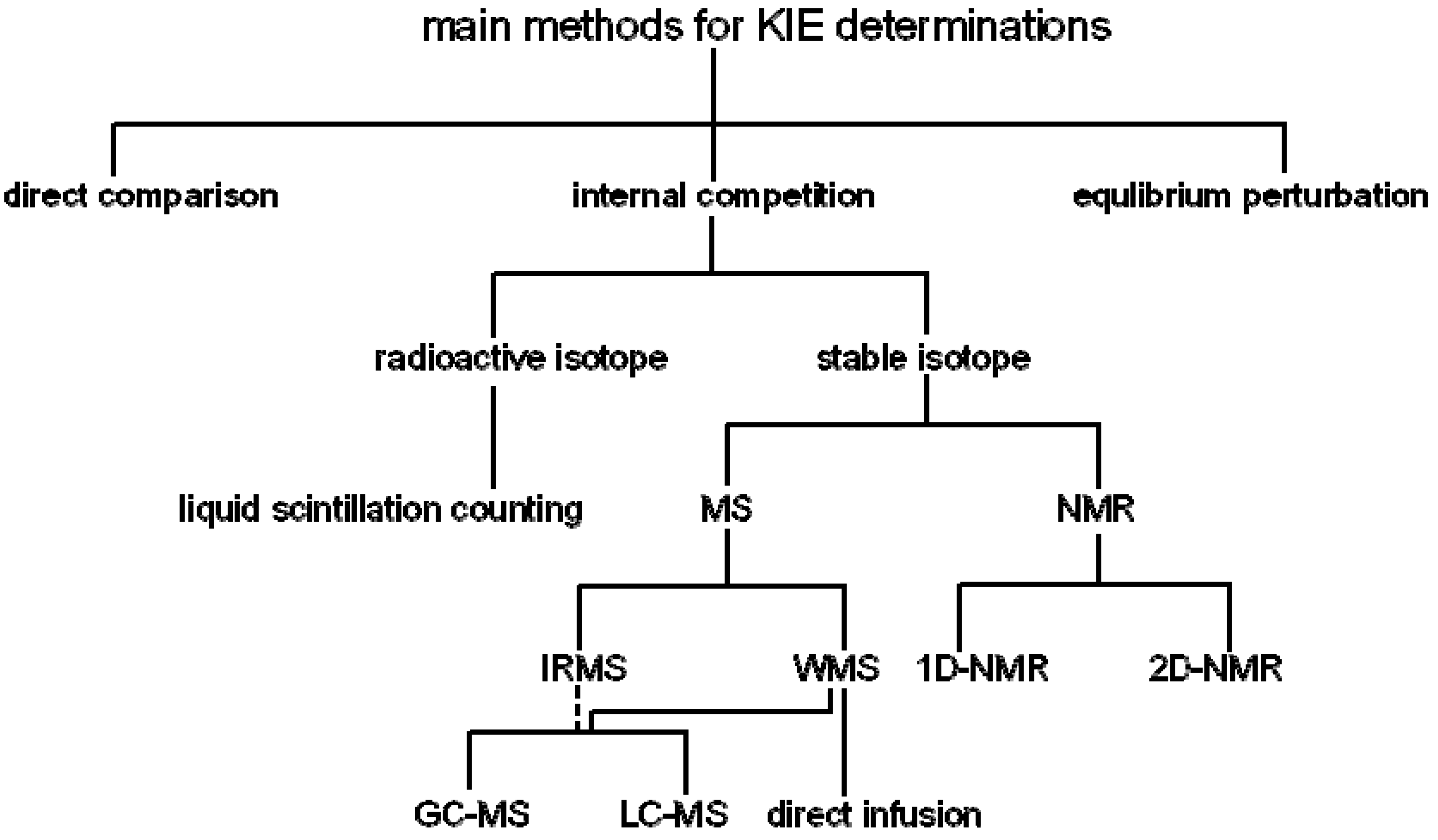
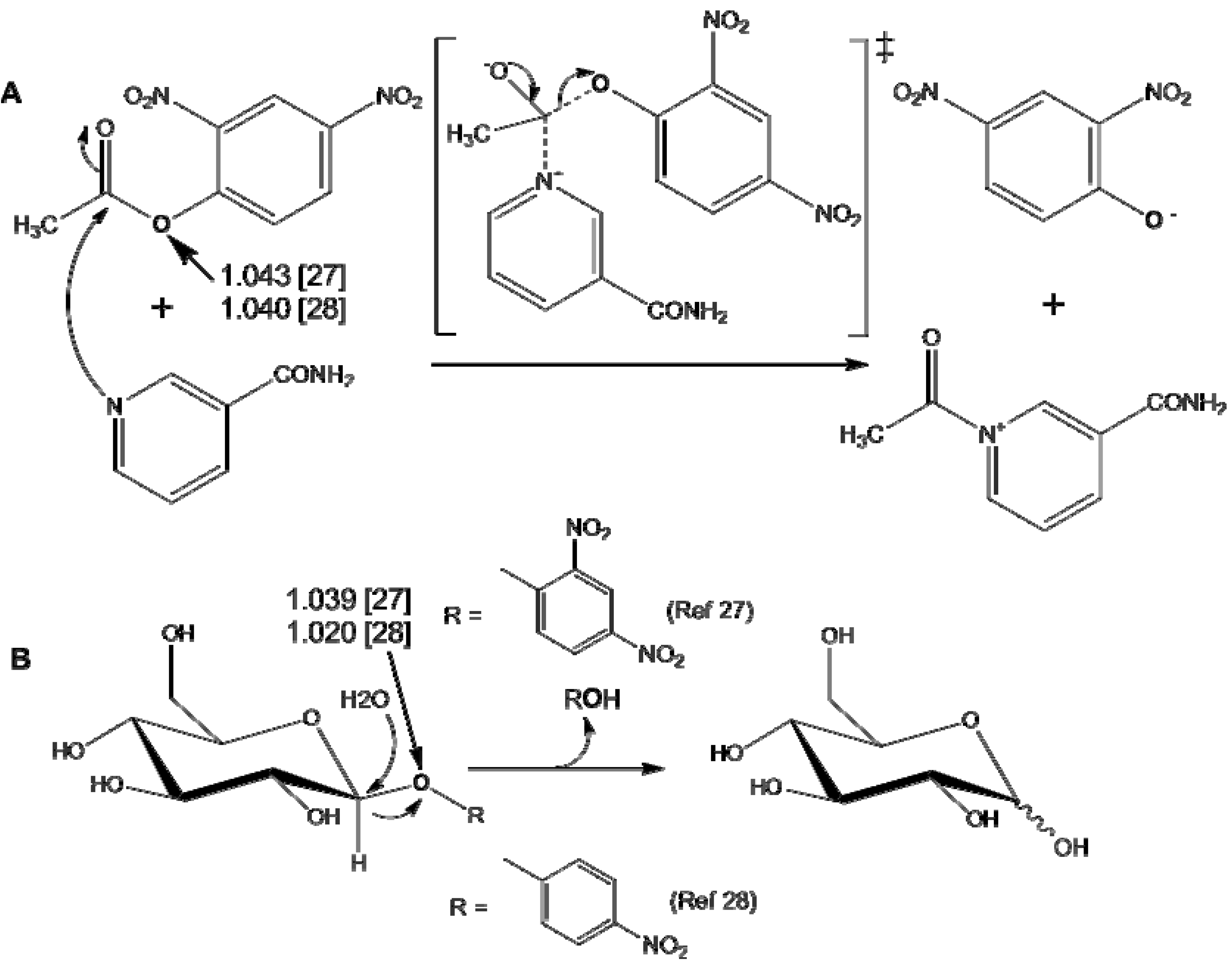
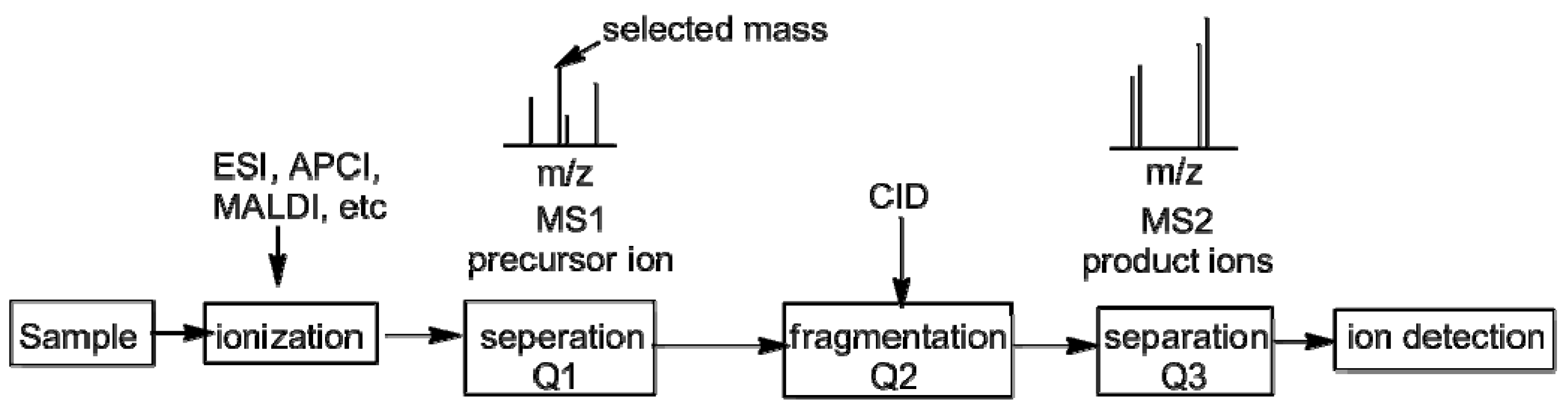
2. Measurement of KIEs by Mass Spectrometry

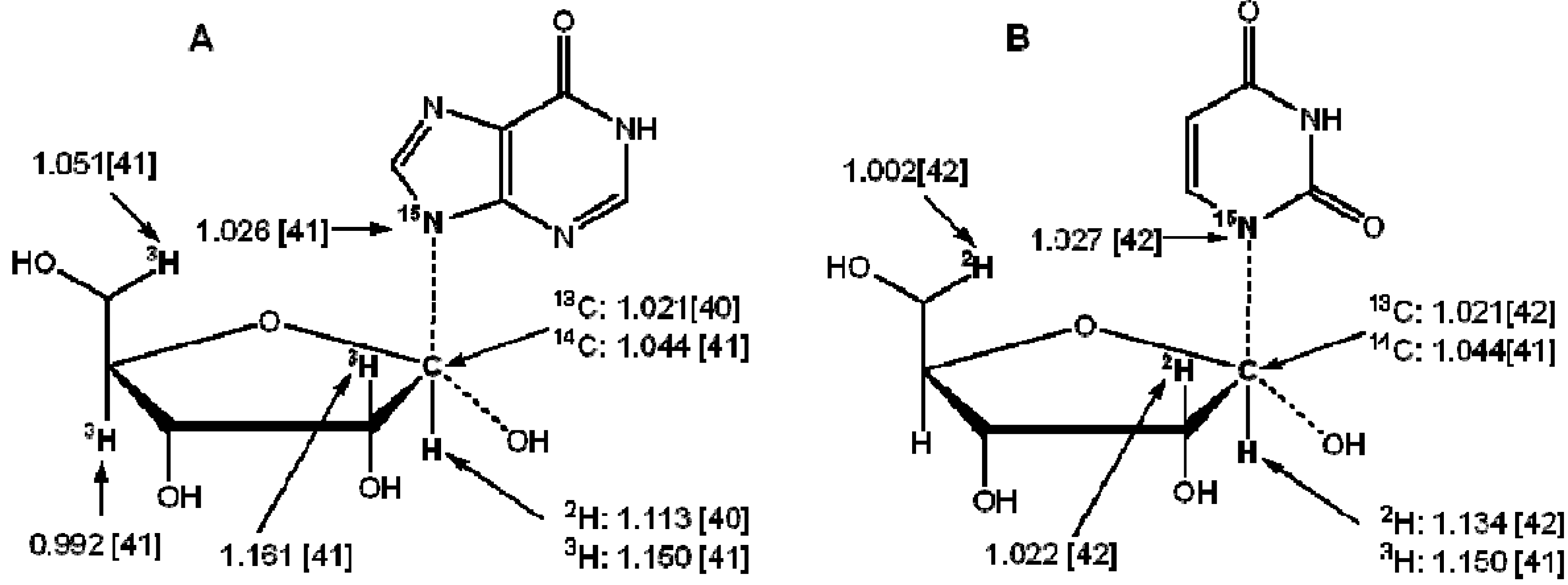
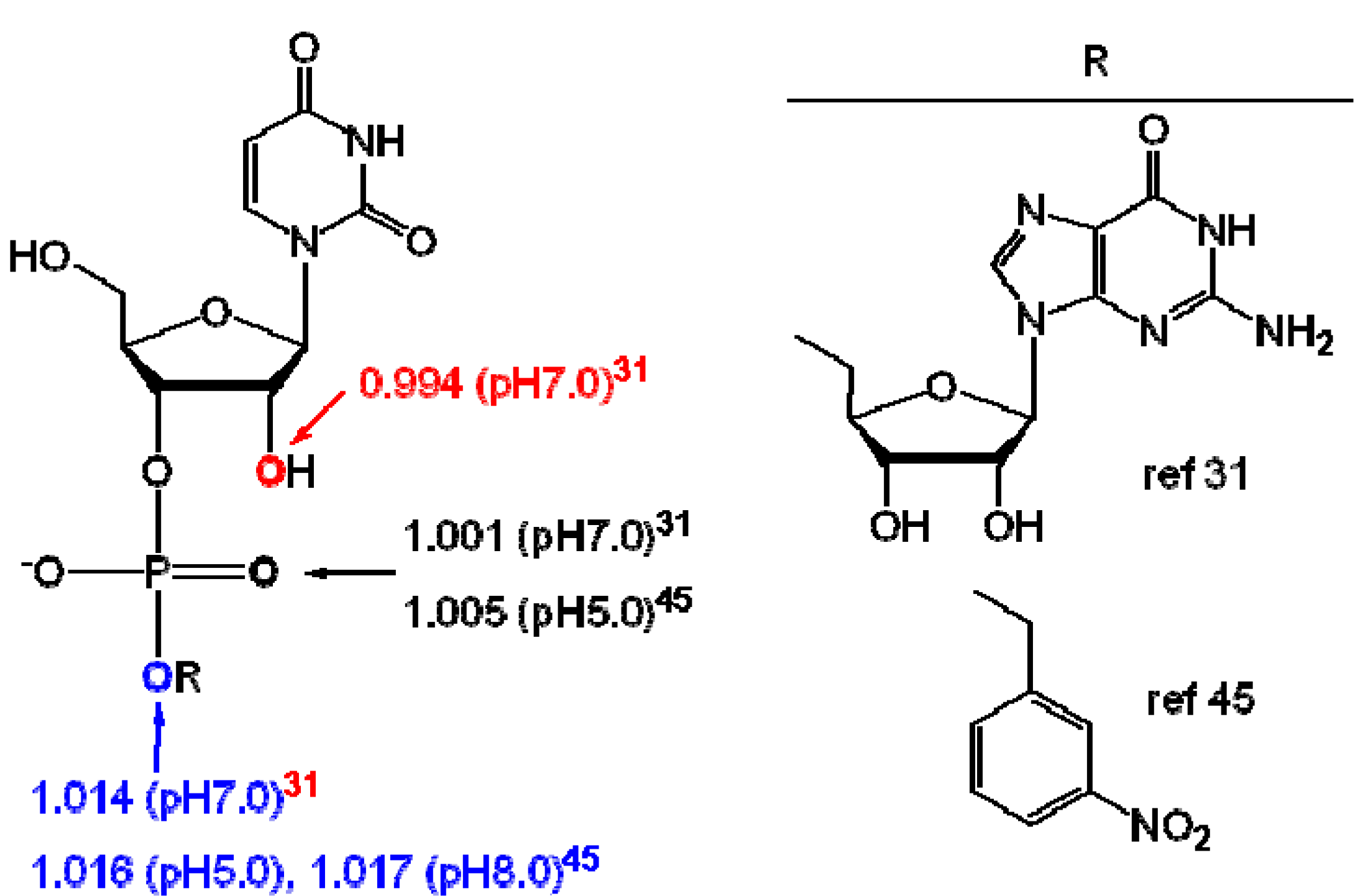
3. Measurement of KIEs by Nuclear Magnetic Resonance Spectrometry

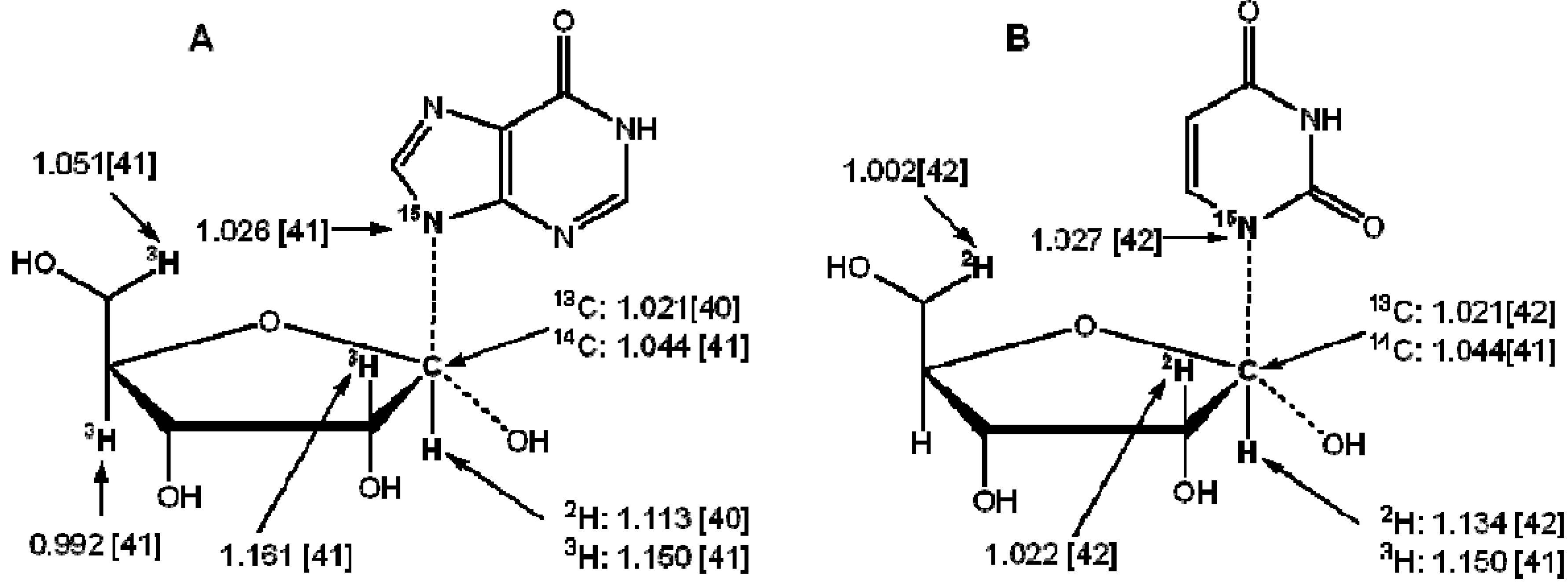{kind=link}
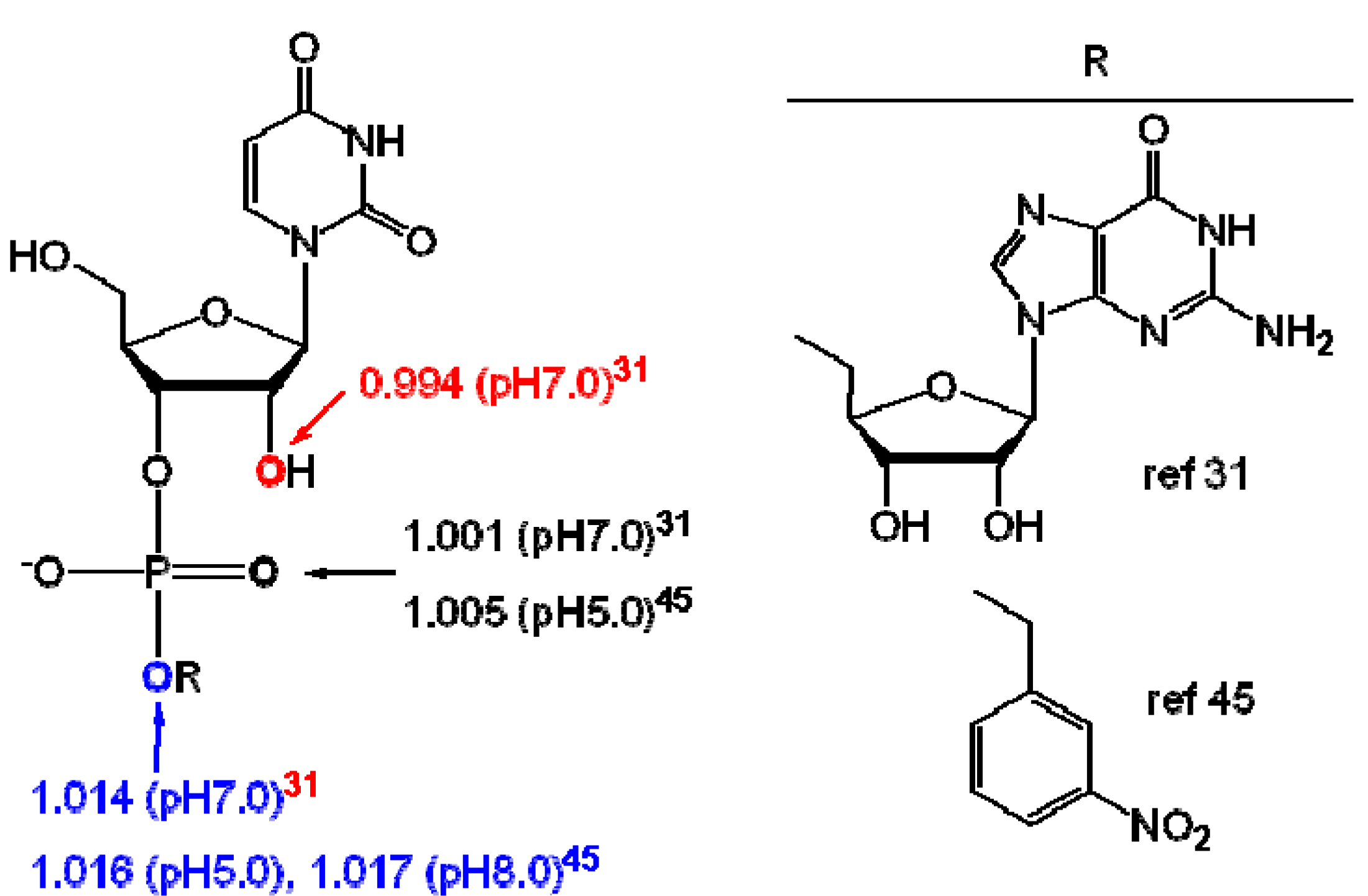{kind=link}
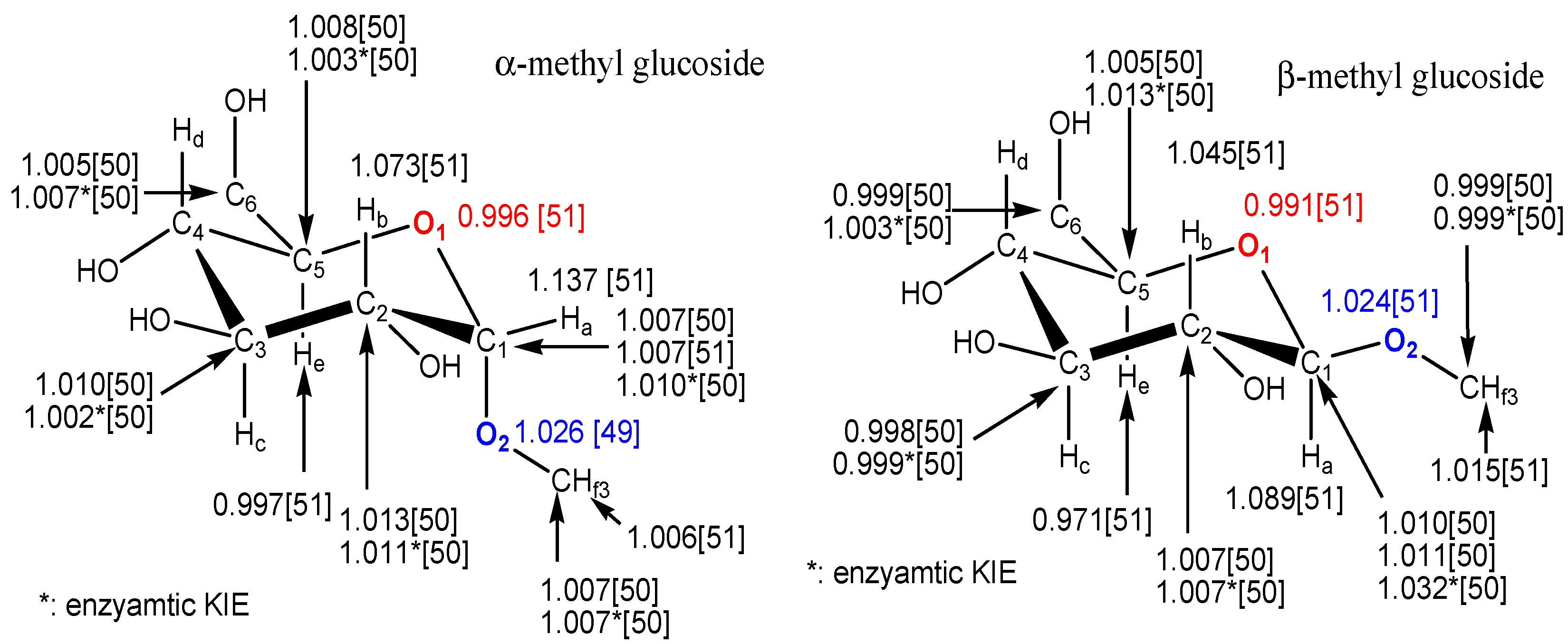{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
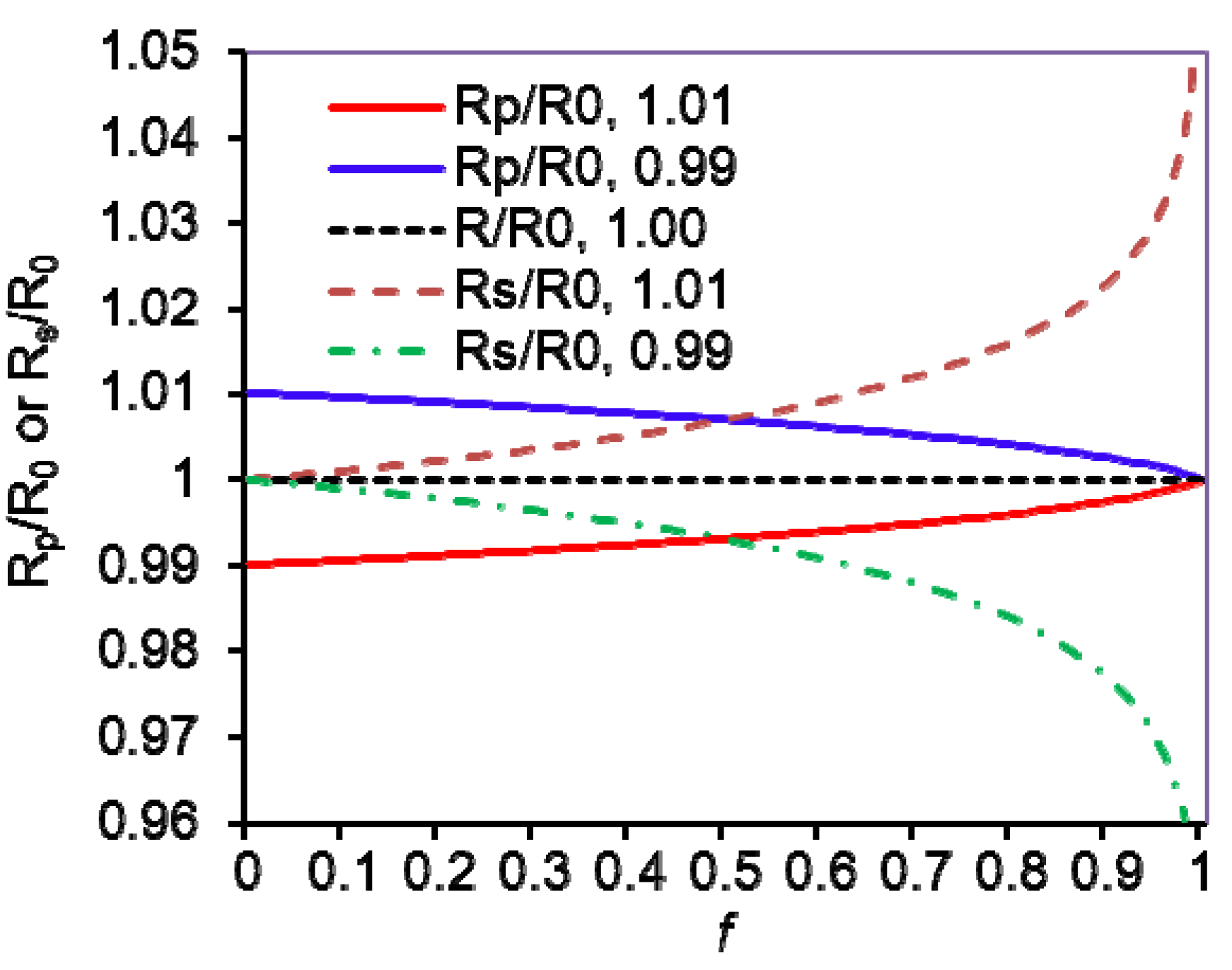
4. KIE Calculations from Isotope Ratios and Their Uncertainties

 (f is variable)
(f is variable)
 (Rs/R0 is variable)
(Rs/R0 is variable)

 (f is variable)
(f is variable)
 (Rp/R0 is variable)
(Rp/R0 is variable)

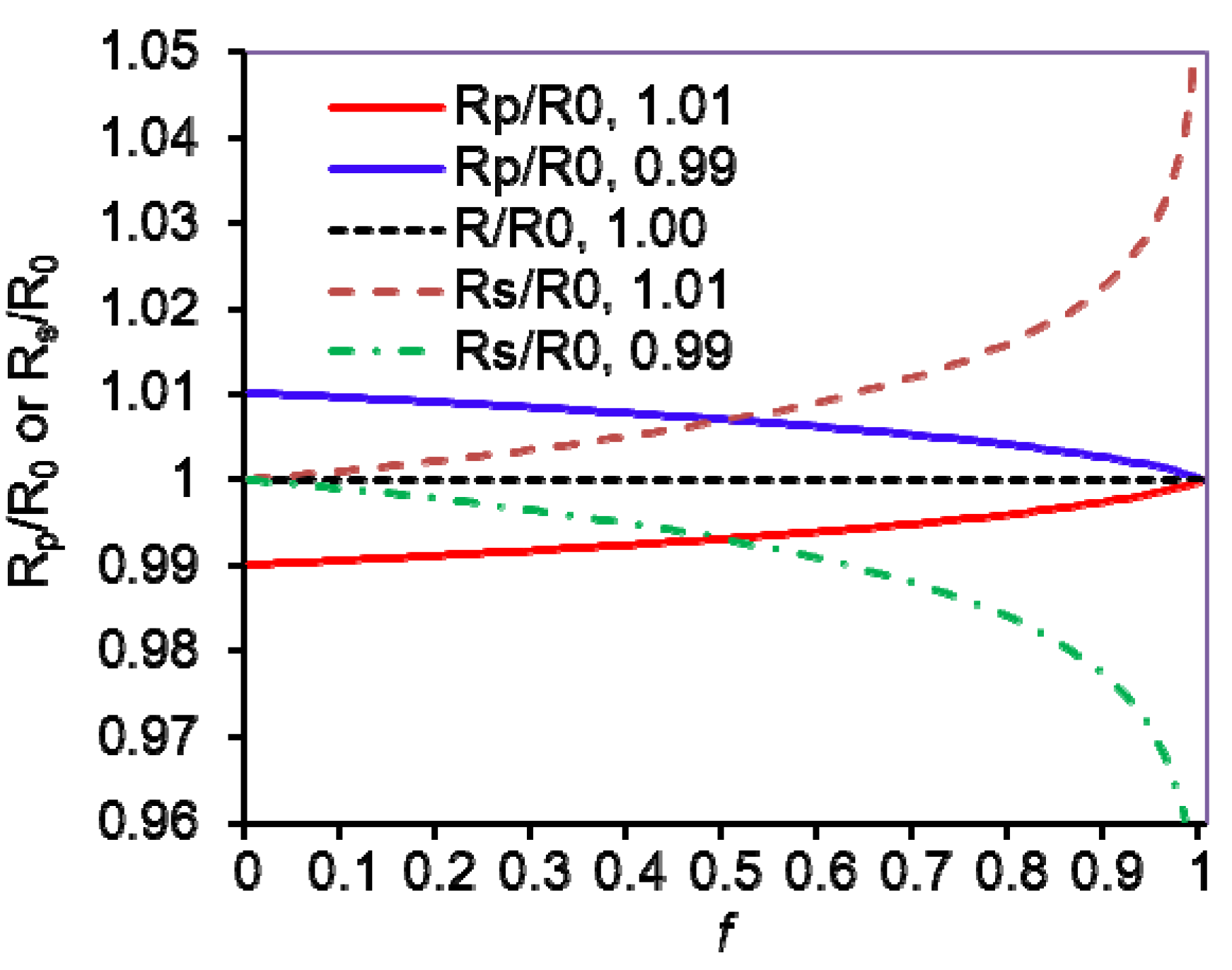


 , Rs =
, Rs =  , Rp =
, Rp =  , and R0 =
, and R0 =  =
=  ; where H0 and L0, H and L represent the concentration of the heavy and light substrates at initio and the fractial reaction, f, respectively.
; where H0 and L0, H and L represent the concentration of the heavy and light substrates at initio and the fractial reaction, f, respectively.5. Measurement of KIEs by Liquid Scintillation Counting
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cleland, W.W. Determining the chemical mechanisms of enzyme-catalyzed reactions by kinetic studies. Adv. Enzymol. Relat. Areas Mol. Biol. 1977, 45, 273–387. [Google Scholar]
- Schramm, V.L. Enzymatic transition states and transition state analog design. Annu. Rev. Biochem. 1998, 67, 693–720. [Google Scholar] [CrossRef]
- Schramm, V.L. Enzymatic transition states, transition-state analogs, dynamics, thermodynamics, and lifetimes. Annu. Rev. Biochem. 2011, 80, 703–732. [Google Scholar] [CrossRef]
- O’leary, M.H. Determination of heavy-atom isotope effects on enzyme-catalyzed reactions. Methods Enzymol. 1980, 64, 83–104. [Google Scholar] [CrossRef]
- Cleland, W.W. The use of isotope effects to determine enzyme mechanisms. Arch. Biochem. Biophys. 2005, 433, 2–12. [Google Scholar] [CrossRef]
- Melander, L.; Saunders, W.H. Isotope Effects on Reaction Rates, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1980. [Google Scholar]
- Isotope Effects in Chemistry and Biology; Kohen, A.; Limbach, H.H. (Eds.) CRC Press: Boca Raton, FL, USA, 2006.
- Fry, A. Heavy Atom effects in organic reaction mechanism studies. In Isotope Effects in Chemical Reactions; Van Nodtrand-Reinhold: Princeton, NJ, USA, 1970; pp. 364–414. [Google Scholar]
- Cook, P.F.; Cleland, W.W. Enzyme Kinetics and Mechanisms; Taylor & Francis Group: New York, NY, USA, 2007. [Google Scholar]
- Enzyme Mechanisms from Isotope Effects; Cook, P.F. (Ed.) CRC Press: Boca Raton, FL, USA, 1991.
- Isotope Effects on Enzyme-Catalyzed Reactions; Cleland, W.W.; O’leary, M.H.; Northrop, D.B. (Eds.) University Park Press: Baltimore, MD, USA, 1977.
- Cleland, W.W. Isotope effects: Determination of enzyme transition state structure. In Methods in Enzymology; Academic Press, Inc.: San Diego, CA, USA, 1995; Volume v249, pp. 341–373. [Google Scholar]
- Simon, H.; Palm, D. Isotope effects in organic chemistry and biochemistry. Angew. Chem. Int. Ed. 1966, 5, 920–933. [Google Scholar] [CrossRef]
- Northrop, D.B. Steady-state analysis of kinetic isotope effects in enzymatic reactions. Biochemistry 1975, 14, 2644–2651. [Google Scholar] [CrossRef]
- Paneth, P. Some analytical aspects of the measurement of heavy-atom kinetic isotope effects. Talanta 1987, 34, 877–883. [Google Scholar] [CrossRef]
- Encyclopaedia of Analytical Science; Townsend, A. (Ed.) Academic Press Limited: London, UK, 1995.
- Brenna, J.T.; Corso, T.N.; Tobias, H.J.; Caimi, R.J. High-precision continuous-flow isotope ratio mass spectrometry. Mass Spectrom. Rev. 1997, 16, 227–258. [Google Scholar] [CrossRef]
- Weiss, P.F. Heavy Atom isotope effects using the isotope ratio mass spectrometer. In Enzyme Mechanism from Isotope Effects; Cook, P.F., Ed.; CRC Press: Boca Raton, FL, USA, 1991; pp. 291–312. [Google Scholar]
- Muccio, Z.; Jacjson, G.P. Isotope ratio mass spectrometry. Analyst 2009, 134, 213–222. [Google Scholar] [CrossRef]
- O’Leary, M.H.; Marlier, J.F. Application of a novel double-labeling procedure to measurement of carbonyl oxygen isotope effects on the alkaline hydrolysis and hydrazinolysis of methyl benzoate. J. Am. Chem. Soc. 1978, 100, 2582–2583. [Google Scholar] [CrossRef]
- Hermes, J.D.; Cleland, W.W. Evidence from multiple isotope effect determinations for coupled hydrogen motion and tunneling in the reaction catalyzed by glucose-6-phosphate dehydrogenase. J. Am. Chem. Soc. 1984, 106, 7263–7264. [Google Scholar] [CrossRef]
- Hermes, J.D.; Roeske, C.A.; O’Leary, M.H.; Cleland, W.W. Use of multiple isotope effects to determine enzyme mechanisms and intrinsic isotope effects. Malic enzyme and glucose-6-phosphate dehydrogenase. Biochemistry 1982, 21, 5106–5114. [Google Scholar] [CrossRef]
- Weiss, P.M.; Garcia, G.A.; Kenyon, G.L.; Cleland, W.W.; Cook, P.F. Kinetic and mechanism of benzoylformate decarboxylase using 13C and solvent deuterium isotope effects on benzoylformate and benzoylformate analogues. Biochemistry 1988, 27, 2197–2205. [Google Scholar] [CrossRef]
- Weiss, P.M.; Cook, P.F.; Hermes, J.D.; Cleland, W.W. Evidence from nitrogen-15 and solvent deuterium isotope effects on the chemical mechanism of adenosine deaminase. Biochemistry 1987, 26, 7378–7384. [Google Scholar] [CrossRef]
- Weiss, P.M.; Chen, C.Y.; Cook, P.F.; Cleland, W.W. Use of primary deuteriumand 15N isotope effects to deduce the relative rates of steps in the mechanisms of alanine and glutamate dehydrogenase. Biochemistry 1988, 27, 4814–4822. [Google Scholar] [CrossRef]
- Zakett, D.; Flynn, R.G.A.; Cooks, R.G. Chlorine isotope effects in mass spectrometry by multiple reaction monitoring. J. Phys. Chem. 1978, 82, 2359–2362. [Google Scholar] [CrossRef]
- Rosenberg, S.; Kirsch, J.F. Measurement of heavy atom kinetic isotope effects by direct mass spectrometric analysis. Anal. Chem. 1979, 51, 1375–1379. [Google Scholar] [CrossRef]
- Rosenberg, S.; Kirsch, J.F. Direct spectrophotometric measurement of small kinetic isotope effects. Anal. Chem. 1979, 51, 1379–1383. [Google Scholar] [CrossRef]
- Harris, M.E.; Dai, Q.; Gu, H.; Kellerman, D.; Piccirilli, J.A.; Anderson, V.E. kinetic isotope effects for RNA cleavage by 2’-O-transphosphorylation: Nucleophilic ativation by specific base. J. Am. Chem. Soc. 2010, 132, 11613–11621. [Google Scholar]
- Wong, K.Y.; Gu, H.; Zhang, S.; Piccirilli, J.A.; Harris, M.E.; York, D.M. Characterization of the reaction path and transition states for RNA transphosphorylation models from theory and experiment. Angew. Chem. Int. Ed. 2012, 51, 647–651. [Google Scholar]
- Gu, H.;Zhang, S.; Wong, K.-Y.; Radak, B.K.; Dissanayake, T.; Kellerman, D.L.; Dai, Q.; Miyagi, M.; Anderson, V.E.; York, D.M.; Piccirilli, J.A.; Harris, M.E. Experimental and computational analysis of the transition state for ribonuclease A - catalyzed RNA 2′-O-transphosphorylation. Proc. Nat. Am. Soc. USA 2013. [Google Scholar] [CrossRef]
- Bahnson, B.J.; Anderson, V.E. Crotonase-catalyzed β-elimination is concerted: A double isotope effect study. Biochemistry 1991, 30, 5894–5906. [Google Scholar] [CrossRef]
- Gawlita, E.; Paneth, P.; Anderson, V.E. Equilibrium isotope effect on ternary complex formation of [1-18O]Oxamate with NADH and Lactate Dehydrogenase. Biochemistry 1995, 34, 6050–6058. [Google Scholar] [CrossRef]
- Berti, P.J.; Blanke, S.R.; Schramm, V.L. Transition state structure for the hydrolysis of NAD+ catalyzed by diphtheria toxin. J. Am. Chem. Soc. 1997, 119, 12079–12088. [Google Scholar] [CrossRef]
- Cassano, A.G.; Anderson, V.E.; Harris, M.E. Evidence for direct attack by hydroxide in phosphodiester hydrolysis. J. Am. Chem. Soc. 2002, 124, 10964–10965. [Google Scholar] [CrossRef]
- Cassano, A.G.; Anderson, V.E.; Harris, M.E. Analysis of solvent nucleophile isotope effects: Evidence for concerted mechanisms and nucleophilic activation by metal coordination in nonenzymatic and ribozyme-catalyzed phosphodiester hydrolysis. Biochemistry 2004, 43, 10547–10559. [Google Scholar] [CrossRef]
- Cassano, A.G.; Wang, B.; Anderson, D.R.; Previs, S.; Harris, M.E.; Anderson, V.A. Inaccuracies in selected ion monitoring determination of isotope ratios obviated by profile acquisition: Nucleotide 18O/16O measurements. Anal. Biochem. 2007, 367, 28–39. [Google Scholar] [CrossRef]
- Kudo, F.; Yamauchi, N.; Suzuki, R.; Kakinuma, K. Kinetic isotope effect and reaction mechanism of 2-deoxy-scyllo-inosose synthase derived from butirosin-producing Bacillus circulans. J. Antibiot. 1997, 50, 424–428. [Google Scholar] [CrossRef]
- Yamauchi, N.; Kakinuma, K. Enzymatic carbocycle formation in microbial secondary metabolism. The mechanismof 2-deoxy-scyllo-inosose synthase reaction as a crucial step in the 2-deoxystreptamine biosynthesis in Streptomyces fradiae. J. Org. Chem. 1995, 60, 5614–5619. [Google Scholar] [CrossRef]
- Kline, P.C.; Rezaee, M.; Lee, T.A. Determination of kinetic isotope effects for nucleoside hydrolases using gas chromatography/mass spectrometry. Anal. Biochem. 1999, 275, 6–10. [Google Scholar] [CrossRef]
- Horenstein, B.A.; Parkin, D.W.; Estupinan, B.; Schramm, V.L. Transition-state analysis of nucleoside hydrolase from Crithidia fasciculate. Biochemistry 1991, 30, 10788–10795. [Google Scholar] [CrossRef]
- Hunt, C.; Gillani, N.; Farone, A.; Rezaei, M.; Kline, P.C. Kinetic isotope effects of nucleoside hydrolase from Escherichia coli. Biochim. Biophys. Acta 2005, 1751, 140–149. [Google Scholar]
- Wilde, T.C.; Blotny, G.; Pollack, R.M. Experimental evidence for enzyme-enhanced coupled motion/quantum mechanical hydrogen tunneling by ketosteroid isomerase. J. Am. Chem. Soc. 2008, 130, 6577–6585. [Google Scholar] [CrossRef]
- Numazawa, M.; Takahashi, M.; Nagaoka, M.; Handa, W.; Yamashita, K. Mass spectrometric analysis of oxygenations in aromatization of androst-4-ene-3,6,17-trione, a suicide substrate of aromatase, by placental microsome. Isotope effect and stereochemistry. J. Steroid Biochem. Mol. Biol. 2007, 107, 220–227. [Google Scholar] [CrossRef]
- Sowa, G.A.; Hengge, A.C.; Cleland, W.W. 18O isotope effects support a concerted mechanism for ribonuclease A. J. Am. Chem. Soc. 1997, 119, 2319–2320. [Google Scholar] [CrossRef]
- Liuni, P.; Olkhov-Mitsel, E.; Orellana, A.; Wilson, D.J. Measuring kinetic isotope effects in enzyme reactions using time-resolved electrospray mass spectrometry. Anal. Chem. 2013, 85, 3758–3764. [Google Scholar] [CrossRef]
- Pascal, R.A.; Baum, M.W.; Wagner, C.K.; Rodgers, L.R.; Huang, D.S. Measurement of deuterium kinetic isotope effects in organic and biochemistry reactions by natural abundance deuterium NMR spectroscopy. J. Am. Chem. Soc. 1986, 108, 6477–6482. [Google Scholar] [CrossRef]
- Singleton, D.A.; Thomas, A.A. High-precision simultaneous determination of multiple small kinetic isotope effects at natural abundance. J. Am. Chem. Soc. 1995, 117, 9357–9358. [Google Scholar] [CrossRef]
- Gajewski, J.J.; Peterson, K.B.; Kagel, J.R.; Huang, Y.C.J. Transition-state structure variation in the Diels-Alder reaction from secondary deuterium kinetic isotope effects: The reaction of nearly symmetrical dienes and dienophiles is nearly synchronous. J. Am. Chem. Soc. 1989, 111, 9078–9081. [Google Scholar] [CrossRef]
- Lee, J.K.; Bain, A.D.; Berti, P.J. Probing the transition states of four glucoside hydrolyses with 13C kinetic isotope effects measured at natural abundance by NMR spectrometry. J. Am. Chem. Soc. 2004, 126, 3769–3776. [Google Scholar] [CrossRef]
- Bennet, A.J.; Sinnott, M.L. Complete kinetic isotope effect description of transition state for acid-catalyzed hydrolyses of methyl α- and β-glucopyranoside. J. Am. Chem. Soc. 1986, 108, 7287–7294. [Google Scholar] [CrossRef]
- Huang, M.; Garrett, G.E.; Birlirakis, N.; Bohe, L.; Pratt, D.A.; Crich, D. Dissecting the mechanisms of a class of chemical glycosylation using primary 13C kinetic isotope effects. Nat. Chem. 2012, 4, 663–667. [Google Scholar] [CrossRef]
- Chan, J.; Lewis, A.R.; Gilbert, M.; Karwaski, M.-F.; Bennet, A.J. A direct NMR method for the measurement of competitive kinetic isotope effects. Nat. Chem. Biol. 2010, 6, 405–407. [Google Scholar] [CrossRef]
- Chan, J.; Tang, A.; Bennet, A.J. A stepwise solvent-promoted SNi reaction of α-D-glucopyranosyl fluoride: Mechanistic implications for retaining glycosyltransferases. J. Am. Chem. Soc. 2012, 134, 1212–1220. [Google Scholar] [CrossRef]
- Pabis, A.; Kaminski, R.; Ciepielowski, G.; Jankowski, S.; Paneth, P. Measurements of heavy-atom isotope effects using 1H NMR spectroscopy. J. Org. Chem. 2011, 76, 8033–8035. [Google Scholar] [CrossRef]
- Manning, K.A.; Sathyamoorthy, B.; Elesky, A.; Szyperski, T.; Murkin, A.S. Highly precise measurement of kinetic isotope effects using 1H-detected 2D [13C, 1H]-HSQC NMR spectroscopy. J. Am. Chem. Soc. 2012, 134, 20589–20592. [Google Scholar] [CrossRef]
- Cleland, W.W. Enzyme mechanisms from isotope effects. In Isotope Effects in Chemistry and Biology; Kohen, A., Limbach, H.H., Eds.; CRC Press: Boca Raton, FL, USA, 2006; pp. 915–930. [Google Scholar]
- Parkin, D.W. Methods for the determination of competitive and noncompetitive kinetic isotope effects. In Enzyme Mechanism from Isotope Effects; Cook, P.F., Ed.; CRC Press: Boca Raton, FL, USA, 1991; pp. 269–290. [Google Scholar]
- Cassera, M.B.; Zhang, Y.; Hazleton, K.Z.; Schramm, V.L. Purine and pyrimidine pathways as targets in Plasmodium falciparium. Curr. Top. Med. Chem. 2011, 11, 2103–2115. [Google Scholar]
- Schwartz, P.A.; Vetticatt, M.J.; Schramm, V.L. Transition state analysis of the arsenolytic depyrimidination of thymidine by human thymidine phosphorylase. Biochemistry 2011, 50, 1412–1420. [Google Scholar] [CrossRef]
- Knapp, M.J.; Klinman, J.P. Environmentally coupled hydrogen tunneling. Linking catalysis to dynamics. Eur. J. Biochem. 2002, 269, 3113–3121. [Google Scholar] [CrossRef]
- Sen, A.; Yahashiri, A.; Kohen, A. Triple isotope labeling and kinetic isotope effects: Exposing H-transfer steps in enzymatic systems. Biochemistry 2011, 50, 6462–6468. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gu, H.; Zhang, S. Advances in Kinetic Isotope Effect Measurement Techniques for Enzyme Mechanism Study. Molecules 2013, 18, 9278-9292. https://doi.org/10.3390/molecules18089278
Gu H, Zhang S. Advances in Kinetic Isotope Effect Measurement Techniques for Enzyme Mechanism Study. Molecules. 2013; 18(8):9278-9292. https://doi.org/10.3390/molecules18089278
Chicago/Turabian StyleGu, Hong, and Shuming Zhang. 2013. "Advances in Kinetic Isotope Effect Measurement Techniques for Enzyme Mechanism Study" Molecules 18, no. 8: 9278-9292. https://doi.org/10.3390/molecules18089278