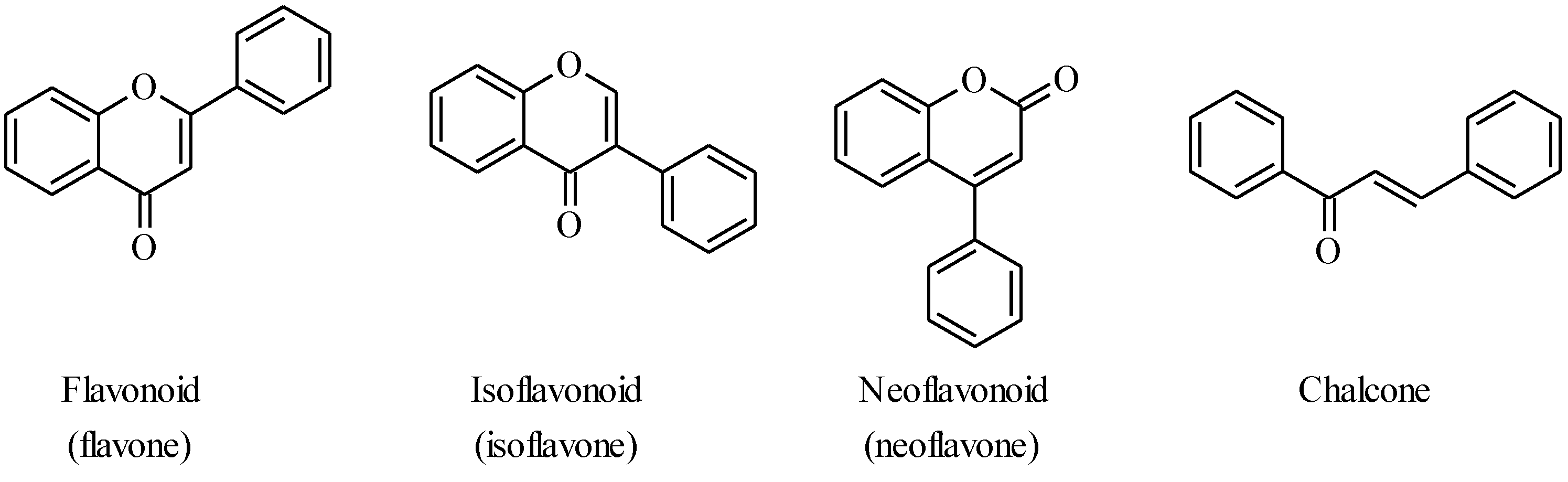
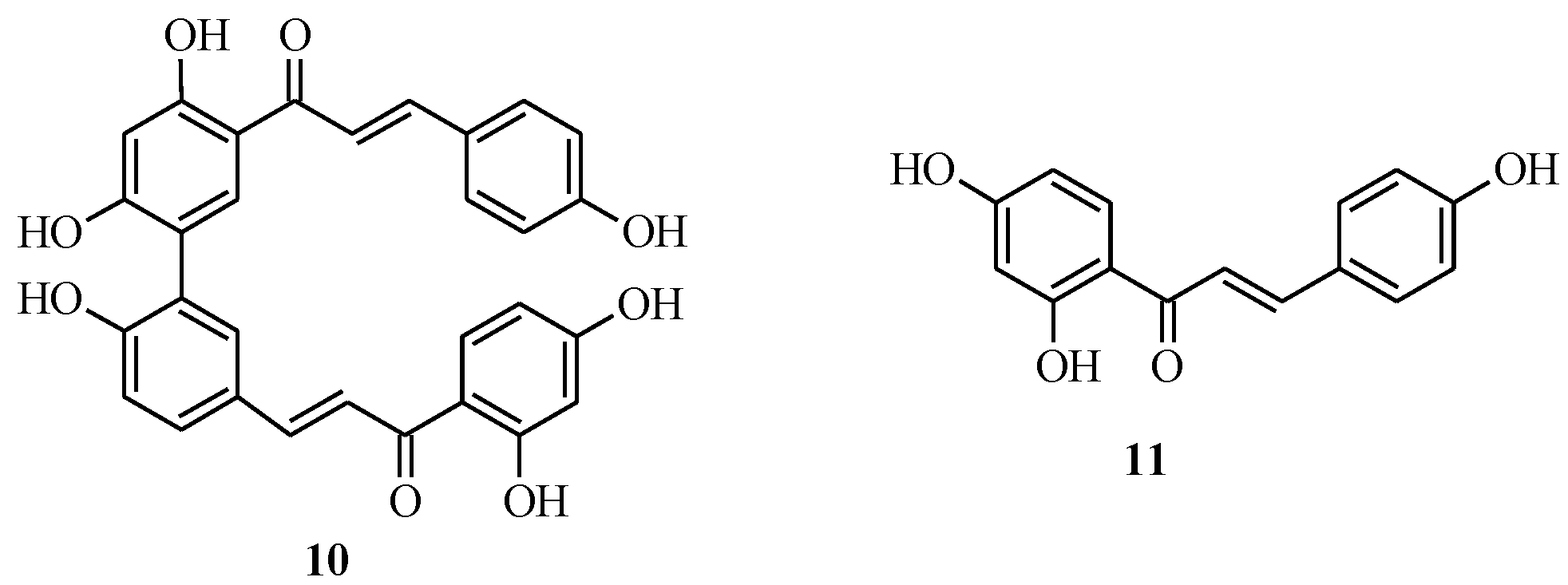
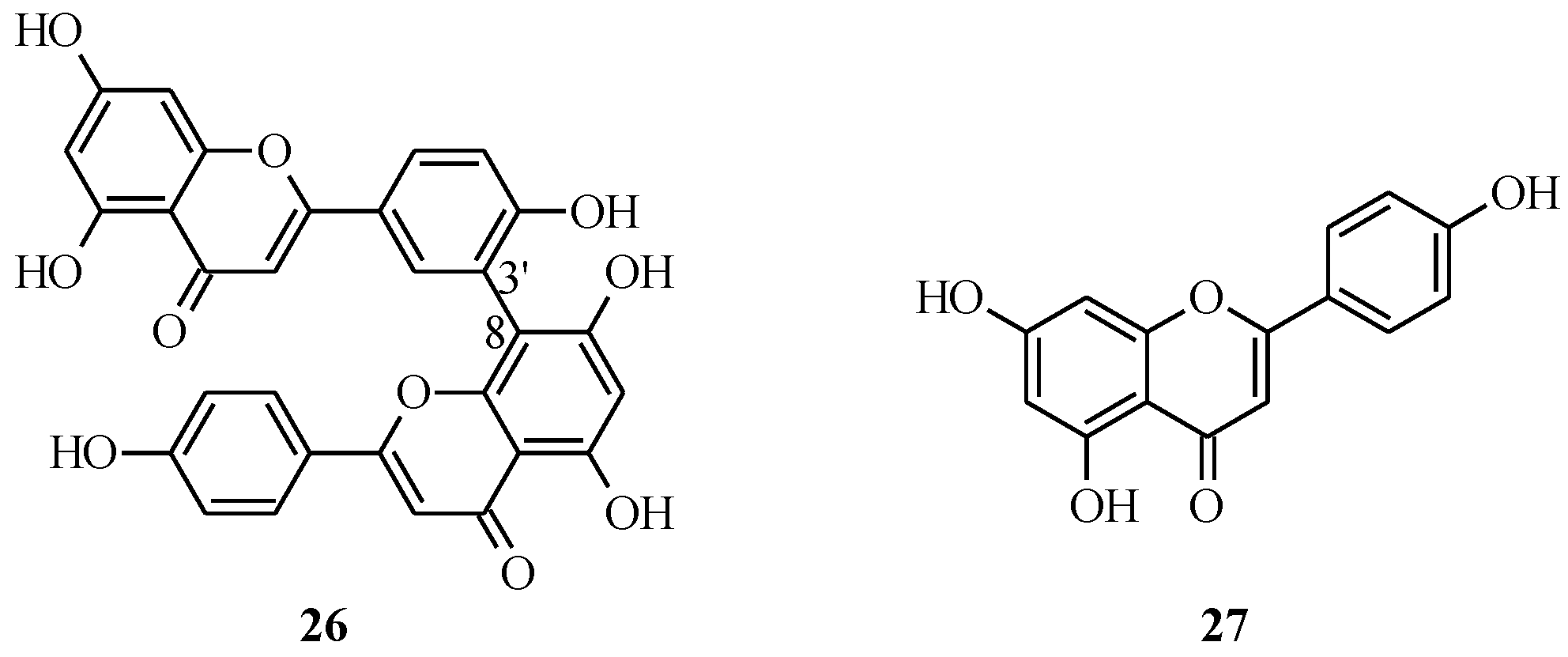
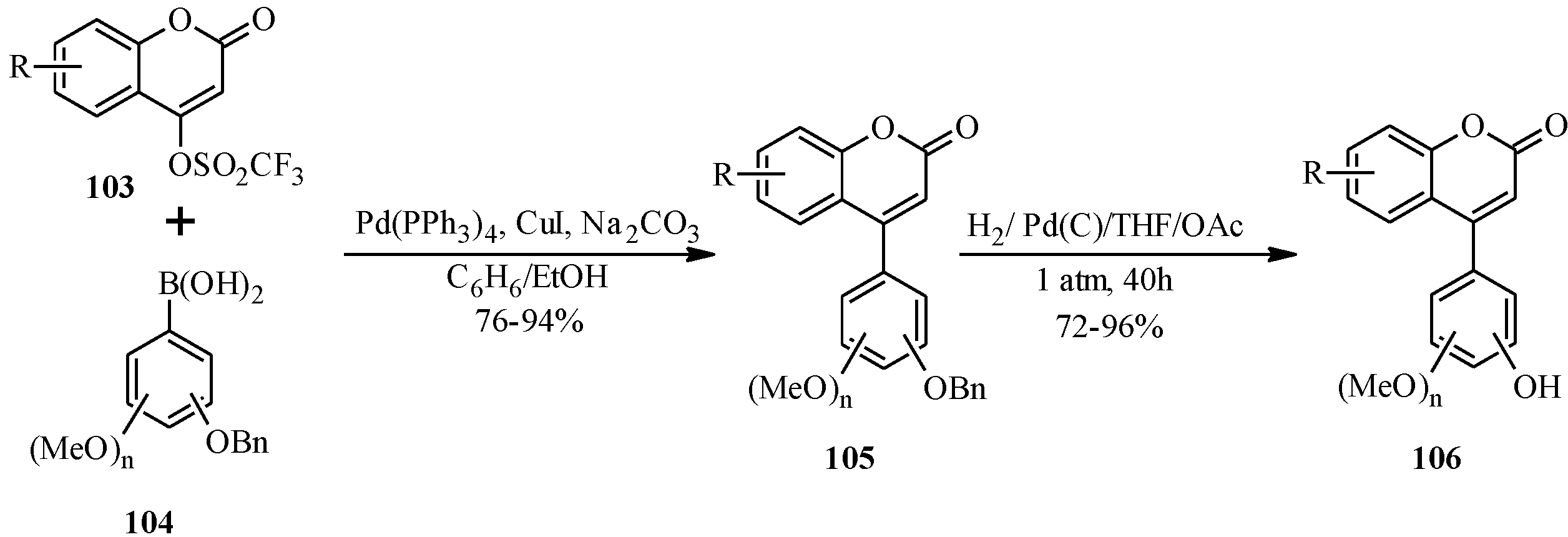
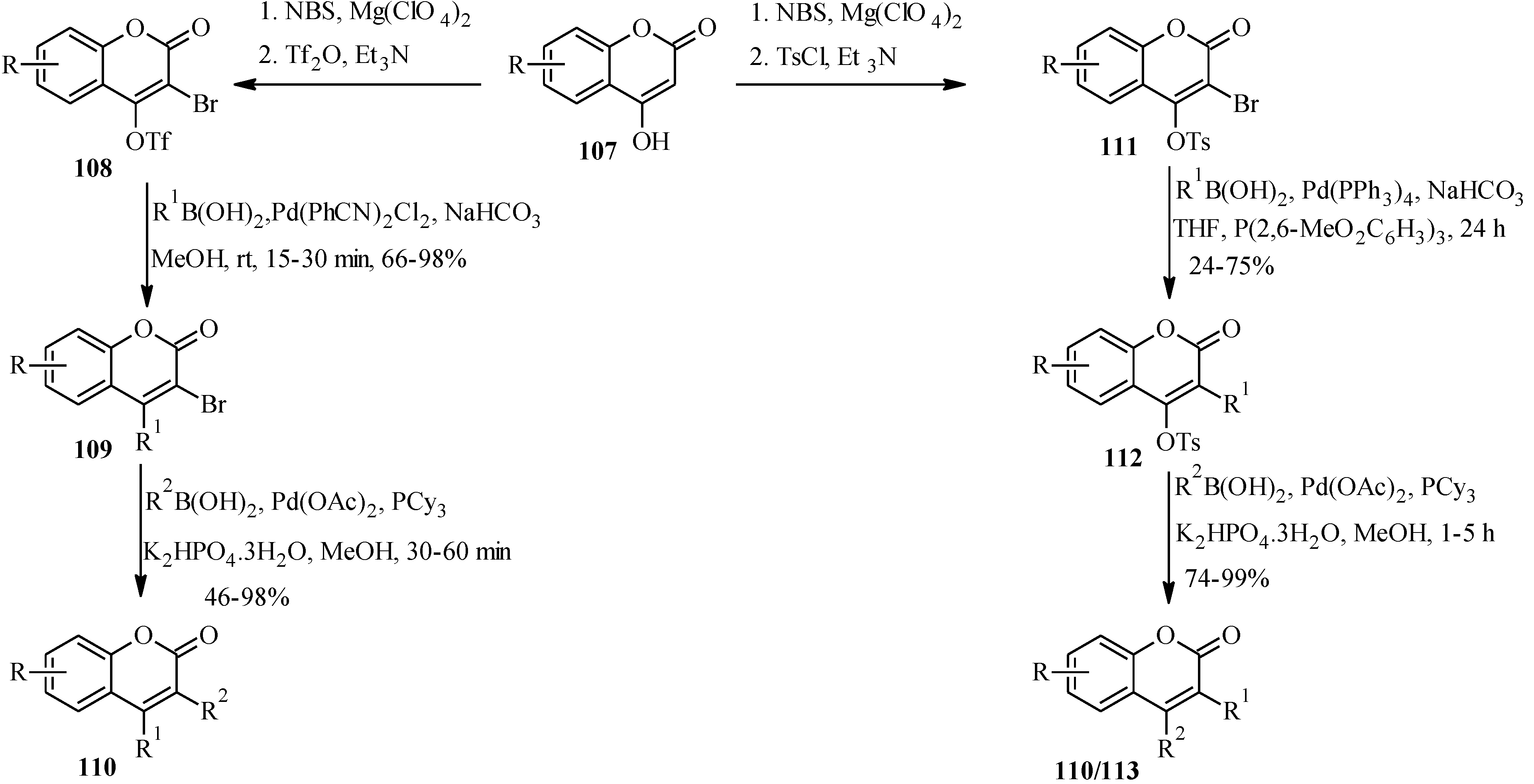
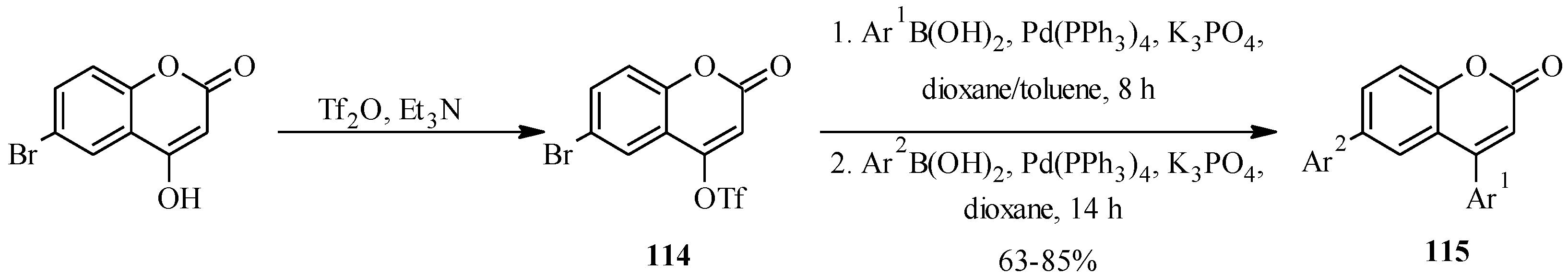
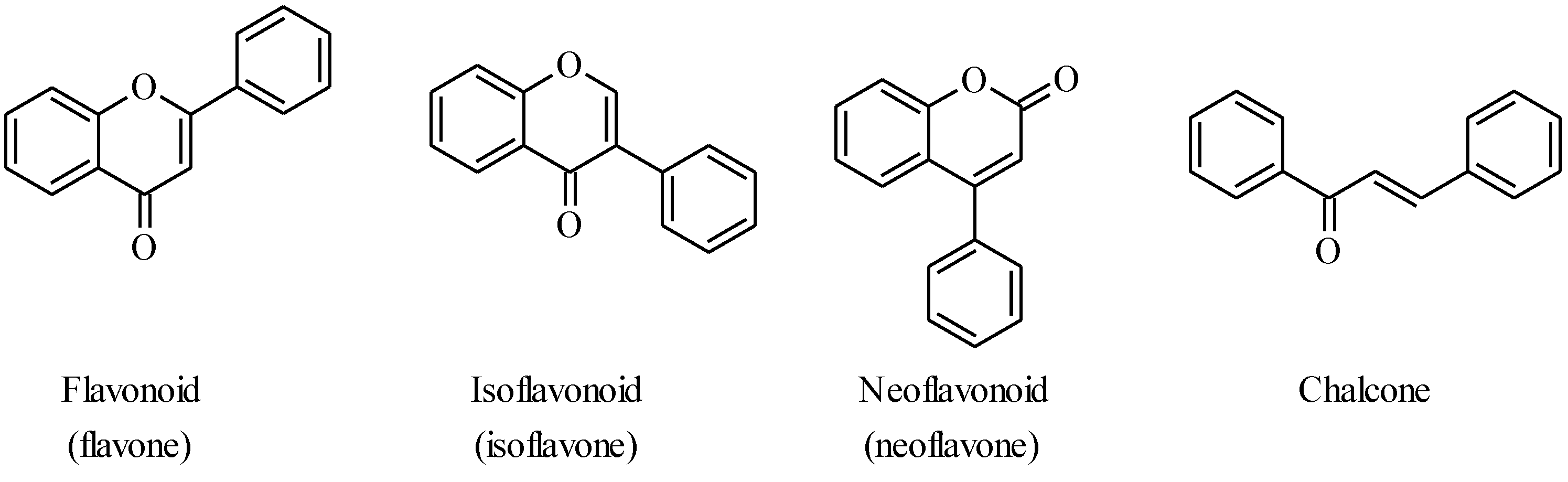
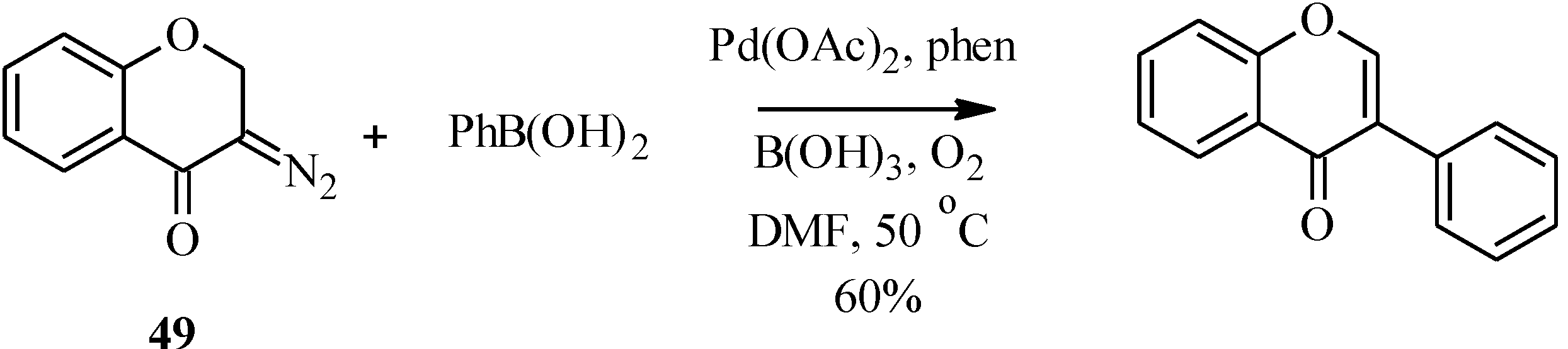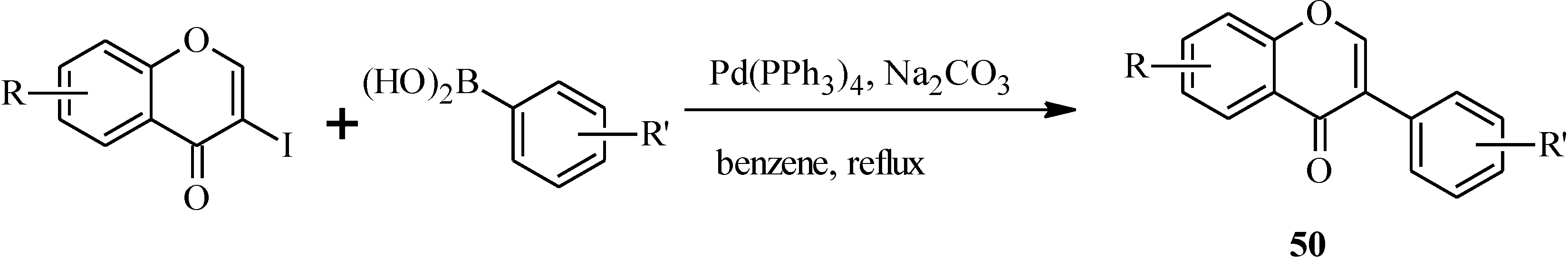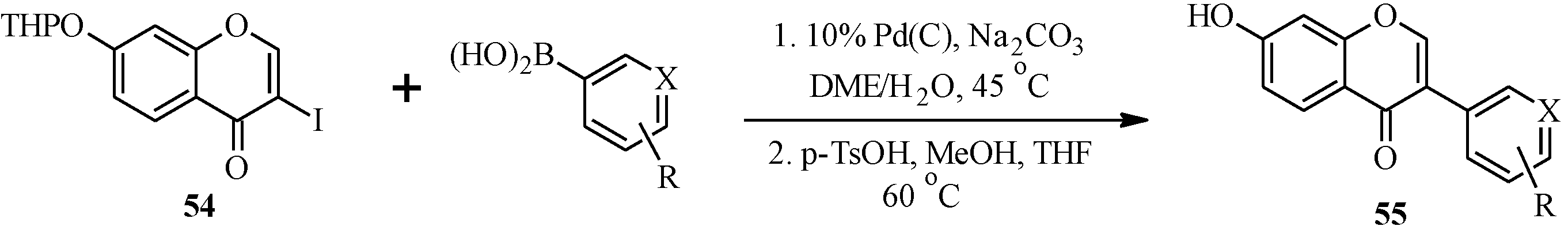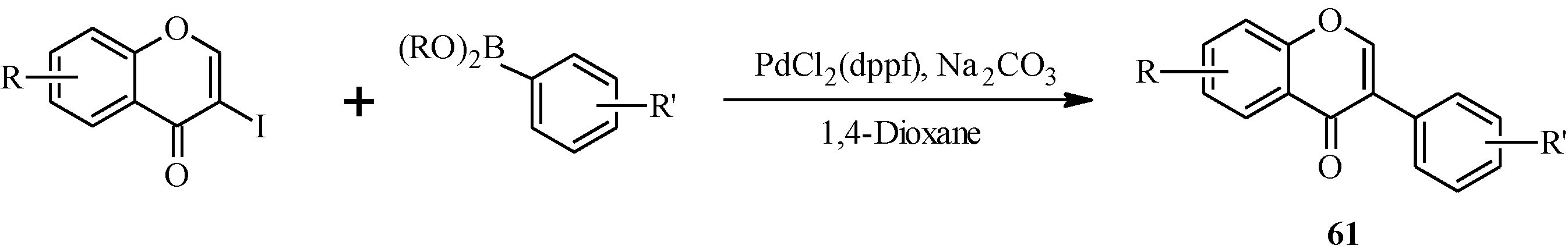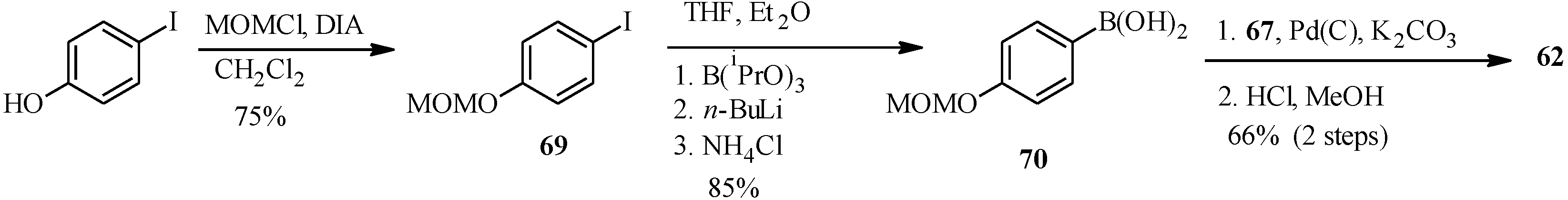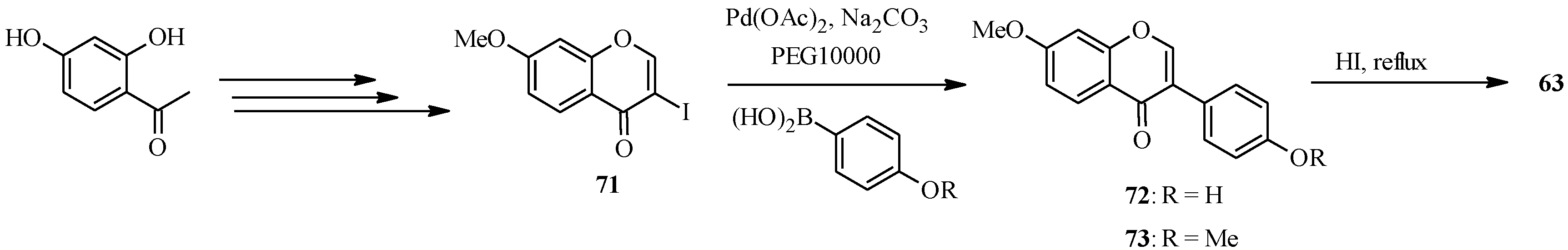4.1. Isoflavones
There are two long-established procedures for the preparation of isoflavones which are still widely used,
i.e., the deoxybenzoin route and the chalcone route [
47,
48,
49]. Other methods which have been developed include reductive cleavage of isoxazoles, intramolecular ketene cycloaddition followed by decarboxylation, rearrangement and cyclisation of chalcone epoxides and rearrangement of flavanones [
49,
50]. The methods that have been developed more recently are the Wacker-Cook tandem conversion of α-methylene deoxybenzoins into isoflavones [
51] and the Cu(I)-mediated cyclization of 3-(2-bromophenyl)-3-oxopropanal [
52]. Regardless of the many new synthetic approaches presented, the application of many of them has not been demonstrated in the synthesis of polyhydroxylated isoflavones and isoflavones bearing other naturally-occurring substitution patterns.
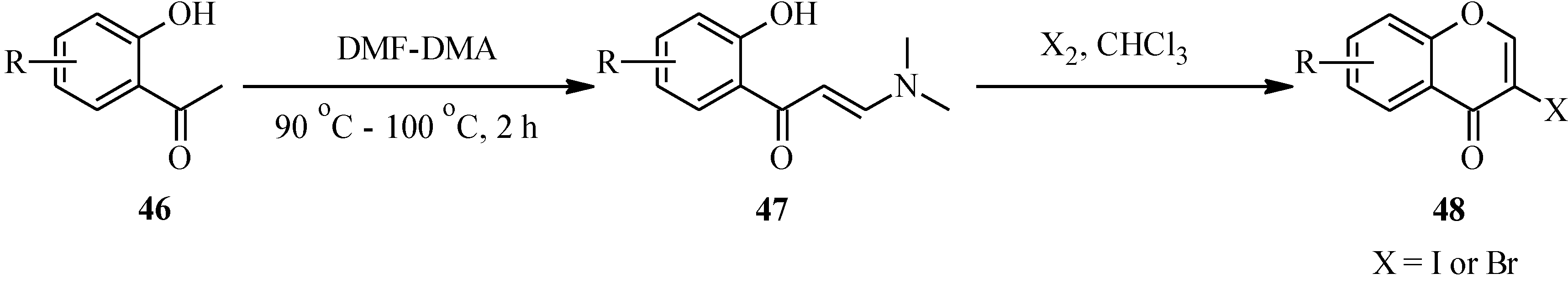
In 1988, Suzuki and co-workers were the first to demonstrate the versatility of the palladium-catalyzed cross-coupling reactions in the synthesis of isoflavones from 3-bromochromones
45 and arylboronic acids/esters (
Scheme 12) [
53]. Following this, the Suzuki-Miyaura cross-coupling reaction has been applied in several instances to the synthesis of isoflavones in the presence of palladium(0) or palladium(II) catalysts [
3,
11,
12,
54,
55]. Examples of catalysts which have successfully facilitated this C-C bond formation are Pd(PPh
3)
4[
1,
53], (C) [
11,
12],
trans-[PdCl
2(2-ethyl-2-oxazoline-к
1N)
2] [
55], Pd(dppf)
2Cl
2 [
3,
56], benzothiazole-oxime-based Pd(II) catalyst [
54], and Pd(OAc)
2 in the presence of poly(ethyleneglycol) [
56,
57] or 2-(2,6-dimethoxybiphenyl)dicyclohexylphosphane (SPhos) [
4].
Scheme 12.
Preparation of isoflavones by the Suzuki reaction [
53].
Scheme 12.
Preparation of isoflavones by the Suzuki reaction [
53].
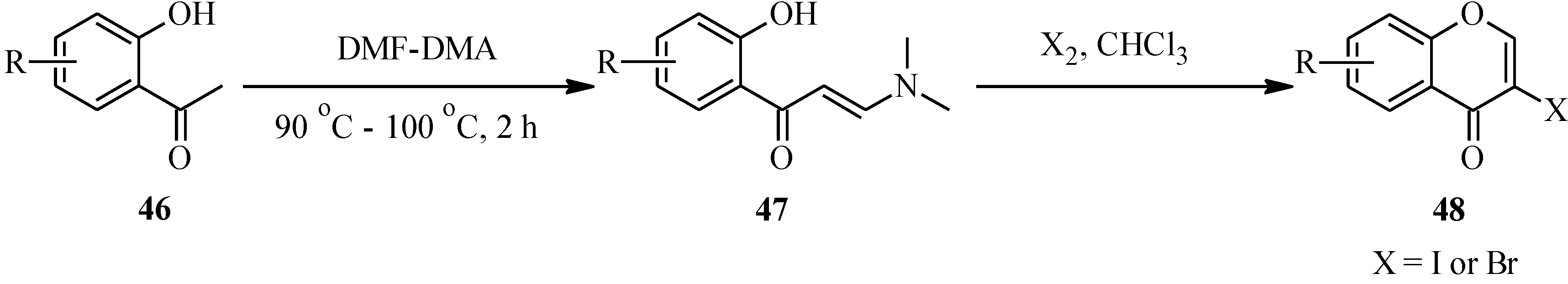
Unlike the 2-halochromone derivatives required in the synthesis of flavones by the Suzuki reaction, the 3-halochromone precursors for isoflavones can be conveniently prepared by Gammill’s protocol [
58], which involves condensation of appropriately substituted 2'-hydroxyacetophenones
46 with DMF-DMA to form enaminoketones
47. Halogen-mediated ring closure of the enaminoketones gives the corresponding 3-halochromones
48 [
58] (
Scheme 13). Alternatively, the 3-halochromones can be obtained by direct halogenation of chromones [
4].
Scheme 13.
Preparation of 3-halochromones by Gammill’s protocol [
58].
Scheme 13.
Preparation of 3-halochromones by Gammill’s protocol [
58].
In a variation of the Suzuki-Miyaura reaction, Tsoi
et al. reported the formation of an unsubstituted isoflavone by the palladium-catalyzed oxidative cross-coupling of a diazochromone (
49) with phenylboronic acid in 60% yield (
Scheme 14) [
59].
Scheme 14.
Oxidative cross coupling of phenylboronic acid with a diazochromone [
59].
Scheme 14.
Oxidative cross coupling of phenylboronic acid with a diazochromone [
59].
4.2. Preparation of Isoflavone Analogues for Biological Activity Studies
As a result of the readily availability of 3-halochromones and their potential to be elaborated into a wide range of compounds by coupling with different boronic acids in the final stages of the synthesis, several groups have taken advantage of the Suzuki coupling reaction for the synthesis of libraries of novel compounds based on the isoflavone scaffold for biological activity studies [
4,
60,
61]. For example, Vasselin
et al. synthesized a series of fluoro, methoxy, nitro and amino isoflavones (
50a–
o) from appropriately substituted 3-iodochromones and arylboronic acids (
Scheme 15) [
61]. However, an attempt to prepare 5,7-dibenzyloxy-3-iodochromone from 4,6-dibenzyloxy-2-hydroxyacetophenone resulted in an inseparable mixture of products while a poor yield (29%) was obtained for 3-iodo-5,7-dimethoxychromone from the corresponding enaminoketone.
The synthesized isoflavones were tested for
in vitro growth inhibition of human breast (MDA-MB-468 and MCF-7) and colon (HT29 and HT-116) cancer cell lines. The isoflavones
50d,
50f,
50h,
50k,
50l and
50o showed pronounced growth inhibition of MDA-MD-468 cells co-incubated with TBDD, a powerful inducer of cytochrome P450 (CYP)-1A1 activity. This suggested that the isoflavone derivatives were potential substrates for (CYP)-1A1 bioactivation [
61].
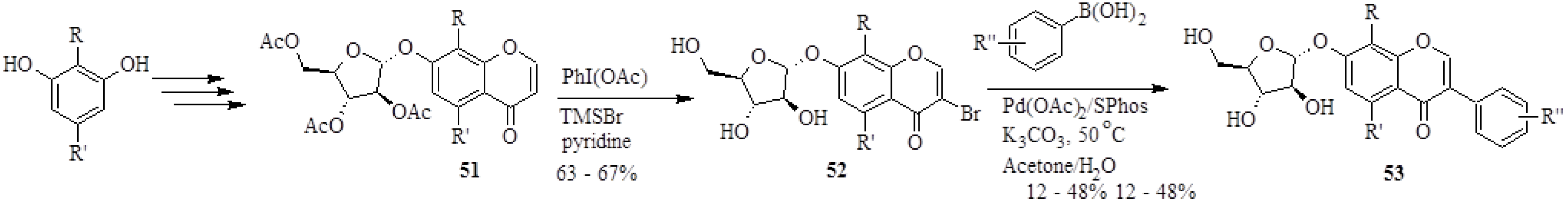
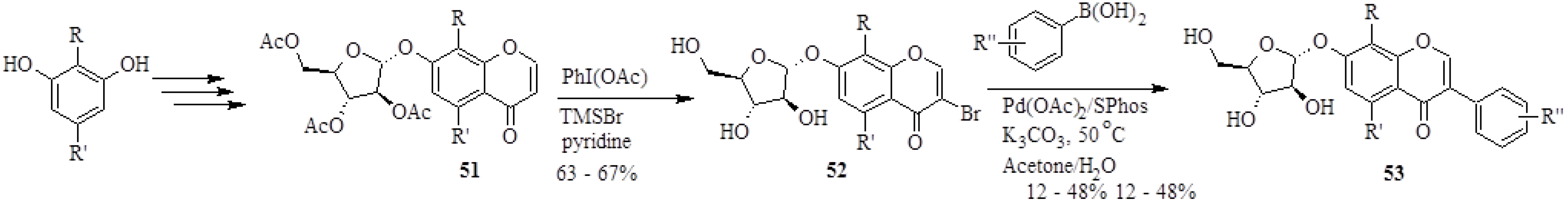
In 2008, Wei and Yu synthesized 26 isoflavone glycoside derivatives based on the structure of a potent inhibitor of α-glucosidases of rat liver microsomes, 7-
O-α-D-arabinofuranosyloxy-4',8-dihydroxyisoflavone (
53a), also called A-76202 [
4]. The analogues of
53a were prepared by coupling 3-bromochromones
52 to differently substituted arylboronic acids in the presence of Pd(OAc)
2, SPhos and K
2CO
3 in a water-based solvent (H
2O/acetone) (
Scheme 16). However, in certain instances the reaction conditions were slightly altered and coupling was performed in the absence of a ligand, with NaOAc as base and MeOH as solvent to accommodate substrates which were more susceptible to degradation.
Scheme 15.
Preparation of a series of isoflavones by Vasselin and co-workers [
61].
Scheme 15.
Preparation of a series of isoflavones by Vasselin and co-workers [
61].
| 50 | R | R' |
|---|
| a | H | 4-Cl |
| b | H | 4-Br |
| c | H | 3-NO2 |
| d | H | 3-OMe |
| e | H | 4-OMe |
| f | H | 3,4-di-OMe |
| g | 6-F | 4-OMe |
| h | 6-F | 3,4-di-OMe |
| i | 6-F | 3,4-OCH2O |
| j | 7-F | 4-OMe |
| k | 7-F | 3,4-di-OMe |
| l | 8-F | 3,4-di-OMe |
| m | 6,7-di-F | 3,4-di-OMe |
| n | 6,8-di-F | 3,4-di-OMe |
| o | 7,8-di-F | 3,4-di-OMe |
The 3-bromochromones
52 were in turn prepared by direct bromination of the chromones
51 at C-3 by treatment with PhI(OAc), TMSBr and pyridine [
4]. The synthetic natural product
53a and its derivatives were evaluated for α-glucosidase inhibition [
4]. The results showed that the stereochemistry of the α-D-arabinofuranosyl unit and the 8-hydroxy group in the A-ring are essential for the activity, whereas modifications at the B-ring did not adversely affect the α-glucosidase inhibitory activity of the isoflavone 7-
O-glycosides [
4]. It is noteworthy that the 3-OMe and 4-NMe
2 derivatives (
53e and
53k, respectively) were three fold more active than the parent compound
53a (
Scheme 16) [
4].
Scheme 16.
Synthesis of isoflavone glycosides by the Suzuki-Miyaura reaction [
4].
Scheme 16.
Synthesis of isoflavone glycosides by the Suzuki-Miyaura reaction [
4].
| 62 | R | R' | R'' | IC50 (μM) * |
|---|
| a | OH | H | 4-OH | 0.018 |
| b | OH | H | H | 0.040 |
| c | OH | H | 4-OMe | 0.022 |
| d | OH | H | 2-OMe | 0.015 |
| e | OH | H | 3-OMe | 0.006 |
| f | OH | H | 4-Me | 0.035 |
| g | OH | H | 2-Me | 0.020 |
| h | OH | H | 3-Me | 0.050 |
| i | OH | H | 3-OH | 0.017 |
| j | OH | H | 4-F | 0.015 |
| k | OH | H | 4-NMe2 | 0.008 |
| l | OH | H | 4-NHBoc | 0.020 |
| m | OH | H | 4-CF3 | 0.030 |
| n | OH | H | 4-SiMe3 | (35% at 0.050) |
| o | H | OH | H | 10 |
| p | H | OH | 4-OMe | 8 |
| q | H | OH | 2-OMe | 10 |
| r | H | OH | 3-OMe | 10 |
| s | H | OH | 4-Me | 7 |
| t | H | OH | 3-Me | 5 |
| u | H | OH | 4-OH | 15 |
| v | H | OH | 3-OH | (33% at 50) |
Matin and co-workers [
5] also prepared a series of isoflavones by coupling of 3-iodochromone
54 and a variety of arylboronic acids in the presence of Pd(C) [
11], followed by cleavage of the THP protecting group (
Scheme 17). The isoflavones were screened together with other compounds (chalcones, flavones, flavanones, isoflavones and pyrazole derivatives) for dual PPARα and γ agonism. Of the 77 tested compounds, the isoflavones
55a,
55c,
55e and
55i were identified as novel potent dual PPARα and γ agonists, which could serve as future leads in PPAR-related disorders that include type II diabetes mellitus and metabolic syndrome [
5].
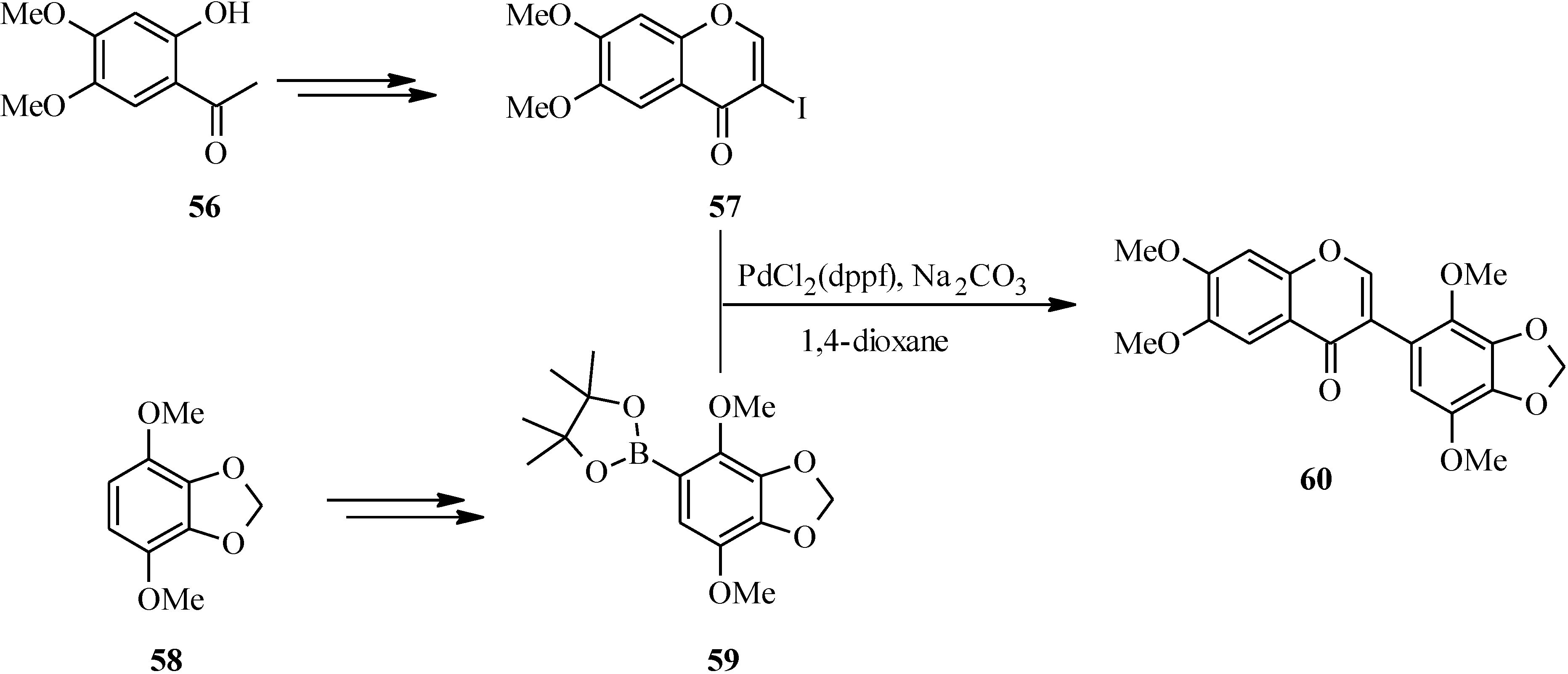
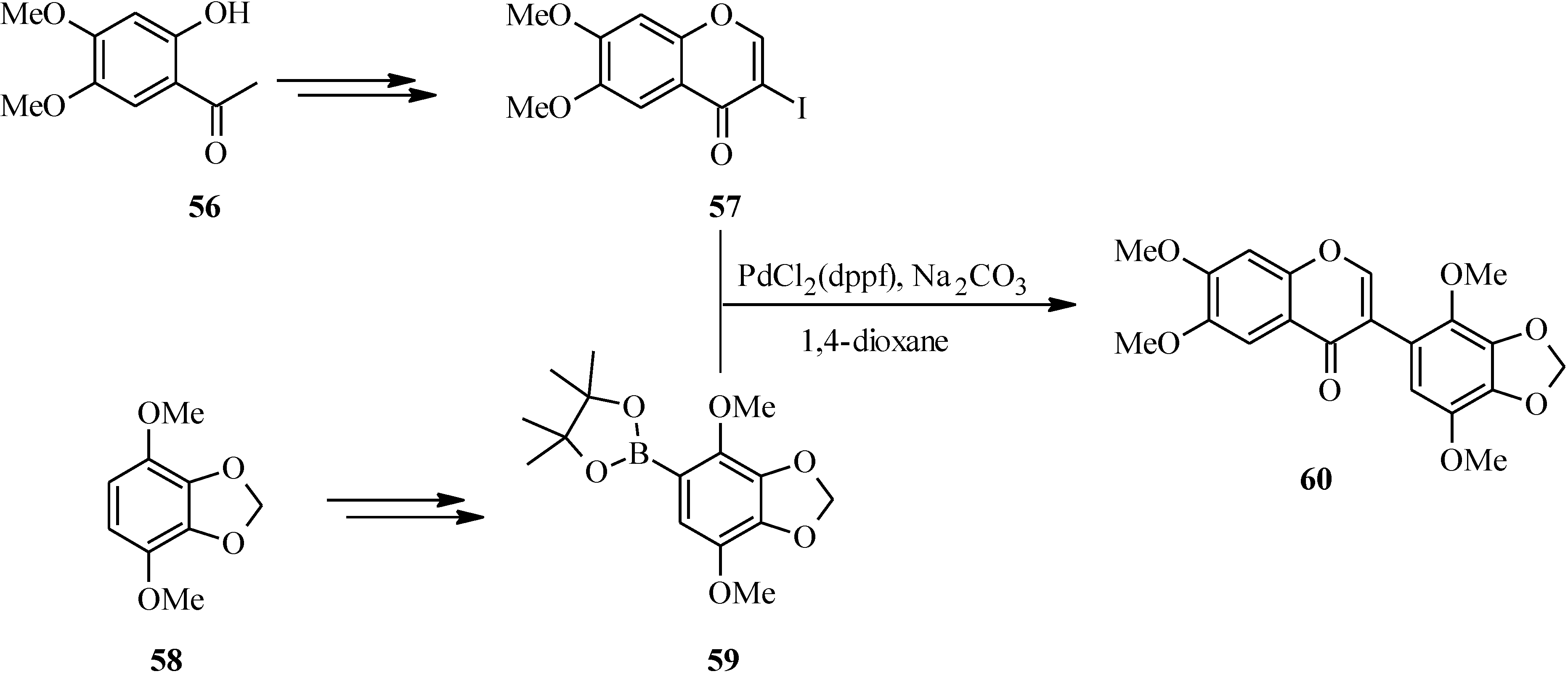
In 2007, Kigoshi’s group reported the total synthesis glaziovianin A (
60), a metabolite of
Astelia glazioviana that exhibited cytotoxicity against HL-60 cells [
60,
62]. As illustrated in
Scheme 18, the synthesis of glaziovianin A (
60) commenced by preparing the 3-iodochrome
57 from acetophenone
56 by Gammill’s procedure [
58], and the boronic acid
59 from aryl derivative
58. Coupling of 3-iodochromone
57 with phenylboronic acid
59 gave glaziovianin A (
60) [
60,
62].
Scheme 17.
Preparation of isoflavones by Matin and co-workers [
5].
Scheme 17.
Preparation of isoflavones by Matin and co-workers [
5].
| 55 | X | R | 55 | X | R |
|---|
| a | C | 3,5-di-OMe | j | C | 2-OMe, 3,5-di-F |
| b | C | 4-OMe | k | C | 3,4,5-tri-F |
| c | C | 3,4-OCH2O- | l | N | 2,4-di-OMe |
| d | C | 4-CF3 | m | C | 4-Cl |
| e | C | 4-F | n | C | 3-F |
| f | C | 3,4-di-OMe | o | C | 2-OMe |
| g | C | 3,4-O(CH2)2-O | p | C | 3-OCF3 |
| h | C | 2,4-di-F | q | C | 3-OBn |
| i | C | 3-OMe | r | C | 3,4,5-tri-OMe |
Scheme 18.
Total synthesis of glaziovianin A (
60) [
62].
Scheme 18.
Total synthesis of glaziovianin A (
60) [
62].
Thereafter, they employed the same strategy to prepare glaziovianin analogues
61a–
i by altering the substituents on the A- and B-rings (
Scheme 19) [
60]. The synthesized compounds were tested for cytotoxicity against HeLa S
3 cells. Of the screened compounds, the 7-
O-allyl derivative
61i (IC
50 = 0.19 μM) was found to be more cytotoxic than the parent compound
60 (IC
50 = 0.59 μM) [
60]. Moreover,
61i was found to be a more potent M-phase inhibitor [
60].
Scheme 19.
Preparation of glaziovianin A derivatives [
60].
Scheme 19.
Preparation of glaziovianin A derivatives [
60].
| 61 | R | R' |
|---|
| a | 6,7-di-OMe | 2',5'-di-OMe, 3',4'-OC(CH3)2O- |
| b | 6,7-di-OMe | 2',3',4',5'-tetra-OMe |
| c | 6,7-di-OMe | 3,4-OCH2O- |
| d | 6-OMe, 7-OTHP | 2',5'-di-OMe, 3',4'-OCH2O- |
| e | 5,6,7-tri-OMe | 2',5'-di-OMe, 3',4'-OCH2O- |
| f | 6-OMe, 7-OH | 2',5'-di-OMe, 3',4'-OCH2O- |
| g | 6-OMe, 7-OBn | 2',5'-di-OMe, 3',4'-OCH2O- |
| h | 6-OMe, 7-O-propargyl | 2',5'-di-OMe, 3',4'-OCH2O- |
| i | 6-OMe, 7-O-allyl | 2',5'-di-OMe, 3',4'-OCH2O- |
4.3. Synthesis of Soy Isoflavones
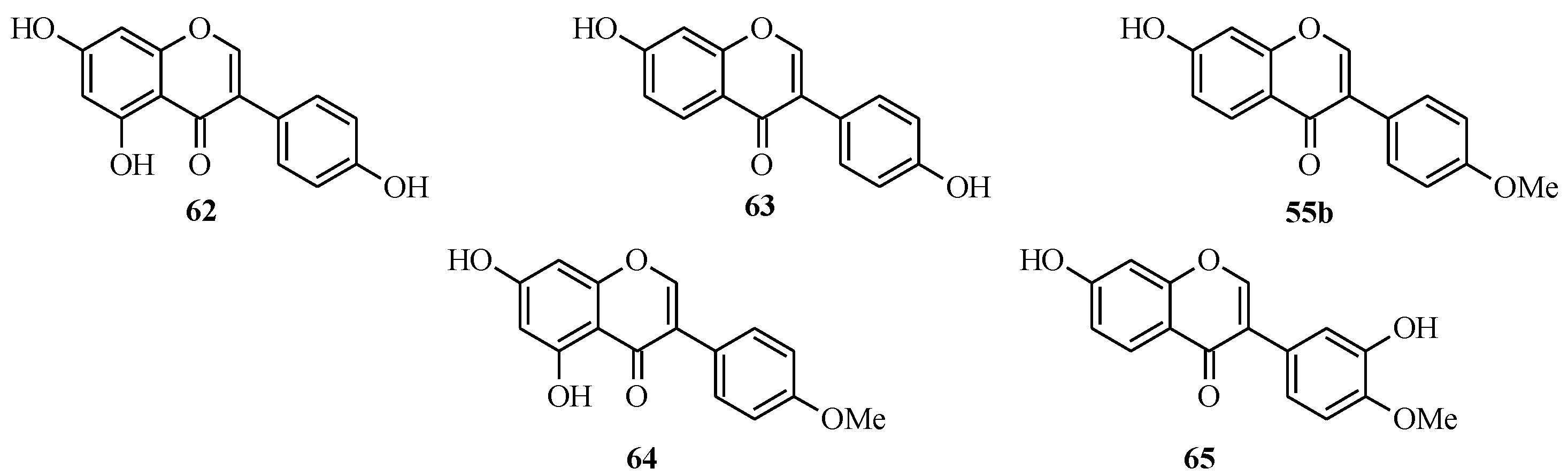
The soy isoflavones consist of genistein (
62), daidzein (
63), formononetin (
55b), biochanin A (
64) and the less common glycitein (
65) [
50,
57,
63,
64] (
Figure 5). They normally exist as 7-
O-glycosides, which are metabolized into the aglycones [
50,
65]. These isoflavones, particularly genistein, are frequently referred to as phytoestrogens, because they are non-steroidal plant-derived compounds with the ability to bind to estrogen receptors and modulate estrogenic responses [
57,
63,
65,
66]. The estrogenic properties of the soy isoflavonoids have been extensively studied with regards to health benefits [
50,
63,
65,
66,
67]. The consumption of phytoestrogen-rich food has been linked to protection against hormone-dependent breast cancer and prostate cancer, alleviation of postmenopausal disorders, osteoporosis as well as cardiovascular protection [
50,
63,
66,
67].
Figure 5.
Structures of soy isoflavones.
Figure 5.
Structures of soy isoflavones.
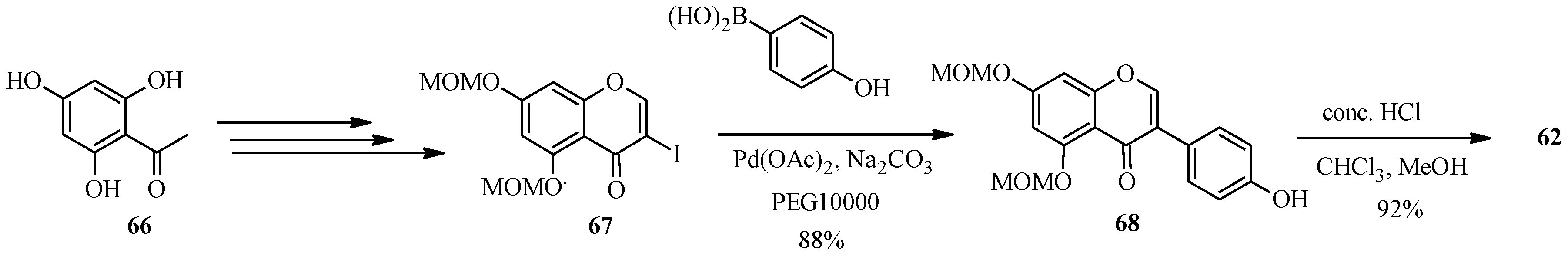
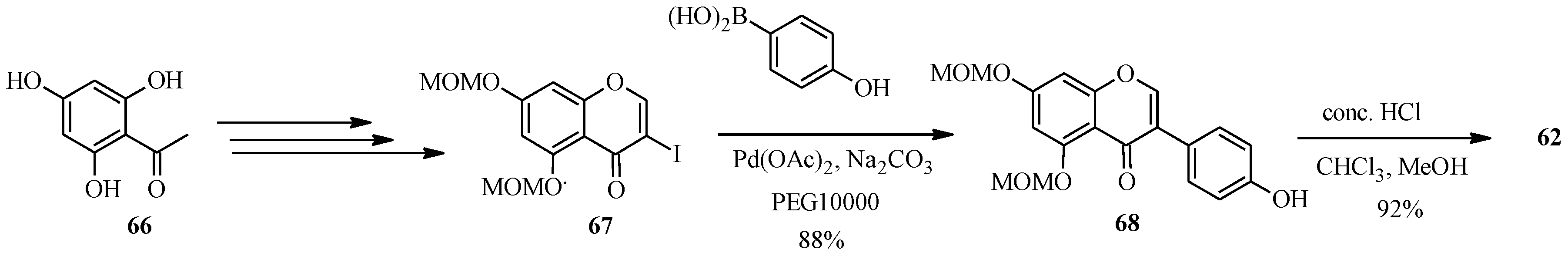
The synthesis of genistein (
62) by the Suzuki-Miyaura reaction was first reported in 2010 [
2,
64], while that of formononetin was reported by Matin and co-workers in 2009 in the preparation of a series of isoflavones for biological activity studies [
5]. Priefer’s group synthesized genistein (
62) in five steps as shown in
Scheme 20, which involved preparation of 3-iodo-5,7-dimethoxymethoxychromone (
67) from phloroacetophenone (
66) by Gammill’s procedure and coupling of the 3-iodochromone
67 with commercially available 4-hydroxyphenylboronic acid using Pd(OAc)
2, poly(ethylene glycol) (PEG10000) and Na
2CO
3. The MOM-protecting groups on the resulting isoflavone
68 were removed with HCl to give genistein (
62) [
64].
Scheme 20.
Preparation of genistein (
62) by Priefer’s group [
64].
Scheme 20.
Preparation of genistein (
62) by Priefer’s group [
64].
In the same year Selepe
et al. concurrently reported the synthesis of genistein from the MOM-protected 3-iodochromone
67 (
Scheme 21) [
2]. Their synthesis involved preparation of the boronic acid
70 from aryl iodide
69 in a one-pot sequence that involved addition of
n-BuLi to a solution of aryl iodide
69 and triisopropyl borate in THF/Et
2O (1:2), followed by the hydrolysis of the boronate ester with an NH
4Cl solution. Heterogeneous Pd(C)-assisted cross-coupling of boronic acid
70 with 3-iodochromone
67, prepared by Gammill’s protocol and removal of the MOM protecting groups gave genistein (
62).
Scheme 21.
Preparation of genistein (
62) by Selepe
et al. [
2].
Scheme 21.
Preparation of genistein (
62) by Selepe
et al. [
2].
The syntheses of daidzein (
63) and its methyl derivatives isoformononetin (
72) and dimethyldaidzein (
73) were also reported by Priefer’s group under similar conditions to those employed for genistein (
62) (
Scheme 22) [
57]. However, the main precursor for the C-C bond formation was 3-iodo-7-methoxychromone (
71), which was coupled to 4-hydroxyphenylboronic acid and 4-methoxyphenylboronic acid to give isoformononetin (
72) and dimethyldaidzein (
73), respectively. Demethylation of
72 and
73 with HI in refluxing chloroform gave daidzein (
63) [
57].
Scheme 22.
Preparation of daidzein and its methyl derivatives by Priefer’s group [
57].
Scheme 22.
Preparation of daidzein and its methyl derivatives by Priefer’s group [
57].
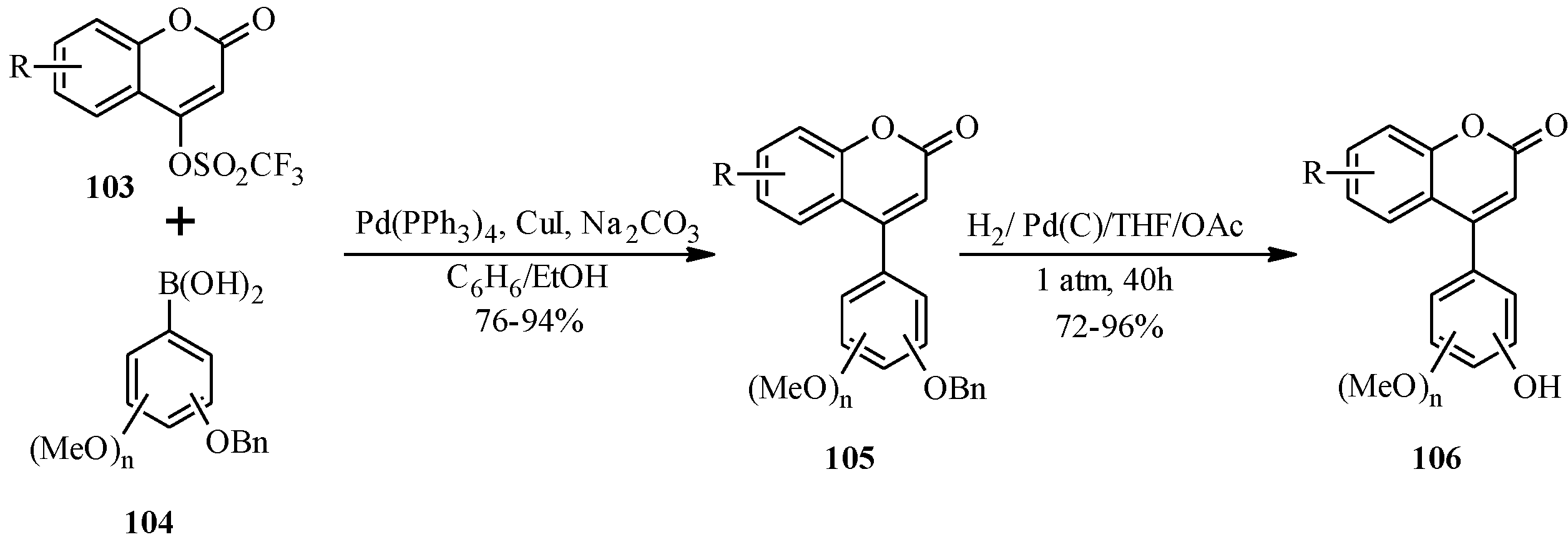
4.4. Synthesis of Prenylated Isoflavonoids
Prenylated isoflavonoids are attained by
C- or
O-prenylation. Prenylation is often carried out after construction of the isoflavonoid framework [
68,
69,
70]. More complex prenylated isoflavonoids are obtained by cyclization of the prenyl side chains to adjacent hydroxy groups to give furano or pyrano rings, or by modification of phenolic A- and B-rings as well as the C-ring via oxidation or incorporation of other additional substituents [
2,
3,
71,
72,
73,
74].
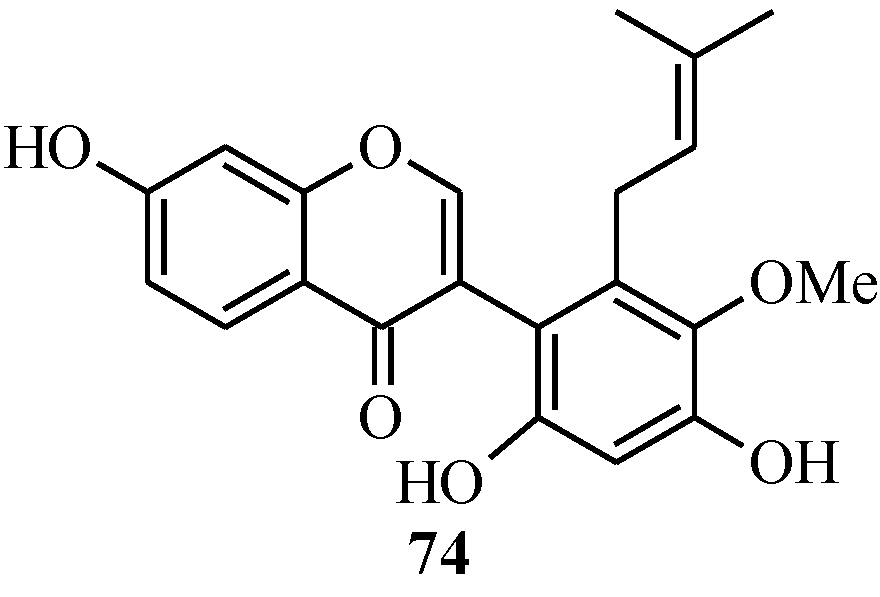
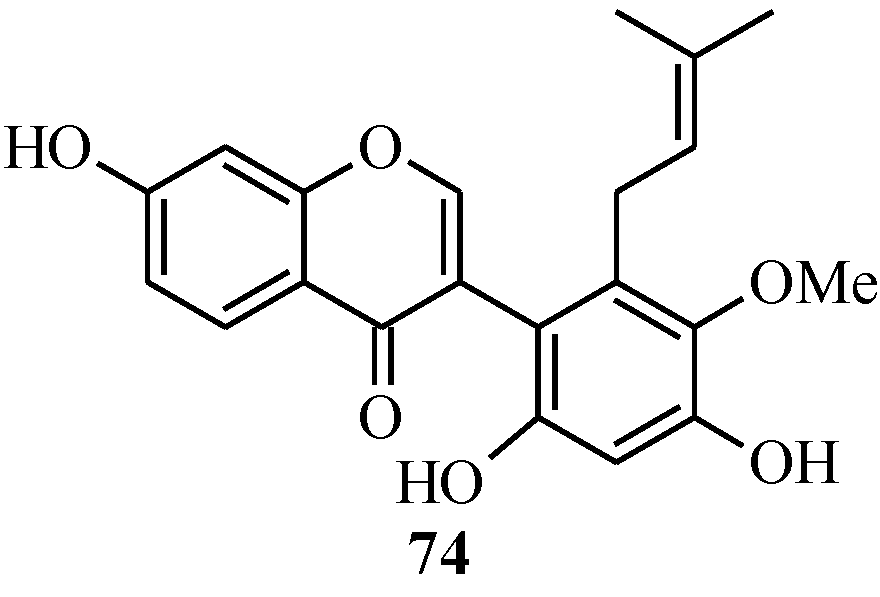
In 2005, Ito and colleagues reported the first total synthesis of kwakhurin (
74) (
Figure 6), a 6'-prenyl-phytoestrogen that was isolated from
Pueraria mirifica (Leguminosae) [
1]. The initial synthetic strategy for
74, which was based on the deoxybenzoin route, failed to give the targeted compound in the last step that involved deprotection of the isopropyl protecting groups. Thus, an alternative route which enabled the use of easily removable protecting groups was sought [
1].
Figure 6.
Structure of kwakhurin.
Figure 6.
Structure of kwakhurin.
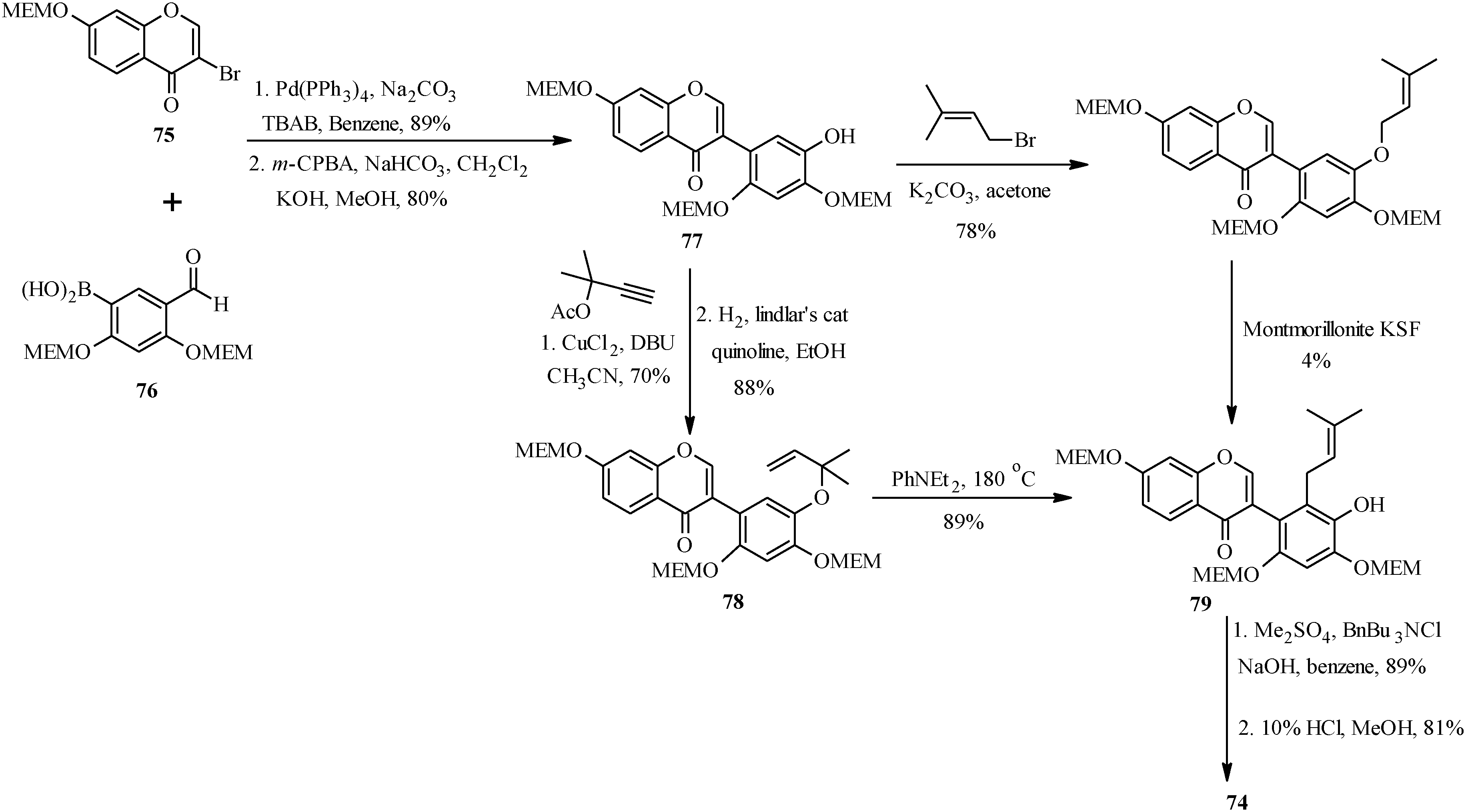
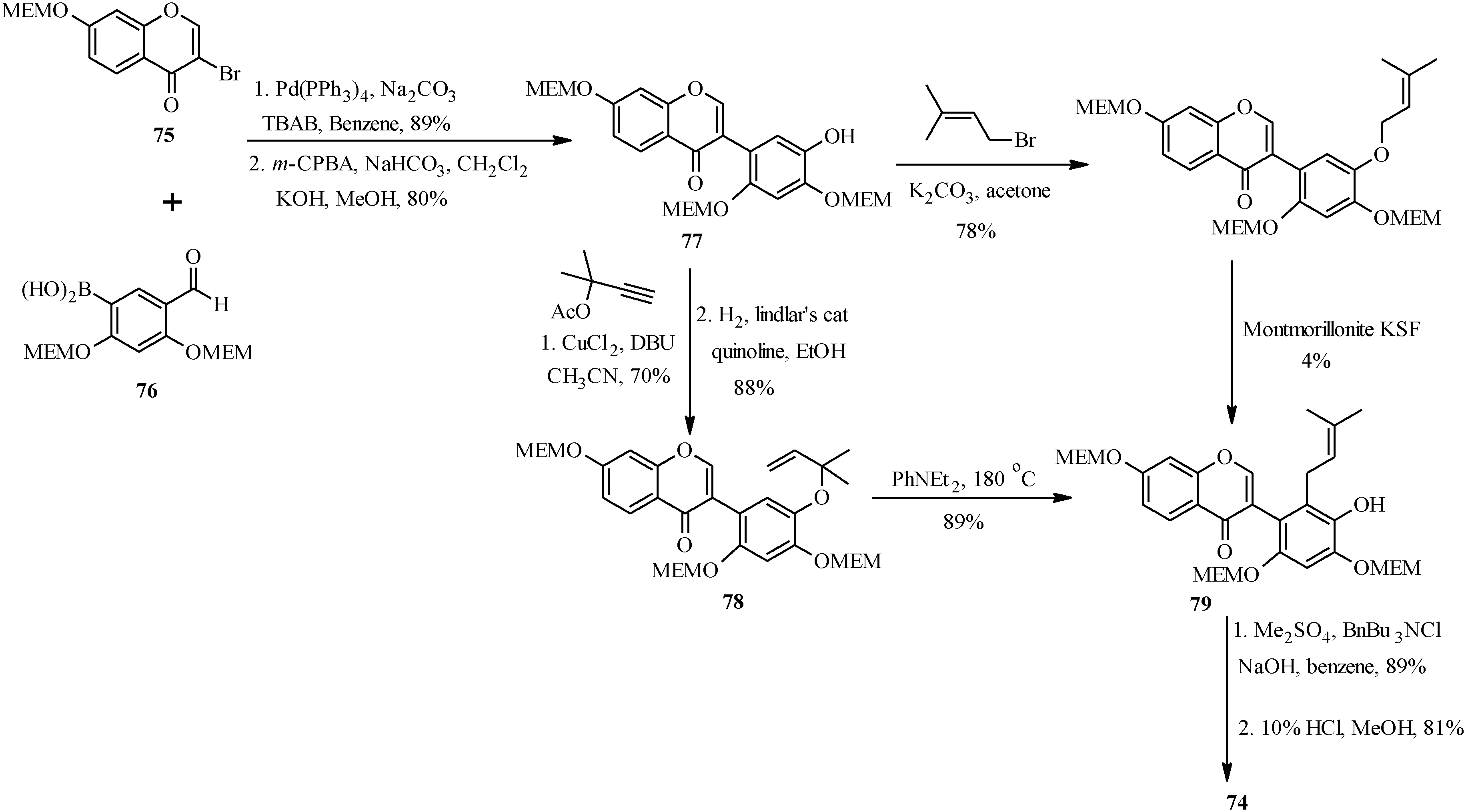
As shown in
Scheme 23, the first steps in the alternative route were preparation of the MEM-protected 3-bromochromone
75 and boronic acid
76. These were coupled using Pd(PPh
3)
4 and TBAB in benzene and the formyl group was converted into a hydroxy group by Baeyer-Villiger oxidation and alkaline hydrolysis to give isoflavone
77 [
1]. The prenyl group was introduced on the isoflavone
77 via two pathways. The first one involved
O-prenylation and montmorillonite KSF-assisted 1,3-migration of the prenyl group. This gave the expected
C-prenylated isoflavone
79 in a poor yield of 4%. The
C-prenylated isoflavone
79 was obtained in a good yield upon
O-propargylation of
77 followed by reduction of the propargyl ether and thermal rearrangement of the resulting 1,1-dimethylallyl ether
78. The last steps were methylation of the 5'-hydroxy group and deprotection of the 2'- and 4'-hydroxy groups under acid conditions to give kwakhurin (
74) [
1].
Scheme 23.
Total synthesis of kwakhurin (74).
Scheme 23.
Total synthesis of kwakhurin (74).
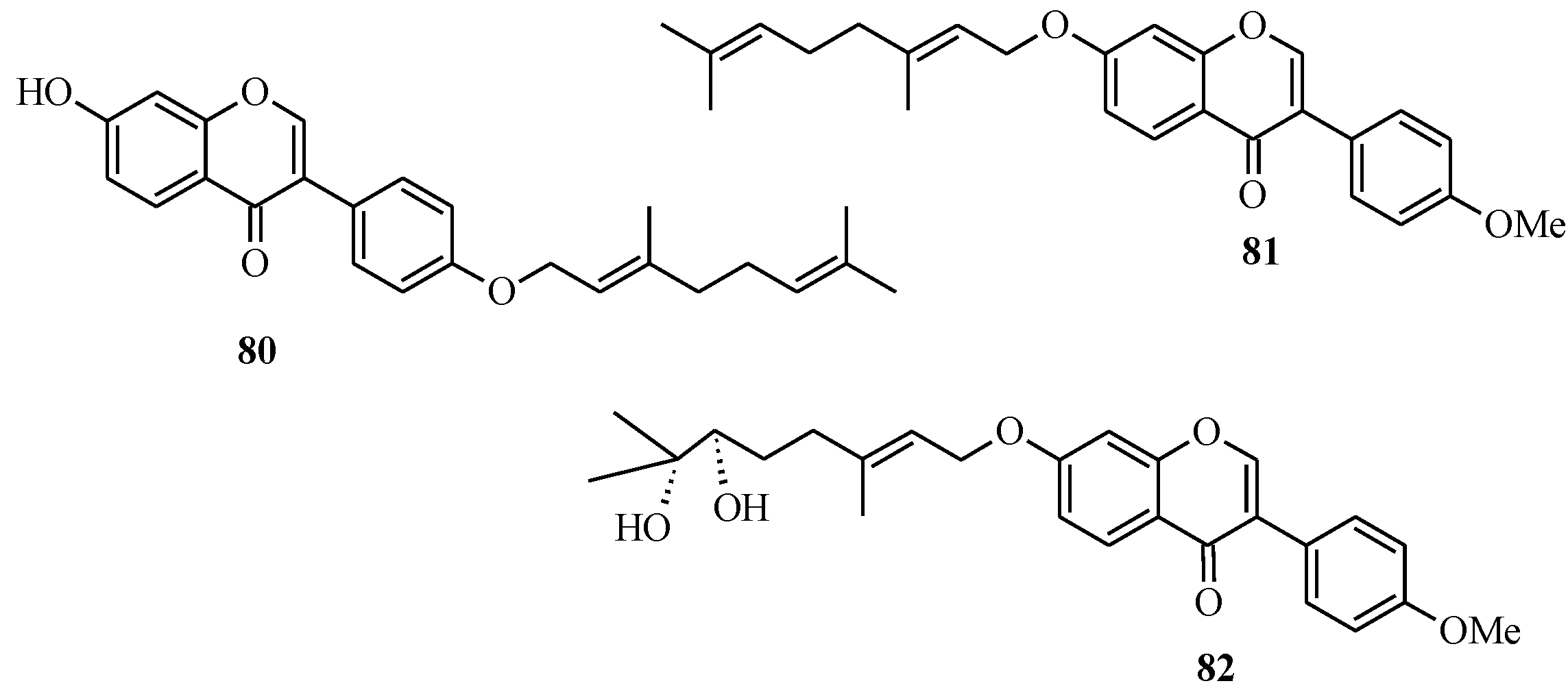
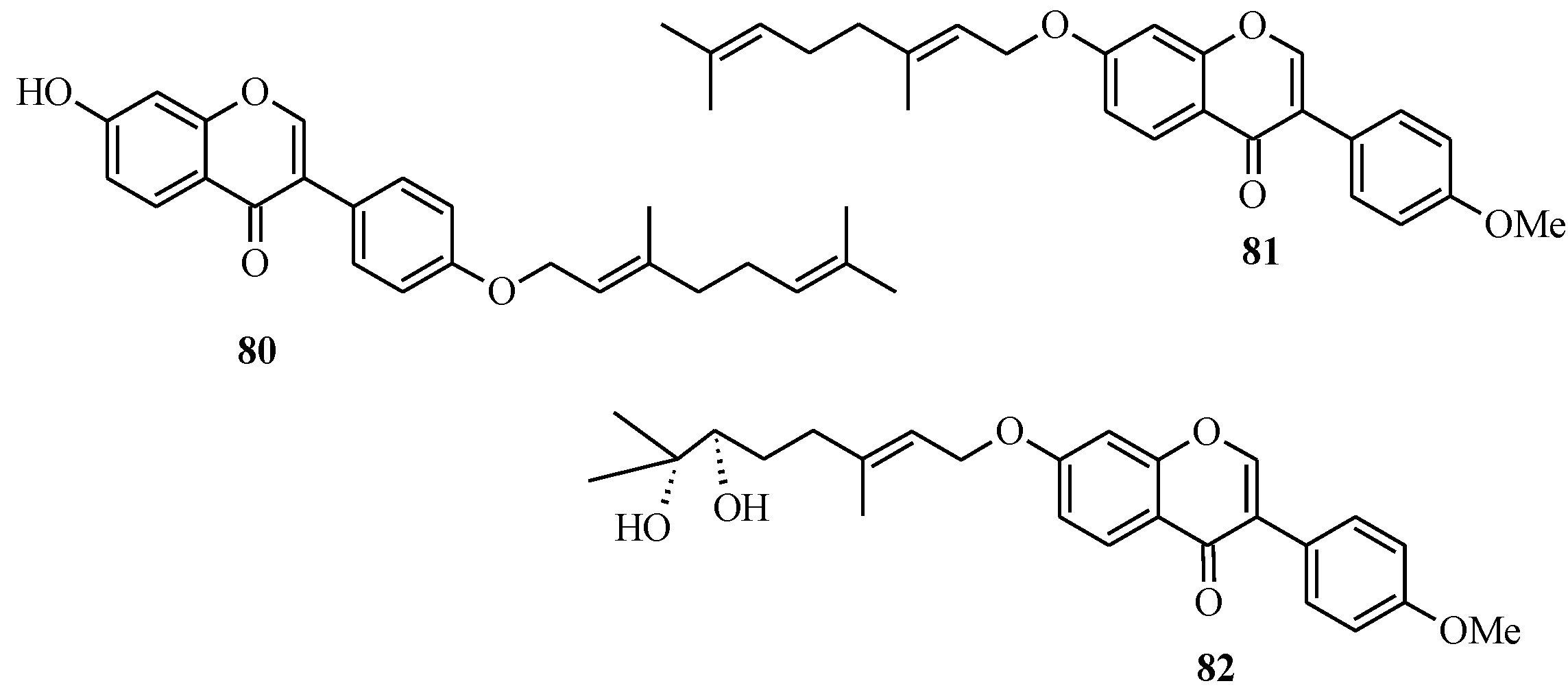
In 2005, Felpin and co-workers developed a method for the Pd(C)-catalyzed Suzuki-Miyaura cross-coupling of iodocycloenones with arylboronic acids [
11]. The use of the heterogeneous Pd(C) presents many advantages compared to other palladium-based catalysts. The reaction is conducted in the absence of additives and ligands, which are often expensive, and air and moisture sensitive. Furthermore, the catalyst can be easily removed from the reaction mixture by filtration, can be reused and is also compatible with water based solvents [
11]. Felpin
et al. further demonstrated the versatility of the Pd(C) chemistry in C-C bond forming reactions that led to the synthesis of geranylated isoflavones conrauinone D (
80), 7-
O-geranylformononentin (
81) and griffonianone D (
82) [
12] (
Figure 7).
Figure 7.
Structures of geranylated isoflavones.
Figure 7.
Structures of geranylated isoflavones.
As illustrated in
Scheme 24 [
12], Pd(C)-catalyzed reaction of THP protected 3-iodochromone
54 with an 4-hydroxyphenylboronic acid or 4-methoxyphenylboronic acid gave isoflavone derivatives which could be used as precursors for the synthesis of the targeted compounds.
Scheme 24.
Synthesis of conrauinone D (
80), 7-
O-geranylformononentin (
81) and griffonianone D (
82) by Felpin
et al. [
12].
Scheme 24.
Synthesis of conrauinone D (
80), 7-
O-geranylformononentin (
81) and griffonianone D (
82) by Felpin
et al. [
12].
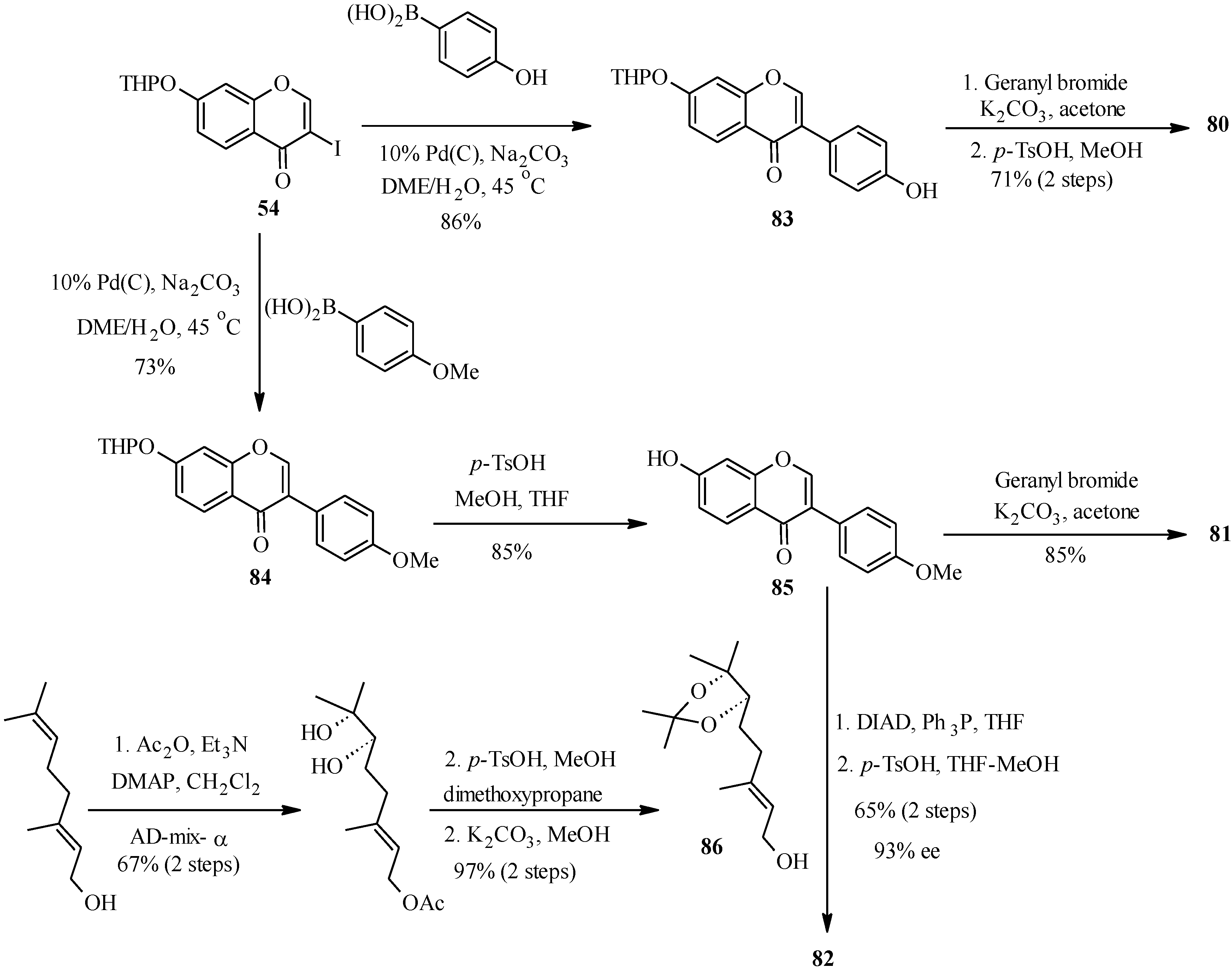
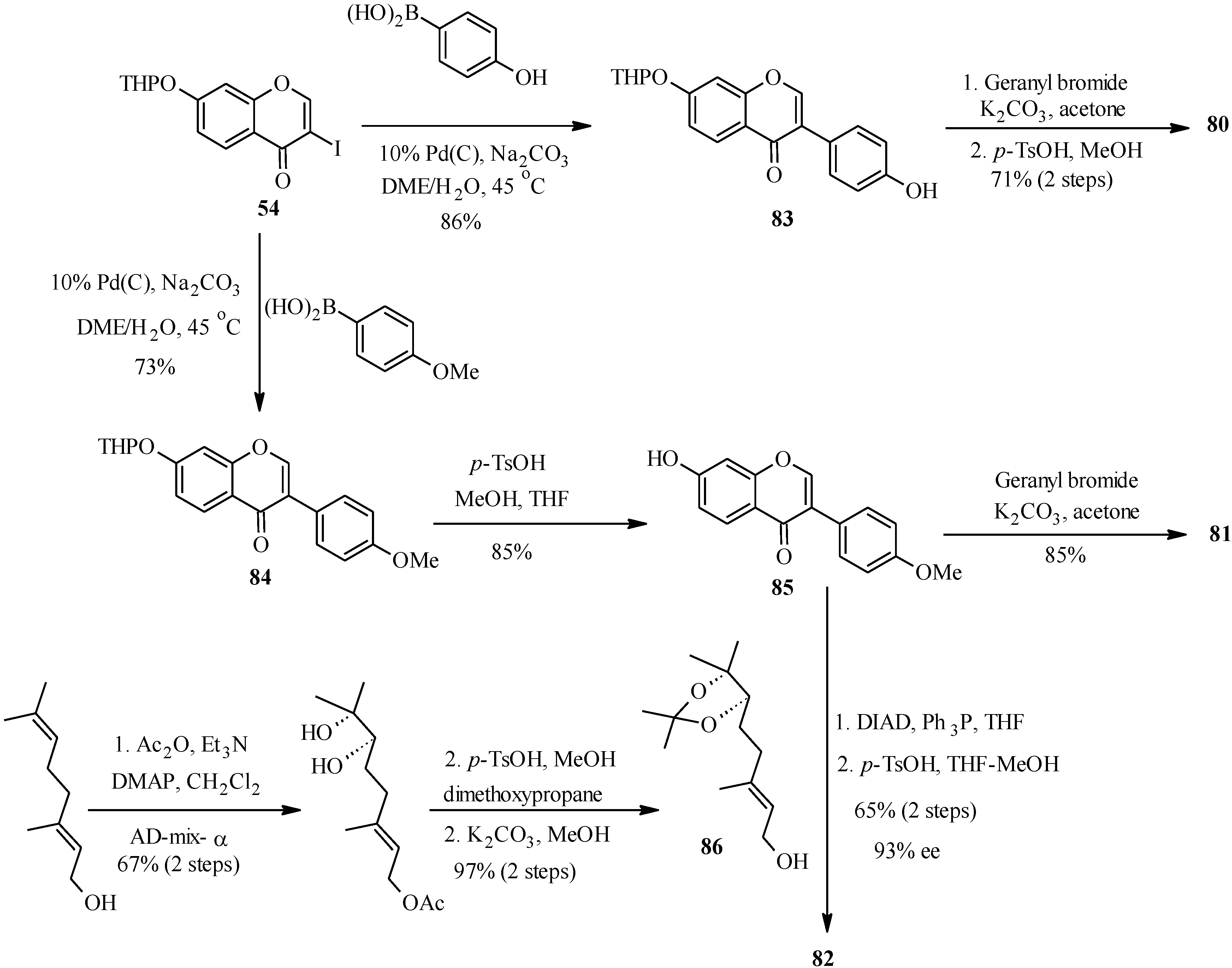
Thus, conrauinone D (
80) was prepared by
O-alkylation of
83 with geranyl bromide using K
2CO
3 as base, followed by cleavage of the THP protecting group under mild conditions.
O-Geranylation of the isoflavone
85, obtained by the THP-deprotection of
84 gave 7-
O-geranylformononentin (
81). The last target compound griffonianone D (
82) was prepared by Mitsunobu reaction of dihydroxylated geraniol derivative
86 and subsequent cleavage of the acetonide [
12].
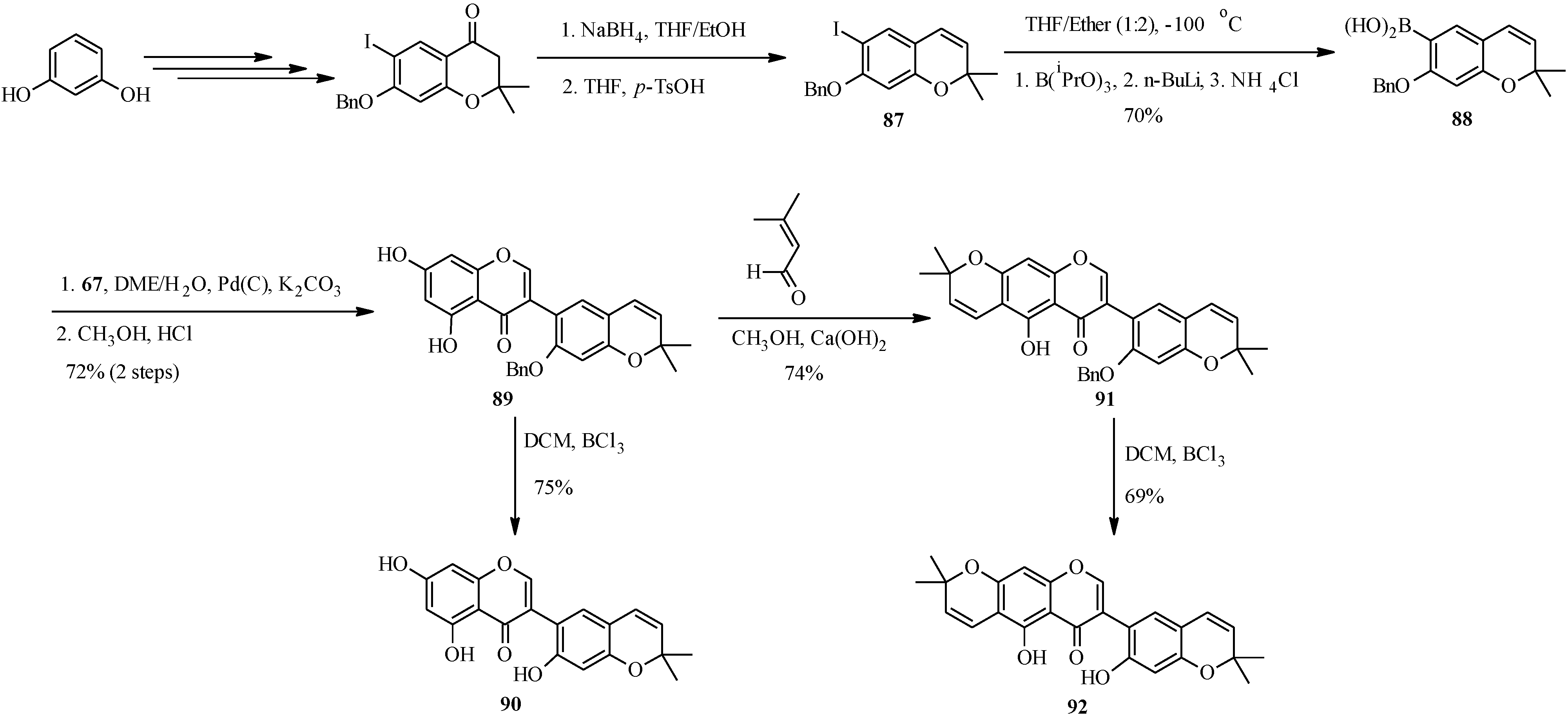
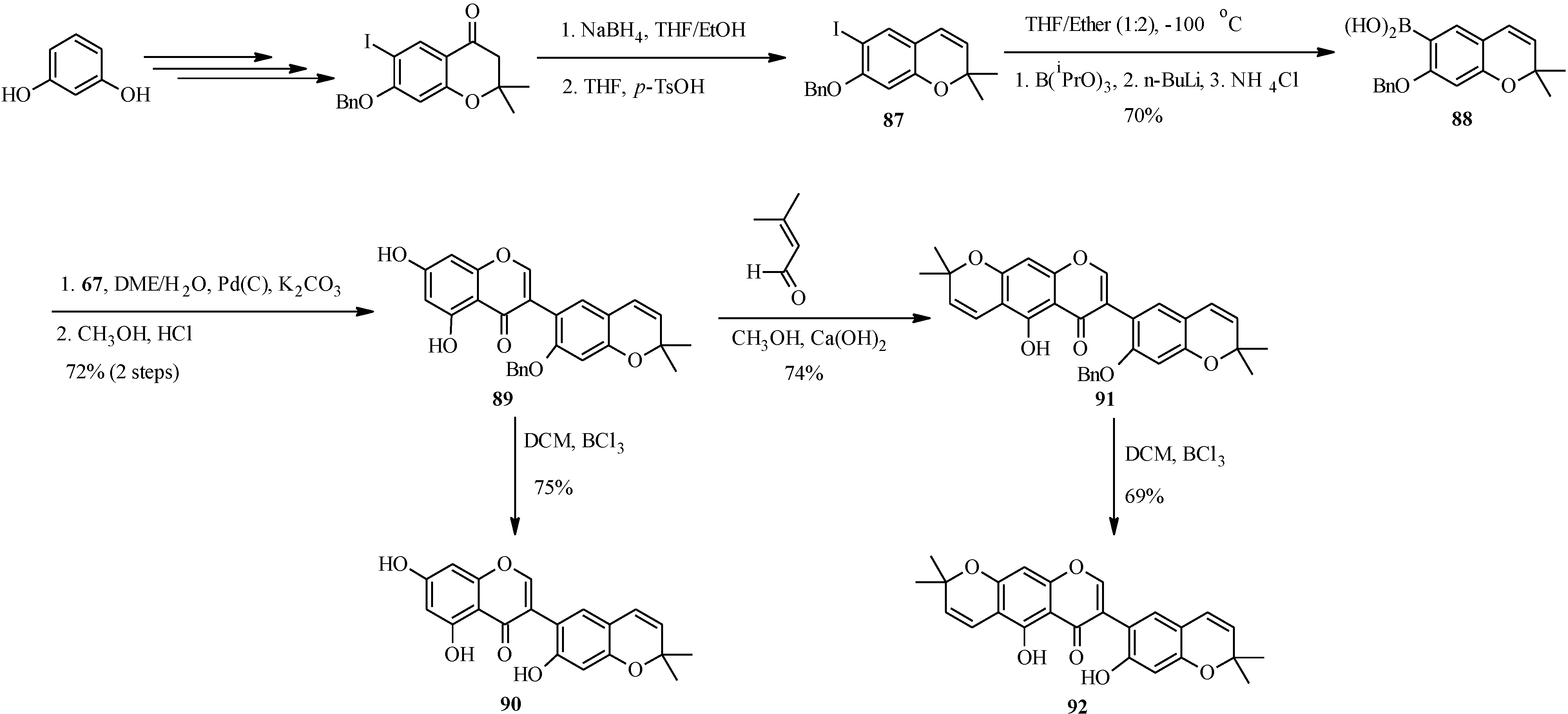
The first total syntheses two biologically-active pyranoisoflavones, the anti-impotence pyranoisoflavone kraussianone 1 (
92) and the anti-fungal pyranoisoflavone eriosemaone D (
90) was reported by Selepe
et al. in 2010 [
2]. The key steps involved the Suzuki-Miyaura reaction for the construction of the isoflavone core and the regioselective formation of the dimethylpyran scaffolds to the phloroglucinol (A-ring) and resorcinol (B-ring) moieties (
Scheme 25). The synthesis commenced by preparing the boronic acid
88 from aryl iodide
87 by an “
in situ quench” procedure described for boronic acid
70. Coupling of boronic acid
88 with 3-iodochromone
67 under Felpin’s conditions and subsequent removal of the MOM protecting groups gave a pyranoisoflavone
89, which was transformed into eriosemaone D (
90) by debenzylation with BCl
3. Kraussianone 1 (
92) was prepared by base-catalyzed aldol-type condensation of
91 with prenal followed by removal of the benzyl protecting group with BCl
3 (
Scheme 25) [
2].
Scheme 25.
Total synthesis of kraussianone 1 (
92) and eriosemaone D (
90) [
2].
Scheme 25.
Total synthesis of kraussianone 1 (
92) and eriosemaone D (
90) [
2].
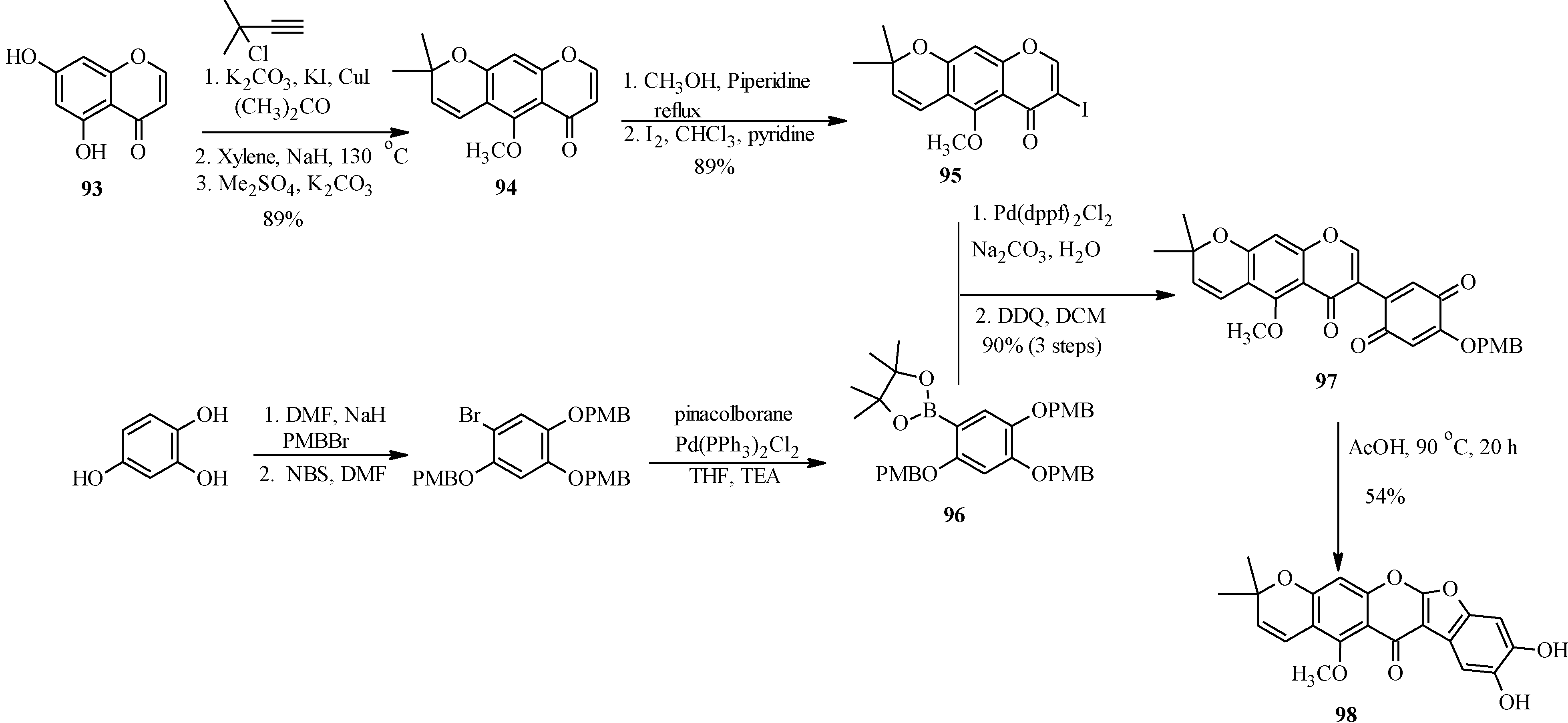
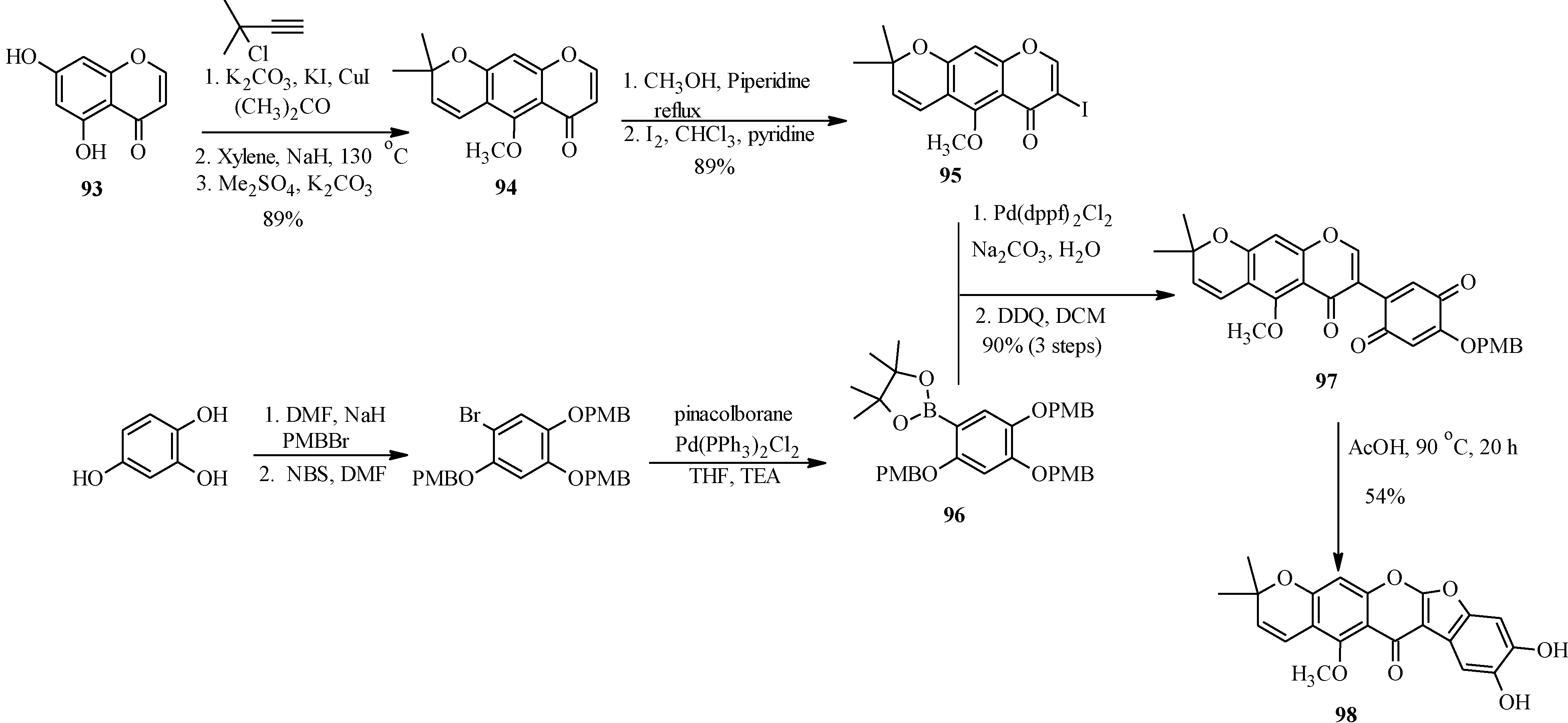
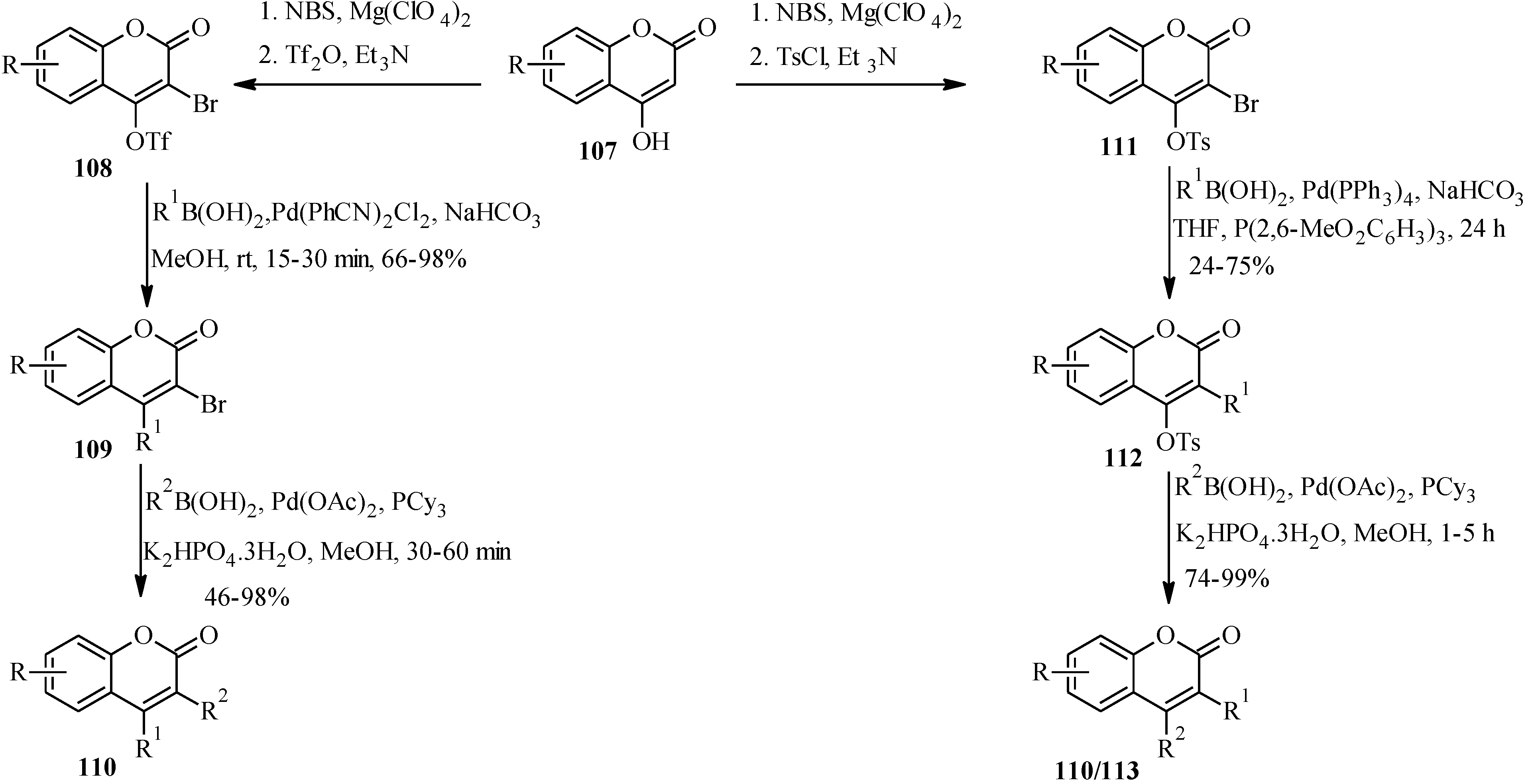
The application of Suzuki-Miyaura reaction has also been demonstrated in the synthesis of other subclasses of isoflavonoids that include coumaronochromones. Zheng and Shen reported the total synthesis of hirtellanine A (
98) [
3], a coumaronochromone derivative that exhibits immunosuppresive activity (
Scheme 26) [
75]. The main precursors were 3-iodochromone bearing dimethylchromene scaffold
95 and a boronic ester
96. The 3-iodochromone
95 was prepared in a sequence of steps, which involved regioselective formation the chromene scaffold by a Thom-Harfenist [
76] rearrangement of the propargyl ether [
77], prepared by
O-alkylation of the 7-hydroxy group of chromone
93 with 3-chloro-3-methylbut-1-yne. Iodination of the resulting pyranochromone
94 at C-3 following Gammill’s protocol rendered
95. The boronic ester
96 on the other hand was prepared in three steps from 1,2,4-trihydroxybenzene. The Suzuki coupling of
95 with
96 and subsequent oxidative cleavage of the
p-methoxybenzyl protecting groups gave a quinone
97, which upon treatment with acetic acid gave hirtellanine A (
98) [
3].
Scheme 26.
Total synthesis of hirtellanine A (
98) [
3].
Scheme 26.
Total synthesis of hirtellanine A (
98) [
3].
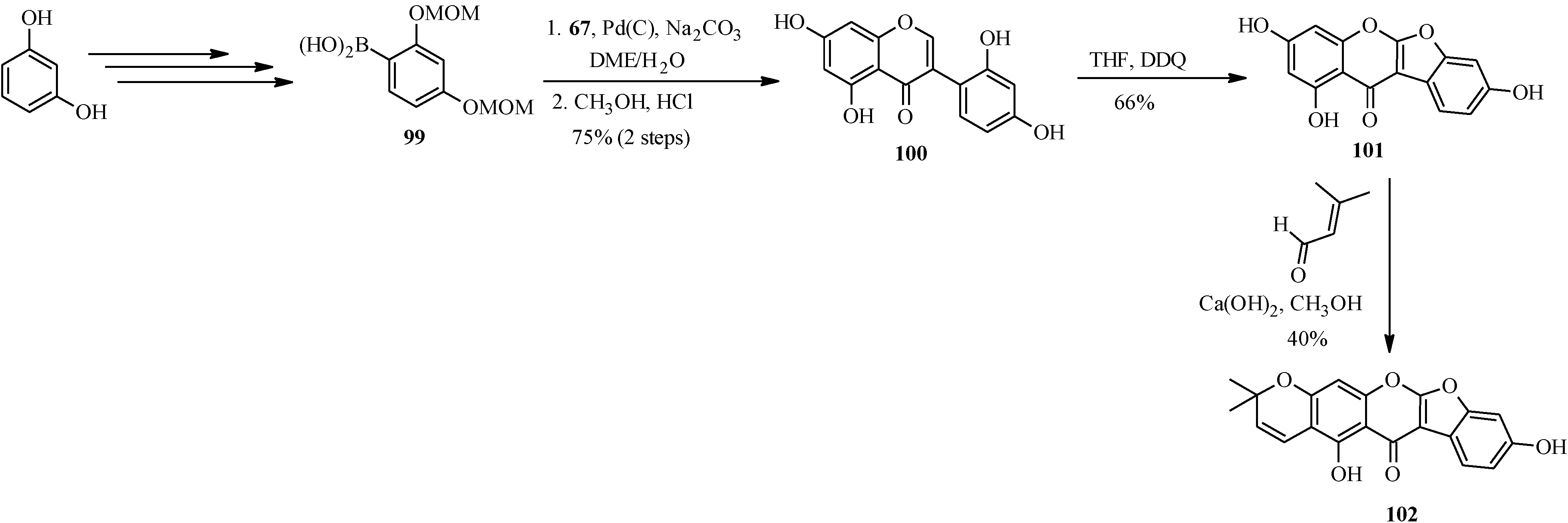
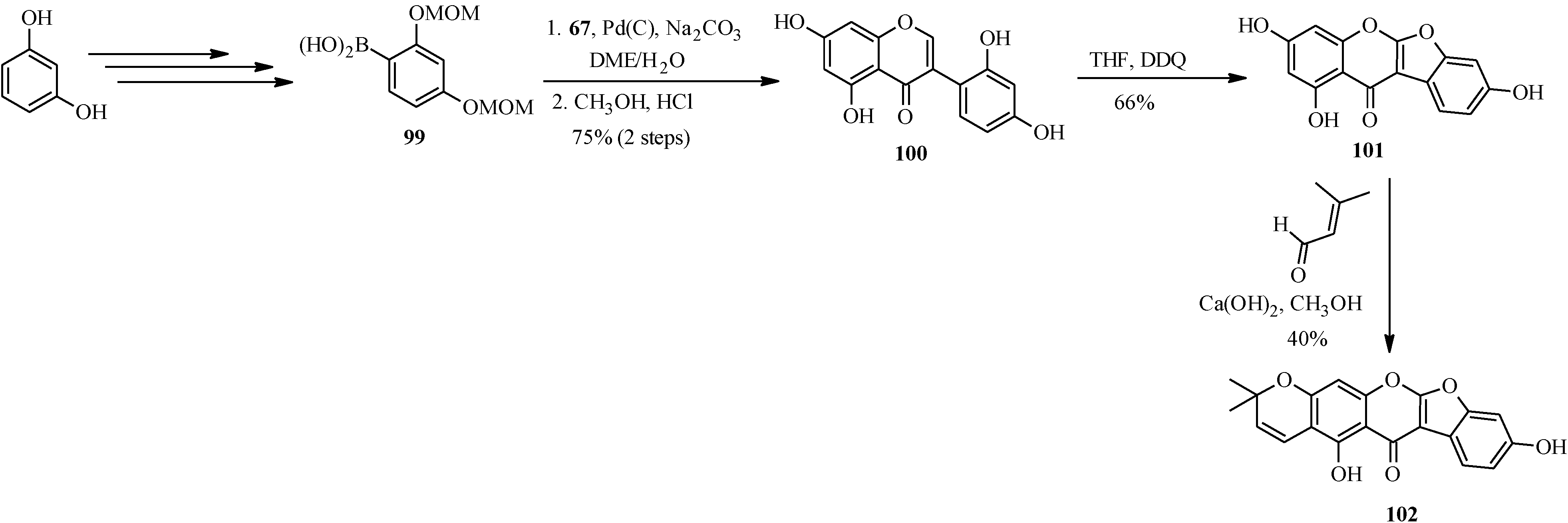
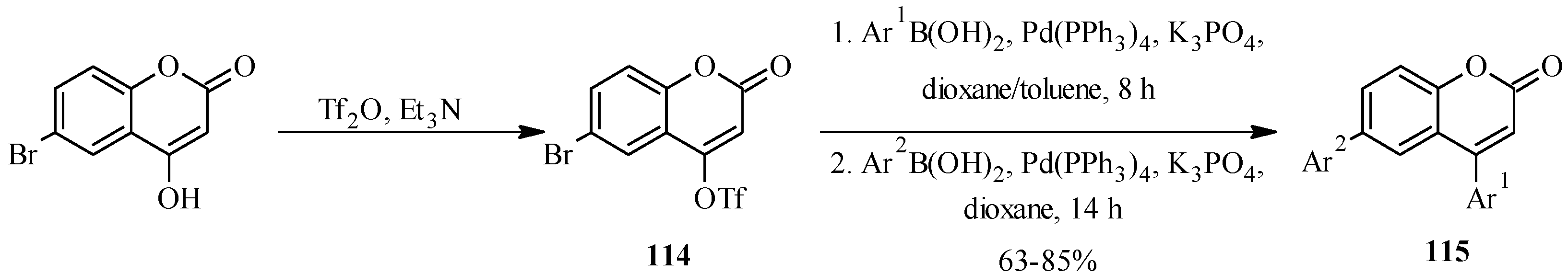
Another pyranocoumaronochromone lupinalbin H (
102) was synthesized by Selepe and co-workers in steps that involved preparation of 2'-hydroxygenistein (
100) from 3-iodochromone
67 and a boronic acid
99, followed by cyclodehydrogenation to lupinalbin A (
101) [
72]. The final step was the regioselective introduction of the dimethylpyran moiety to the A-ring of 101 via an aldol-type condensation with 3-methyl-2-butenal and 6π-electrocyclization (
Scheme 27) [
72].
Scheme 27.
Total synthesis of lupinalbin H (
102) [
72].
Scheme 27.
Total synthesis of lupinalbin H (
102) [
72].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
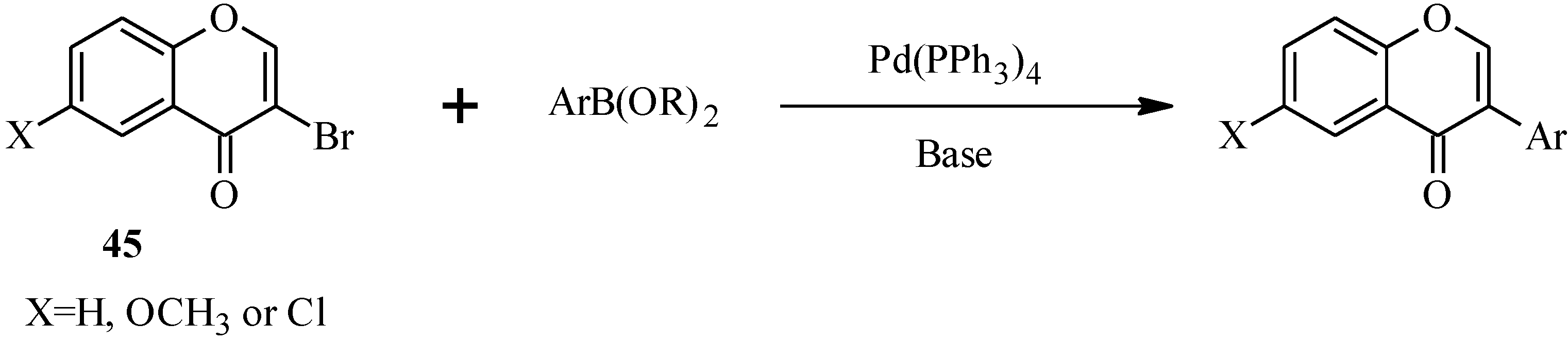{kind=link}
{kind=link}
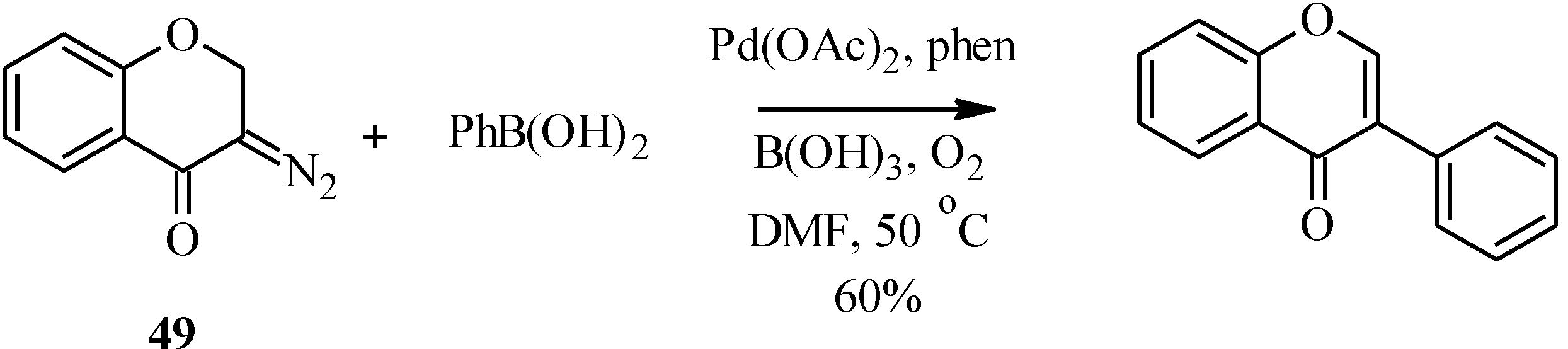{kind=link}
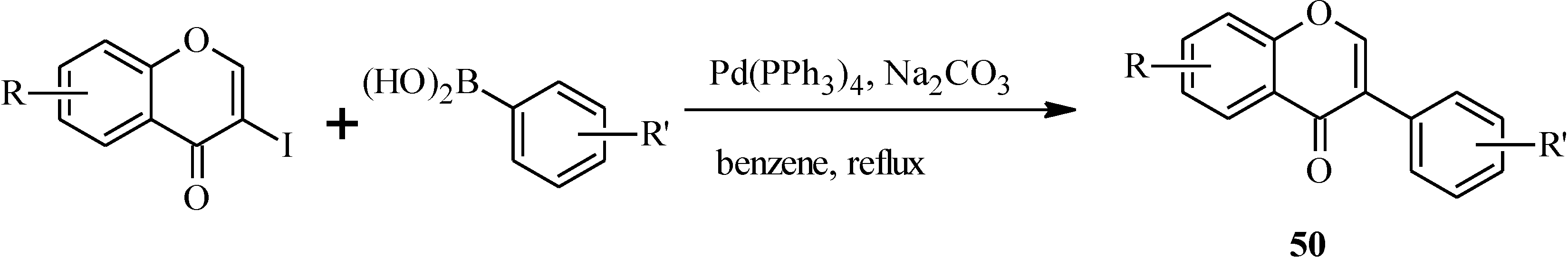{kind=link}
{kind=link}
{kind=link}
{kind=link}
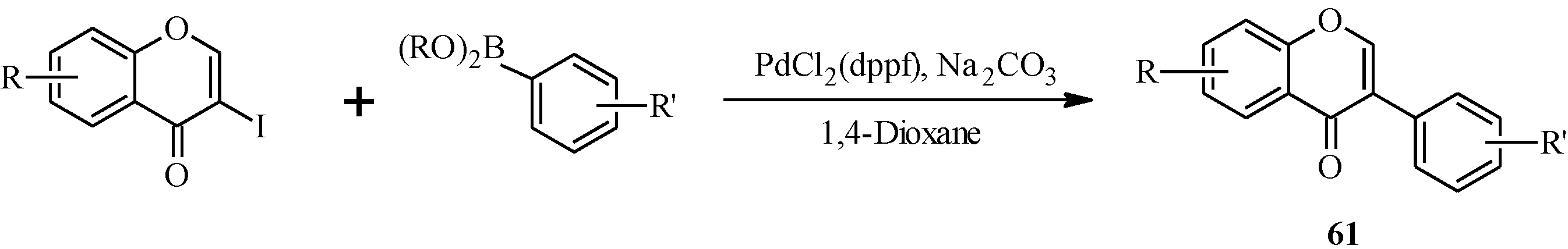{kind=link}
{kind=link}
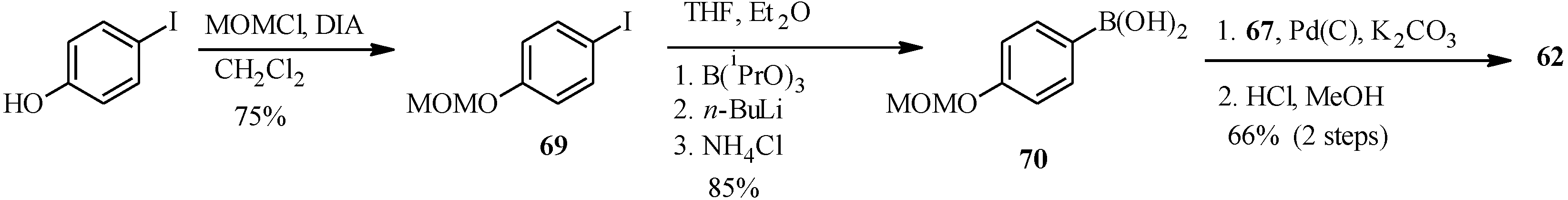{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}