Phenylmethimazole Suppresses dsRNA-Induced Cytotoxicity and Inflammatory Cytokines in Murine Pancreatic Beta Cells and Blocks Viral Acceleration of Type 1 Diabetes in NOD Mice

Abstract
:1. Introduction
2. Results and Discussion
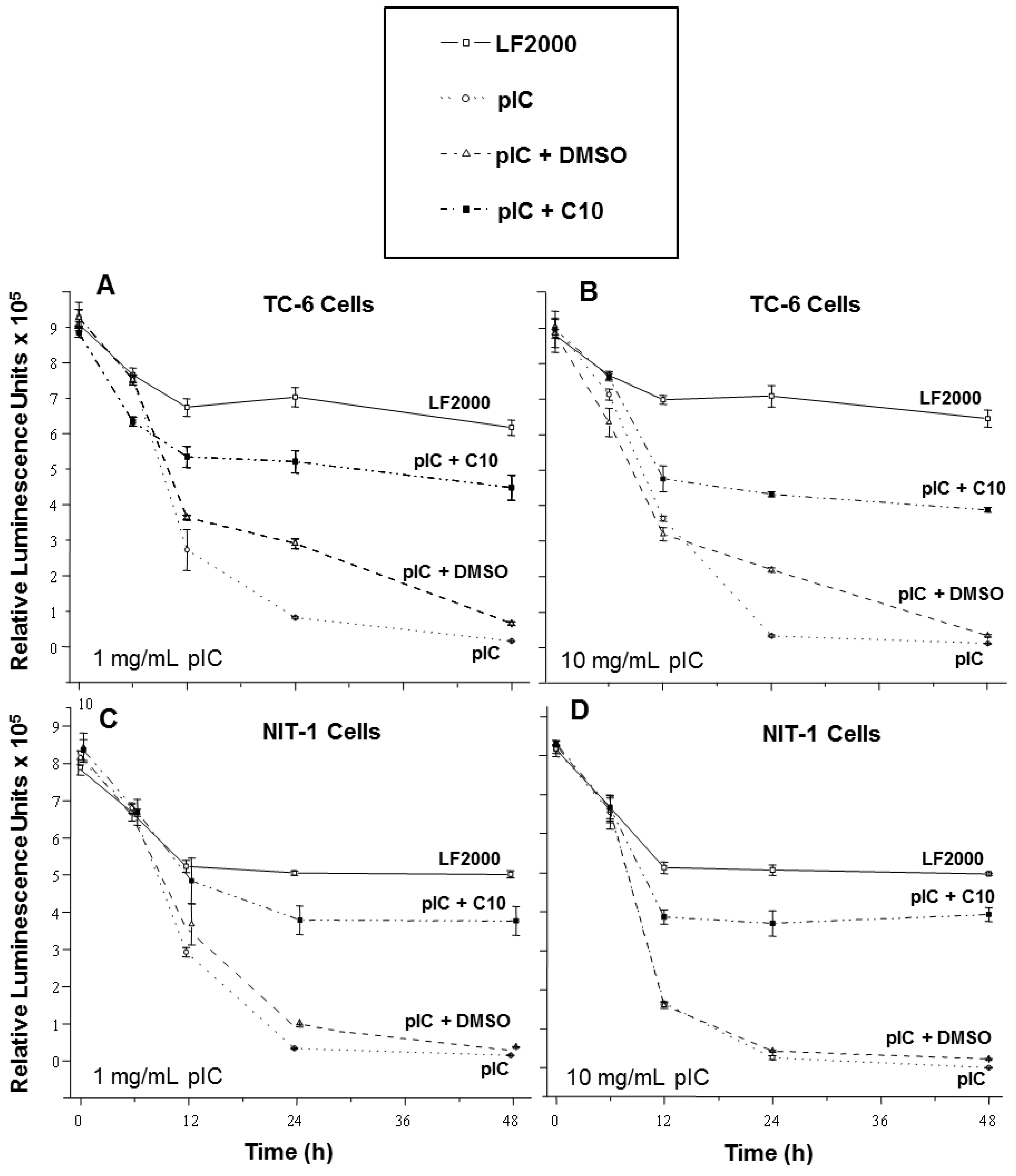
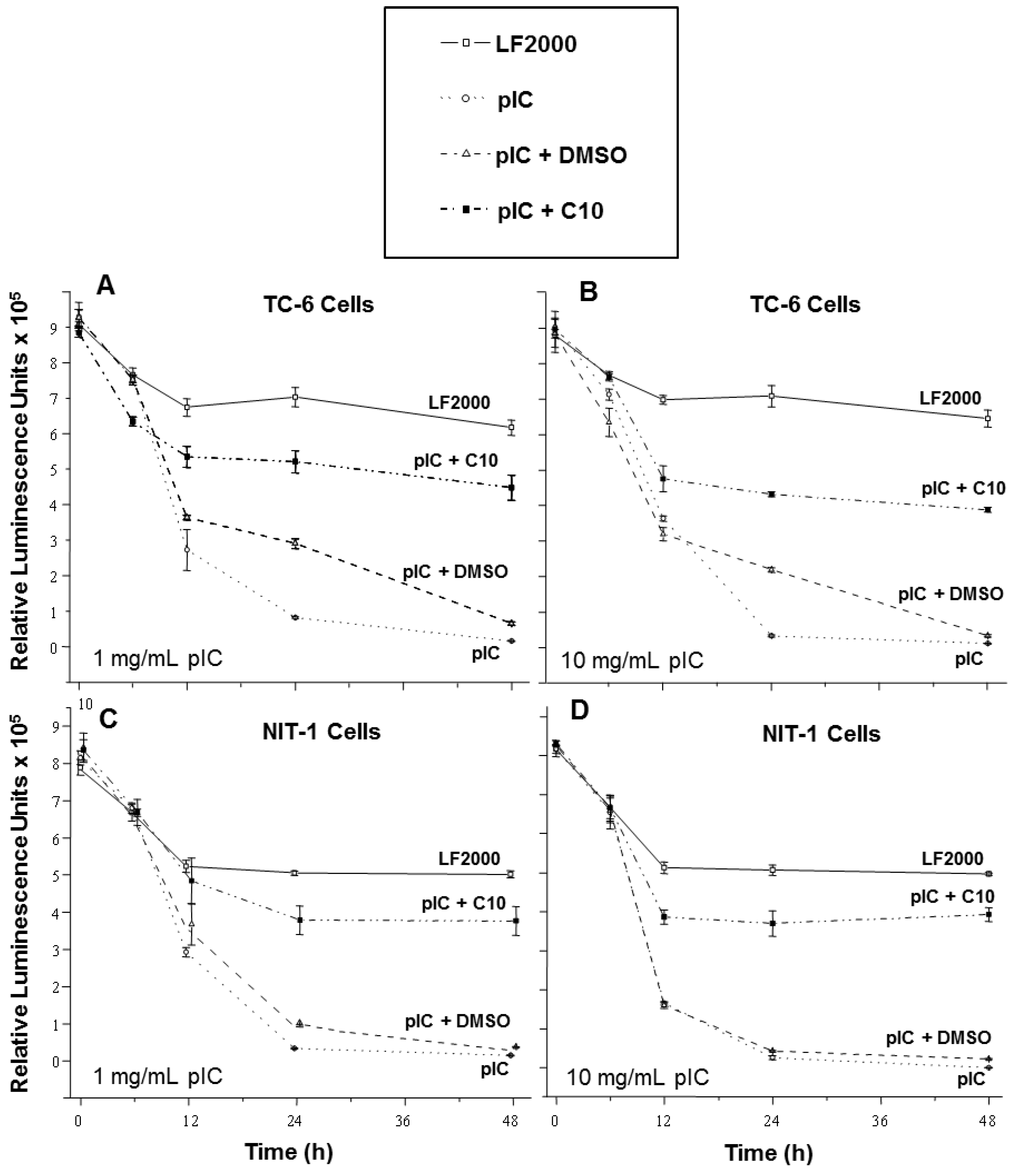
2.1. C10 Protects Pancreatic Beta Cells from dsRNA-Induced Cytotoxicity
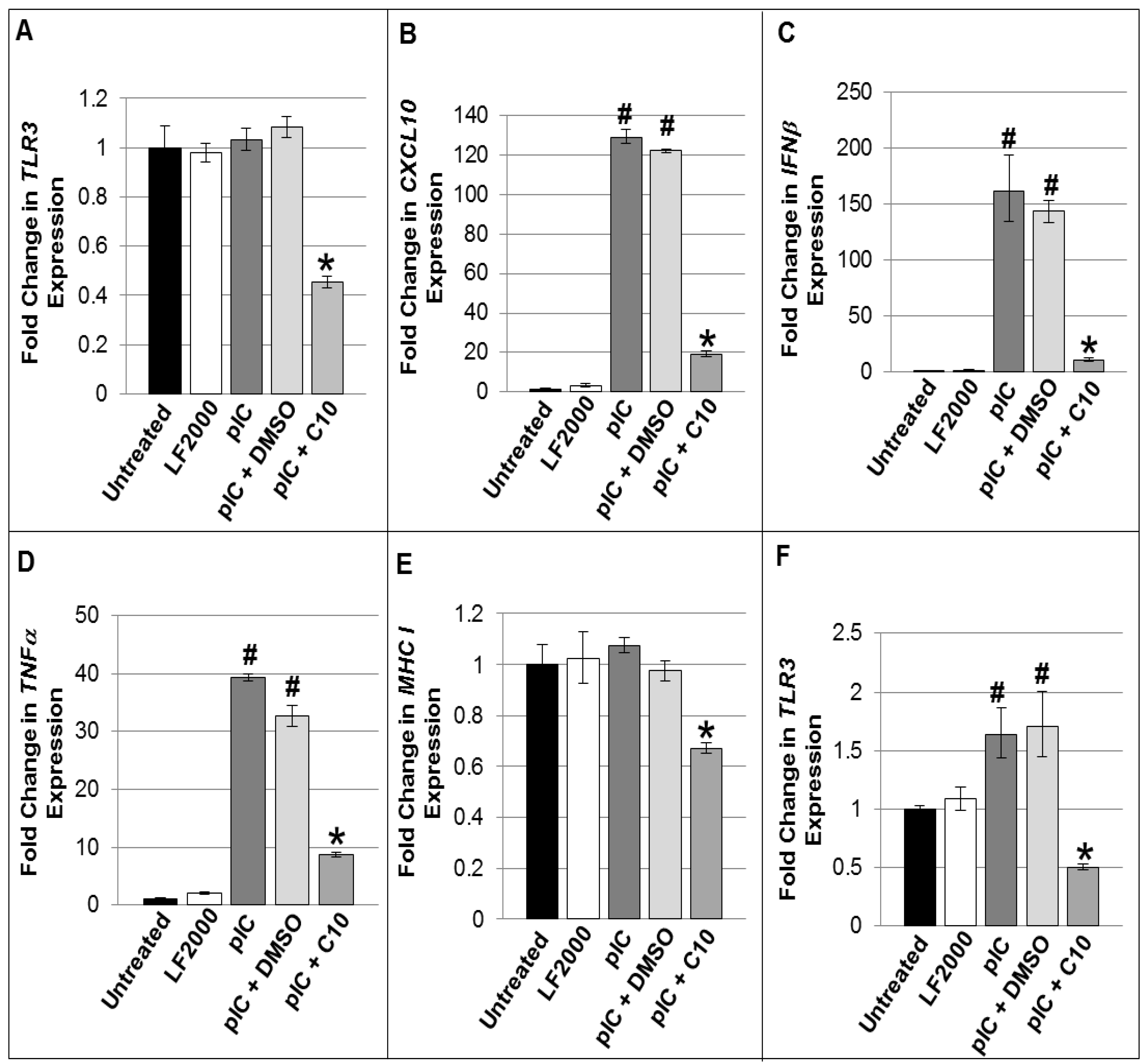
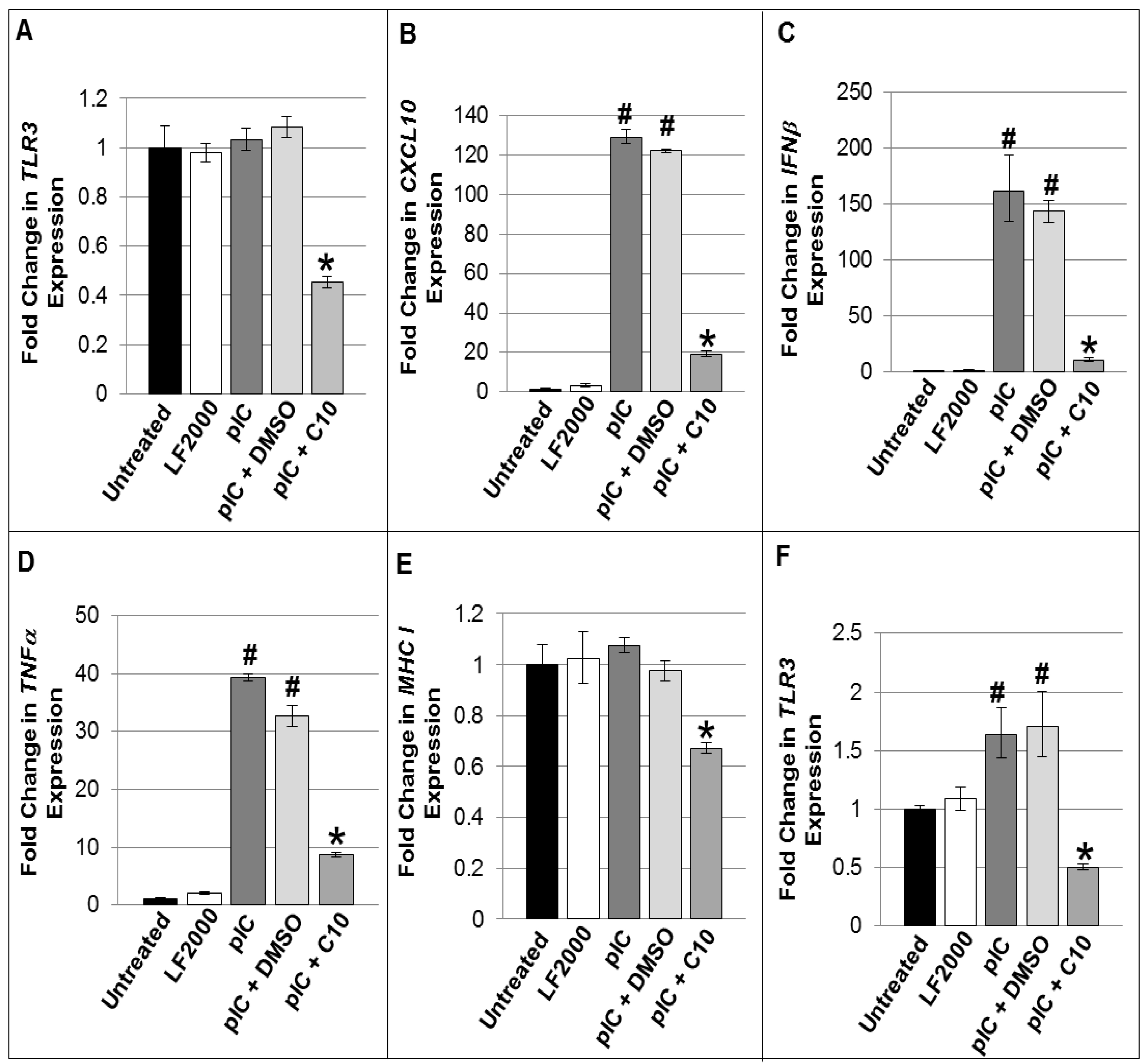
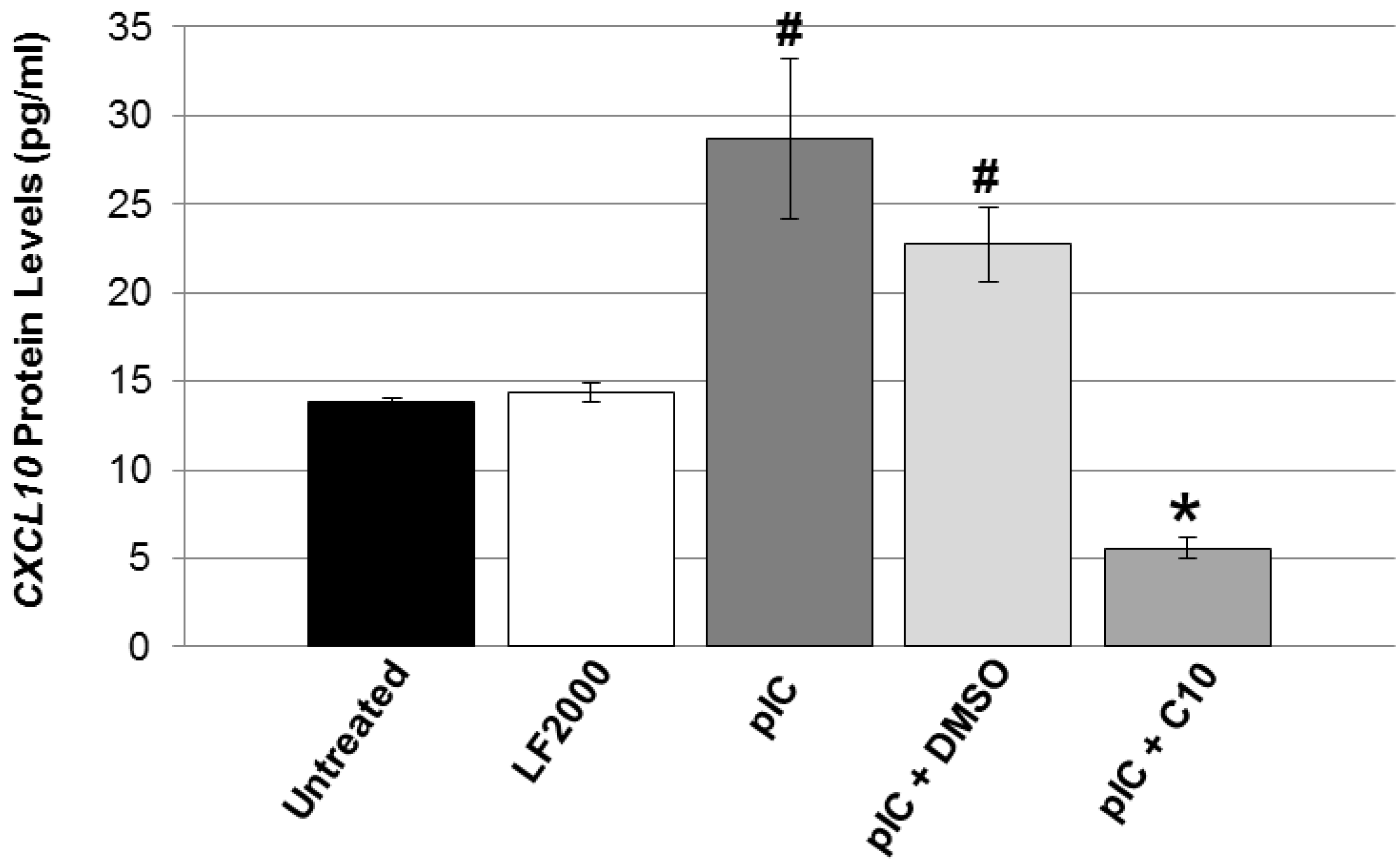
2.2. C10 Blocks dsRNA-Induced Upregulation of TLR3 Expression and Signaling Products in Pancreatic Beta Cells

{kind=link}
{kind=link}
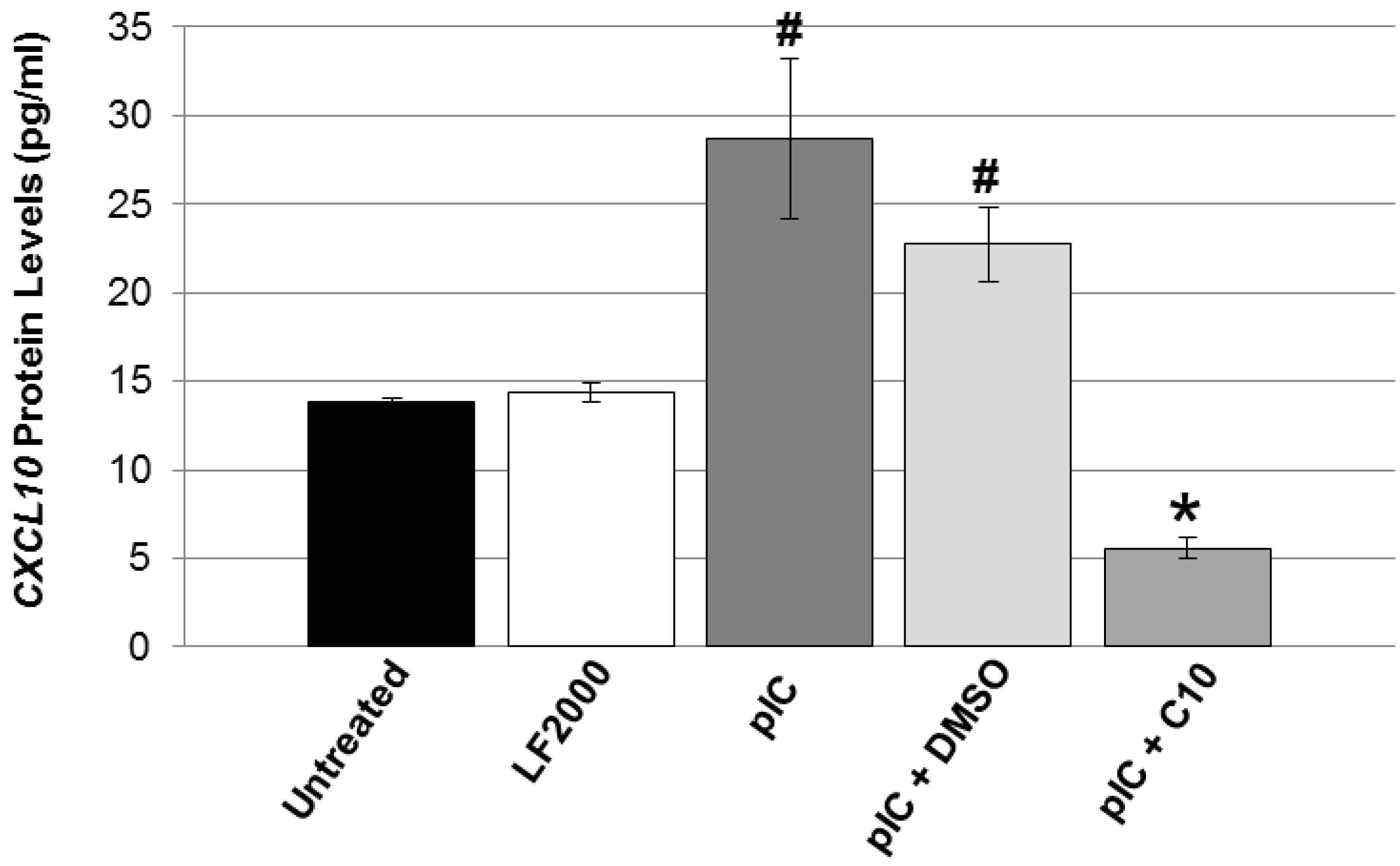{kind=link}
{kind=link}
| Functional Gene Groupings/Gene Name | TC-6 cells | NIT-1 Cells | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pattern Recognition Receptors | pIC | pIC + DMSO | pIC + C10 | pIC | pIC + DMSO | pIC + C10 | |||
| Toll-Like Receptor 1 | 5.78 | <5 | <5 | <5 | <5 | <5 | |||
| Toll-Like Receptor 2 | 8.20 | <5 | <5 | 26.17 | 11.59 | <5 | |||
| Toll-Like Receptor 3 | 66.7 | 23.2 | <5 | 176.0 | 64.2 | <5 | |||
| Toll-Like Receptor 4 | 5.78 | <5 | <5 | <5 | <5 | <5 | |||
| Toll-Like Receptor 6 | <5 | <5 | <5 | 22.24 | <5 | <5 | |||
| Toll-Like Receptor 7 | 5.74 | <5 | <5 | <5 | <5 | <5 | |||
| Toll-Like Receptor 8 | 5.74 | <5 | <5 | <5 | <5 | <5 | |||
| Functional Gene Groupings/Gene Name | TC-6 cells | NIT-1 Cells | ||||
|---|---|---|---|---|---|---|
| pIC | pIC + DMSO | pIC + C10 | pIC | pIC + DMSO | pIC + C10 | |
| TLR adaptor proteins | ||||||
| Heat shock 70kDa protein 1A (Hspa1a) | <5 | <5 | <5 | 20.32 | <5 | 8.30 |
| TICAM2 | <5 | <5 | <5 | 9.58 | 6.57 | <5 |
| Transcription factors | ||||||
| c-Fos | <5 | <5 | <5 | 5.45 | <5 | <5 |
| c-Jun | <5 | <5 | <5 | 14.37 | <5 | <5 |
| CCAAT/enhancer binding protein β (Cebpb) | <5 | <5 | <5 | 9.22 | <5 | <5 |
| NF-κB pathway | ||||||
| CCL2 | 21.50 | 22.25 | <5 | 172.45 | 30.70 | <5 |
| CD80 | 6.03 | 6.69 | <5 | 40.22 | 20.97 | <5 |
| CD86 | <5 | <5 | <5 | 68.12 | 35.38 | <5 |
| Granulocyte macrophage colony-stimulating factor (Csf2) | <5 | <5 | <5 | 5.92 | <5 | <5 |
| Granulocyte colony-stimulating factor (Csf3) | <5 | <5 | <5 | 6.80 | <5 | <5 |
| IKKα (Chuk) | <5 | <5 | <5 | 5.37 | <5 | <5 |
| IL-1α | <5 | <5 | <5 | 7.31 | <5 | <5 |
| IL-2 | 5.78 | <5 | <5 | <5 | <5 | <5 |
| IL-6 | 8.91 | 8.98 | <5 | 580.04 | 143.01 | <5 |
| IL-6 Receptor α (IL-6ra) | <5 | <5 | <5 | 25.46 | <5 | <5 |
| IL-10 | <5 | <5 | <5 | 57.68 | 22.24 | <5 |
| IL-12A | 6.62 | <5 | <5 | 10.78 | 22.09 | <5 |
| NF-κB2 | 8.52 | <5 | <5 | 22.86 | 16.56 | <5 |
| IκBα (Nfkbia) | <5 | <5 | <5 | 13.50 | <5 | <5 |
| IκBβ (Nfkbib) | <5 | <5 | <5 | 14.47 | <5 | <5 |
| Prostaglandin-endoperoxide synthase 2 (Ptgs2) | <5 | <5 | <5 | 48.34 | 17.69 | <5 |
| TNF-α | 5.78 | <5 | <5 | 26.72 | 6.70 | <5 |
| TNF-β (Lta) | NS | <5 | <5 | 45.57 | 38.59 | <5 |
| Tumor necrosis factor, alpha-induced protein 3 (Tnfaip3) | 7.87 | <5 | <5 | <5 | <5 | <5 |
| IRF pathway | ||||||
| IP-10 (CXCL10) | 189.49 | 74.85 | <5 | 543.07 | 141.53 | <5 |
| IFN-β1 | 135.86 | 72.81 | <5 | 2778.33 | 196.04 | <5 |
| IRF-1 | 6.85 | 6.32 | <5 | 12.04 | 11.24 | <5 |
| Functional Gene Groupings/Gene Name | TC-6 Cells | NIT-1 Cells | ||||||
|---|---|---|---|---|---|---|---|---|
| pIC | pIC + DMSO | pIC + C10 | pIC | pIC + DMSO | pIC + C10 | |||
| Chemokines | ||||||||
| CCL2 | 13.60 | 6.96 | <5 | 160.12 | 25.05 | <5 | ||
| CCL4 | 39.15 | 5.94 | <5 | 25.33 | 25.58 | <5 | ||
| CCL5 | 320.9 | 112.21 | <5 | 1245.92 | 384.54 | <5 | ||
| CCL7 | 18.2 | 14.42 | <5 | 107.49 | 91.90 | <5 | ||
| CCL9 | <5 | <5 | <5 | 6.86 | <5 | <5 | ||
| CCL12 | 8.76 | <5 | <5 | 19.95 | 8.80 | <5 | ||
| CCL19 | <5 | <5 | <5 | 26.32 | 32.38 | 64.40 | ||
| CCL20 | 183.67 | 54.19 | <5 | 76.27 | 43.17 | <5 | ||
| CCL25 | 23.36 | 21.33 | <5 | <5 | <5 | <5 | ||
| CXCL1 | 7.57 | <5 | <5 | <5 | <5 | <5 | ||
| CXCL5 | 5.86 | <5 | <5 | <5 | 10.14 | <5 | ||
| CXCL9 | 58.32 | 17.94 | <5 | 344.41 | 275.71 | <5 | ||
| CXCL10 | 150.23 | 133.9 | <5 | 522.03 | 170.31 | <5 | ||
| CXCL11 | <5 | <5 | <5 | 74.70 | 67.51 | <5 | ||
| Chemokine receptors | ||||||||
| CCR10 | 179.27 | 57.48 | <5 | <5 | <5 | <5 | ||
| Cytokines | ||||||||
| IL-1F6 | <5 | <5 | <5 | 38.94 | 15.97 | <5 | ||
| IL-4 | 41.53 | 11.84 | <5 | <5 | <5 | <5 | ||
| IL-10 | <5 | <5 | <5 | 28.11 | 10.87 | <5 | ||
| IL-11 | 33.61 | 10.67 | <5 | 19.00 | 8.65 | <5 | ||
| IL-15 | 139.68 | 43.26 | <5 | 36.45 | 35.19 | <5 | ||
| Osteopontin (Spp1) | 18.33 | NS | <5 | <5 | <5 | <5 | ||
| TGF-β1 | 62.51 | 20.32 | <5 | <5 | <5 | <5 | ||
| TNF-α | <5 | <5 | <5 | 21.38 | 5.76 | <5 | ||
| TNF-β (Lta) | <5 | <5 | <5 | 38.27 | 34.82 | <5 | ||
| Lymphotoxin-β (Ltb) | 299.41 | 13.83 | <5 | <5 | <5 | <5 | ||
| Cytokine receptors | ||||||||
| Glycoprotein 130 (IL-6st) | 324.26 | 110.66 | <5 | <5 | <5 | <5 | ||
| IL-1R2 | 21.27 | 5.82 | <5 | <5 | <5 | <5 | ||
| IL-5Rα | 11.88 | <5 | <5 | <5 | <5 | <5 | ||
| Tumor necrosis factor receptor superfamily 1B (Tnfrsf1b) | −23.65 | −62.47 | <5 | <5 | <5 | <5 | ||
| Integrin β2 (Itgb) | <5 | <5 | <5 | 10.54 | <5 | <5 | ||
| Other inflammatory genes | ||||||||
| Caspase 1 (Casp1) | 17.52 | 6.06 | <5 | 15.98 | 7.02 | −6.15 | ||
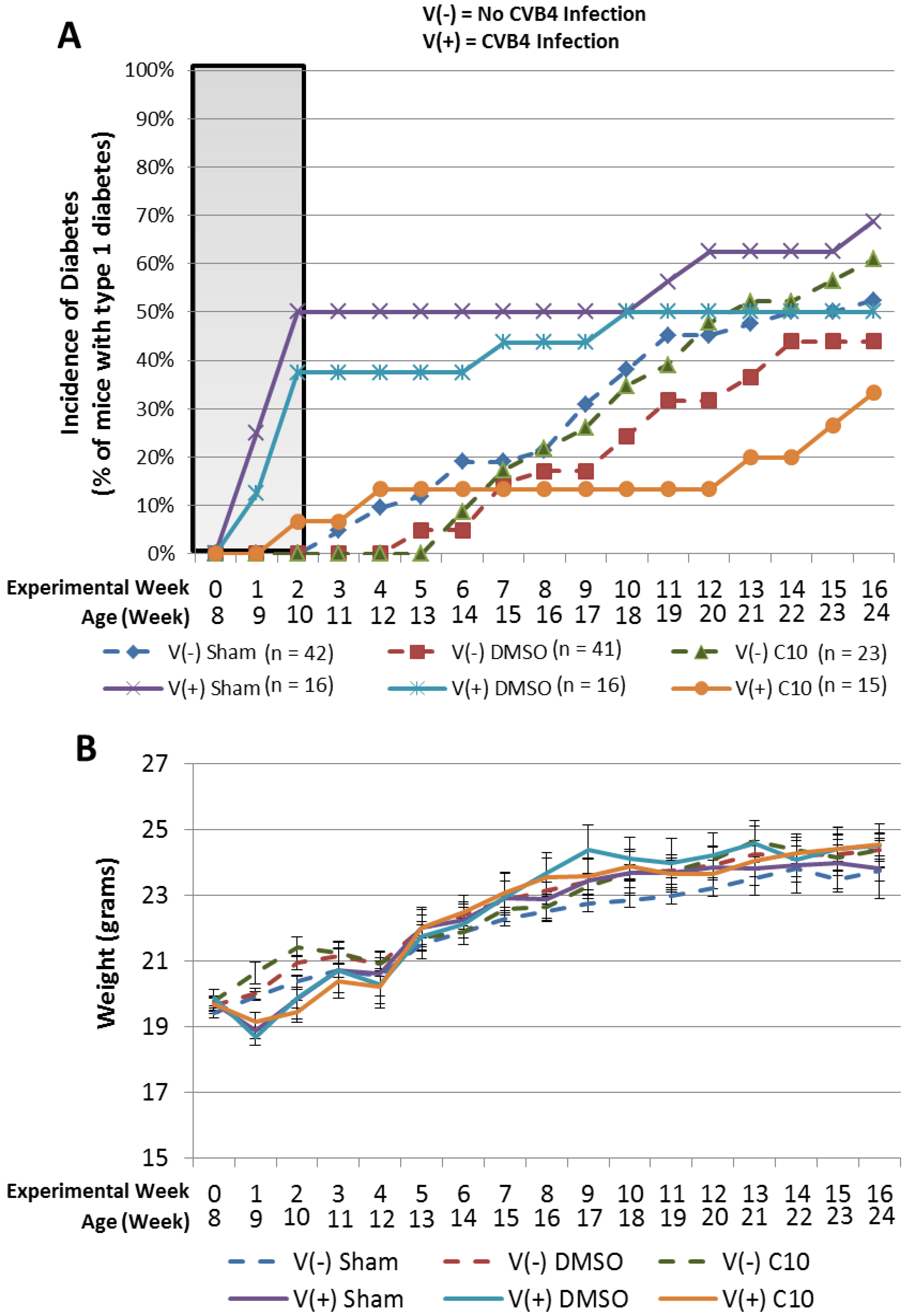
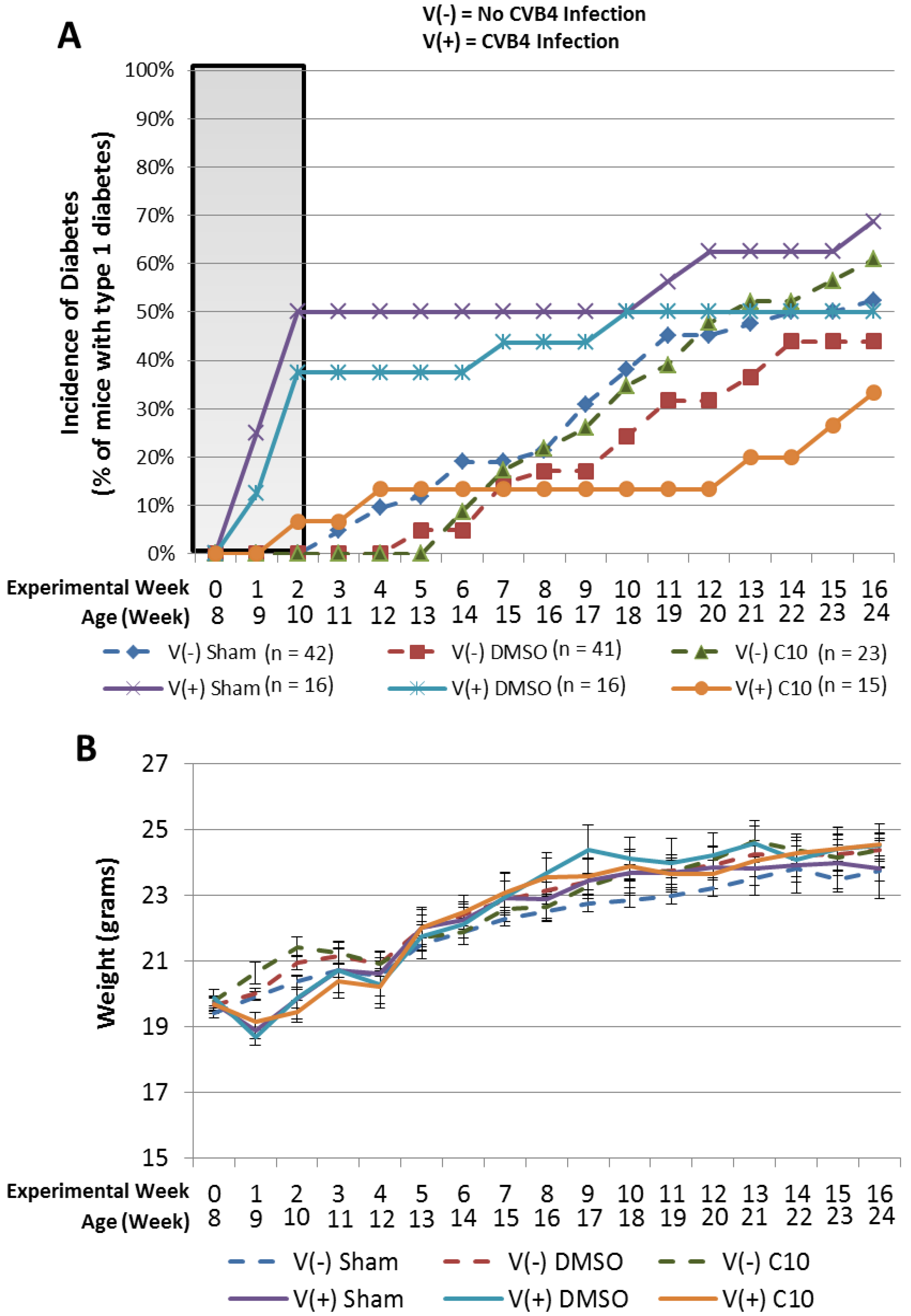
2.3. C10 Attenuates Coxsackievirus B4 (CVB4) Acceleration of T1DM in Non-Obese Diabetic (NOD) Mice
3. Experimental
3.1. Reagents
3.2. Cell Culture and Transfections
3.3. Cellular Viability Assays
3.4. Quantitative Real Time PCR
3.5. Quantitative Real Time PCR Arrays
3.6. CXCL10 ELISA
3.7. NOD Mouse Experiment
4. Conclusions
Acknowledgments
References
- Stene, L.C.; Rewers, M. Immunology in the clinic review series; focus on type 1 diabetes and viruses: The enterovirus link to type 1 diabetes: Critical review of human studies. Clin. Exp. Immunol. 2012, 168, 12–23. [Google Scholar] [CrossRef]
- Viskari, H.; Knip, M.; Tauriainen, S.; Huhtala, H.; Veijola, R.; Ilonen, J.; Simell, O.; Surcel, H.M.; Hyoty, H. Maternal enterovirus infection as a risk factor for type 1 diabetes in the exposed offspring. Diabetes Care 2012, 35, 1328–1332. [Google Scholar] [CrossRef]
- Jaidane, H.; Sane, F.; Hiar, R.; Goffard, A.; Gharbi, J.; Geenen, V.; Hober, D. Immunology in the clinic review series; focus on type 1 diabetes and viruses: Enterovirus, Thymus and type 1 diabetes pathogenesis. Clin. Exp. Immunol. 2012, 168, 39–46. [Google Scholar] [CrossRef]
- Hober, D.; Sane, F.; Jaidane, H.; Riedweg, K.; Goffard, A.; Desailloud, R. Immunology in the clinic review series; focus on type 1 diabetes and viruses: Role of antibodies enhancing the infection with Coxsackievirus-B in the pathogenesis of type 1 diabetes. Clin. Exp. Immunol. 2012, 168, 47–51. [Google Scholar] [CrossRef]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 2009, 52, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Dotta, F.; Censini, S.; van Halteren, A.G.; Marselli, L.; Masini, M.; Dionisi, S.; Mosca, F.; Boggi, U.; Muda, A.O.; Del Prato, S.; et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. USA 2007, 104, 5115–5120. [Google Scholar] [CrossRef]
- Willcox, A.; Richardson, S.J.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Immunohistochemical analysis of the relationship between islet cell proliferation and the production of the enteroviral capsid protein, VP1, in the islets of patients with recent-onset type 1 diabetes. Diabetologia 2011, 54, 2417–2420. [Google Scholar] [CrossRef]
- In't Veld, P. Insulitis in the human endocrine pancreas: Does a viral infection lead to inflammation and beta cell replication? Diabetologia 2011, 54, 2220–2222. [Google Scholar] [CrossRef]
- Chistiakov, D.A. Interferon induced with helicase C domain 1 (IFIH1) and virus-induced autoimmunity: A review. Viral Immunol. 2010, 23, 3–15. [Google Scholar] [CrossRef]
- Ylipaasto, P.; Klingel, K.; Lindberg, A.M.; Otonkoski, T.; Kandolf, R.; Hovi, T.; Roivainen, M. Enterovirus infection in human pancreatic islet cells, islet tropismin vivo and receptor involvement in cultured islet beta cells. Diabetologia 2004, 47, 225–239. [Google Scholar] [CrossRef]
- Jun, H.S.; Yoon, J.W. A new look at viruses in type 1 diabetes. Diabetes Metab. Res. Rev. 2003, 19, 8–31. [Google Scholar] [CrossRef]
- Dahlquist, G.; Mustonen, L. Childhood onset diabetes--time trends and climatological factors. Int. J. Epidemiol. 1994, 23, 1234–1241. [Google Scholar] [CrossRef]
- Brown, D.W.; Welsh, R.M.; Like, A.A. Infection of peripancreatic lymph nodes but not islets precedes Kilham rat virus-induced diabetes in BB/Wor rats. J. Virol. 1993, 67, 5873–5878. [Google Scholar]
- Guberski, D.L.; Thomas, V.A.; Shek, W.R.; Like, A.A.; Handler, E.S.; Rossini, A.A.; Wallace, J.E.; Welsh, R.M. Induction of type I diabetes by Kilham's rat virus in diabetes-resistant BB/Wor rats. Science 1991, 254, 1010–1013. [Google Scholar]
- Yoon, J.W.; Kominek, H.I. Role of Coxsackie B Virus in the pathogenesis of diabetes mellitus. In Microorganisms,Autoimmune Diseases; Rose, N.R., Friedman, H., Eds.; Plenum Press: New York, NY, USA, 1996; pp. 129–158. [Google Scholar]
- Yoon, J.W.; McClintock, P.R.; Onodera, T.; Notkins, A.L. Virus-induced diabetes mellitus. XVIII. Inhibition by a nondiabetogenic variant of encephalomyocarditis virus. J. Exp. Med. 1980, 152, 878–892. [Google Scholar] [CrossRef]
- Yoon, J.W.; Austin, M.; Onodera, T.; Notkins, A.L. Isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med. 1979, 300, 1173–1179. [Google Scholar] [CrossRef]
- Menser, M.A.; Forrest, J.M.; Bransby, R.D. Rubella infection and diabetes mellitus. Lancet 1978, 1, 57–60. [Google Scholar]
- Craighead, J.E.; McLane, M.F. Diabetes mellitus: Induction in mice by encephalomyocarditis virus. Science 1968, 162, 913–914. [Google Scholar]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Morgan, N.G.; Foulis, A.K. Immunopathology of the human pancreas in type-I diabetes. Semin Immunopathol. 2011, 33, 9–21. [Google Scholar] [CrossRef]
- Horwitz, M.S.; Ilic, A.; Fine, C.; Rodriguez, E.; Sarvetnick, N. Presented antigen from damaged pancreatic beta cells activates autoreactive T cells in virus-mediated autoimmune diabetes. J. Clin. Invest. 2002, 109, 79–87. [Google Scholar] [CrossRef]
- Liu, D.; Cardozo, A.K.; Darville, M.I.; Eizirik, D.L. Double-stranded RNA cooperates with interferon-gamma and IL-1 beta to induce both chemokine expression and nuclear factor-kappa B-dependent apoptosis in pancreatic beta-cells: Potential mechanisms for viral-induced insulitis and beta-cell death in type 1 diabetes mellitus. Endocrinology 2002, 143, 1225–1234. [Google Scholar]
- Garcia, M.; Dogusan, Z.; Moore, F.; Sato, S.; Hartmann, G.; Eizirik, D.L.; Rasschaert, J. Regulation and function of the cytosolic viral RNA sensor RIG-I in pancreatic beta cells. Biochim. Biophys. Acta 1793, 1768–1775. [Google Scholar]
- Dogusan, Z.; Garcia, M.; Flamez, D.; Alexopoulou, L.; Goldman, M.; Gysemans, C.; Mathieu, C.; Libert, C.; Eizirik, D.L.; Rasschaert, J. Double-stranded RNA induces pancreatic beta-cell apoptosis by activation of the toll-like receptor 3 and interferon regulatory factor 3 pathways. Diabetes 2008, 57, 1236–1245. [Google Scholar] [CrossRef]
- Ewel, C.H.; Sobel, D.O.; Zeligs, B.J.; Bellanti, J.A. Poly I:C accelerates development of diabetes mellitus in diabetes-prone BB rat. Diabetes 1992, 41, 1016–1021. [Google Scholar] [CrossRef]
- Sobel, D.O.; Newsome, J.; Ewel, C.H.; Bellanti, J.A.; Abbassi, V.; Creswell, K.; Blair, O. Poly I:C induces development of diabetes mellitus in BB rat. Diabetes 1992, 41, 515–520. [Google Scholar] [CrossRef]
- Serreze, D.V.; Hamaguchi, K.; Leiter, E.H. Immunostimulation circumvents diabetes in NOD/Lt mice. J. Autoimmun. 1989, 2, 759–776. [Google Scholar] [CrossRef]
- Wen, L.; Peng, J.; Li, Z.; Wong, F.S. The effect of innate immunity on autoimmune diabetes and the expression of Toll-like receptors on pancreatic islets. J. Immunol. 2004, 172, 3173–3180. [Google Scholar]
- Moriyama, H.; Wen, L.; Abiru, N.; Liu, E.; Yu, L.; Miao, D.; Gianani, R.; Wong, F.S.; Eisenbarth, G.S. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc. Natl. Acad. Sci. USA 2002, 99, 5539–5544. [Google Scholar] [CrossRef]
- Baldeon, M.E.; Chun, T.; Gaskins, H.R. Interferon-alpha and interferon-gamma differentially affect pancreatic beta-cell phenotype and function. Am. J. Physiol. 1998, 275, C25–C32. [Google Scholar]
- Foulis, A.K.; Farquharson, M.A.; Meager, A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet 1987, 2, 1423–1427. [Google Scholar]
- Stewart, T.A.; Hultgren, B.; Huang, X.; Pitts-Meek, S.; Hully, J.; MacLachlan, N.J. Induction of type I diabetes by interferon-alpha in transgenic mice. Science 1993, 260, 1942–1946. [Google Scholar]
- Lang, K.S.; Recher, M.; Junt, T.; Navarini, A.A.; Harris, N.L.; Freigang, S.; Odermatt, B.; Conrad, C.; Ittner, L.M.; Bauer, S.; et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat. Med. 2005, 11, 138–145. [Google Scholar] [CrossRef]
- Fabris, P.; Betterle, C.; Floreani, A.; Greggio, N.A.; de Lazzari, F.; Naccarato, R.; Chiaramonte, M. Development of type 1 diabetes mellitus during interferon alfa therapy for chronic HCV hepatitis. Lancet 1992, 340, 548. [Google Scholar]
- Alba, A.; Puertas, M.C.; Carrillo, J.; Planas, R.; Ampudia, R.; Pastor, X.; Bosch, F.; Pujol-Borrell, R.; Verdaguer, J.; Vives-Pi, M. IFN beta accelerates autoimmune type 1 diabetes in nonobese diabetic mice and breaks the tolerance to beta cells in nondiabetes-prone mice. J. Immunol. 2004, 173, 6667–6675. [Google Scholar]
- Huang, X.; Yuang, J.; Goddard, A.; Foulis, A.; James, R.F.; Lernmark, A.; Pujol-Borrell, R.; Rabinovitch, A.; Somoza, N.; Stewart, T.A. Interferon expression in the pancreases of patients with type I diabetes. Diabetes 1995, 44, 658–664. [Google Scholar] [CrossRef]
- Harii, N.; Lewis, C.; Vasko, V.; McCall, K.; Benavides-Peralta, U.; Sun, X.; Ringel, M.; Saji, M.; Kohn, L. Thyrocytes express a functional toll-like receptor 3(TLR3): Overexpression can be induced by viral infection, Reversed by Phenylmethimazole, and is associated with Hashimoto’s autoimmune thyroiditis. Mol. Endocrinol. 2005, 19, 1231–1250. [Google Scholar] [CrossRef]
- McCall, K.D.; Harii, N.; Lewis, C.J.; Malgor, R.; Kim, W.B.; Saji, M.; Kohn, A.D.; Moon, R.T.; Kohn, L.D. High Basal Levels of Functional Toll-Like Receptor 3 (TLR3) and Non-Cannonical Wnt5a Are Expressed in Papillary Thyroid Cancer (PTC) and Are Coordinately Decreased by Phenylmethimazole Together with Cell Proliferation and Migration. Endocrinology 2007, 148, 4226–4237. [Google Scholar] [CrossRef]
- Schwartz, A.L.; Malgor, R.; Dickerson, E.; Weeraratna, A.T.; Slominski, A.; Wortsman, J.; Harii, N.; Kohn, A.D.; Moon, R.T.; Schwartz, F.L.; et al. Phenylmethimazole decreases Toll-like receptor 3 and noncanonical Wnt5a expression in pancreatic cancer and melanoma together with tumor cell growth and migration. Clin. Cancer Res. 2009, 15, 4114–4122. [Google Scholar] [CrossRef]
- Courreges, M.C.; Kantake, N.; Goetz, D.J.; Schwartz, F.L.; McCall, K.D. Phenylmethimazole blocks dsRNA-induced IRF3 nuclear translocation and homodimerization. Molecules 2012, 17, 12365–12377. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef]
- Hamaguchi, K.; Gaskins, H.R.; Leiter, E.H. NIT-1, A pancreatic beta-cell line established from a transgenic NOD/Lt mouse. Diabetes 1991, 40, 842–849. [Google Scholar] [CrossRef]
- Efrat, S.; Linde, S.; Kofod, H.; Spector, D.; Delannoy, M.; Grant, S.; Hanahan, D.; Baekkeskov, S. Beta-cell lines derived from transgenic mice expressing a hybrid insulin gene-oncogene. Proc. Natl. Acad. Sci. USA 1988, 85, 9037–9041. [Google Scholar] [CrossRef]
- Poitout, V.; Stout, L.E.; Armstrong, M.B.; Walseth, T.F.; Sorenson, R.L.; Robertson, R.P. Morphological and functional characterization of beta TC-6 cells—An insulin-secreting cell line derived from transgenic mice. Diabetes 1995, 44, 306–313. [Google Scholar] [CrossRef]
- Gianani, R.; Campbell-Thompson, M.; Sarkar, S.A.; Wasserfall, C.; Pugliese, A.; Solis, J.M.; Kent, S.C.; Hering, B.J.; West, E.; Steck, A.; et al. Dimorphic histopathology of long-standing childhood-onset diabetes. Diabetologia 2010, 53, 690–698. [Google Scholar] [CrossRef]
- Rowe, P.A.; Campbell-Thompson, M.L.; Schatz, D.A.; Atkinson, M.A. The pancreas in human type 1 diabetes. Semin. Immunopathol. 2011, 33, 29–43. [Google Scholar] [CrossRef]
- Robbins, M.A.; Maksumova, L.; Pocock, E.; Chantler, J.K. Nuclear factor-kappaB translocation mediates double-stranded ribonucleic acid-induced NIT-1 beta-cell apoptosis and up-regulates caspase-12 and tumor necrosis factor receptor-associated ligand (TRAIL). Endocrinology 2003, 144, 4616–4625. [Google Scholar] [CrossRef]
- Pelegrin, M.; Devedjian, J.C.; Costa, C.; Visa, J.; Solanes, G.; Pujol, A.; Asins, G.; Valera, A.; Bosch, F. Evidence from transgenic mice that interferon-beta may be involved in the onset of diabetes mellitus. J. Biol. Chem. 1998, 273, 12332–12340. [Google Scholar] [CrossRef]
- Frigerio, S.; Junt, T.; Lu, B.; Gerard, C.; Zumsteg, U.; Hollander, G.A.; Piali, L. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat. Med. 2002, 8, 1414–1420. [Google Scholar] [CrossRef]
- Giarratana, N.; Penna, G.; Amuchastegui, S.; Mariani, R.; Daniel, K.C.; Adorini, L. A vitamin D analog down-regulates proinflammatory chemokine production by pancreatic islets inhibiting T cell recruitment and type 1 diabetes development. J. Immunol. 2004, 173, 2280–2287. [Google Scholar]
- Shimada, A.; Morimoto, J.; Kodama, K.; Suzuki, R.; Oikawa, Y.; Funae, O.; Kasuga, A.; Saruta, T.; Narumi, S. Elevated serum IP-10 levels observed in type 1 diabetes. Diabetes Care 2001, 24, 510–515. [Google Scholar] [CrossRef]
- Nicoletti, F.; Conget, I.; Di Mauro, M.; Di Marco, R.; Mazzarino, M.C.; Bendtzen, K.; Messina, A.; Gomis, R. Serum concentrations of the interferon-gamma-inducible chemokine IP-10/CXCL10 are augmented in both newly diagnosed Type I diabetes mellitus patients and subjects at risk of developing the disease. Diabetologia 2002, 45, 1107–1110. [Google Scholar] [CrossRef]
- Rhode, A.; Pauza, M.E.; Barral, A.M.; Rodrigo, E.; Oldstone, M.B.; von Herrath, M.G.; Christen, U. Islet-specific expression of CXCL10 causes spontaneous islet infiltration and accelerates diabetes development. J. Immunol. 2005, 175, 3516–3524. [Google Scholar]
- Rewers, M. Challenges in diagnosing type 1 diabetes in different populations. Diabetes Metab. J. 2012, 36, 90–97. [Google Scholar] [CrossRef]
- Serreze, D.V.; Ottendorfer, E.W.; Ellis, T.M.; Gauntt, C.J.; Atkinson, M.A. Acceleration of type 1 diabetes by a coxsackievirus infection requires a preexisting critical mass of autoreactive T-cells in pancreatic islets. Diabetes 2000, 49, 708–711. [Google Scholar] [CrossRef]
- Benavides, U.; Gonzalez-Murguiondo, M.; Harii, N.; Lewis, C.J.; Schwartz, A.L.; Giuliani, C.; Napolitano, G.; Dagia, N.M.; Malgor, R.; McCall, K.D.; et al. Phenylmethimazole inhibits production of proinflammatory mediators and is protective in an experimental model of endotoxic shock*. Crit. Care. Med. 2011, 40, 886–894. [Google Scholar]
- McCall, K.D.; Holliday, D.; Dickerson, E.; Wallace, B.; Schwartz, A.L.; Schwartz, C.; Lewis, C.J.; Kohn, L.D.; Schwartz, F.L. Phenylmethimazole blocks palmitate-mediated induction of inflammatory cytokine pathways in 3T3L1 adipocytes and RAW 264.7 macrophages. J. Endocrinol. 2010, 207, 343–353. [Google Scholar] [CrossRef]
- Webb, S.R.; Loria, R.M.; Madge, G.E.; Kibrick, S. Susceptibility of mice to group B coxsackie virus is influenced by the diabetic gene. J. Exp. Med. 1976, 143, 1239–1248. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compound (C10) are available from the Interthyr Corporation.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McCall, K.D.; Schmerr, M.J.; Thuma, J.R.; James, C.B.L.; Courreges, M.C.; Benencia, F.; Malgor, R.; Schwartz, F.L. Phenylmethimazole Suppresses dsRNA-Induced Cytotoxicity and Inflammatory Cytokines in Murine Pancreatic Beta Cells and Blocks Viral Acceleration of Type 1 Diabetes in NOD Mice. Molecules 2013, 18, 3841-3858. https://doi.org/10.3390/molecules18043841
McCall KD, Schmerr MJ, Thuma JR, James CBL, Courreges MC, Benencia F, Malgor R, Schwartz FL. Phenylmethimazole Suppresses dsRNA-Induced Cytotoxicity and Inflammatory Cytokines in Murine Pancreatic Beta Cells and Blocks Viral Acceleration of Type 1 Diabetes in NOD Mice. Molecules. 2013; 18(4):3841-3858. https://doi.org/10.3390/molecules18043841
Chicago/Turabian StyleMcCall, Kelly D., Martin J. Schmerr, Jean R. Thuma, Calvin B. L. James, Maria C. Courreges, Fabian Benencia, Ramiro Malgor, and Frank L. Schwartz. 2013. "Phenylmethimazole Suppresses dsRNA-Induced Cytotoxicity and Inflammatory Cytokines in Murine Pancreatic Beta Cells and Blocks Viral Acceleration of Type 1 Diabetes in NOD Mice" Molecules 18, no. 4: 3841-3858. https://doi.org/10.3390/molecules18043841